

Abstract
Ras has been found to be mutated in 30% of non‐small cell lung cancers, and its mutation has been regarded as a causal factor underlying tumorigenesis. However, no successful medicine has been developed so far to inhibit Ras for lung cancer treatment. We have previously identified DHX33 as a Ras downstream effector, promoting cell cycle progression and cell growth. In this study, with the K‐Ras (G12D);DHX33 (lox/lox) mouse model, we discovered that genetic ablation of DHX33 inhibited tumor development. We further found that ablation of DHX33 altered the expression of nearly 2000 genes which are critical in cancer development such as cell cycle, apoptosis, glycolysis, Wnt signaling, and cell migration. Our study for the first time demonstrates the pivotal role of the DHX33 in Ras‐driven lung cancer development in vivo and highlights that pharmacological targeting DHX33 can be a feasible option in treating Ras‐mutant lung cancers.
Keywords: DHX33, lung cancer, oncogene, Ras, RNA helicase
Our study provides the initial evidence in vivo to reveal the critical role of DHX33 in Ras‐driven lung tumorigenesis. This study highlights a novel therapeutic target in treating human cancers.
1. INTRODUCTION
Proto‐oncogene Ras encodes a small GTPase of approximately 20 kDa, which can be tethered to plasma membrane via farnesylation. 1 Upon extracellular signal stimulation, Ras‐GDP is activated by Guanine Exchange Factor (GEF) into GTP binding form, which then activates multiple downstream signaling pathways in order to regulate cell growth, proliferation, survival, and differentiation. 2 Three major signaling pathways including PI3K/Akt, Raf/MAP, and Rho GTPase have been found to be activated by Ras as its downstream effectors. 3 Activated Ras can be turned off by GTP hydrolysis into the Ras‐GDP form. However, in human cancers, this is frequently disrupted by mutations on Ras. Ras mutation most frequently occurs on G12, which can be mutated into Val, Cys, Ser, or Asp with different frequency in human cancers. 4 The mutation leads to the locking of Ras in the GTP binding form. Although Ras has been found to be frequently mutated in human cancers for a long time, no effective drugs have been discovered and applied in clinics yet. Multiple downstream effectors can be activated by Ras‐GTP, allowing cancer cells to circumvent one single pathway inhibition to confer the resistance of cancer cells to many therapeutic drugs. Combinatorial inhibitors have been found to be effective in treating many human cancers. For instance, simultaneous inhibition of c‐Raf and epidermal growth factor receptor (EGFR) have been effective to reduce cancer growth in pancreatic ductal adenocarcinoma (PDAC) and patient‐derived xenograft (PDX) models. 5 SHP2 inhibition has been found to augment the antitumor effects of MEK inhibition in Ras‐mutated lung, pancreatic, and gastric carcinoma cells. 6 , 7 Additionally, lipophilic bisphosphonates and rapamycin compensate with each other to inhibit KRAS‐G12D–induced mouse lung tumorigenesis. 8 Other combinatorial drugs such as PI3K and MEK inhibitors, 9 mTOR and HDAC inhibitors, 10 and PLK1 and ROCK pair, 11 have all been shown to be effective towards Ras‐driven tumors. Recently significant progress has been made in treating Ras‐G12C–mutant cancers as conjugating inhibitors for Cysteine have been developed and analyzed in clinical trials to treat Ras‐G12C–mutant cancers. 12 Despite all these efforts, majority of Ras‐mutant cancers are still difficult to be treated.
Our previous work has shown that DHX33 is a downstream transcriptional and translational target of Ras in cells. 13 As a member of Asp‐Glu‐Ala‐His (DEAH) box RNA helicase, DHX33 has previously been identified as a key factor in ribosome biogenesis 14 and a cytosolic RNA sensor to activate the NLRP3 inflammasome. 15 DHX33 protein is further found to be regulated by multiple cancer‐critical genes and is deregulated in many human cancers, such as lung cancer, liver cancer, glioblastoma, and lymphoma. 16 , 17 , 18 , 19 Upregulation of DHX33 has been found to be important for Ras to enhance cell growth through promoting ribosome biogenesis and protein translation. 13 Like many members of RNA helicase family, DHX33 is a multifunctional protein. DHX33 is further found to be actively involved in mRNA translation 20 and mRNA transcription via associating with gene promoters. In lung cancer cells, DHX33 was found to regulate a subset of genes though altering epigenetic marks. 18 , 19 , 21 , 22
It remains unknown whether DHX33 is important for Ras‐driven cancer development in vivo, or whether DHX33 might serve as molecular target in treating Ras‐driven cancer. In the current study, taking advantage of the Cre/LoxP recombination system to deactivate DHX33 in mouse, we generated K‐Ras (G12D‐LSL);DHX33 (lox/lox) mice. Genetic ablation of DHX33 significantly blocks lung tumorigenesis. Mechanistically, multiple cancer‐associated genes were found to be regulated by DHX33 in Ras‐activated lung cancers, and DHX33 modulated cellular transcriptome to favor tumorigenesis. Our results implicate that pharmacological targeting DHX33 might provide a feasible option for treating Ras‐mutated lung cancers.
