

Abstract
Wilms tumor 1 (WT1) is an intracellular tumor‐associated antigen that remains inaccessible to antibodies. Recently, T‐cell receptor (TCR) mimic antibodies (TCRm‐Abs), which recognize peptides loaded on human leukocyte antigen (HLA) with higher specificity and affinity than TCR, have been developed as a new antibody class that can target intracellular antigens. To expand the therapeutic targets in tumors with WT1, we developed TCRm‐Abs targeting a novel HLA‐A*02:01‐restricted peptide, WT1C (ALLPAVPSL), and validated their specificity using multiple techniques. Screening of these antibodies by ELISA with a panel of peptide/HLA complexes and by glycine scanning of peptide‐pulsed T2 cells identified one specific clone, #25‐8. Despite the low risk for eliciting broad cross–reactivity of this TCRm‐Ab, analysis of a panel of cell lines, in conjunction with exogenous expression of either or both the HLA‐A*02:01 and WT1 genes in HeLa cells, revealed that #25‐8 reacts with WT1C but also with unknown peptides in the context of HLA‐A*02:01. This potentially dangerous cross–reactivity was confirmed through analysis using chimeric antigen receptor T‐cells carrying the single‐chain variable fragment of #25‐8, which targets WT1‐negative HeLa/A02 cells. To determine the cross–reactive profiles of #25‐8, we applied the PresentER antigen presentation platform with the #25‐8‐recognition motif, which enables the identification of potential off–target peptides expressed in the human proteome. Our results demonstrate the potential of TCRm‐Abs to target a variety of peptides in the context of HLA but also depict the need for systematic validation to identify the cross–reactive peptides for the prediction of off–target toxicity in future clinical translation.
Keywords: chimeric antigen receptor T‐cells, off–target, T‐cell receptor mimic antibody, tumor‐associated antigen, Wilms tumor 1
Development of T‐cell receptor mimic antibodies targeting a novel WT1 peptide.

1. INTRODUCTION
Wilms tumor 1 (WT1) is a promising cancer therapeutic target that is aberrantly expressed in myeloid and lymphoid leukemia and several solid tumors, but its expression is limited in normal adult tissues. 1 , 2 Because WT1 is an intracellular tumor‐associated antigen that is inaccessible to conventional antibodies, T‐cell recognition of peptides loaded on major histocompatibility complex/human leukocyte antigen (HLA) has been the basis for WT1‐targeted therapy. 3 To date, a limited number of immunodominant peptides preferentially targeted by T‐cells of multiple individuals, such as RMFPNAPYL for HLA‐A*02 and CYTWNQMNL for HLA‐A*24, have been used for clinical application as vaccines. 4 , 5 However, they have shown limited therapeutic efficacy. 6 To enhance antitumor efficacy, adoptive immunotherapy with T‐cells genetically modified to express an antigen‐specific T‐cell receptor (TCR) has been developed. However, the clinical application of TCR‐engineered T‐cells remains in its infancy. 7 , 8
A T‐cell receptor mimic antibody (TCRm‐Ab) can recognize epitopes comprising both the peptide and the HLA molecule, similar to the recognition of such complexes by the TCR on T‐cells. 9 , 10 , 11 , 12 Thus, it can target intracellular or extracellular antigens presented on HLA. TCRm‐Abs targeting the two WT1‐derived immunodominant peptides have been developed and used as targeting agents of chimeric antigen receptor‐engineered (CAR) T‐cells or bispecific T‐cell engager (BiTE) antibodies. 13 , 14 , 15 In the search for TCR epitopes of WT1, WT1C (ALLPAVPSL) showed a high binding score for HLA‐A*02:01 but elicited low frequencies of T‐cells in individuals. 16 , 17 This result suggests that WT1C can be processed from WT1 and presented on HLA‐A*02, but its cognate TCR are eliminated during the course of negative selection of thymocytes in nonresponding individuals. Because a TCRm‐Ab can target any peptides loaded on HLA regardless of their immunogenicity against T‐cells, WT1C may represent a promising target for TCRm‐Ab development. 18 , 19 , 20
One of the major concerns with T‐cell‐based therapeutics is cross–reactivity against structurally related or unrelated peptides loaded on HLA on normal tissues, which has been documented in multiple clinical trials using TCR gene‐modified T‐cells. 21 , 22 For example, a clinical trial of T‐cells genetically modified to express an affinity‐enhanced MAGE‐specific TCR resulted in lethal adverse effects due to cross–reactivity with titin peptide partly homologous to MAGE sequences, highlighting a major challenge for the development of safe T‐cell‐based therapeutics. 23 To date, strategies for selecting TCRm‐Abs have included a binding assay using a panel of cell lines and peptide‐pulsed T2 cells. However, their ability to validate TCRm‐Ab specificity and detect off–target toxicity has been limited. Recently, novel technologies have emerged that enable the prediction of the cross–recognition potential of TCRm‐Abs. A detailed biochemical evaluation of a well‐characterized TCRm‐Ab, ESK1, by crystallographic studies explained the mechanism of its recognition of other peptides that share sequence similarity with the target peptide. 24 Gejman et al 25 , 26 developed the PresentER antigen presentation platform to identify the precise peptide epitope recognized by a TCRm‐Ab and TCR. These results demonstrate the nature of TCRm‐Abs, as well as TCR, having cross–reactivity with nontarget peptides. Thus, it is crucial to identify the cross–reactive peptides and to address the tissue expressing the parental proteins for the prediction of off–target toxicity in future clinical translation.
We recently reported a FACS‐based strategy combined with a single‐cell immunoglobulin heavy chain variable (VH) and light chain variable (VL) gene cloning method for efficient development of TCRm‐Abs targeting a Survivin 2B‐derived peptide loaded on HLA‐A*24. 27 By using this approach, we generated antibody clones recognizing WT1C loaded on HLA‐A*02:01 (WT1C/HLA‐A*02). In screening these clones by their affinity and ability to bind key amino acid residues within the target peptide by T2 cells, one specific clone, #25‐8, was selected as a candidate TCRm‐Ab. Analysis of a small panel of cell lines revealed that #25‐8 binding was restricted to the HLA‐A*02+ and WT1+ cancer cell line THP1. However, a considerable level of off–target binding was found on JY cells that express HLA‐A*02 but not WT1.
