

Abstract
Oncogenic mutations often trigger antitumor cellular response such as induction of apoptosis or cellular senescence. Studies in the last decade have identified the presence of the third guardian against mutation‐induced tumorigenesis, namely “cell competition.” Cell competition is a context‐dependent cell elimination whereby cells with higher fitness eliminate neighboring cells with lower fitness by inducing cell death. While oncogene‐induced apoptosis or oncogene‐induced senescence acts as a cell‐autonomous tumor suppressor, cell competition protects the tissue from tumorigenesis via cell‐cell communication. For instance, in Drosophila epithelium, oncogenic cells with cell polarity mutations overproliferate and develop into tumors on their own but are eliminated from the tissue when surrounded by wild‐type cells. Genetic studies in flies have unraveled that such tumor‐suppressive cell competition is regulated by at least three mechanisms: direct cell‐cell interaction between polarity‐deficient cells and wild‐type cells, secreted factors from epithelial cells, and systemic factors from distant organs. Molecular manipulation of tumor‐suppressive cell competition could provide a novel therapeutic strategy against human cancers.
Keywords: cell competition, Drosophila, tumor suppression
Cell competition is a context‐dependent cell elimination whereby cells with higher fitness eliminate neighboring cells with lower fitness by inducing cell death. In Drosophila epithelium, oncogenic cells with cell polarity mutations overproliferate and develop into tumors on their own but are eliminated from the tissue when surrounded by wild‐type cells. Genetic studies in flies have unraveled that such tumor‐suppressive cell competition is regulated by at least three mechanisms: direct cell‐cell interaction between polarity‐deficient cells and wild‐type cells, secreted factors from epithelial cells, and systemic factors from distant organs.
1. INTRODUCTION
Oncogenic mutations not only confer cells with proliferative advantage but also trigger antiproliferative effects that suppress tumorigenesis, a phenomenon called “intrinsic tumor suppression.” 1 One such mechanism is oncogene‐induced apoptosis, which is triggered by upregulation of oncogenes such as Myc and E1A. 2 Another important mechanism of intrinsic tumor suppression is oncogene‐induced cellular senescence, 3 an irreversible cell cycle arrest induced by the activation of oncogenes such as Ras, Braf, Akt, E2F1, mos, and Cdc6 or inactivation of tumor suppressor genes such as PTEN and NF1. 1 , 4 , 5 , 6 , 7 These tumor‐suppressive machineries eliminate or inactivate premalignant cells emerged in the tissue in a cell‐autonomous manner. Apart from these classical tumor‐suppressive mechanisms, studies in the last decade have identified a prominent role of surrounding wild‐type cells to eliminate premalignant mutant cells: the third machinery of intrinsic tumor suppression via cell‐cell interaction. This phenomenon is called cell competition, a context‐dependent cell elimination whereby cells with higher fitness eliminate neighboring cells with lower fitness by inducing cell death (Figure 1).
Figure 1.
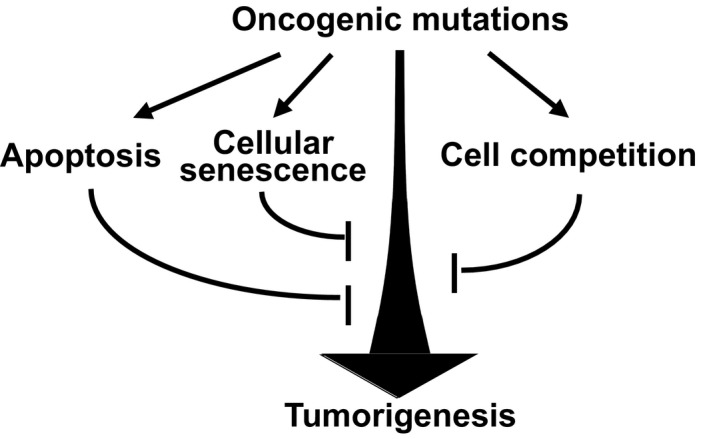
Oncogenic mutations trigger intrinsic tumor‐suppressive programs. A variety of oncogenic mutations not only promote tumorigenesis but simultaneously activate intrinsic tumor‐suppressive mechanisms such as induction of apoptosis, cellular senescence, and cell competition
Cell competition was first reported in 1975 in the wing imaginal epithelia of Drosophila melanogaster, where mutant cells heterozygous for the ribosomal protein (Rp) gene are eliminated from the tissue during development when surrounded by wild‐type cells. 8 Notably, Rp/+ animals develop into essentially normal flies with a slight delay in developmental time as well as thinner bristles (thus called “Minute” mutant) compared with wild‐type flies. 8 , 9 Thus, viable but less fit Rp/+ cells (“losers”) are eliminated when surrounded by fitter, wild‐type cells (“winners”). Twenty years later, it was found in Drosophila epithelium that oncogenic polarity‐deficient cells such as scribble (scrib) mutant cells overproliferate on their own but are eliminated by cell competition when surrounded by wild‐type cells. 10 This underscores the significance of cell competition in epithelial tumor suppression. Notably, similar elimination of oncogenic mutant cells via cell‐cell interaction has been observed in mammalian systems, a phenomenon called epithelial defense against cancer (EDAC). 11 In this review, we summarize recent studies on the mechanism of tumor‐suppressive cell competition in flies, which is regulated by at least three factors including direct cell‐cell interaction, secreted factors from epithelial cells, and systemic factors from distant organs (Figure 2). We also discuss how it is relevant to cancer regulation.