2. MATERIALS AND METHODS
2.1. Generation of DHX33 conditional knockout mice
The DHX33 gene in mouse genome spans a region of about 20.5 kb on the reverse strand of chromosome 11. We used Crispr/Cas9 system to introduce two Lox P sites flanking Exon 2‐4, in order to conditionally knock out Exon 2‐4 in DHX33 gene by the Cre‐lox P system. Intron 2‐3 and intron 4‐5 are big, and the insertion of the lox element will not interfere with mRNA splicing. To minimize the possibility of disruption of the DHX33 gene expression, both lox P sites were inserted into nonconserved regions. We used the following two guide RNA sequences for gene editing: forward‐sgRNA (from 5′‐ to 3′): CATGTATAATGGTAGGTAC and reverse‐sgRNA: ATGTACCATGCATAGAGGCC. Both guide RNAs were cloned into pT7 plasmids and sequenced for confirmation. The targeting vector contains two lox P sites flanking Exon 2‐4 with a 5′‐homologous arm and a 3′‐homologous arm (each arm has ~1400 bp). Zygote microinjection was performed following standard protocol. Founder mice genotyping was identified by PCR. For the 5′‐Lox P site, the primers were as follows (from 5′‐ to 3′): FP: TGAATGGACATCTCGTCGTATCAGT and RF: AATTCCACTTGCCGAAATTGAGTCT. The PCR product for wild‐type mice had a size of 208 bp, while for the mutant mice, it had a size of 287 bp. For the 3′‐Lox P site, the primers were as follows (from 5′ to 3′): FP: TGTGAGCTCTAGATTCAGTGAGAAC and RP: TGTTTACCTCTAAATTGTCTC AGGA. For wild‐type mice, the PCR product was 212 bp, while for the mutant mice, the PCR product had a size of 298 bp. The mutant allele was further confirmed by Southern blot analysis. The 5′‐probe was amplified from forward primer (5′‐GTGCCTGTCTTTCTGTGTTGAATT‐3′) and reverse primer (5′‐GAGACTAAAGTCATTTCGGATGCTG‐3′). After StuI digestion, Southern blot was performed with the labeled probe. For wild‐type allele, it had a size of 7.5 kb, while for the mutant allele, it had a size of 5.6 kb. The 3′‐probe was amplified by forward primer (5′‐TAAATAGATTCAACAGTTGCTG‐3′) and reverse primer (5′‐TGGGTTCAGCTCATTTTTGAAAGG‐3′). After NcoI digestion, Southern blot was performed with the labeled probe. For wild‐type allele, it had a size of 6.1 kb, while for the mutant allele, it had a size of 5.1 kb.
2.2. Adenovirus infection
Adenovirus was purchased from Vigene (Shangdong, China); adeno‐LacZ or adeno‐Cre has a titer of 1.0 × 1011 pfu/mL. Mouse embryonic fibroblasts (MEFs) at early passages were trypsinized, and approximately 2.0 × 1106 cells were resuspended into 3 mL of fresh DMEM media containing 10% FBS/penicillin and streptomycin (PS). Adenovirus was added at a MOI of 150 and incubated for 4 hours at 37°C in CO2 incubators. The cells were then supplemented with 6 mL of fresh and complete DMEM medium and further incubated overnight. The cells were then replenished with fresh complete medium. To set up a lung cancer model with K‐Ras (G12D‐LSL) mice, we followed the protocol described previously. 23 Briefly, approximately 1 × 1107 pfu Ade‐Cre virus was mixed with MEM and CaCl2 for precipitation. After the mice had been anesthetized by avertin through introperitoneal injection, the viruses were directly administered through nasal inhalation into their lungs.
2.3. Cell Culture
MEF cells were isolated as described previously 24 and maintained in DMEM medium containing 10% FBS, 2 mmol/L l‐Glutamine, streptomycin, and penicillin.
2.4. RNA sequencing
Tumor tissues were cut into small pieces for total RNA extraction. RNeasy Mini Kit (Qiagen) was applied for isolating RNA from tissues according to the manufacturer's recommendation. RNA samples were then further purified with magnetic oligo‐dT beads after denaturation. Purified mRNA samples were reverse‐transcribed into first‐strand cDNA, and second complementary DNA was further synthesized. Fragmented DNA samples were blunt‐ended and adenylated at 3′ ends. Adaptors were ligated to construct library. DNA was quantified by Qubit (Invitrogen). After cBot cluster generation, DNA samples were then sequenced by Ilumina HiSeq2500 SBS from Genergy Bio (Shanghai). Raw data were converted into Fastq format. The amount of transcripts in each sample was calculated based on FPKM (fragments per kilobase Million reads) of transcript per million fragments mapped. Cuffnorm software was used to calculate the Fragments Per Kilobase Million reads (FPKM) value for each sample, and log 2 value was applied. Cuffdiff software was used to calculate the differential gene transcripts between different samples.
2.5. Western blot
Cells were lysed by RIPA buffer supplemented with protease and phosphatase inhibitors (Thermo fisher). After incubation for 10 minutes on ice, cell lysates were further disrupted by sonication. The whole‐cell extract was then subjected to a SDS‐PAGE gel with a loading amount of 50 µg protein per sample. Proteins were then transferred onto a polyvinylidene difluoride membrane. Membranes were blocked in 5% nonfat milk which was diluted in 1× Tris buffered saline with Tween‐20 (TBST) buffer for 1 hour at room temperature. Primary antibodies, which were diluted in 5% FBS (diluted in 1× TBST), were incubated with the membrane at 4°C overnight. Membranes were then rinsed with 1× TBST buffer for multiple times and incubated with HRP‐labeled secondary antibodies in 5% FBS (diluted in 1× TBST) at room temperature for 2 hours. The blots were visualized with an ECL kit (Thermo fisher). The antibodies were sourced as follows: anti‐GAPDH, Absin (abs830030); anti‐Aurora A, Santa Cruz Biotechnology (sc‐56881); anti‐MCM2, Santa Cruz Biotechnology (sc‐9839); anti‐MCM3, Santa Cruz Biotechnology (sc‐9850); anti‐MCM4, Santa Cruz Biotechnology (sc‐22779); anti‐MCM6, Santa Cruz Biotechnology (sc‐9843); anti‐MCM7, Santa Cruz Biotechnology (sc‐9960); anti‐cyclin D1, Santa Cruz Biotechnology (sc‐753); anti‐cyclin E2, Santa Cruz Biotechnology (sc‐9566); anti‐cyclin B2, Santa Cruz Biotechnology (sc‐22776).
