

Abstract
Precision medicine is a promising strategy for cancer treatment. In this study, we developed an in‐house clinical sequencing system to perform a comprehensive cancer genomic profiling test as a clinical examination and analyzed the utility of this system. Genomic DNA was extracted from tumor tissues and peripheral blood cells collected from 161 patients with different stages and types of cancer. A comprehensive targeted amplicon exome sequencing for 160 cancer‐related genes was performed using next‐generation sequencing (NGS). The sequencing data were analyzed using an original bioinformatics pipeline, and multiple cancer‐specific gene alterations were identified. The success rate of our test was 99% (160/161), while re‐biopsy was required for 24% (39/161) of the cases. Potentially actionable and actionable gene alterations were detected in 91% (145/160) and 46% (73/160) of the patients, respectively. The actionable gene alterations were frequently detected in PIK3CA (9%), ERBB2 (8%), and EGFR (4%). High tumor mutation burden (TMB) (≥10 mut/Mb) was observed in 12% (19/160) of the patients. The secondary findings in germline variants considered to be associated with hereditary tumors were detected in 9% (15/160) of the patients. Seventeen patients (11%, 17/160) were treated with genotype‐matched therapeutic agents, and the response rate was 47% (8/17). The median turnaround time for physicians was 20 days, and the median survival time after the initial visit was 8.7 months. The results of the present study prove the feasibility of implementing in‐house clinical sequencing as a promising laboratory examination technique for precision cancer medicine.
Keywords: actionable gene alteration, clinical sequencing, genomic testing, genotype‐matched treatment, precision medicine
In‐house clinical sequencing system is a promising laboratory examination for precision cancer medicine.

Abbreviations
- ACMG
American College of Medical Genetics and Genomics
- CLHURC
Clinical Sequencing System in Hokkaido University Hospital for Cancer Individualized Medicine
- CNAs
copy number alterations
- DIN
DNA integrity number
- FDA
Food and Drug Administration
- ECOG
Eastern Cooperative Oncology Group
- FFPE
formalin‐fixed paraffin‐embedded
- ISO
International Organization for Standardization
- NGS
next‐generation sequencing
- NSCLC
non‐small cell lung cancer
- PBMCs
peripheral blood mononuclear cells
- PD‐1
programmed cell death 1
- SNVs
single nucleotide variants
- TMB
tumor mutation burden
1. INTRODUCTION
Implementation of genomic sequencing assays for clinical cancer analyses is a recent development in the field of cancer treatment. 1 , 2 The wider availability of next‐generation sequencing (NGS) has promoted the analysis of genomic biomarkers in recent years. Precision medicine based on genomic biomarkers has especially advanced in the treatment of lung adenocarcinoma. 3 Genomic testing for EGFR mutations, 4 ALK fusions, 5 ROS1 fusions, 6 and BRAF mutations 7 is universally utilized as a laboratory examination, and the molecular targeted therapies against these gene alterations are accepted as one of the standard treatments for patients with lung cancer. Moreover, microsatellite instability (MSI)‐high or deficient mismatch repair (dMMR) for programmed cell death 1 (PD‐1) inhibitor 8 and NTRK fusions for TRK inhibitor 9 , 10 are utilized as tumor‐agnostic genomic biomarkers.
While traditional anticancer drugs act on all rapidly dividing normal and cancerous cells, new approaches have shifted to gene alteration strategies in pursuit of potential therapeutic targets with enhanced specificity. Therefore, it is essential to undertake genotype‐matched clinical trials, such as basket and umbrella trials, to confirm the utility of precision cancer medicine. 11 , 12 , 13 Some large basket trials aimed at studying different cancer types, like TAPUR (Targeted Agent and Profiling Utilization Registry) trial (NCT02693535) and NCI‐MATCH (National Cancer Institute‐Molecular Analysis for Therapy Choice) trial (NCT02465060), are currently in progress and are expected to result in genotype‐matched treatment strategies. 14 , 15
Alternatively, comprehensive cancer genomic profiling service providers, such as MSK‐IMPACT 16 , 17 and FoundationOne, 18 have innovated routine laboratory investigation techniques for a variety of biological samples in clinical settings to assist everyday cancer care for different types of cancer. In the present study, we developed a novel, in‐house clinical sequencing system called “Clinical Sequencing System in Hokkaido University Hospital for Cancer Individualized Medicine (CLHURC)” and launched an outpatient department from April 2016 that is specialized in clinical sequencing for patients with cancer. Our division implements precision medicine in clinical practice and explores suitable individualized treatment strategies, including referral enrollment for appropriate clinical trials.
Sequencing systems for molecular characterization of cancers have been increasingly developed in recent years; however, the clinical utility of in‐house comprehensive cancer genomic profiling systems for patients with cancer, as a part of medical services, has never been reported in detail. Therefore, in this study, we report the utility of our in‐house clinical sequencing system as a potential tool for precision cancer medicine in clinical practice.
2. MATERIALS AND METHODS
2.1. Clinical Sequencing System in Hokkaido University Hospital for Cancer Individualized Medicine (CLHURC)
In April 2016, we launched an outpatient division specialized for sequencing samples obtained from cancer patients. Since then, we have started operating an original in‐house clinical sequencing system called CLHURC. All working processes of genome sequencing were completed in our hospital. We received tumor specimens from patients with different types of cancer, who agreed to undergo a comprehensive cancer genomic profiling test, with mainly formalin‐fixed paraffin‐embedded (FFPE) blocks or 10‐20 pieces of undyed 5 μm pathology specimens. In re‐biopsy cases, we collected specimens that were fixed using the PAXgene Tissue System (Qiagen) that is intended for high quality nucleic acid purification, to achieve high success rates in sequencing. In general, the quality of nucleic acids extracted from PAXgene‐fixed paraffin‐embedded specimens is better than that of nucleic acids extracted from FFPE specimens. 19
In our system, genomic DNA was extracted and purified from tumor tissues as well as peripheral blood mononuclear cells (PBMCs) obtained from patients. The minimum amount of DNA extracted was 50 ng. We checked the quality of DNA based on the DNA integrity number (DIN) score, which was calculated using the Agilent 2000 TapeStation (Agilent Technologies), and DNA libraries were prepared for genome sequencing if the quality of DNA had a DIN score above 3.1. Subsequently, we performed a targeted amplicon exome sequencing for 160 cancer‐related genes using the Illumina MiSeq sequencing platform (Illumina) (Table S1). All the above processes were performed in an Internatinal Organization for Standardization (ISO) 15189‐certified laboratory at our hospital.