In investigating the extent of off–target toxicity, we found that forced expression of a given HLA in a tumor cell line not expressing both the HLA and a target protein in combination with interferon gamma (IFN‐γ) treatment can be an efficient alternative approach to screening of a panel of tumor cell lines whose HLA expression is downregulated. Exogenous expression of either or both the HLA‐A*02:01 and WT1 genes in HLA‐A*02:01− WT1− HeLa cells revealed that this antibody reacts with WT1C but also with unknown peptides in the context of HLA‐A*02:01. The observed off–target reactivity was reflected in the cytotoxicity of T‐cells expressing CAR carrying the single‐chain variable fragment (ScFv) of #25‐8. Screening of #25‐8 cross–reactive peptides by the PresentER antigen presentation platform with the #25‐8‐recognition motif allowed us to identify candidate peptides derived from the human proteome.
These results showed the potential of TCRm‐Abs to target a variety of peptides in the context of HLA but also depicted the need for systematic validation to characterize the cross–reactivity and specificity of TCRm‐Ab.
2. MATERIALS AND METHODS
2.1. Generation of T‐cell receptor mimic antibodies
Isolation of TCRm‐specific plasma cells (PC) was performed as previously described, with slight modifications. 27 Mouse splenocytes (1 × 107/mL) were suspended in 1 mL of PBS and stained with PE‐labeled WT1C/HLA*A02‐tetramer (WT1C/HLA*A02) (0.1 µg/mL), APC‐labeled tyrosinase/HLA*A02‐tetramer (tyrosinase/HLA*A02) (0.25 µg/mL), and DyLight 488‐labeled antibody against mouse IgG (anti–IgG) at 4°C for 30 minutes with gentle agitation. After washing with PBS, the cells were suspended in 3 mL of PBS containing ER‐tracker (0.25 µmol/L) and subsequently analyzed by FACS. Single‐cell sorting was performed using a JSAN Cell Sorter equipped with an automatic cell deposition unit (Bay Bioscience). Molecular cloning of the immunoglobulin genes from single cells and recombinant antibody expression were performed as previously described. 28 , 29 Antibodies were produced via the Expi293 cell culture system and purified with protein G column chromatography.
2.2. Detection of the peptide/human leukocyte antigen complex on target cells by FACS
The ability of the TCRm‐Ab to bind to cells was assessed by FACS according to a previously described method. 27 Briefly, cells were labeled with 1 µg/mL #25‐8 for 30‐60 minutes at 4°C and then stained with a goat anti–mouse IgG (H + L) DyLight 650 antibody (Thermo Fisher Scientific) for 30 minutes at 4°C. JY cells were treated with ice‐cold citric acid buffer (pH 3.2) for 90 seconds and were immediately suspended in IMDM in the presence of 1.5 mg/mL β2‐microglobulin and 1 µg/mL peptide. After incubation for 2 hours at 4°C, cells were stained with #25‐8 and BB7.2 anti–HLA‐A*02 monoclonal antibodies. FACS data were collected on a JSAN Cell Sorter or FACS Melody (BD Biosciences) and analyzed with FlowJo software (BD Biosciences).
2.3. In vitro T‐cell‐dependent cellular cytotoxicity assay
T‐cells were prepared as previously described. 27 The CAR was constructed using CD19 or the #25‐8 scFv, which was fused at the C‐terminus directly to the CD8 hinge and transmembrane domain and the intracellular portions of the 3rd generation CAR incorporating the cytoplasmic domains of 4‐1BB, CD28, and CD3ζ. Each CAR construct was subcloned into the pLVSIN‐EF1α Pur vector, and lentiviral particles were prepared as previously described. 27 T‐cells stably expressing the CD19 CAR or #25‐8 CAR were generated by infection with the lentiviral particles. The transduction efficiency was approximately 45%. The CAR T‐cells were mixed with either HeLa/A24, HeLa/A02, or HeLa/A02/WT cells that stably expressed luciferase at an E:T ratio of 10:1 (1 × 105:1 × 104) in 96‐well plates and cultured in GT‐T551 culture medium that contained 10% FBS and 2.5 ng/mL IL2 for 8‐12 hours. Bioluminescence was measured as previously described. 27 The assay measures lytic activity by calculating the number of viable luciferase‐positive cells. The 100% viability reference point (maximal RLU) was determined by plating target HeLa cells (T). The percent cell viability was calculated from the data using the following equation: % cell viability = 100 × (E + T)/(maximal RLU).
2.4. PresentER
A DNA fragment encoding a signal peptide from mouse mammary tumor virus envelope protein followed by the WT1C peptide was synthesized in accordance with data reported by Gejiman et al. 25 The DNA fragment was subcloned into the XbaI/BamHI sites of the pLVSIN vector to generate the PresentER‐WT1C plasmid. This plasmid was digested with SfiI to remove the DNA fragment encoding the WT1C peptide and used as the PresentER cassette. A library of DNA fragments encoding #25‐8 cross–reactivity motifs, (A/V)LXX(S/A)VPX(L/V), was synthesized by PCR with primers (binding motif and PresentER AS2). The amplified DNA fragments were digested with SfiI, gel purified, and inserted into the PresentER cassette. The ligation products were electroporated into competent cells, approximately 1 × 105 colonies were harvested from agar plates, and plasmids were extracted. A lentiviral minigene library was produced by transfecting the plasmids and ViraPower Packaging Mix (Takara Bio) into 293FT cells with Lipofectamine 3000 (Thermo Fisher Scientific). T2 cells (1 × 106) were transduced with the lentiviral minigene library with a multiplicity of infection of 2 and were selected with 0.5 μg/mL puromycin for 5 days to establish the 1st‐round T2 cell library. This protocol enables the 9‐mer peptide to be transported into the endoplasmic reticulum, loaded on HLA, and expressed on the T2 cell surface. The minigene sequences integrated into genomic DNA of the 1st‐round T2 cell library (2.5 × 106 cells) were recovered by PCR with primers (P7 barcode 1 and P5) and used as input minigenes. The 1st‐round T2 cell library (2.5 × 106 cells) was stained with #25‐8, and positively stained cells (#25‐8 High) were sorted. The sorting gate was set at the mean fluorescent intensity of the stained T2 cells expressing PresentER‐WT1C as a control. The minigene sequences recovered from the sorted cells by PCR with primers (PresentER S and PresentER AS) were digested with SfiI and inserted into the PresentER cassette, and the 2nd‐round T2 cell library was produced as described above. #25‐8 High cells were sorted from the 2nd‐round T2 cell library, and the minigene sequences recovered by PCR with primers (P7 barcode 2 and P5) were used as the 2nd minigene. PCR was performed using PrimeSTAR HS DNA polymerase (Takara Bio) with 25 cycles of denaturation (95°C, 30 seconds), annealing (60°C, 20 seconds), and extension (72°C, 20 seconds). PCR products were subsequently purified, and nucleotide sequencing was performed on an Illumina MiSeq sequencer. The PCR primers are listed in Table S1.