2.6. Quantitative RT‐PCR
The primers were all designed by IDT (http://sg.idtdna.com/site) online “Realtime PCR Tool” and purchased from BGI (Shenzhen). Total RNA was extracted by High Pure RNA Isolation kit (Roche) and then transcribed into cDNA using PrimeScript mix kit (Takara). RT‐PCR is performed with an ABI One step plus cycler that is managed with the corresponding software. To analyze mRNA levels, SYBR green Supermix (Bio‐Rad) was used, and transcript quantification was calculated by ΔΔCT value after normalized to GAPDH values. Melting curve was used to confirm the amplification of single products. The primer sequences for this study have been described previously. 18 Primers for some newly added genes are shown below (all start from 5′ to 3′):
| mouse E2F1 Forward | GGATCTGGAGACTGACCATCAG |
| mouse E2F1 Reverse | GGTTTCATAGCGTGACTTCTCCC |
| mouse vimentin Forward | CGGAAAGTGGAATCCTTGCAGG |
| mouse vimentin Reverse | AGCAGTGAGGTCAGGCTTGGAA |
| mouse Cyclin E2 Forward | GTACTGTCTGGAGGAATCAGCC |
| mouse Cyclin E2 Reverse | CCAAACCTCCTGTGAACATGCC |
| mouse Cyclin D1 Forward | GCAGAAGGAGATTGTGCCATCC |
| mouse Cyclin D1 Reverse | AGGAAGCGGTCCAGGTAGTTCA |
| mouse β‐catenin Forward | GTTCGCCTTCATTATGGACTGCC |
| mouse β‐catenin Reverse | ATAGCACCCTGTTCCCGCAAAG |
| mouse Wnt3a Forward | AACTGCACCACCGTCAGCAACA |
| mouse Wnt3a Reverse | AGCGTGTCACTGCGAAAGCTAC |
| mouse MCM4 Forward | TTCAGCCTTGGCTCCCAGCATT |
| mouse MCM4 Reverse | GAAGGATGTTGATCTCAGCACGG |
| mouse GADPH Forward | CAAGGAGTAAGAAACCCTGGAC |
| mouse GADPH Reverse | GGATGGAAATTGTGAGGGAG |
| mouse Aldh2 Forward | GCTGTTGTACCGATTGGCGGAT |
| mouse Aldh2 Reverse | GCGGAGACATTTCAGGACCATG |
| mouse Eno4 Forward | AGACTCGGAGTCCTCTGATGAC |
| mouse Eno4 Reverse | GGCGGTTGTATTTGGTCATCCG |
| mouse LDHA Forward | CAAAGACTACTGTGTAACTGCGA |
| mouse LDHA Reverse | TGGACTGTACTTGACAATGTTGG |
| mouse PDK1 Forward | GGACTTCGGGTCAGTGAATGC |
| mouse PDK1 Reverse | TCCTGAGAAGATTGTCGGGGA |
| mouse PKM2 Forward | GTGGCTCGGCTGAATTTCTCT |
| mouse PKM2 Reverse | CACCGCAACAGGACGGTAG |
| mouse CDC6 Forward | GACACAAGCTACCATCGGTTT |
| mouse CDC6 Reverse | CAGGCTGGACGTTTCTAAGTTT |
| mouse DHX33 Forward | CAGCTCCGAAACCTGGACAA |
| mouse DHX33 Reverse | CCCCTTCATAGAGGTACTGTGG |
2.7. Immunohistochemistry
Mouse lung tissues were dissected and dehydrated, paraffin‐embedded and further sliced based on standard protocols. Tissues were deparaffinized in xylene and rehydrated in a series of alcohol solution with decreasing concentration. The antigen was retrieved in Tris buffer (pH 9.0) in a steamer. Tissues were then incubated in 1% H2O2 in methanol to extinguish endogenous peroxidase. After blocking with 10% FBS for 1 hour at room temperature, tissues were incubated with primary antibody at 4°C overnight. Standard protocol was then followed with DAKO kit according to the manufacturer's recommendation.
To quantify the immunohistochemistry signals, normally five mice were chosen from each group. For each mouse, three different tumors were selected for quantification. For each tumor, five different fields were selected, and approximately 50 cells were counted from each field for evaluation by a pathologist. The data are presented as average with standard deviation from five different mice.
The antibodies used were sourced as follows: anti‐Ki67, Abcam (ab15580); anti‐pERK1/2, Abcam (ab50011); anti‐pAkt (T308), Abcam (ab38449); anti‐vimentin, Abcam (ab92547); anti‐cyclin D1, Abcam (ab16663); anti‐cyclin E2, Abcam (ab32103); anti‐β‐catenin, Abcam (ab16051); anti‐PDK1, Abcam (ab52893); anti‐LDHA, Abcam (ab101562); anti‐cdc6, Abcam (ab125195); anti‐E2F1, Abcam (ab179445); anti‐MMP9, Abcam (ab119906).
Data are presented as the mean ± SD. Statistical significance was determined using the Student's t test, with a P value < .05 considered significant.
3. RESULTS
3.1. DHX33 deletion reduced the transformation potency of Ras V12 in vitro
DHX33 was found to be upregulated by oncogenic Ras in cells. 13 To fully understand the significance of DHX33 in Ras‐induced transformation, we knocked down DHX33 gene in wild‐type mouse embryonic fibroblasts. First, in order to immortalize the cells, they were infected by lentivirus encoding shARF. 25 ARF deletion caused DHX33 protein levels to be increased due to translational control. 13 These cells were then infected with lentivirus encoding shDHX33 to knock down endogenous DHX33, with scrambled as a control (shSCR). Cellular transformation analysis was then performed on these MEFs after enforced RasV12 expression. As shown in Figure 1A, DHX33 protein levels were markedly reduced, while Ras was efficiently expressed. The transformation activity of Ras V12 was significantly reduced when DHX33 was ablated prior to Ras V12 overexpression (Figure 1B‐C). Colony numbers were significantly decreased.
Figure 1.