2.2. Bioinformatics analysis
Genome annotation and curation for analyzing the sequencing data were performed using an original bioinformatics pipeline called GenomeJack (Mitsubishi Space Software) (http://genomejack.net/english/index.html) within three working days. In the GenomeJack pipeline, mapping of the NGS reads to the human reference genome (UCSC human genome 19) was performed using Burrows‐Wheeler Aligner, 20 and the reads were realigned with ABRA. 21 For identification of single nucleotide variants (SNVs), SAMtools 22 was used to pile up the sequencing reads and defective SNVs showing conflict between pairwise reads were abandoned. The criteria for calling mutations are as follows. We determined the noise distributions arising from random sequencing errors. Each mutation was evaluated by binomial test (P < 0.05) to reduce random sequencing errors. We called somatic mutations by comparing the number of mismatch bases in tumor with normal control using Fisher’s exact test (P < 0.001). Possible germline SNPs were excluded using Single Nucleotide Polymorphism Database (dbSNP) (https://www.ncbi.nlm.nih.gov/snp/), the ExAC database (https://gnomad.broadinstitute.org/), Human Genetic Variation Database (HGVD) (http://www.hgvd.genome.med.kyoto‐u.ac.jp/), and the ToMMo 2KJPN database (https://jmorp.megabank.tohoku.ac.jp/ijgvd/). The copy number of each gene was calculated as the median value of all the sequencing reads covering the target genes and compared with the median value of control samples. In calling copy number alterations (CNAs), we defined more than three‐fold copy number increases as “gain” and less than two‐fold decreases as “loss.” We identified cancer‐specific somatic gene alterations, such as SNVs, insertions/deletions (Indels), and CNAs. Moreover, tumor mutation burden (TMB) was measured as a potential biomarker for immunotherapy. In our test, TMB was defined as the number of nonsynonymous and synonymous mutations in the target regions per megabase of tumor genome (the total size of targeted region in our test was 0.74 Mb), and high TMB was defined as at least 10 mutations per megabase (≥10 mut/Mb). The TMB values measured by our system showed a strong correlation (R 2 = 0.92) with those assessed by whole exome sequencing in the comparative test using 46 trial samples. We chose 10 mut/Mb as a threshold of high TMB, which has been recently proposed by reports about the association between TMB and the efficacy of immune checkpoint inhibitors. 23 All the detected gene alterations in 160 cancer‐related genes were annotated and curated using the COSMIC database (https://cancer.sanger.ac.uk/cosmic), the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), the CIViC database (https://civicdb.org/home), SnpEff, 24 and the Clinical Knowledgebase (CKB) database (https://ckb.jax.org/).
2.3. Definition of actionable gene alteration
We added a level of evidence to each detected gene alteration based on procedures outlined in the following: ‘‘A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists’’ 25 and ‘‘Clinical Practice Guidance for Next‐generation Sequencing in Cancer Diagnosis and Treatment (Edition 1.0).’’ 26
In ‘‘A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists,’’ it cites the following levels of evidence for each gene alteration: Level A, biomarkers that predict response or resistance to US Food and Drug Administration (FDA)‐approved therapies for a specific type of tumor or have been included in professional guidelines as therapeutic, diagnostic, and/or prognostic biomarkers for specific types of tumors; Level B, biomarkers that predict response or resistance to a therapy based on well‐powered studies with consensus from experts in the field, or have diagnostic and/or prognostic significance of certain diseases based on well‐powered studies with expert consensus; Level C, biomarkers that predict response or resistance to therapies approved by the FDA or professional societies for a different tumor type (ie, off‐label use of a drug), serve as inclusion criteria for clinical trials, or have diagnostic and/or prognostic significance based on the results of multiple small studies; and Level D, biomarkers that show plausible therapeutic significance based on preclinical studies, or may assist disease diagnosis and/or prognosis themselves or along with other biomarkers based on small studies or multiple case reports with no consensus.
In ‘‘Clinical Practice Guidance for Next‐generation Sequencing in Cancer Diagnosis and Treatment (Edition 1.0),’’ it cites the following levels of evidence for each gene alteration: Level 1A, biomarker (gene mutation) that is approved by regulatory authority as a companion diagnostic for the said cancer type; Level 1B, biomarker (gene mutation) that is approved by the FDA as a companion diagnostic (or complementary diagnostic) for the said cancer type, or biomarker (gene abnormality), with which consistent results have been obtained to support clinical usefulness of an anticancer agent in a prospective clinical study with biomarker‐based patient selection, or in meta‐analysis data for the specific cancer; Level 2A, biomarker (gene abnormality), with which results have been obtained to support clinical usefulness of an anticancer agent in subgroup analysis of a prospective clinical study for the said cancer type; Level 2B, biomarker (gene abnormality) approved by regulatory authority for other cancer type(s), or with which results have been obtained to support clinical usefulness of an anticancer agent; Level 3A, biomarker (gene abnormality), for which a correlation with clinical usefulness of an anticancer agent has been reported in scientific knowledge‐based case reports, etc.; Level 3B, biomarker (gene abnormality), with which a correlation with therapeutic efficacy of an anticancer agent has been reported by pharmacodynamic evaluation in vitro and in vivo; and Level 4, gene abnormality that is known to be involved in cancer.
We defined gene alterations that had evidence levels A–C in ‘‘A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists’’ or evidence levels 1A–3A in ‘‘Clinical Practice Guidance for Next‐generation Sequencing in Cancer Diagnosis and Treatment (Edition 1.0)’’ as actionable gene alterations. Furthermore, we defined gene alterations that had evidence levels A–D in the former or evidence levels 1A–3B in the latter as potentially actionable gene alterations.
2.4. Panel of genome experts (expert panel)
The analysis reports were discussed and reviewed by a panel of genome experts including medical oncologists, molecular oncologists, pathologists, medical geneticists, clinical laboratory technicians, bioinformaticians, genetic counselors, pharmacists, and nurses. The final judgement on actionable gene alterations was made by a panel of genome experts. The final report, including information regarding the recommended treatments based on genomic profiling, was confirmed after approval of the panel, after which the report was returned to physicians and patients. The expected turnaround time of this system was approximately 14 days.