2.5. Bioinformatics and data analysis
Sequences encoding 9‐mer peptides were extracted from Illumina sequencing reads, and enrichment values (EV) were calculated as EV = Abundance of each 9‐mer peptide in the 2nd minigene/abundance of each 9‐mer peptide in the input minigene. The sequence logo was generated using the Weblogo algorithm (https://weblogo.berkeley.edu/logo.cgi) to generate the #25‐8 binding motif. Prediction of peptide binding to HLA‐A*02 was performed with NetMHCpan 4.1 (http://www.cbs.dtu.dk/services/NetMHCpan/). Statistically significant differences between groups were determined by ANOVA followed by Tukey’s multiple comparison test or Dunnett’s test by using JMP Pro14 software (SAS Institute). A peptide search against the human proteome was performed using protein‐protein BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Materials and other methods are presented in Appendix S1.
3. RESULTS
3.1. Selection of monoclonal antibodies recognizing WT1C/HLA‐A*02:01
For the production of TCRm‐Abs targeting WT1C/HLA‐A*02, we immunized mice with the WT1C/HLA‐A*02 monomer as an antigen. To isolate WT1C/HLA‐A*02‐specific PC, splenocytes taken from the mice were stained with anti–IgG, ER‐tracker, tyrosinase/HLA‐A*02, and WT1C/HLA‐A*02; the WT1C/HLA‐A*02‐specific PC gated as anti–IgG Medium ER‐tracker High, tyrosinase/HLA‐A*02 low, and WT1C/HLA‐A*02 High (R3 gate) were single‐sorted by FACS (Figure 1A). Antibodies were expressed by transfecting cognate pairs of heavy and light chain genes into 293FT cells and were assayed by ELISA with a panel of peptide/HLA molecules. As shown in Figure 1B, 25 out of 67 antibodies were WT1C/HLA‐A*02 binders, among which 16 binders specifically reacted with WT1C but not with 11 irrelevant peptides, demonstrating the high efficiency of FACS subtraction with tyrosinase/HLA‐A*02 for the removal of PC expressing nonspecific antibodies. DNA sequencing of these clones revealed that the antibodies were divided into four phylogenetic clusters (Figure 1C). We selected four antibodies from each cluster: #25‐8, #26‐23, and #26‐22 readily bound to WT1C pulsed‐T2 cells but not to HIV‐pulsed cells, except for #201‐14 (Figure 1D). The antibody binding affinity and kinetics were determined by surface plasmon resonance: the highest equilibrium dissociation constant (K D) was found for #26‐22 (5 nmol/L), followed by #25‐8 and #201‐14 (11 and 18 nmol/L), while #26‐22 had the lowest (38 nmol/L; Figure 2A). To map key amino acid residues involved in the antibody interaction, each residue on WT1C was replaced with glycine (except the canonical anchor residues at positions 2 and 9), and antibody binding was assessed on T2 cells. As shown in Figure 2B, the four antibodies possessed different molecular recognition patterns, among which #25‐8 and #26‐22 showed the widest epitope coverage. Because TCRm‐Abs possess the ability to recognize epitopes that share sequence homology with the chosen 9‐mer amino acid sequences, potential off–target peptides with similar sequences to WT1C carrying anchor residue motifs for HLA‐A*02 were searched using the BLAST program. Six peptides sharing high sequence similarity with WT1C were identified: zinc finger MYM‐type containing 6 (ZMYM6), NEDD4‐binding protein 1 (N4BP1), lysophospholipid acyltransferase 7 (MBOAT7), ankyrin repeat and KH domain‐containing 1 (ANKHD1), Wingless‐type MMTV integration site family member 6 (WNT6) and killer‐cell Ig‐like receptors (KIR). These peptides were used to pulse T2 cells, and the ability of the antibodies to bind these targets was analyzed. As shown in Figure 2C, each of the antibodies showed different levels of off–target reactivity, among which #25‐8 moderately bound to the ZMYM6 peptide but not to other peptides. All other antibodies appeared to bind with more than two peptides. N4BP1, WNT6, and KIR peptide did not stabilize HLA‐A*02 on T2 cells. Based on glycine substitution and off–target peptide analysis, #25‐8, recognizing the putative binding sequence, (A/V)LXX(S/A)VPX(L/V), showed the highest specificity and was, thus, selected and subjected to further analysis. #25‐8 was tested for a dose response regarding its binding to WT1C/HLA‐A*02 on T2 cells. T2 cells pulsed with the WT1C peptide showed increased #25‐8 binding when the antibody concentration was increased. Increasing WT1C peptide concentrations also increased #25‐8 binding (Figure S1). Competitive ELISA performed with WT1C/HLA‐A*02 at 25 nmol/L completely abolished #25‐8 binding to immobilized WT1C/HLA‐A*02, but WT1C peptide at a 20‐fold higher concentration did not inhibit binding, indicating that #25‐8 recognizes conformational epitopes consisting of the HLA‐A*02 molecule with the WT1C peptide embedded within it (Figure S1).
FIGURE 1.