Figure 2.
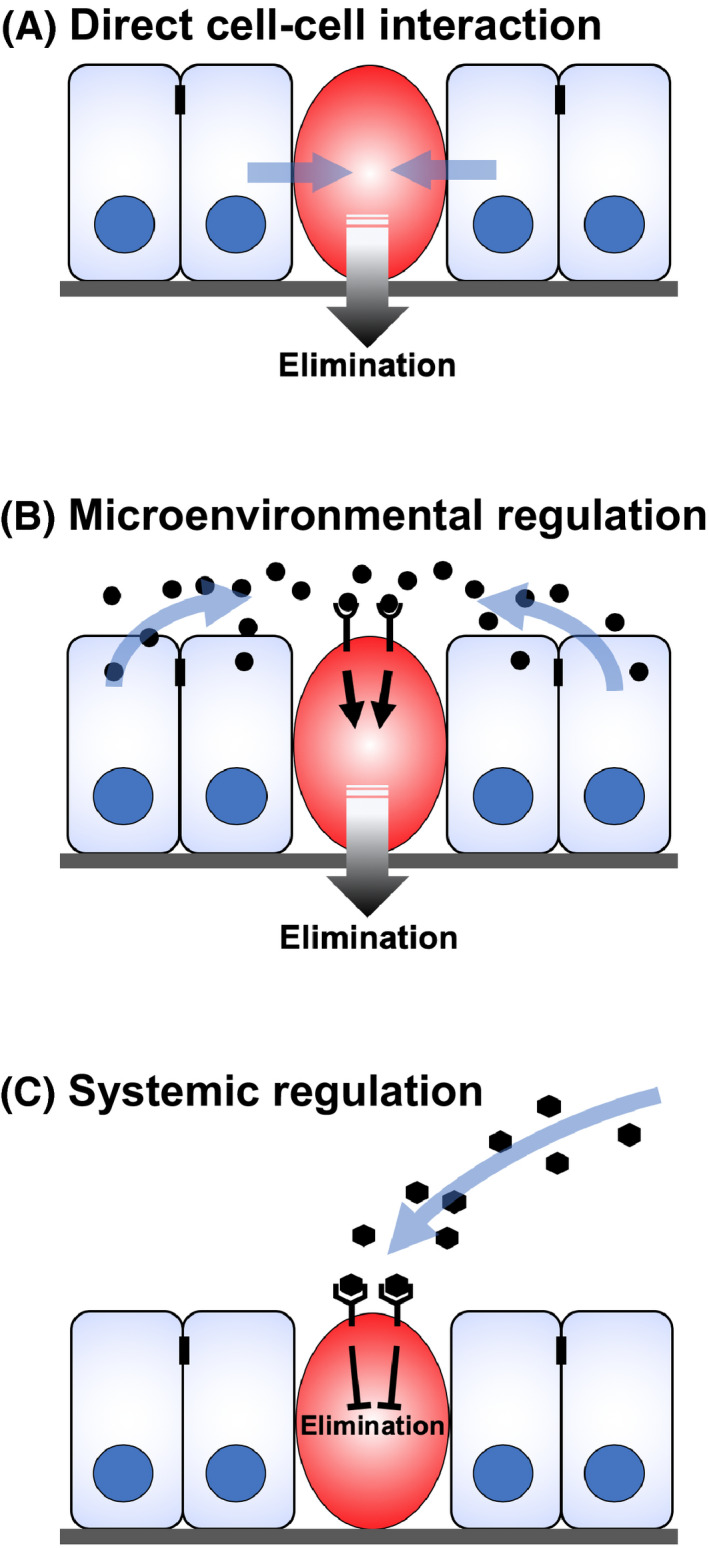
Factors that regulate scrib cell elimination by cell competition. scrib mutant cell (red) is eliminated when surrounded by wild‐type cells (blue) via at least three mechanisms including (1) direct cell‐cell interaction with neighboring wild‐type cells, (2) microenvironmental regulation by locally provided secreted factors such as Slit and Spz, and (3) systemic regulation by factors such as Drosophila insulinlike peptides (Dilps)
2. TUMORIGENIC POLARITY‐DEFICIENT CELLS ARE ELIMINATED BY CELL COMPETITION
Tumor‐suppressive cell competition has been best characterized in the studies of the phenomenon whereby polarity‐deficient scrib mutant cells are eliminated from Drosophila imaginal epithelium when surrounded by wild‐type cells. The protein product of scrib localizes to the epithelial septate junction, the analogue of the vertebrate tight junction, and regulates the apico‐basal polarity. 12 Deregulation or mislocalization of human Scrib or other polarity regulators such as Dlg1 and Lgl2 has been associated with human cancer development. 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 In flies, loss of scrib in the epithelium causes unrestricted localization of an apically localized membrane protein Crumbs (Crb), resulting in strongly disorganized, overgrown tissue. 22 Developing scrib tumors show characteristic transition from growth arrest to proliferation state, which is regulated by dynamic change in intrinsic MAPK signaling activity. 23 Thus, scrib is called a Drosophila “neoplastic tumor suppressor” gene. 22 Interestingly, however, when clones of scrib mutant cells are induced in wild‐type imaginal discs in a mosaic manner using the mitotic recombination technique (hereafter referred to as scrib clones), 24 mutant cells do not overgrow but cause cell death. 10 This suggests that surrounding wild‐type cells exert antitumor effects against nearby polarity‐deficient cells. Similar tumor‐suppressive cell elimination is observed when mutant clones for dlg, whose protein product functions as “Scrib module” together with Scrib and Lethal (2) giant larvae (Lgl), 25 as well as clones defective in the endocytic avl/syx7, rab5, vps22, vps25, or vps36 gene are induced in the imaginal disc. 26 , 27 , 28 , 29 , 30 , 31 Notably, epithelial cells mutant for these genes show diffusion of apically localized proteins to the basolateral domain. 