2.5. Patients
In this system, we included patients who were diagnosed with any type of cancer and who had an Eastern Cooperative Oncology Group (ECOG) performance status score of 0‐2, regardless of age, staging, or treatment history. We targeted the outpatients who visited our division between April 2016 and April 2017. Written informed consent was obtained from all patients regarding the use of genomic and clinical data for research. The study protocol was approved by the ethics committee of Hokkaido University Hospital (016‐0260).
2.6. Reporting secondary findings from germline variants
In our system, secondary findings in germline variants could be identified by comparing the genomic profile of tumor tissues with that of PBMCs. Therefore, we instituted a policy based on patients’ consent to disclose secondary findings revealed by comprehensive cancer genomic profiling test. If the patients agreed to receive the germline information, such information was disclosed only if the detected germline variants were confirmed as pathogenic or likely pathogenic by the global cancer genome database, such as ClinVar, and were also listed in the recommendations of the American College of Medical Genetics and Genomics (ACMG) for reporting incidental findings in clinical exome and genome sequencing. 27
2.7. Statistical analyses
Survival analyses for estimating the median survival time was calculated using the Kaplan‐Meier method considering the time‐to‐event period, ie, from the time of initial visit to death. All statistical analyses were undertaken using the IBM SPSS Statistics software (version 21.0 for Windows; IBM).
3. RESULTS
3.1. Patient characteristics
Between April 2016 and April 2017, 169 patients with different types of cancer enquired for comprehensive genome sequencing at our division. Of these patients, 161 applied for a comprehensive cancer genomic profiling test. The characteristics of 161 cancer patients are summarized in Table 1. Most of the patients had an ECOG performance status score of 0 or 1, which was an ideal indicator for undergoing the comprehensive cancer genomic profiling test that would help to decide their future course of treatment. Although the majority of patients had unresectable tumors, a small number of patients presented with cancer in the early stage. The top five primary sites of cancer were colorectal (18%), pancreas (13%), breast (10%), stomach (7%), and non‐small cell lung cancer (NSCLC) (6%) (Table 2). Moreover, most patients had received chemotherapy for their cancer and many patients had one or more standard chemotherapy sessions pending at the time of their initial visit. Upon enquiring each patient’s desire to know secondary findings at the time of the initial visit, only one‐third of the patients indicated that they wanted to know about secondary findings from germline information.
TABLE 1.
Characteristics of 161 patients recruited in the study
| Characteristics | Patients (N = 161) |
|---|---|
| Age, years | |
| Median (range) | 65 (2‐85) |
| Sex, n (%) | |
| Male | 83 (52) |
| Female | 78 (48) |
| ECOG performance status, n (%) | |
| 0 | 106 (66) |
| 1 | 34 (21) |
| 2 | 19 (12) |
| 3 | 2 (1) |
| UICC stage, n (%) | |
| Non‐stage IV or recurrence | 20 (12) |
| Stage IV or recurrence | 141 (88) |
| Prior treatment, n (%) | |
| Yes | 137 (85) |
| No | 24 (15) |
| Unadministered standard chemotherapy, n (%) | |
| Yes | 118 (73) |
| No | 43 (27) |
UICC (Union for International Cancer Control) TNM Classification of Malignant Tumors, 8th Edition.
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
TABLE 2.
Tumor types among 161 patients recruited in the study. (A) Primary sites. (B) Detail of the other sites of tumors
| (A) | |
|---|---|
| Primary sites | Patients (N = 161), n (%) |
| Colorectal | 29 (18) |
| Pancreas | 21 (13) |
| Breast | 16 (10) |
| Stomach | 11 (7) |
| NSCLC | 10 (6) |
| Endometrial | 9 (6) |
| Biliary | 9 (6) |
| Ovary | 8 (5) |
| Head and neck | 6 (4) |
| Esophagus | 6 (4) |
| Others | 36 (22) |
| (B) | |
|---|---|
| Primary sites | Patients (n = 36) |
| Urothelial | 4 |
| Neuroendocrine carcinoma | 4 |
| Kidney | 3 |
| Sarcoma | 3 |
| Small intestine | 3 |
| Liver | 2 |
| Acute lymphocytic leukemia | 1 |
| Cervix | 1 |
| Inflammatory myofibroblastic tumor | 1 |
| Intraductal papillary neoplasm of the bile duct | 1 |
| Mantle cell lymphoma | 1 |
| Mesothelioma | 1 |
| Mixed acinar‐neuroendocrine carcinoma | 1 |
| Peritoneal | 1 |
| Polycythemia vera | 1 |
| Prostate | 1 |
| Seminoma | 1 |
| Skin | 1 |
| Solid pseudopapillary neoplasm | 1 |
| Solitary fibrous tumor | 1 |
| Thymoma | 1 |
| Unknown primary | 1 |
| Vagina | 1 |
Abbreviation: NSCLC, non‐small cell lung cancer.
3.2. Results of genome sequencing
In our sequencing system, the quality and quantity of DNA were checked before the library was constructed for genome sequencing. Re‐biopsy was required for patients whose samples yielded insufficient or poor‐quality DNA. Although re‐biopsy was required for 24% (39/161) of the patients, the comprehensive cancer genomic profiling test was carried out successfully in almost all patients and was complemented by re‐biopsy. We sequenced genomic DNA from 160 (99%, 160/161) patients with different types of cancer, with a mean sequencing depth of 860× (range, 518‐3250×). The mean tumor cellularity of the samples, calculated by their pathological review, was 46% (range, 5%‐90%; Figures S1 and S2, Tables S2‐S5).
Potentially actionable gene alterations were detected in 91% (145/160) of the patients, and they were frequently detected in TP53 (55%, 88/160), KRAS (29%, 46/160), APC (16%, 26/160), and BRCA2 (12%, 20/160; Table 3). On the contrary, actionable gene alterations were detected in 46% (73/160) of the patients, and they were frequently detected in PIK3CA (9%, 15/160), ERBB2 (8%, 12/160), and EGFR (4%, 7/160) (Table 4, Tables S6‐S8). The median TMB calculated from our pipeline was 5.4 mut/Mb (range, 1.3‐120.8 mut/Mb). In our cohort, 11% (19/160) of the patients were TMB‐high (≥10 mut/Mb) (Figure 1, Table 5). The distribution of the maximum evidence level for gene alterations detected in each patient is shown in Table 6.