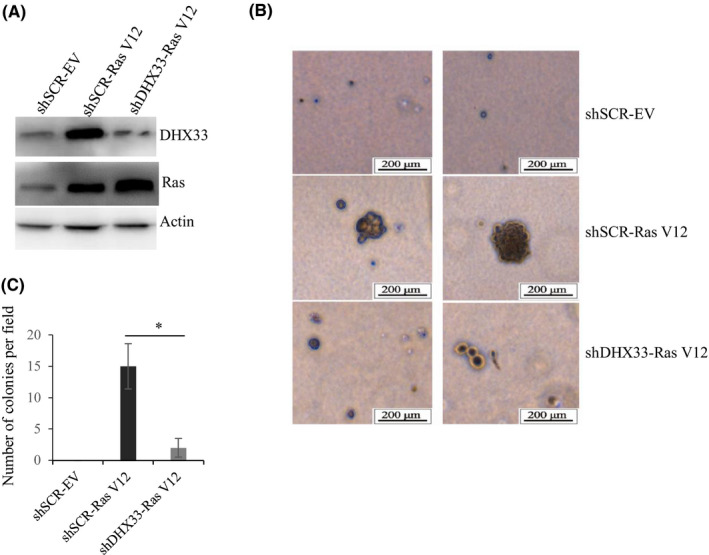
DHX33 knockdown abolished the transformation capacity of RasV12 in vitro. A, Wild‐type MEFs were immortalized by infecting with lentivirus encoding shARF. Cells were then infected with lentivirus encoding either shSCR (scrambled RNA control) or shDHX33. Cells were then further infected by lentivirus encoding RasV12. Western blot analysis was performed to check the expression of indicated proteins. Actin was used as a control. B, Abovementioned cells were then subjected to soft agar analysis; typical images are shown. C, Quantitation was performed to analyze colony numbers for the samples. Typically, five fields were selected to count colony numbers; data represent the average and standard deviation from these counts. *P < .05, n = 3
3.2. Construction of conditional knockout mice for DHX33
For its functional studies in vivo, we generated DHX33 conditional knockout mice. DHX33 gene is located in Chromosome 11 of mouse genome encompassing 12 exons. Exons 2‐4 span a region of approximately 2.3 kb, which encode the pivotal helicase domain important for RNA helicase activity. Several conserved peptide motifs such as GETGSGKT, PRRVAA, TDGM, and DEAH are all in this region. As shown in Figure 2A, a Crispr/Cas9‐based experimental protocol was used to introduce two lox P sites flanking exon 2 to exon 4. Further Southern blot analysis confirmed that no off‐target occurred in the mouse genome. We then crossed these heterozygote mice to generate the homozygous DHX33 lox/lox mice. To confirm that DHX33 can be really deleted, we isolated mouse embryo fibroblasts from these mice and then treated the cells with Ade‐Cre at a MOI of 150. As shown in Figure 2B, we found that upon Cre treatment, DHX33 protein was deleted in a dose‐dependent manner. We further analyzed the mRNA levels of DHX33 and confirmed that the reduction of DHX33 is at the level of mRNA transcription due to gene deletion (Figure 2C).
Figure 2.
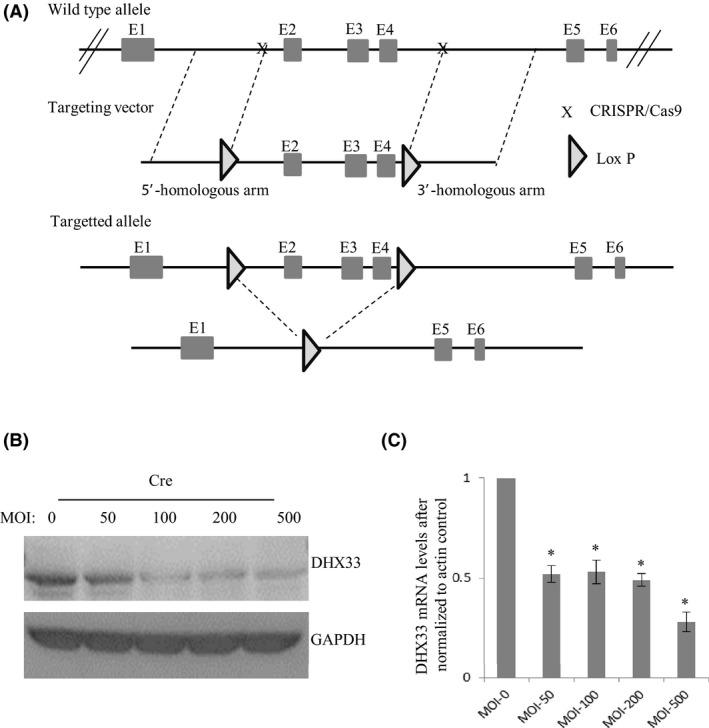
Establishment of DHX33 conditional knockout mice. A, Diagram for the construction of DHX33 conditional knockout. Two lox P sites were introduced into the introns flanking Exon 2‐4. B, Mouse embryo fibroblasts were isolated from DHX33 conditional knockout mice. They were infected by Adenovirus encoding Cre recombinase with different doses. Three days post infection, Western blot was performed to analyze the protein levels of DHX33; GAPDH served as a control. DHX33 protein was decreased. C, quantitative PCR (Q‐PCR) analysis for the abovementioned cells after Cre treatment. DHX33 mRNA was downregulated. *P < .05, n = 3
3.3. DHX33 is induced by activated Ras in vivo
To investigate whether DHX33 is a key gene in Ras‐induced tumorigenesis in vivo, we first analyzed the expression of DHX33 in Ras‐driven lung cancer tissues. Although DHX33 has been found to be induced by Ras V12 in vitro, 13 this has never been established in vivo. As shown in Figure 3, Ras‐driven tumors demonstrated significant upregulation of DHX33 in lung adenoma compared with the wild‐type lung tissues. Ras activation was also analyzed as shown by phosphorylated Akt and phosphorylated ERK, as well as cell proliferation marker Ki‐67, as shown in Figure 3A‐B. All were significantly increased when Ras was activated.
Figure 3.