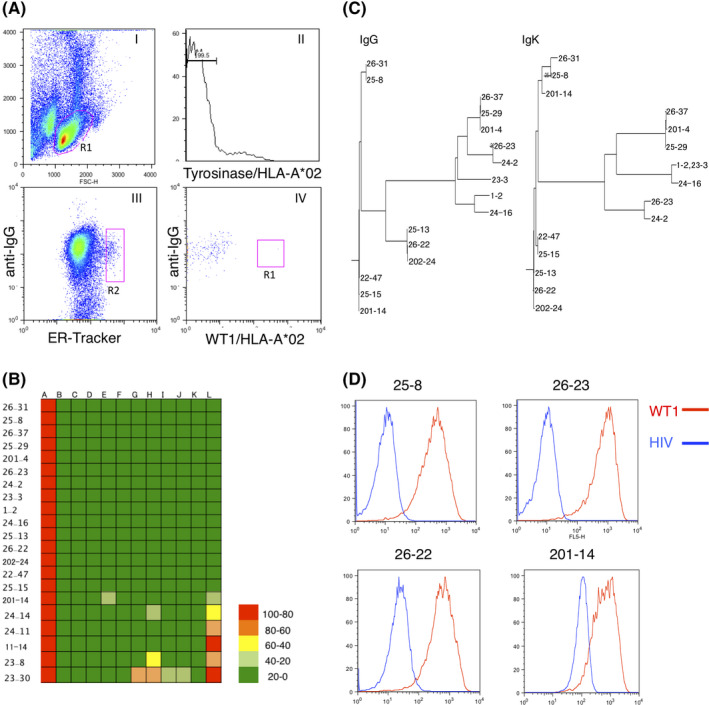
Development of monoclonal antibodies against WT1C/HLA‐A*02. A, FACS gating strategy for the isolation of WT1C/HLA‐A*02‐specific plasma cells (PC). Splenocytes were stained with anti–IgG, ER‐tracker, tyrosinase/HLA‐A*02, and WT1C/HLA‐A*02 antibodies. Plots (I–VI) represent the sequential gating strategy. (I) FSC vs SSC with gate R1 represents lymphocytes. (II) Cells nonspecifically binding to the HLA tetramer were subtracted. (III) The anti–IgG Low ER‐Tracker High fraction was defined as PC. (IV) The WT1C/HLA‐A*02‐specific PC were defined as anti–IgG Medium, ER‐Tracker High, Tyrosinase/HLA‐A*02 low, and WT1C/HLA‐A*02 High (R3 gate). A total of 50 000 events were recorded. B, Binding specificity of antibodies by ELISA with a panel of peptide/HLA‐A*02 monomers. A colored heat map shows the relative immunoreactivity of each antibody clone against the WT1C/HLA‐A*02 monomer compared to that against irrelevant peptide/HLA‐A*02 monomers. A: WT1C, B: Survivin, C: Tyrosinase, D: HCV NS3, E: HIV pol, F: Mart‐1, G: HTLV‐1 Tax, H: WT1126‐134, I: Her2/neu, J: PR1, K: HPV16 E7, and L: MUC1. Signal intensities are color coded as follows: light green (<0%), green (>0%‐25%), yellow (>25‐50), orange (>50%‐75%), and red (>75%). Values are represented as the means of two replicates. C, Phylogenetic analysis of heavy chain variable (VH) and light chain variable (VL) amino acid sequences of WT1C/HLA‐A*02‐specific antibody clones. D, FACS analysis of antibody clones by T2 cells pulsed with peptides. T2 cells pulsed with either WT1C or HIV were stained with the indicated antibody. The binding ability of each antibody was evaluated by the mean fluorescent intensity of stained T2 cells. Representative data from two independent experiments are shown.
FIGURE 2.
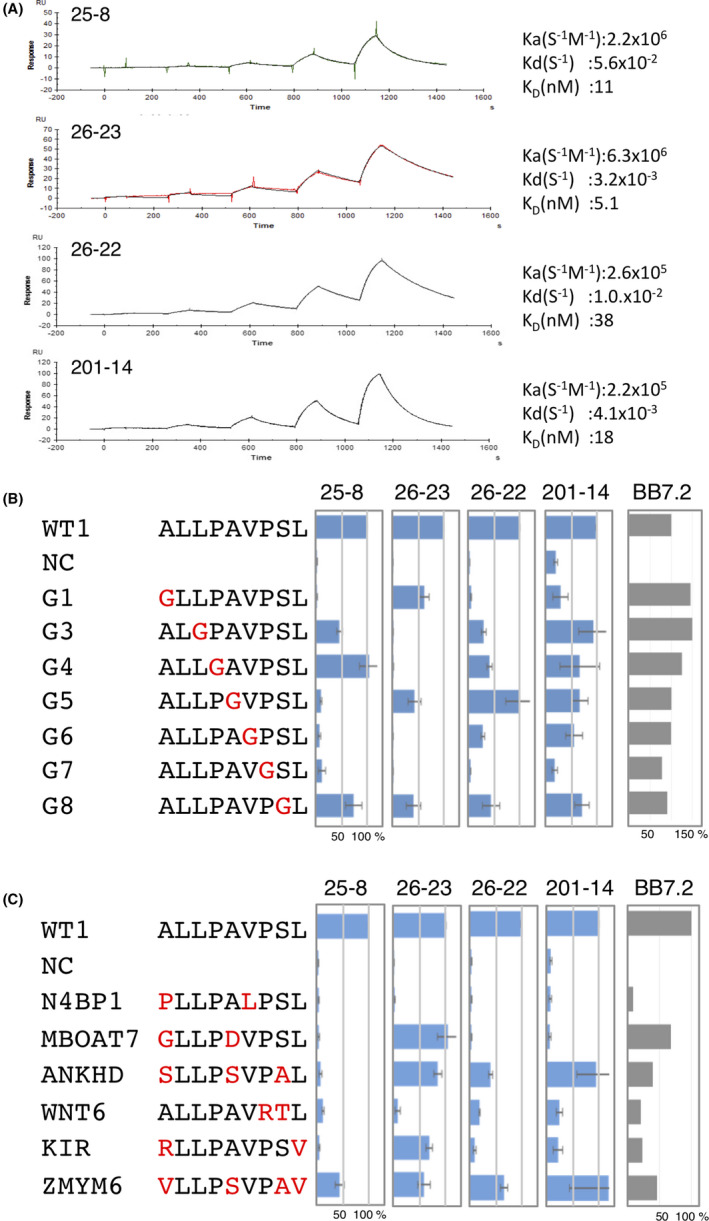
Biochemical analysis of candidate T‐cell receptor mimic antibodies (TCRm‐Abs). A, Surface plasmon resonance analysis of the candidate TCRm‐Abs for affinity to WT1C/HLA‐A*02. Calculated affinity constants are displayed. Data are representative of two independent experiments. B, Epitope mapping of the candidate TCRm‐Abs based on glycine‐substituted WT1C peptides. T2 cells pulsed with either WT1C or glycine‐substituted WT1C peptides (1.0 μg/mL) were incubated with the selected antibodies (1.0 μg/mL) and the PE‐labeled anti–HLA‐A*02 antibody (BB7.2). Antibody binding was determined by FACS relative to that for native WT1C‐pulsed T2 cells. C, Antibody binding to a panel of WT1C‐like peptides found in the human proteome. The indicated peptides were pulsed onto T2 cells, and the antibody binding was determined by FACS relative to that for WT1C‐pulsed T2 cells. The positions with mismatched amino acids are shown in red. Data are expressed as the average ±SD for three (#25‐8) or two (BB7.2) replicates/group.