TABLE 3.
Potentially actionable gene alterations found in 160 patients who underwent cancer genomic profiling test. (A) Potentially actionable gene mutations. (B) Potentially actionable copy number alterations. (C) Potentially actionable gene alterations (gene mutations + copy number alterations)
| (A) | |
|---|---|
| Top 10 genes | Patients (n = 160), n (%) |
| TP53 | 86 (53) |
| KRAS | 41 (26) |
| APC | 37 (23) |
| SMAD4 | 11 (7) |
| FBXW7 | 10 (6) |
| ARID1A | 9 (6) |
| BRAF | 7 (4) |
| PIK3CA | 7 (4) |
| PTEN | 7 (4) |
| BRCA2 | 6 (4) |
| (B) | ||
|---|---|---|
| Top 14 genes | Copy numbers | Patients (n = 160), n (%) |
| BRCA2 | Loss | 15 (9) |
| MYC | Amplification | 14 (9) |
| MLH1 | Loss | 12 (8) |
| ATM | Loss | 11 (7) |
| ERBB2 | Amplification | 10 (6) |
| TP53 | Loss | 10 (6) |
| BRCA1 | Loss | 8 (5) |
| FBXW7 | Loss | 8 (5) |
| PIK3CA | Amplification | 8 (5) |
| ARID1A | Loss | 7 (4) |
| CHEK2 | Loss | 7 (4) |
| FLT3 | Amplification | 7 (4) |
| JAK2 | Amplification | 7 (4) |
| KRAS | Amplification | 7 (4) |
| (C) | |
|---|---|
| Top 10 genes | Patients (n = 160), n (%) |
| TP53 | 88 (55) |
| KRAS | 46 (29) |
| APC | 26 (16) |
| BRCA2 | 20 (12) |
| ATM | 16 (10) |
| FBXW7 | 16 (10) |
| ARID1A | 15 (9) |
| PIK3CA | 15 (9) |
| MYC | 14 (9) |
| BRCA1 | 13 (8) |
TABLE 4.
Actionable gene alterations found in 160 patients who underwent cancer genomic profiling test. (A) Actionable gene mutations. (B) Actionable copy number alterations. (C) Actionable gene alterations (gene mutations + copy number alterations)
| (A) | ||
|---|---|---|
| Top 11 genes | Agents | Patients (n = 160), n (%) |
| PIK3CA | PI3K/AKT/MTOR inhibitor | 7 (4) |
| ATM | PARP inhibitor, platinum | 5 (3) |
| BRAF | BRAF inhibitor | 5 (3) |
| BRCA1 | PARP inhibitor, platinum | 5 (3) |
| BRCA2 | PARP inhibitor, platinum | 5 (3) |
| KRAS G12C | KRAS G12C inhibitor | 5 (3) |
| AKT1 | AKT/MTOR inhibitor | 3 (2) |
| EGFR | EGFR inhibitor | 3 (2) |
| ERBB2 | HER2 inhibitor | 2 (1) |
| ESR1 | Estrogen blocker | 2 (1) |
| PBRM1 | PD‐1/PD‐L1 inhibitor | 2 (1) |
| (B) | |||
|---|---|---|---|
| Top six genes | Copy numbers | Agents | Patients (n = 160), n (%) |
| BRCA2 | Loss | PARP inhibitor, platinum | 10 (6) |
| PIK3CA | Amplification | PI3K/AKT/MTOR inhibitor | 8 (5) |
| CDK4 | Amplification | CDK4/6 inhibitor | 4 (3) |
| EGFR | Amplification | EGFR inhibitor | 4 (3) |
| MDM2 | Amplification | MDM2 inhibitor | 2 (1) |
| MET | Amplification | MET inhibitor | 1 (1) |
| (C) | ||
|---|---|---|
| Top 13 genes | Agents | Patients (n = 160), n (%) |
| PIK3CA | PI3K/AKT/MTOR inhibitor | 15 (9) |
| ERBB2 | HER2 inhibitor | 12 (8) |
| EGFR | EGFR inhibitor | 7 (4) |
| ATM | PARP inhibitor, platinum | 5 (3) |
| BRAF | BRAF inhibitor | 5 (3) |
| BRCA1 | PARP inhibitor, platinum | 5 (3) |
| BRCA2 | PARP inhibitor, platinum | 5 (3) |
| KRAS G12C | KRAS G12C inhibitor | 5 (3) |
| CDK4 | CDK4/6 inhibitor | 4 (3) |
| AKT1 | AKT/MTOR inhibitor | 3 (2) |
| ESR1 | Estrogen blocker | 2 (1) |
| MDM2 | MDM2 inhibitor | 2 (1) |
| PBRM1 | PD‐1/PD‐L1 inhibitor | 2 (1) |
FIGURE 1.
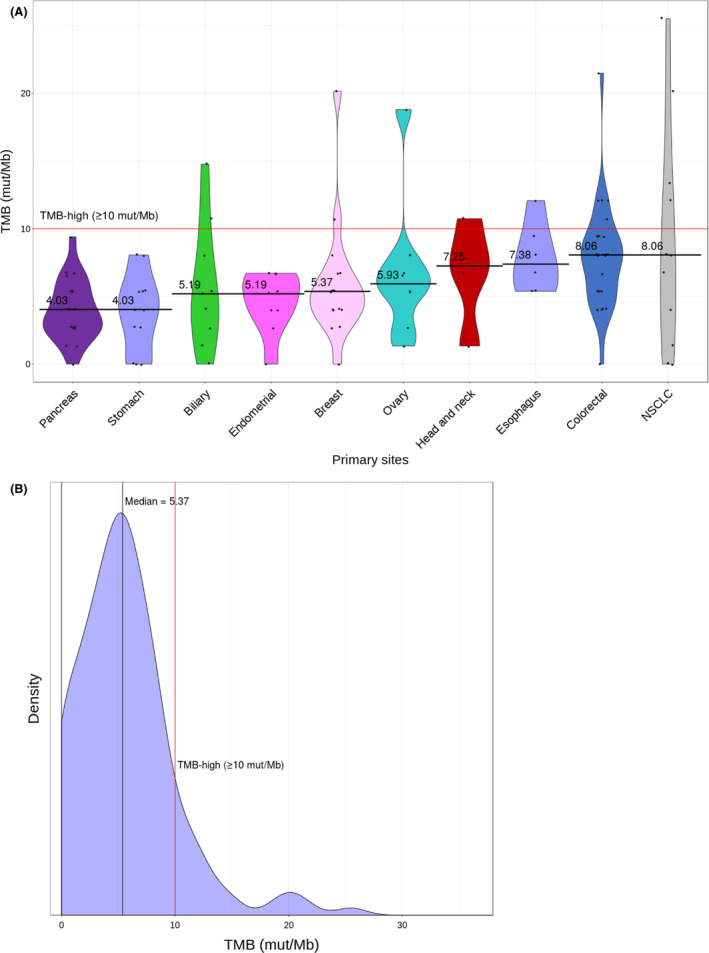
Tumor mutation burden (TMB) profile of 160 cancer patients. A, TMB spectrum across tumor types. Violin plots show the distribution of TMB for the top 10 tumor types. Tumor types are arranged from right to left with decreasing median TMB. The width of each plot indicates the frequency of cases with a given TMB. The red line indicates the threshold for cases with TMB‐high status (10 mutations [mut]/Mb). B, Distribution of TMB observed across all patients. NSCLC, non‐small cell lung cancer.