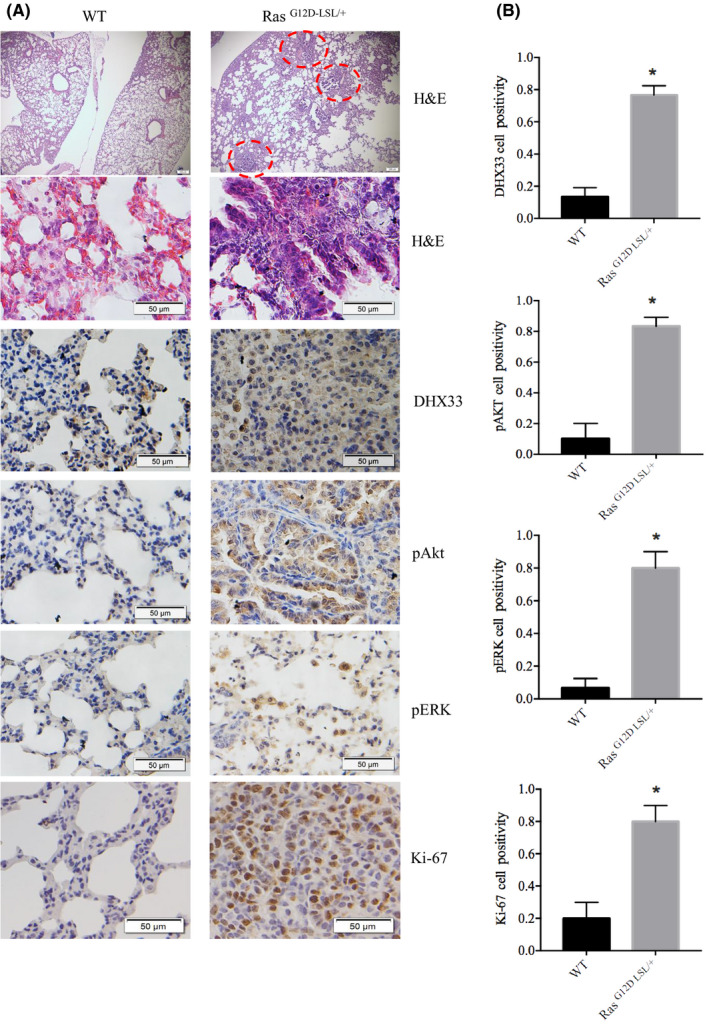
DHX33 is upregulated in Ras‐driven lung cancer development. A, Lung tumor was dissected from RasG12D‐LSL/+ model after Ade‐Cre treatment, with wild‐type lungs as controls. Immunohistochemistry staining is shown for the wild‐type and the RasG12D‐LSL/+ mouse lungs. DHX33 protein expression is elevated when Ras signaling is activated, as demonstrated by increased expression of pERK and pAkt. Ki‐67 serves as a proliferation marker. Tumors have a higher Ki‐67 index. B, Quantitation for the IHC results. *P < .05, n > 3
3.4. Genetic deletion of DHX33 blocks lung tumorigenesis in a Ras‐driven lung cancer model
We then crossed DHX33 lox/lox mice with K‐Ras (G12D‐LSL) strain and generated cohorts of four different genotypes. They are K‐Ras (G12D‐LSL);DHX33 (lox/lox), K‐Ras (G12D‐LSL);DHX33 (lox/+), K‐Ras (G12D‐LSL);DHX33 (+/+), and wild‐type mice. We delivered Ade‐Cre virus through nasal inhalation into these mice when they were 8 weeks old. After virus administration, the mice were sacrificed 2 months later for lung dissection. As these mice were at the same age, we roughly evaluated the development of tumors by the size of lungs. As shown in Figure 4A, all the wild‐type mice exhibited no tumor development (lung weight ~0.17‐0.24 g), while all K‐Ras (G12D‐LSL);DHX33 (+/+) mice demonstrated lung tumor development as shown by the enlarged lungs with multiple visible tumor lesions (lung weight ~0.4‐0.8 g). Five out of 14K‐Ras (G12D‐LSL);DHX33 (lox/+) mice developed lung tumors, too. In contrast, most K‐Ras (G12D‐LSL);DHX33 (lox/lox) mice appeared to have normal lungs (lung weight 0.16‐0.26 g). Ten out of eleven appeared to have normal lungs; no lesions were visible by the looking at the lungs, except one special case with an enlarged lung (lung weight ~0.53). The existence of tumors cannot be solely judged by the appearance and size of lungs. Therefore, the whole lung tissue sections were further analyzed by Hematoxylin staining. The representative images are shown in Figure 4B. Clearly, DHX33 deletion decreased the development of adenomas but did not completely block them. Several small neoplastic regions were still visible in the lung tissues of K‐Ras (G12D‐LSL);DHX33 (lox/lox) mice but with dramatic reduction not only in size but also in number, as compared with the K‐Ras (G12D‐LSL);DHX33 (+/+) group (Figure 4C‐D). We then quantitated the lung weight, the body weight, and the pulmonary ratio for these mice. As shown by Figure 4E‐F, we found that deletion of DHX33 caused a significant inhibition for tumor development statistically.
Figure 4.
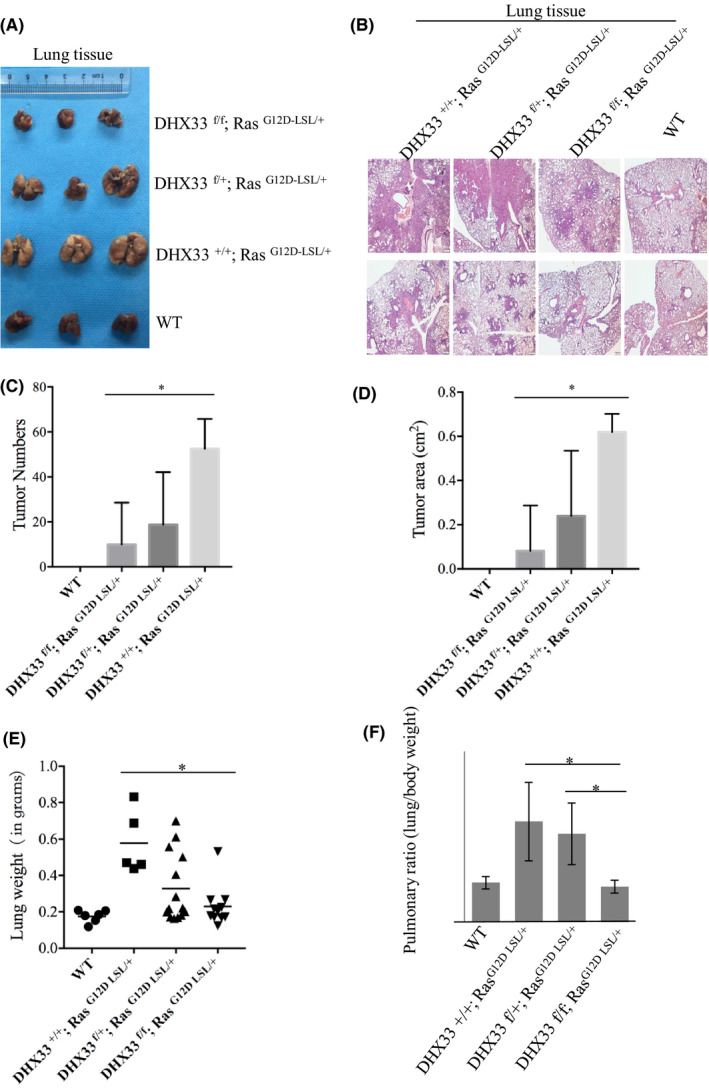
DHX33 deletion markedly decreased lung cancer development. A, Mice with the four designated genotypes were treated with Ade‐Cre at the age of 2 months old. Each group had 5‐15 mice with the same genotypes. Lungs were dissected 2 month later and photographed. B, H&E staining for the whole cross‐section of mouse lungs with different genotypes. C, Quantitation of adenoma numbers in mouse lungs from different groups. *P < .05, n > 5. D, Quantitation of total tumor area per mouse lung from different groups. *P < .05, n > 5. E, Lung weight was monitored and calculated. The quantitation of lung weights is shown for each group. *P < .05, n > 5, statistically significant difference. F, The ratio between lung weight and body weight was calculated for each group. *P < .05, n > 5, statistically significant difference
3.5. DHX33 is pivotal for the expression of Ras‐induced cell‐cycle–promoting genes
To investigate the underlying mechanism for DHX33 in promoting lung tumorigenesis, we first analyzed a few genes involved in cell cycle as they have been found to be regulated by DHX33 previously. 18 When DHX33 was knocked out, although ERK and Akt were still activated, the expression of the previously identified genes downstream of DHX33 was decreased. We chose a few representative genes for analysis, including cdc6, cyclin D1, and cyclin E2 (Figure 5A). The quantitation of the signals from immunohistochemistry is shown in the right panel of Figure 5B. The expression of these target genes was reduced in lung tissues from the K‐Ras (G12D‐LSL);DHX33 (lox/+) group as compared with that of the K‐Ras (G12D‐LSL);DHX33 (+/+) group, indicating a dose‐dependent response.