3.2. #25‐8 recognizes WT1C but also cross–reacts to unknown peptides loaded on HLA‐A*02
To provide direct confirmation that #25‐8 binds the WT1C peptide loaded on HLA‐A*02, we analyzed a small panel of cell lines to assess their ability to be recognized by #25‐8. As shown in Figure 3A,B, #25‐8 binding was restricted to the HLA‐A*02+ WT1+ cancer cell line THP1. #25‐8 labeling was not observed on HLA‐A*02+ WT1− tumor cell lines (MCF7, NALM‐6 and HEK293) and normal cells taken from HLA‐A*02+ healthy donors (NB1 fibroblasts and PBMC). In addition, #25‐8 did not bind with HLA‐A*02− WT1+ cells (K562, OVCAR‐3, and SK‐OV‐3) or HLA‐A*02− WT1− tumor cell lines (Jurkat and NK92 MI; Figure S2). However, a considerable level of #25‐8 binding was observed on JY cells that express high levels of HLA‐A*02 but not WT1. When JY cells were treated with an acid wash to remove the peptides from the HLA molecules, #25‐8 no longer reacted with them. Pulsing WT1C on the washed JY cells recovered #25‐8 binding, indicating that #25‐8 reacted to unknown peptide/HLA‐A*02 complexes expressed on JY cells (Figure S3). Thus, we investigated the cross–reactive potential of this TCRm‐Ab using different screening methods and characterized its fine specificity. HeLa cells (WT1−, HLA‐A*02−) were transfected with HLA‐A*02:01 (HeLa/A02) or both the WT1 and HLA‐A*02:01 genes (HeLa/A02/WT1), and #25‐8 binding to these cells was analyzed by FACS. #25‐8 stained HeLa/A2/WT1 cells, while HeLa/A2 cells were also stained by #25‐8, indicating that 43% of the #25‐8 binding to HeLa/A2/WT1 cells originated from off–target binding (Figure 3C). It has been shown that IFN‐γ alters proteasome catalytic functions, leading to the increased production of certain TCR epitopes. 30 , 31 Therefore, we analyzed the effect of IFN‐γ on the cross–reactive potential of #25‐8. IFN‐γ treatment of HeLa/A2/WT1 cells led to a sixfold increase in #25‐8 binding compared to that of untreated HeLa/A2/WT1 cells. Enhanced binding of #25‐8 was also found on HeLa/A2 cells (ninefold) compared to untreated HeLa/A2 cells, indicating that that 62% of the #25‐8 binding to HeLa/A2/WT1 cells originated from off–target binding under IFN‐γ stimulation. These results indicate that WT1C is processed and loaded on HLA‐A*02 and that #25‐8 recognizes the WT1C/HLA‐A*02 complex but also reacts to unknown peptides loaded on HLA‐A*02. We next sought to assess whether the potential cross–reactive ZMYM6 peptide for #25‐8 is actually processed and presented on HLA‐A*02:01+ cells and whether #25‐8 binds it. As shown in Figure S4, forced expression of the ZMYM6 gene in HeLa/A2 cells did not induce #25‐8 binding, suggesting that the ZMYM6 epitope may not be properly processed or presented on HLA‐A*02. Thus, we could rule out cross–reactivity of this peptide.
FIGURE 3.
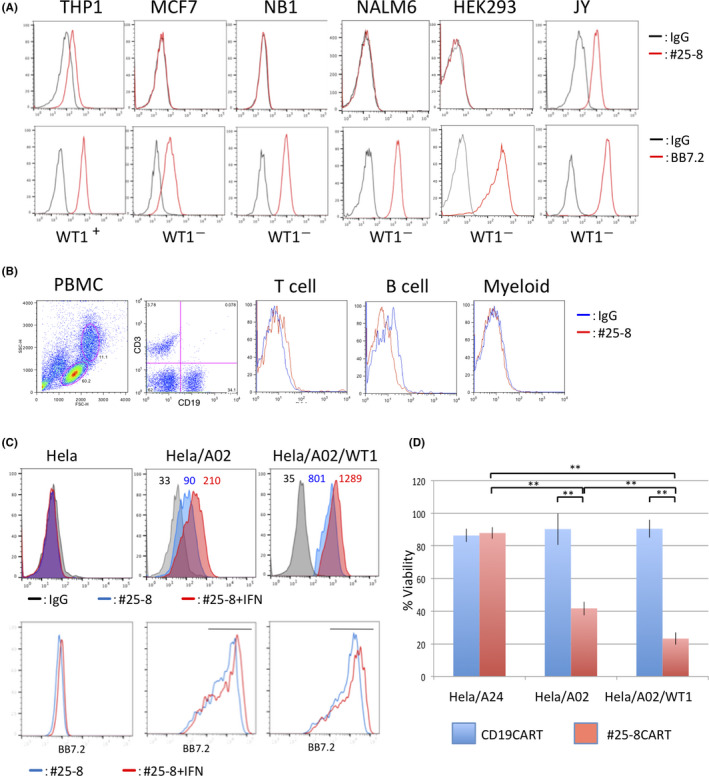
#25‐8 recognizes WT1C but cross–reacts to unknown peptides. A, Binding of #25‐8 to a panel of human cell lines. The indicated human cell lines were stained with 1.0 μg/mL #25‐8 or control mouse IgG, and binding was analyzed by FACS. The integrity of the peptide/HLA‐A*02 expression was confirmed with an antibody (BB7.2) against HLA‐A*02. B, PBMC from HLA‐A*24+ healthy donors stained with #25‐8. A representative gating strategy and #25‐8 histogram compared with a control IgG are shown. C, HLA‐A*02 high cells stained with #25‐8 or control IgG. HeLa, HeLa/A02, or HeLa/A2/WT1 cells were treated with or without IFN‐γ for 12 h, and HLA‐A*02 high cells were gated. The binding ability of #25‐8 was evaluated according to mean fluorescent intensity. The bar in each histogram represents the gate defined as HLA‐A*02 high cells (lower panel). Data are representative of two independent experiments. D, The specificity and cross–reactivity of #25‐8 reflects the cytotoxic activity of #25‐8 CAR T‐cells. #25‐8 CAR or CD19 CAR T‐cells were mixed with HeLa, HeLa/A02, or HeLa/A2/WT1 cells, and the cytotoxic activity of the CAR T‐cells was measured. The values for cell viability are represented as the average ± SD of three replicates (n = 3) of two independent experiments (**P < 0.01).