TABLE 5.
Patients with high tumor mutation burden (TMB; ≥10 mut/Mb)
| Patients | Primary sites | Smoking history (Brinkman index) | TMB (mut/Mb) |
|---|---|---|---|
| CLHURC‐148 | Neuroendocrine carcinoma | 0 | 120.8 |
| CLHURC‐093 | NSCLC | 1840 | 25.5 |
| CLHURC‐004 | Colorectal | 0 | 21.5 |
| CLHURC‐133 | NSCLC | 380 | 20.1 |
| CLHURC‐136 | Breast | 0 | 20.1 |
| CLHURC‐059 | Ovary | 0 | 18.8 |
| CLHURC‐138 | Biliary | 0 | 14.8 |
| CLHURC‐151 | Small intestine | 0 | 14.8 |
| CLHURC‐058 | NSCLC | 3060 | 13.4 |
| CLHURC‐073 | Colorectal | 0 | 12.1 |
| CLHURC‐095 | Colorectal | 440 | 12.1 |
| CLHURC‐104 | Colorectal | 800 | 12.1 |
| CLHURC‐130 | Esophagus | 1800 | 12.1 |
| CLHURC‐154 | NSCLC | 940 | 12.1 |
| CLHURC‐042 | Head and neck | 1260 | 10.7 |
| CLHURC‐043 | Neuroendocrine carcinoma | 430 | 10.7 |
| CLHURC‐074 | Breast | 100 | 10.7 |
| CLHURC‐076 | Biliary | 0 | 10.7 |
| CLHURC‐113 | Colorectal | 1750 | 10.7 |
Abbreviation: NSCLC, non‐small cell lung cancer.
TABLE 6.
Distribution of the maximum evidence level for gene alterations detected in each patient. (A) Evidence levels according to ‘‘A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists’’. (B) Evidence levels according to ‘‘Clinical Practice Guidance for Next‐generation Sequencing in Cancer Diagnosis and Treatment (Edition 1.0)’’
| (A) | ||
|---|---|---|
| Evidence levels | Evidence level classifications | Patients (n = 160), n (%) |
| A | Biomarkers that predict response or resistance to US FDA‐approved therapies for a specific type of tumor or have been included in professional guidelines as therapeutic, diagnostic, and/or prognostic biomarkers for specific types of tumors | 13 (8) |
| B | Biomarkers that predict response or resistance to a therapy based on well‐powered studies with consensus from experts in the field, or have diagnostic and/or prognostic significance of certain diseases based on well‐powered studies with expert consensus | 13 (8) |
| C | Biomarkers that predict response or resistance to therapies approved by FDA or professional societies for a different tumor type (ie, off‐label use of a drug), serve as inclusion criteria for clinical trials, or have diagnostic and/or prognostic significance based on the results of multiple small studies | 47 (29) |
| D | Biomarkers that show plausible therapeutic significance based on preclinical studies, or may assist disease diagnosis and/or prognosis themselves or along with other biomarkers based on small studies or multiple case reports with no consensus | 72 (45) |
| None | None of the above | 15 (9) |
| (B) | ||
|---|---|---|
| Evidence levels | Evidence level classifications | Patients (n = 160), n (%) |
| 1A | Biomarker (gene mutation) that is approved by regulatory authority as a companion diagnostic for the said cancer type | 7 (4) |
| 1B | Biomarker (gene mutation) that is approved by FDA as a companion diagnostic (or complementary diagnostic) for the said cancer type, or biomarker (gene abnormality), with which consistent results have been obtained to support clinical usefulness of an anticancer agent in a prospective clinical study with biomarker‐based patient selection, or in meta‐analysis data for the specific cancer | 6 (4) |
| 2A | Biomarker (gene abnormality), with which results have been obtained to support clinical usefulness of an anticancer agent in subgroup analysis of a prospective clinical study for the said cancer type | 13 (8) |
| 2B | Biomarker (gene abnormality) approved by regulatory authority for other cancer type(s), or with which results have been obtained to support clinical usefulness of an anticancer agent | 14 (9) |
| 3A | Biomarker (gene abnormality), for which a correlation with clinical usefulness of an anticancer agent has been reported in scientific knowledge‐based case reports, etc | 33 (21) |
| 3B | Biomarker (gene abnormality), with which a correlation with therapeutic efficacy of an anticancer agent has been reported by pharmacodynamic evaluation in vitro and in vivo | 45 (28) |
| 4 | Gene abnormality that is known to be involved in cancer | 27 (17) |
| None | None of the above | 15 (9) |
Abbreviation: FDA, Food and Drug Administration.
On the basis of genomic information, 18% (28/160) of the patients received some kind of treatment after genome sequencing (Table S9). Most importantly, only 11% (17/160) of the patients received genotype‐matched treatments based on the actionable gene alterations. This indicates that approximately three‐quarters of patients who had tumors with actionable gene alterations could not eventually undergo genotype‐matched treatments, mainly because of the deterioration in their physical condition and the inaccessibility of off‐label drugs. Detailed information regarding genotype‐matched treatments is shown in Table 7. Tumor assessment related to the best overall response rate was analyzed using the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Accordingly, the response and disease control rate of the genotype‐matched treatments in our cohort were 47% (8/17) and 76% (13/17), respectively. The median turnaround time from initial visit for physicians and patients was 20 days (range, 9‐65 days) and 28 days (range, 10‐78 days), respectively. Moreover, the median survival time from the time of initial visit to death (post‐sequencing survival time) was 8.7 months (95% confidence interval, 5.0‐12.4 months).