Figure 5.
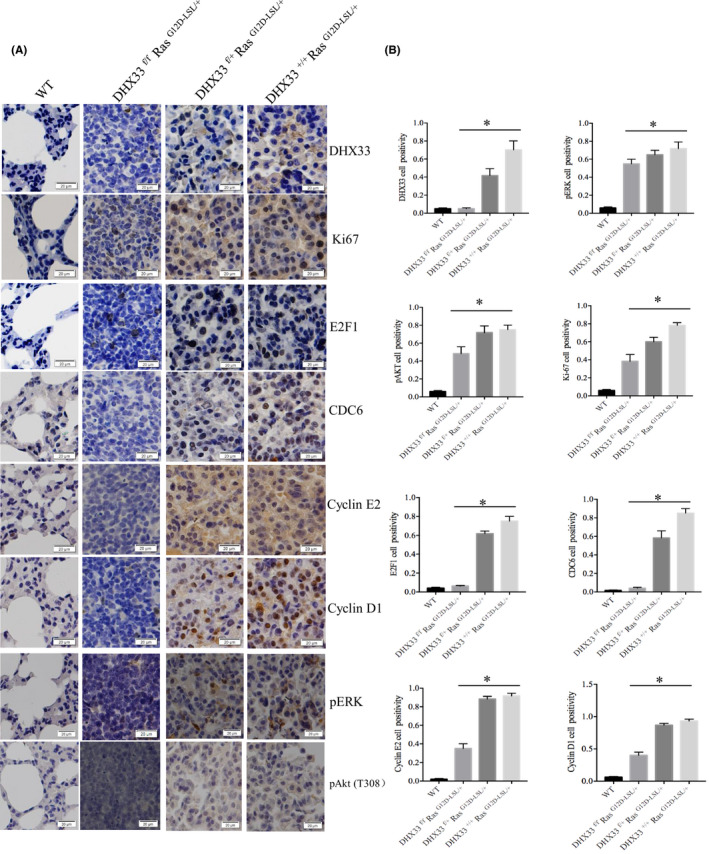
DHX33 deletion caused significant downregulation of proteins involved in cell proliferation. A, Tumor tissues from the four groups were analyzed for the indicated gene expression by immunohistochemistry staining. Representative images were shown with calibration on the bottom of the image. B, Quantitation is shown in the right panel. *P < .05, n > 3, statistically significant difference
3.6. DHX33 influences cancer gene signature in Ras‐driven cancers
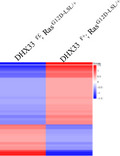
We further analyzed the impact of DHX33 knockout on Ras‐regulated gene network by RNA sequencing. We extracted total RNA from representative lung tissues (for lungs with tumors, we primarily dissect tumor area for RNA extraction) and compared global gene expression changes between the K‐Ras (G12D‐LSL);DHX33 (lox/+) and K‐Ras (G12D‐LSL);DHX33 (lox/lox) groups. Downregulation of DHX33 mRNA level in K‐Ras (G12D‐LSL);DHX33 (lox/lox) mice was confirmed by the RT‐PCR analysis in Figure 6A. As shown in Figure 6B, the global gene expression patterns were compared between these two samples, and dramatic gene expression changes were observed. Further analysis for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways is shown in Figure 6C. Typical pathways involved in cell proliferation, cell adhesion, and cell migration were altered after DHX33 knockout. Pathways favoring cancer development were downregulated after DHX33 deficiency. The FPKM values for representative genes from the RNA‐sequencing analysis are listed in Figure 6D. These genes were categorized based on their functions. Other than known genes involved in cell cycle and migration were found to be downregulated, and genes involved in Wnt signaling and glycolysis were also decreased after DHX33 knockout.
Figure 6.