We next analyzed whether the observed cross–reactivity and specificity of #25‐8 influence the ability of #25‐8 CAR T‐cells to kill target and nontarget cells in vitro. To ascertain the specificity of #25‐8 ScFv fragments, HEK293 cells expressing #25‐8 CAR were labeled with a panel of peptide/HLA molecules and examined by FACS. As shown in Figure S5, #25‐8 ScFv retains the binding specificity of the parent antibody, for which the greatest binding was observed to WT1C/HLA‐A*02, whereas moderate binding to ZMYM6 and no binding to other peptide/HLA molecules were observed. T‐cells expanded from PBMCs were transduced with either the #25‐8 CAR or CD19 CAR construct, and the capacity of the CAR T‐cells to lyse target cells was analyzed. The #25‐8 CAR T‐cells killed a significantly increased percentage of HeLa/A2/WT1 cells but not HeLa cells expressing HLA‐A*24 (HeLa/A24). However, a substantial level of cytotoxicity was also found in HeLa/A2 cells, and approximately 77% of the observed cytotoxicity of #25‐8 CAR T‐cells originated from off–target recognition of #25‐8.
3.3. Identification of cross–reactive peptides by the PresentER antigen presentation platform
To uncover the origin of this cross–reactive binding, we constructed a minigene lentiviral library encoding 9‐mer peptides based on the putative #25‐8 binding sequence and transduced it into T2 cells (Figure 4A). After two rounds of FACS for #25‐8, T2 cells expressing #25‐8‐cross–reactive peptides were enriched (Figure 4B). The minigenes integrated into genomic DNA recovered from the T2 cells before (input minigene) and after the two rounds of FACS (2nd minigene) were sequenced with Illumina. Among 6803 peptides in the input minigene, 397 peptides were recovered in the 2nd minigene. ZMYM6 was not found in the 2nd minigene. A total of 134 peptides were more enriched than WT1C (Figure 4C and Table S2). However, none of those enriched peptides matched exactly with the human proteome by BLAST search. Thus, we selected the top 100 enriched peptides, and the most common amino acids were visualized via a sequence logo (Figure 4D). A search for the most similar sequence in the human proteome with high binding affinity to HLA‐A*02 identified three HLA‐A*02 restricted peptides: VLCSSLPTV from small G protein signaling modulator 3 (SGSM3), VLIASVPVV from potassium voltage‐gated channel subfamily Q member 3 (KCNQ3) and VLVSEVPLV from titin (TTN). When we assayed these peptides for binding to #25‐8 by pulsing T2 cells, KCN and SGSM3 were recognized by #25‐8, but TTN was not (Figure 4E and Table 1). Analysis of protein expression profiles for tissues and cell types using The Human Protein Atlas (https://www.proteinatlas.org) revealed that SGSM3 is expressed across a wide range of tissues, while the expression of KCNQ3 is limited to neuronal cells. Thus, these peptides, if processed and presented on HLA in normal tissues, could potentially cause off–target toxicity.
FIGURE 4.
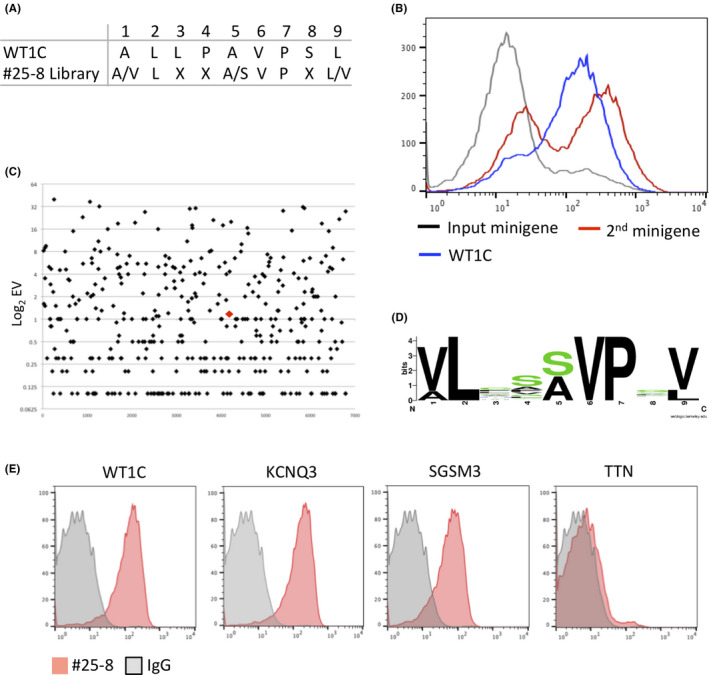
Discovery of cross–reactive peptides of #25‐8 by PresentER. A, #25‐8 recognition motif. X indicates any amino acid. B, FACS histograms depicting #25‐8 binding to T2 cells expressing minigenes. T2 cells expressing the indicated minigene library were stained with #25‐8 and analyzed by FACS. C, Manhattan plots showing the enrichment value (EV) of each peptide in the 2nd minigene library. Log2 EV on the y‐axis against the input minigene peptides on the x‐axis is shown. The red diamond indicates WT1C. D, Sequence logos showing the amino acid frequency for a given position in the #25‐8 binding sequence. E, The indicated peptides were pulsed onto T2 cells and #25‐8 binding was determined by FACS. Data are representative of two independent experiments.
TABLE 1.
Candidate cross–reactive peptides for #25‐8
| Protein | Peptide | Affinity (nmol/L) | Relative MFI (%) | Protein accession number (NCBI) |
|---|---|---|---|---|
| WT1 | ALLPAVPSL | 6.01 | 100 ± 3 | NP_000369 |
| SGSM3 | VLCSSLPTV | 9.90 | 41 ± 5 | NP_056520 |
| KCNQ3 | VLIASVPVV | 7.16 | 128 ± 5 | NP_004510 |
| TTN | VLVSEVPLV | 7.96 | 5 ± 3 | NP_035782 |
| VIM | RLRSSVPGV | 105.98 | ND | NP_003371 |
| RTTN | ILESAVPLL | 69.67 | ND | NP_775901 |
Relative MFI relative to that for WT1C‐pulsed T2 cells is expressed as the average ± SD for three replicates/group.
Abbreviations: MFI, mean fluorescent intensity; ND, not determined.