TABLE 7.
Detailed information regarding genotype‐matched treatments
| Patients | Primary sites | Targeted gene alterations | Treatments | Best responses |
|---|---|---|---|---|
| CLHURC‐030 | Breast | PIK3CA H1047R | Everolimus + S‐1 | PD |
| CLHURC‐048 | NSCLC | EGFR E746_S752V | Gefitinib | CR |
| CLHURC‐051 | Urothelial | ERBB2 amp | Trastuzumab | PR |
| CLHURC‐074 | Breast | AKT1 E17K | AKT inhibitor + fulvestrant | SD |
| CLHURC‐081 | Breast | AKT1 E17K | AKT inhibitor | SD |
| CLHURC‐083 | Ovary | PIK3CA amp | Everolimus | SD |
| CLHURC‐096 | Breast | ERBB2 amp | Trastuzumab + carboplatin | PD |
| CLHURC‐100 | Stomach | BRCA1 E1754G | Capecitabine + cisplatin + trastuzumab | PR |
| CLHURC‐110 | Esophagus | ERBB2 amp | Capecitabine + cisplatin + trastuzumab | PR |
| CLHURC‐133 | NSCLC | ERBB2 G778_P780dup | Trastuzumab | PR |
| CLHURC‐136 | Breast | PIK3CA amp | Everolimus + exemestane | PD |
| CLHURC‐141 | Head and neck | ERBB2 S310Y | Trastuzumab + docetaxel | PR |
| CLHURC‐142 | Endometrial | PIK3CA H1047R | Everolimus + letrozole | SD |
| CLHURC‐148 | Neuroendocrine carcinoma | TMB‐high (120.8 mut/Mb) | PD‐1 inhibitor | CR |
| CLHURC‐149 | Vagina | Germline BRCA2 G3355Vfs*28 | Olaparib | SD |
| CLHURC‐150 | Esophagus | BRCA2 K2538T | 5‐Fluorouracil + cisplatin | PR |
| CLHURC‐157 | Colorectal | BRCA2 S2670L | mFOLFOX6 | PD |
Abbreviations: CR, complete response; mFOLFOX6, 5‐fluorouracil, leucovorin, and oxaliplatin; NSCLC, non‐small cell lung cancer; PR, partial response; SD, stable disease; TMB, tumor mutation burden.
3.3. Secondary findings in germline variants
Secondary findings were detected in tumor tissues as well as PBMCs of 9% (15/160) of the patients (Table S10). Detailed information regarding secondary findings considered to be associated with hereditary tumors is shown in Table 8. If germline variants were detected in the genes listed in the ACMG recommendations 27 of secondary findings, and the patients agreed to be informed, the detail was shared with the patients after genetic counseling.
TABLE 8.
Detailed information regarding secondary findings in germline variants considered to be associated with hereditary tumors
| Patients | Primary sites | Sex | Age, years | Genes | AA changes | Phenotypes | Family histories |
|---|---|---|---|---|---|---|---|
| CLHURC‐012 | Breast | Female | 33 | PTEN | R130* | PTEN hamartoma tumor syndrome | Breast: grandmother, grandaunt ×2; Colorectal: grandfather |
| CLHURC‐025 | Kidney | Male | 37 | FH | N415Kfs*37 | Hereditary leiomyomatosis and renal cell carcinoma syndrome | NSCLC: grandmother; Stomach: grandfather |
| CLHURC‐027 | NSCLC | Female | 62 | PMS2 | K580* | Lynch syndrome | Stomach: father; Unknown primary: grandmother |
| CLHURC‐038 | Sarcoma | Female | 41 | TP53 | R196* | Li‐Fraumeni syndrome | None |
| CLHURC‐039 | Breast | Female | 44 | BRCA2 | T630Nfs*6 | Hereditary breast and ovarian cancer | Breast: grandmother ×2; Prostate: father; Colorectal: sister; Stomach: uncle |
| CLHURC‐040 | Pancreas | Male | 67 | ATM | Q2729* | Ataxia telangiectasia | Breast: mother, sister ×3; Head and neck: sister |
| CLHURC‐044 | Ovary | Female | 54 | BRCA1 | E1148Rfs*7 | Hereditary breast and ovarian cancer | Unknown primary: grandmother, grandfather ×2; Colorectal: uncle |
| CLHURC‐053 | Kidney | Male | 30 | FH | V84Dfs*5 | Hereditary leiomyomatosis and renal cell carcinoma syndrome | Kidney: father, grandfather; Pancreas: uncle; Breast: uncle; Childhood cancer: aunt |
| CLHURC‐081 | Breast | Female | 50 | BRCA1 | Q1447Rfs*22 | Hereditary breast and ovarian cancer | Breast: mother, aunt ×2; Ovary: mother; Pancreas: aunt; Stomach: father |
| CLHURC‐096 | Breast | Female | 52 | BRCA1 | L63* | Hereditary breast and ovarian cancer | Colorectal: uncle |
| CLHURC‐134 | Skin | Male | 42 | ATM | E376lfs*2 | Ataxia telangiectasia | Unknown primary: grandfather |
| CLHURC‐136 | Breast | Female | 38 | BRCA2 | D252Vfs*24 | Hereditary breast and ovarian cancer | Breast: mother; Endometrial: aunt |
| CLHURC‐149 | Breast | Female | 67 | BRCA2 | G3355Vfs*28 | Hereditary breast and ovarian cancer | Liver: mother |
| CLHURC‐152 | Breast | Female | 44 | BRCA1 | Q81* | Hereditary breast and ovarian cancer | Breast: mother, sister |
| CLHURC‐153 | Cervix | Female | 56 | BRIP1 | c.508‐1G > A | Hereditary breast and ovarian cancer | Breast: aunt |
Abbreviations: AA, amino acid; NSCLC, non‐small cell lung cancer
4. DISCUSSION
In this study, we report the clinical utility of an in‐house targeted amplicon sequencing system for cancer patients. Clinical implementation of precision cancer medicine has been considered a promising treatment strategy for cancer, and a variety of studies in experimental settings have been performed and reported. However, only few reports have focused on an in‐house clinical sequencing system as a laboratory examination. Our cohort consisted of patients with different types of cancer who were willing for genotype‐matched treatments in clinical practice. The sequencing reports were returned within a reasonable turnaround time with a very high success rate of sequencing, and we concluded that our in‐house sequencing system was feasible and promising. However, undertaking in‐house comprehensive cancer genomic profiling tests in real‐world medical settings remains challenging, mainly due to the need for highly qualified staff and the high cost involved.