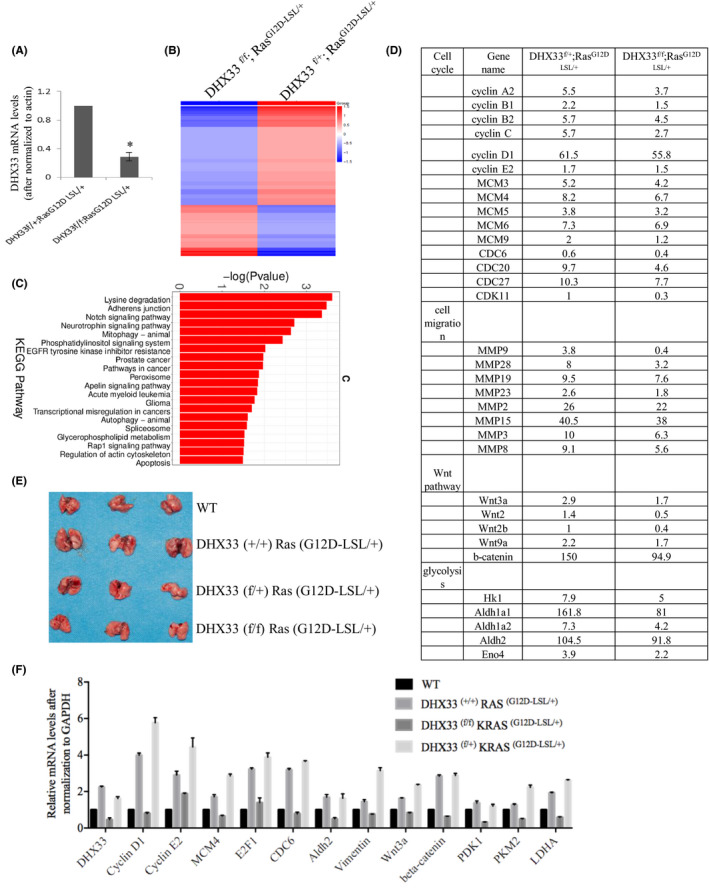
DHX33 knockout caused a global change in the gene expression pattern. A, Total RNA was extracted from lung tissues of the DHX33f/+;RasG12D‐LSL/+ and DHX33f/f;RasG12D‐LSL/+ groups. The mRNA levels of DHX33 were analyzed by RT‐PCR analysis. Actin served as an internal control. B, Heat map of global gene expression levels from RNA‐sequencing analysis of the abovementioned two groups. C, KEGG pathway analysis for expression changes in mouse lung tissues after DHX33 knockdown. Differential gene expression changes are shown between the DHX33f/+;RasG12D‐LSL/+ and DHX33f/f;RasG12D‐LSL/+ groups. Many are involved in cancer development. D, Comparison for selected gene expression levels in FPKM values for the DHX33f/+;RasG12D‐LSL/+ and DHX33f/f;RasG12D‐LSL/+ groups. Typical genes are categorized into cell cycle, migration, Wnt pathway, and glycolysis. E, To validate the expression changes of these genes at an early time point, mice with the abovementioned four designated genotypes were treated with Ade‐Cre to induce tumor formation. Lungs were dissected at an early point, 6 weeks post viral infection, and photographed. F, Total RNA was isolated from each of the lung tissues from the indicated group (Figure 6D). The mRNA levels for the designated genes were evaluated by RT‐PCR analysis. GAPDH served as an internal control. *P < .05, n = 3
In order to validate the results from RNA‐seq analysis, we repeated the tumor induction experiment and harvested lung tissue at an early timepoint, 6 weeks post Ade‐Cre administration (Figure 6E). Notably, we still observed a specific case of mouse with tumor development. Total RNA from the four representative tissues was extracted. RT‐PCR experiments were performed to analyze representative genes involved in cell cycle, cell migration, glycolysis, and Wnt signaling. As shown in Figure 6F, Ras activation normally caused the upregulation of many genes involved in cancer development; however, they were all downregulated after DHX33 was knocked out. To confirm the expression changes for representative genes at the protein level, we performed immunohistochemistry staining as shown in Figure 7A‐B. We found that these genes were all downregulated after DHX33 was knocked out. These genes included lactate dehydrogenase A (LDHA), pyruvate kinase muscle isoform 2 (PKM2), pyruvate dehydrogenase kinase 1 (PDK1), matrix metalloproteinase 9 (MMP9), vimentin, and β‐catenin.
Figure 7.
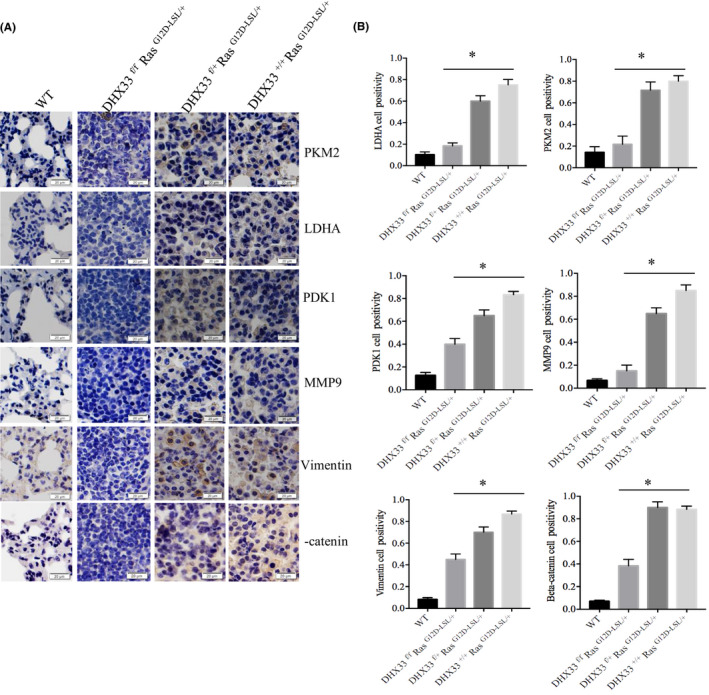
DHX33 knockout caused significant reduction for the protein expression of genes involved in glycolysis, migration, and Wnt signaling. A, Immunohistochemistry staining for lung tissue sections with four different genotypes by the designated antibodies. B, Quantitation data are shown on the right, *P < .05, n > 3
4. DISCUSSION
In this study, we revealed the previously unappreciated function of DHX33 in Ras‐driven lung cancer development. DHX33 is critical to the function of Ras. DHX33 knockdown caused deregulation for thousands of genes important in cancer development. The results in our study validate a highly promising cancer target, RNA helicase DHX33.
Genetic ablation of DHX33 abolished most lung adenocarcinoma development driven by Ras. But in a few cases, we still observed adenoma developed in DHX33 knockout mice, albeit at much lower efficiency and low speed, possibly due to the activation of alternative upstream signaling. It should be worthwhile to identify the molecular mechanism when DHX33 was deleted in this case. These results strengthen the point that Ras‐driven cancer can be highly heterogeneous. Although tumor cells without DHX33 expression proliferated as shown by the increased Ki67 index, these cells expressed much less CDC6, Cyclin E2, and cyclin D1. Importantly, β‐catenin in the Wnt pathway was significantly downregulated by DHX33 knockout, implicating the pivotal role of DHX33 in regulating Wnt signaling. This is worthwhile to be further studied. Our results validated that DHX33 is pivotal to control a subset of genes important during cancer development.
Notably, we discovered that in a rare case, certain mouse can circumvent DHX33 inhibition to develop cancer with high efficiency in the presence of Ras activation, though the chance is low. In this specific case, we observed that certain cell proliferation genes such as cdc6, cyclin D1, and Ki‐67 were highly expressed (data not shown), possibly due to the activation of other upstream signaling. It would be interesting to reveal the molecular mechanism for this resistance during DHX33 loss. These results strengthen the point that other signaling pathways may be activated to bypass the inhibition on DHX33 in Ras‐driven cancer development.