4. DISCUSSION
Tumor cells continuously display tumor‐associated antigen‐derived peptides on the cell surface in the context of HLA. 32 However, in many cases, most of these peptides do not elicit strong antitumor immune responses because self–reactive T‐cells harboring TCR with high affinity to peptides undergo clonal deletion to maintain self–tolerance. Furthermore, given that different individuals have different TCR repertoires, peptide immunogenicity differs among individuals. 33 For these reasons, TCR‐dependent immunotherapeutic approaches have relied on a limited number of immunodominant peptides preferentially targeted by T‐cells in multiple individuals. In contrast, TCRm‐Abs can target a variety of tumor‐associated antigen‐derived peptides in the form of CAR T‐cells or BiTE antibodies, which may overcome the limitations that hamper TCR‐based immunotherapeutic approaches. In this paper, we found that a nonimmunodominant peptide, WT1C, is processed and presented on the cell surface in the context of HLA‐A*02 and that the #25‐8 TCRm‐Ab can recognize this peptide in the HLA‐A*02 context.
One of the major concerns with TCRm‐Abs for future clinical trials is cross–reactivity against structurally related or unrelated antigens expressed on normal tissues. To date, a variety of TCRm‐Abs have been generated. However, in many cases, little evidence of potential cross–reactivity has been presented. 24 , 34 We performed a series of antibody screenings by ELISA with a panel of peptide/HLA molecules, T2 cells pulsed with glycine‐substituted peptides, and WT1C‐like peptides. Based on these analyses, #25‐8 was selected for its highest affinity and specificity. However, these strategies were not enough to predict the cross–reactivity of TCRm‐Ab; #25‐8 bound to WT1‐negative JY cells that highly expressed HLA‐A*02:01. It has been shown that TCR exhibit substantial cross–reactivity to allogeneic HLA molecules loaded with various peptides. 35 Thus, such phenomena may occur with TCRm‐Ab. JY cells express HLA‐B*07:02 and HLA‐C*07:02. Although those HLA are also expressed on HEK293 (HLA‐ B*07:02+ HLA‐C*07:02+) and OVCAR cells (HLA‐B*07:02+), #25‐8 did not bind to these cell lines. By taking these results along with the higher specificity and affinity of #25‐8 into account, it seems unlikely that #25‐8 cross–reacts to allogeneic HLA molecules on JY cells.
We demonstrated that forced expression of the HLA‐A*02:01 gene in HLA‐A*02:01− WT1− HeLa cells followed by IFN‐γ treatment led to #25‐8 cross–reactive potential with irrelevant peptides. This implies the role of IFN‐γ as a double‐edged sword for TCRm‐Ab: it enhances the expression of the target peptide but also induces the expression of cross–reactive peptides. This potentially dangerous cross–reactivity was confirmed through analysis using #25‐8 CAR T‐cells targeting WT1‐negative HeLa/A02 cells. High affinity antibodies can cause CAR T‐cells to recognize target antigens, but they can also increase the severity of off–target toxicities. A recent study demonstrated that low affinity CAR T‐cells have stronger antitumor activity and lower off‐tumor toxicities than their high affinity counterparts. 36 , 37 Because it is important to balance the relationship between potency and toxicity in CAR T‐cell therapy, the use of high affinity CAR, such as CAR carrying multivalent antibodies, is not necessary as a potential strategy to expand the effectiveness of immunotherapy. Thus, the development of TCRm‐Ab with reduced affinity but enhanced specificity will be needed to achieve superior antitumor efficacy and safety.
We also undertook investigations into the cross–reactive potential of #25‐8 using PresentER and identified candidate cross–reactive peptides. It remains unclear whether those peptides would be naturally processed and presented on HLA‐A*02:01 on tumor and normal cells. Because our PresentER screening was carried out based on the #25‐8 recognition motif, we cannot exclude the possibility that #25‐8 recognizes structurally unrelated peptides. In addition to the methods described in this paper, structure‐guided analysis as well as an HLA peptidome database would provide a framework for predicting potential cross–reactive peptides and the tissues where they are expressed. 38
In conclusion, we generated a TCR‐mAb that recognizes a novel WT1‐derived peptide in the context of HLA‐A*02 with a defined specificity while exhibiting cross–reactivity against a set of peptides. Our findings demonstrate the need for systematic validation by using multiple screening techniques to predict and identify potential cross–reactive epitopes expressed on normal tissues for integrating TCRm‐Abs into future clinical translation.
ETHICAL CONSIDERATIONS
All experiments were performed in accordance to relevant guidelines and regulations. Animal experimental protocols were approved by the Committee on Animal Experimentation at the University of Toyama and conducted using project licence A2017eng‐1. Experimentation with human samples was approved by the Ethics Committee on University of Toyama and conducted using project licence RinNin21‐47.
DISCLOSURE
The authors declare no competing interests for this article.
Supporting information
Fig S1‐S5
Table S1
Table S2
Appendix S1
ACKNOWLEDGMENTS
We thank past and current members of our laboratory for fruitful discussions. We also thank Y. Nohara, M. Nozaki, and T. Kinoshita for technical support. This research was supported in part by grants from the Hokuriku Industrial Advancement Center (HIAC) (H27HIAC122) for N. Kurosawa, and the Basic Science and Platform Technology Program for Innovative Biological Medicine (15657579) and the Emerging/Re‐emerging Infectious Diseases Project of Japan (16768665) from Japan Agency for Medical and Development (AMED) for M. Isobe. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Kurosawa N, Midorikawa A, Ida K, Fudaba YW, Isobe M. Development of a T‐cell receptor mimic antibody targeting a novel Wilms tumor 1‐derived peptide and analysis of its specificity. Cancer Sci. 2020;111:3516–3526. 10.1111/cas.14602
Contributor Information
Nobuyuki Kurosawa, Email: kurosawa@eng.u-toyama.ac.jp.
Masaharu Isobe, Email: isobe@eng.u-toyama.ac.jp.