A recent large cohort study on 10 000 patients with cancer using the MSK‐IMPACT platform reported that 37% of the patients harbored actionable gene alterations, but only 11% of the patients were subsequently enrolled in genotype‐matched clinical trials. 28 In addition, two Japanese retrospective studies that analyzed samples from 85 and 230 cancer patients using the OncoPrime and NCC Oncopanel platforms, reported the identification of actionable gene mutations in 86% and 59% of the patients, respectively, but only 13% of the patients assessed by either platform received subsequent treatments based on the genomic information. 29 , 30 In our cohort, actionable gene alterations were detected in 46% of the patients, and 11% of the patients eventually received the genotype‐matched treatments. The definition of actionable gene alterations differs with every platform; however, the rate of patients who eventually opted for genotype‐matched treatments was 10%‐20% in all platforms. This range is indicative of the current performance of precision cancer medicine using comprehensive cancer genomic profiling, despite the number of targeted cancer‐related genes in the cancer panel.
The turnaround time of genome sequencing is an important factor when implementing comprehensive cancer genomic profiling into clinical practice. The Japanese retrospective study using the OncoPrime and NCC Oncopanel platforms reported that the median turnaround time for physicians was 40 and 32 days, respectively. 29 , 30 In our cohort, the median turnaround time for physicians was 20 days. Although the expected turnaround time of our system was approximately 14 days, it took an additional six days to convene a panel of genome experts. In our system, the entire process of genome sequencing, including extraction of DNA from tumor samples, NGS assay, and genomic analysis, was performed in our hospital. During cancer genome sequencing using pathological tissue samples, the process is mainly divided into three parts: pre‐analysis, analysis, and post‐analysis. The advantages of an in‐house sequencing system depend to a large extent on the pre‐analysis part. Pre‐analysis is the process of handling of tumor samples, consisting of collecting and checking specimens, extraction of nucleic acids, checking the quality and quantity of nucleic acids, and NGS library preparation. In our in‐house sequencing system, all of the processes have been performed by full‐time experts within a week just after the patients agreed to take the examination. Therefore, these steps contributed to shortening of turnaround time, and the short turnaround time with a very high success rate of sequencing was achieved because the in‐house processing was performed as a laboratory examination. In addition, in our case, the median post‐sequencing survival time was 8.7 months, which was sufficient to provide opportunities to patients for receiving genotype‐matched treatments. However, to maximize the benefits of precision cancer medicine, it is essential to develop an efficient system, which is accessible for cancer patients and facilitates enrollment of such patients into genotype‐matched clinical trials.
The therapeutic efficacy of precision cancer medicine remains unknown. A previous prospective randomized controlled phase II trial (the SHIVA trial), designed to evaluate molecularly targeted therapy based on tumor molecular profiling vs. conventional therapy for advanced cancer reported that the use of molecularly targeted agents outside their indications did not improve progression‐free survival compared with treatments at physician’s choice in heavily pretreated patients with cancer. 31 These results provide important insights as they indicate that off‐label use of molecularly targeted agents should not be readily encouraged, even if they were recommended by comprehensive cancer genomic profiling tests. The response rate and disease control rate of the genotype‐matched treatments in our cohort were 47% and 76%, respectively. Furthermore, a previous Japanese comprehensive screening study of targeted molecules by NGS in patients with cancer reported that the response disease control rates of the genotype‐matched phase I clinical trial were 33% and 78%, respectively. 32 The available evidence and our data support the therapeutic efficacy of precision cancer medicine. Additional confirmatory, prospective, genotype‐matched trials are required to validate the clinical utility of precision cancer medicine.
In clinical interpretation of sequencing data, the management of secondary findings is a pivotal issue. In our system, information of secondary findings was disclosed to the patients only after genetic counseling regardless of whether secondary findings were detected or not. Most of the patients did not want to receive genetic counseling at that time and only one‐third of the patients wished to be informed about secondary findings, which seemed to be lower than expected. In our cohort, secondary findings in germline variants considered to be associated with hereditary tumors were identified in 9% (15/160) of all the patients. In particular, secondary findings listed in the cancer‐related genes of the ACMG recommendations were identified in 6% (10/160) of all patients. Based on previous reports, the prevalence of pathogenic or likely pathogenic variants across all the ACMG recommendations was approximately 0.8%‐5%. 33 , 34 , 35 , 36 , 37 Thus, our finding strongly supports these results and paves the way for our future subject aiming at establishing a standard procedure to manage secondary findings, including interpretation of variant of unknown significance (VUS).
The present study had some limitations. First, our cancer panel targeting 160 cancer‐related genes did not cover fusion genes, and therefore we might have underestimated the detection rates of actionable gene alterations. In particular, gene fusions such as ALK, FGFR, NTRK, RET, and ROS1 can be rational actionable targets, although we overlooked the small numbers of patients with these gene fusions. 38 Second, it is very difficult to launch an in‐house clinical sequencing system in every hospital. Not only is the preparation and maintenance of an NGS system costly, but a team of medical experts and trained personnel is required to carry out and interpret genome sequencing. Thus, to promote genome medicine nationwide, an outsourcing system can be helpful. In an in‐house system, the number of inspections in each sequencing run is limited, and therefore, the cost of genome sequencing using an in‐house system is higher than an outsourcing system in general. An outsourcing system using a high‐capacity NGS can process a lot of samples in each sequencing run, and then it can reduce the cost per patient. However, it takes some time to start a sequencing run until a certain number of samples are collected, and therefore, in general, the turnaround time of an outsourcing system is longer than an in‐house system. We have to consider the balance between turnaround time and cost in developing a clinical sequencing system. Therefore, we are developing a novel and low‐cost clinical sequencing system with a short turnaround time, which can be used by nationwide clinical facilities to outsource the cancer genomic profiling test of their patient samples. Finally, our in‐house clinical sequencing was performed in an ISO 15189‐certified laboratory at our hospital. Comprehensive cancer genomic profiling tests as a clinical examination should essentially be carried out in laboratories that are certified in quality control for genome sequencing, such as the Clinical Laboratory Improvement Amendments (CLIA)‐certified and College of American Pathologists‐Laboratory Accreditation Program (CAP‐LAP)‐certified laboratories.