RNA‐sequencing analysis from lung tissues indicated that DHX33 deficiency caused a global change of gene expression, demonstrating the pivotal role of DHX33 in mediating Ras signaling. As these RNAs were extracted from lung tissues with heterogeneous cell origins, we could not exclude the possibility that many of the genes were indirectly regulated by DHX33 or were only regulated by Ras during cancer development. Nonetheless, DHX33 clearly plays a critical and multifaceted role during Ras‐driven lung tumorigenesis.
Recently, RNA‐binding proteins have been found to play important roles in regulating global gene expression through RNA‐based chromatin interaction at hotspots such as gene enhancers and promoters. 26 DHX33 belongs to one of these RNA‐binding proteins. In human lung cancer cell lines, we further confirmed that DHX33 associates with various gene promoters and acts as an important epigenetic regulator to control cancer‐related gene expression. 22 At the transcriptional level, DHX33 knockdown caused dysregulation of approximately 2000‐3000 genes, and epigenetic marks were found to be altered around gene promoters. For a subset of genes that are positively regulated by DHX33, this is believed to be mediated by DNA demethylation proteins such as Gadd45a and Tet enzyme. 22 However, molecular mechanism remains to be revealed for those genes that were negatively influenced by DHX33 at the transcriptional level. Our results highlight the pivotal roles of DHX33 in regulating various aspects of cellular proliferation and migration during cancer development and warrant the therapeutic potential of targeting DHX33 for Ras‐driven lung cancer treatment.
ETHICAL CONSIDERATIONS
All experiments were performed according to the ethical guidelines in China.
DISCLOSURE
We declare that we have no conflict of interest.
ACKNOWLEDGEMENT
Acknowledgement is given to SUSTC core facility. This work was supported by the Science and Technology Innovation Committee of Shenzhen Municipality (CKCY20170721164627271, to Y. Zhang).
Wang X, Feng W, Peng C, et al. Targeting RNA helicase DHX33 blocks Ras‐driven lung tumorigenesis in vivo. Cancer Sci. 2020;111:3564–3575. 10.1111/cas.14601
REFERENCES
- 1. Ahearn IM, Haigis K, Bar‐Sagi D, Philips MR. Regulating the regulator: post‐translational modification of RAS. Nat Rev Mol Cell Biol. 2011;13(1):39‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129(5):865‐877. [DOI] [PubMed] [Google Scholar]
- 3. Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. 2010;1(1):2‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discovery. 2014;13(11):828‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blasco MT, Navas C, Martín‐Serrano G, et al. Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C‐RAF. Cancer Cell. 2019;35(4):573‐87 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wong GS, Zhou J, Bin Liu J, et al. Author Correction: targeting wild‐type KRAS‐amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. 2018;24(10):1627. [DOI] [PubMed] [Google Scholar]
- 7. Wong GS, Zhou J, Liu JB, et al. Targeting wild‐type KRAS‐amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. 2018;24(7):968‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xia Y, Liu Y‐L, Xie Y, et al. A combination therapy for KRAS‐driven lung adenocarcinomas using lipophilic bisphosphonates and rapamycin. Sci Transl Med. 2014;6(263):263ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malone CF, Emerson C, Ingraham R, et al. mTOR and HDAC inhibitors converge on the TXNIP/Thioredoxin pathway to cause catastrophic oxidative stress and regression of RAS‐driven tumors. Cancer Discov. 2017;7(12):1450‐1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J, Hu K, Guo J, et al. Suppression of KRas‐mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat Commun. 2016;7:11363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stalnecker CA, Der CJRAS. wanted dead or alive: advances in targeting RAS mutant cancers. Sci Signal. 2020;13(624):eaay6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y, Saporita AJ, Weber JD. P19ARF and RasV(1)(2) offer opposing regulation of DHX33 translation to dictate tumor cell fate. Mol Cell Biol. 2013;33(8):1594‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y, Forys JT, Miceli AP, Gwinn AS, Weber JD. Identification of DHX33 as a mediator of rRNA synthesis and cell growth. Mol Cell Biol. 2011;31(23):4676‐4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mitoma H, Hanabuchi S, Kim T, et al. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity. 2013;39(1):123‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tian Q‐H, Zhang M‐F, Luo R‐G, et al. DHX33 expression is increased in hepatocellular carcinoma and indicates poor prognosis. Biochem Biophys Res Comm. 2016;473(4):1163‐1169. [DOI] [PubMed] [Google Scholar]
- 17. Wang H, Yu J, Wang X, Zhang Y. The RNA helicase DHX33 is required for cancer cell proliferation in human glioblastoma and confers resistance to PI3K/mTOR inhibition. Cell Signal. 2019;54:170‐178. [DOI] [PubMed] [Google Scholar]
- 18. Yuan B, Wang X, Fan C, et al. DHX33 transcriptionally controls genes involved in the cell cycle. Mol Cell Biol. 2016;36(23):2903‐2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu J, Liu Y, Wang X, Yuan B, Zhang Y. Role of DHX33 in c‐Myc‐induced cancers. Carcinogenesis. 2017;38(6):649‐660. [DOI] [PubMed] [Google Scholar]
- 20. Zhang Y, You J, Wang X, Weber J. The DHX33 RNA helicase promotes mRNA translation initiation. Mol Cell Biol. 2015;35(17):2918‐2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J, Feng W, Yuan Z, Weber JD, Zhang Y. DHX33 interacts with AP‐2beta to regulate Bcl‐2 gene expression and promote cancer cell survival. Mol Cell Biol. 2019;39(17):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feng W, Chen S, Wang J, et al. DHX33 recruits Gadd45a to cause DNA demethylation and regulate a subset of gene transcription. Mol Cell Biol. 2020;40(13):1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4(7):1064‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91(5):649‐659. [DOI] [PubMed] [Google Scholar]
- 25. Lundberg AS, Hahn WC, Gupta P, Weinberg RA. Genes involved in senescence and immortalization. Curr Opin Cell Biol. 2000;12(6):705‐709. [DOI] [PubMed] [Google Scholar]
- 26. Xiao R, Chen J‐Y, Liang Z, et al. Pervasive chromatin‐RNA binding protein interactions enable RNA‐based regulation of transcription. Cell. 2019;178(1):107‐21.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]