REFERENCES
- 1. Inoue K, Ogawa H, Sonoda Y, et al. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood. 1997;89:1405‐1412. [PubMed] [Google Scholar]
- 2. Hastie ND. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development. 2017;144:2862‐2872. [DOI] [PubMed] [Google Scholar]
- 3. Sugiyama H. WT1 (Wilms’ tumor gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol. 2010;40:377‐387. [DOI] [PubMed] [Google Scholar]
- 4. Gao L, Bellantuono I, Elsasser A, et al. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood. 2000;95:2198‐2203. [PubMed] [Google Scholar]
- 5. Oka Y, Udaka K, Tsuboi A, et al. Cancer immunotherapy targeting Wilms’ tumor gene WT1 product. J Immunol. 2000;164:1873‐1880. [DOI] [PubMed] [Google Scholar]
- 6. Oka Y, Tsuboi A, Nakata J, et al. Wilms’ tumor gene 1 (WT1) peptide vaccine therapy for hematological malignancies: from CTL epitope identification to recent progress in clinical studies including a cure‐oriented strategy. Oncol Res Treat. 2017;40:682‐690. [DOI] [PubMed] [Google Scholar]
- 7. Ochi T, Fujiwara H, Okamoto S, et al. Novel adoptive T‐cell immunotherapy using a WT1‐specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood. 2011;118:1495‐1503. [DOI] [PubMed] [Google Scholar]
- 8. Tawara I, Kageyama S, Miyahara Y, et al. Safety and persistence of WT1‐specific T‐cell receptor gene‐transduced lymphocytes in patients with AML and MDS. Blood. 2017;130:1985‐1994. [DOI] [PubMed] [Google Scholar]
- 9. Chang AY, Gejman RS, Brea EJ, et al. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opin Biol Ther. 2016;16:979‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dahan R, Reiter Y. T‐cell‐receptor‐like antibodies ‐ generation, function and applications. Expert Rev Mol Med. 2012;14:e6. [DOI] [PubMed] [Google Scholar]
- 11. He Q, Liu Z, Liu Z, Lai Y, Zhou X, Weng J. TCR‐like antibodies in cancer immunotherapy. J Hematol Oncol. 2019;12:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trenevska I, Li D, Banham AH. Therapeutic antibodies against intracellular tumor antigens. Front Immunol. 2017;8:1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dao T, Pankov D, Scott A, et al. Therapeutic bispecific T‐cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol. 2015;33:1079‐1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao Q, Ahmed M, Tassev DV, et al. Affinity maturation of T‐cell receptor‐like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia. 2015;29:2238‐2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Akahori Y, Wang L, Yoneyama M, et al. Antitumor activity of CAR‐T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood. 2018;132:1134‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doubrovina E, Carpenter T, Pankov D, Selvakumar A, Hasan A, O’Reilly RJ. Mapping of novel peptides of WT‐1 and presenting HLA alleles that induce epitope‐specific HLA‐restricted T cells with cytotoxic activity against WT‐1(+) leukemias. Blood. 2012;120:1633‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pinilla‐Ibarz J, May RJ, Korontsvit T, et al. Improved human T‐cell responses against synthetic HLA‐0201 analog peptides derived from the WT1 oncoprotein. Leukemia. 2006;20:2025‐2033. [DOI] [PubMed] [Google Scholar]
- 18. Li D, Bentley C, Anderson A, et al. Development of a T‐cell receptor mimic antibody against wild‐type p53 for cancer immunotherapy. Cancer Res. 2017;77:2699‐2711. [DOI] [PubMed] [Google Scholar]
- 19. Dao T, Mun SS, Scott AC, et al. Depleting T regulatory cells by targeting intracellular Foxp3 with a TCR mimic antibody. Oncoimmunology. 2019;8 : e1570778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahmed M, Lopez‐Albaitero A, Pankov D, et al. TCR‐mimic bispecific antibodies targeting LMP2A show potent activity against EBV malignancies. JCI insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8:337‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun S, Hao H, Yang G, Zhang Y, Fu Y. Immunotherapy with CAR‐modified t cells: toxicities and overcoming strategies. J Immunol Res. 2018;2018:2386187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross–reactivity of affinity‐enhanced T cells in myeloma and melanoma. Blood. 2013;122:863‐871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ataie N, Xiang J, Cheng N, et al. Structure of a TCR‐mimic antibody with target predicts pharmacogenetics. J Mol Biol. 2016;428:194‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gejman RS, Chang AY, Jones HF, et al. Rejection of immunogenic tumor clones is limited by clonal fraction. eLife. 2018;7:e41090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gejman RS, Jones HF, Klatt MG, et al. Identification of the targets of T‐cell receptor therapeutic agents and cells by use of a high‐throughput genetic platform. Cancer Immunol Res. 2020;8:672‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kurosawa N, Wakata Y, Ida K, Midorikawa A, Isobe M. High throughput development of TCR‐mimic antibody that targets survivin‐2B80‐88/HLA‐A*A24 and its application in a bispecific T‐cell engager. Sci Rep. 2019;9:80‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kurosawa N, Wakata Y, Inobe T, et al. Novel method for the high‐throughput production of phosphorylation site‐specific monoclonal antibodies. Sci Rep. 2016;6:25174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kurosawa N, Yoshioka M, Fujimoto R, Yamagishi F, Isobe M. Rapid production of antigen‐specific monoclonal antibodies from a variety of animals. BMC Biol. 2012;10:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Groettrup M, Ruppert T, Kuehn L, et al. The interferon‐gamma‐inducible 11 S regulator (PA28) and the LMP2/LMP7 subunits govern the peptide production by the 20 S proteasome in vitro. J Biol Chem. 1995;270:23808‐23815. [DOI] [PubMed] [Google Scholar]
- 31. Chang AY, Dao T, Gejman RS, et al. A therapeutic T cell receptor mimic antibody targets tumor‐associated PRAME peptide/HLA‐I antigens. J Clin Investig. 2017;127:3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schuster H, Peper JK, Bosmuller HC, et al. The immunopeptidomic landscape of ovarian carcinomas. Proc Natl Acad Sci USA. 2017;114:E9942‐E9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lorenz FKM, Ellinger C, Kieback E, et al. Unbiased identification of T‐cell receptors targeting immunodominant peptide‐MHC complexes for T‐Cell receptor immunotherapy. Hum Gene Ther. 2017;28:1158‐1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holland CJ, Crean RM, Pentier JM, et al. Specificity of bispecific T cell receptors and antibodies targeting peptide‐HLA. J Clin Investig. 2020;130:2673‐2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kranz DM. Incompatible differences: view of an allogeneic pMHC‐TCR complex. Nat Immunol. 2000;1:277‐278. [DOI] [PubMed] [Google Scholar]
- 36. Park S, Shevlin E, Vedvyas Y, et al. Micromolar affinity CAR T cells to ICAM‐1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. 2017;7:14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ghorashian S, Kramer AM, Onuoha S, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low‐affinity CD19 CAR. Nat Med. 2019;25:1408‐1414. [DOI] [PubMed] [Google Scholar]
- 38. Sarkizova S, Klaeger S, Le PM, et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol. 2020;38:199‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5
Table S1
Table S2
Appendix S1