In conclusion, the present study proves the feasibility of using an in‐house clinical sequencing system as a promising laboratory examination technique in clinical practice. Although outsourcing of cancer genomic profiling tests is currently predominant, in‐house systems are preferable, especially in terms of safe and secure handling of clinical specimens and short turnaround time, and to facilitate research and development. As the cost for genome sequencing drops and genomic medicine gains popularity for cancer treatment, it is anticipated that in‐house clinical sequencing systems will be soon implemented as a clinical laboratory test, particularly in core hospitals for cancer genomic medicine.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank Chikako Sato for clinical assistance. This work was supported in part by the Japan Society for the Promotion of Science KAKENHI (Grant‐in‐Aid for Young Scientists B) Grant Number 17K15910 and AMED under Grant Number JP17kk0205006h000.
Hayashi H, Tanishima S, Fujii K, et al. Clinical impact of a cancer genomic profiling test using an in‐house comprehensive targeted sequencing system. Cancer Sci. 2020;111:3926–3937. 10.1111/cas.14608
REFERENCES
- 1. Hyman DM, Taylor BS, Baselga J. Implementing genome‐driven oncology. Cell. 2017;168:584‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kohno T. Implementation of “clinical sequencing” in cancer genome medicine in Japan. Cancer sci. 2018;109:507‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998‐2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380‐2388. [DOI] [PubMed] [Google Scholar]
- 5. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med. 2013;368:2385‐2394. [DOI] [PubMed] [Google Scholar]
- 6. Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1‐rearranged non‐small‐cell lung cancer. N Engl J Med. 2014;371:1963‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Planchard D, Smit EF, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)‐mutant metastatic non‐small‐cell lung cancer: an open‐label, phase 2 trial. Lancet Oncol. 2017;18:1307‐1316. [DOI] [PubMed] [Google Scholar]
- 8. Le DT, Uram JN, Wang H, et al. PD‐1 Blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372:2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion‐positive cancers in adults and children. N Engl J Med. 2018;378:731‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doebele RC, Drilon A, Paz‐Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion‐positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21:271‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biankin AV, Piantadosi S, Hollingsworth SJ. Patient‐centric trials for therapeutic development in precision oncology. Nature. 2015;526:361‐370. [DOI] [PubMed] [Google Scholar]
- 12. Renfro LA, Sargent DJ. Statistical controversies in clinical research: basket trials, umbrella trials, and other master protocols: a review and examples. Ann Oncol. 2017;28:34‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377:62‐70. [DOI] [PubMed] [Google Scholar]
- 14. Redig AJ, Janne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol. 2015;33:975‐977. [DOI] [PubMed] [Google Scholar]
- 15. Cunanan KM, Gonen M, Shen R, et al. Basket trials in oncology: a trade‐off between complexity and efficiency. J Clin Oncol. 2016;35:271‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering‐integrated mutation profiling of actionable cancer targets (MSK‐IMPACT): a hybridization capture‐based next‐generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hyman DM, Solit DB, Arcila ME, et al. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next‐generation sequencing enabling next‐generation targeted therapy trials. Drug Discov Today. 2015;20:1422‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson DB, Dahlman KH, Knol J, et al. Enabling a genetically informed approach to cancer medicine: a retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next‐generation sequencing panel. Oncologist. 2014;19:616‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanai Y, Nishihara H, Miyagi Y, et al. The Japanese Society of Pathology Guidelines on the handling of pathological tissue samples for genomic research: standard operating procedures based on empirical analyses. Pathol Int. 2018;68:63‐90. [DOI] [PubMed] [Google Scholar]
- 20. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mose LE, Wilkerson MD, Hayes DN, Perou CM, Parker JS. ABRA: improved coding indel detection via assembly‐based realignment. Bioinformatics. 2014;30:2813‐2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campbell BB, Light N, Fabrizio D, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171:1042‐1056.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cingolani P, Platts A, le Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin). 2012;6:80‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sunami K, Takahashi H, Tsuchihara K, et al. Clinical practice guidance for next‐generation sequencing in cancer diagnosis and treatment (Edition 1.0). Cancer sci. 2018;109:2980‐2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249‐255. [DOI] [PubMed] [Google Scholar]
- 28. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature Med. 2017;23:703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kou T, Kanai M, Yamamoto Y, et al. Clinical sequencing using a next‐generation sequencing‐based multiplex gene assay in patients with advanced solid tumors. Cancer Sci. 2017;108:1440‐1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sunami K, Ichikawa H, Kubo T, et al. Feasibility and utility of a panel testing for 114 cancer‐associated genes in a clinical setting: a hospital‐based study. Cancer Sci. 2019;110:1480‐1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open‐label, proof‐of‐concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16:1324‐1334. [DOI] [PubMed] [Google Scholar]
- 32. Tanabe Y, Ichikawa H, Kohno T, et al. Comprehensive screening of target molecules by next‐generation sequencing in patients with malignant solid tumors: guiding entry into phase I clinical trials. Mol Cancer. 2016;15:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Amendola LM, Dorschner MO, Robertson PD, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 2015;25:305‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Olfson E, Cottrell CE, Davidson NO, et al. Identification of medically actionable secondary findings in the 1000 genomes. PLoS One. 2015;10:e0135193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gambin T, Jhangiani SN, Below JE, et al. Secondary findings and carrier test frequencies in a large multiethnic sample. Genome Med. 2015;7:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Natarajan P, Gold NB, Bick AG, et al. Aggregate penetrance of genomic variants for actionable disorders in European and African Americans. Sci Transl Med. 2016;8(364):364ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim J, Luo W, Wang M, et al. Prevalence of pathogenic/likely pathogenic variants in the 24 cancer genes of the ACMG Secondary Findings v2.0 list in a large cancer cohort and ethnicity‐matched controls. Genome Med. 2018;10(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14:735‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material