28 , 32 On the other hand, mutations in other polarity genes such as bazooka/par‐3, which do not cause basal expansion of apical proteins, 28 , 33 do not cause overproliferation and are not subjected to cell competition when surrounded by wild‐type cells. Thus, it is likely that mutant epithelial cells with apical expansion specifically trigger the machinery of tumor‐suppressive cell competition.
3. CELL ELIMINATION BY CELL‐AUTONOMOUS JNK SIGNALING
Although scrib clones surrounded by wild‐type cells in the eye imaginal disc show elevated cell proliferation rate with upregulated CyclinE levels and BrdU incorporation, they do not overgrow but are eliminated from the tissue by apoptosis. 10 This suggests that elimination of scrib clones is led by an active, regulated mechanism rather than passive consequence of impaired cell survival or cell growth. Genetic studies in Drosophila have uncovered the molecular basis for how scrib clones are eliminated from the tissue when surrounded by wild‐type cells. It was first shown that scrib clone elimination is mediated by c‐Jun‐N‐terminal kinase (JNK) as blocking Drosophila JNK Bsk abolished the elimination and led to scrib cell overproliferation. 10 This JNK‐dependent scrib elimination is triggered by Eiger, 34 the sole tumor necrosis factor (TNF) in Drosophila 35 , 36 and its receptor Grindelwald. 37 It was found that scrib clones elevate endocytosis, which translocates Eiger from the plasma membrane to endosomes, thereby leading to activation of downstream JNK signaling (Figure 3). 34 It has also been reported that Eiger expression in the hemocytes attached to the imaginal discs activate JNK signaling in polarity‐deficient imaginal cells. 38
Figure 3.
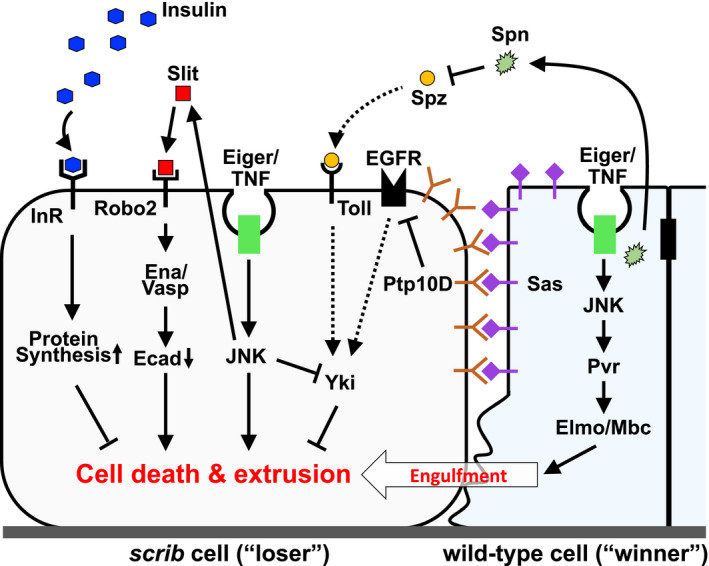
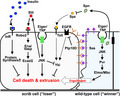
Mechanisms that eliminate scrib cells by cell competition. Sas‐PTP10D signaling activated by direct cell‐cell interaction with neighboring wild‐type cells inhibits EGFR signaling, thereby suppressing oncogenic cooperation between EGFR‐Ras and Eiger/TNF‐JNK signaling that activates the Hippo effector Yki. 41 Slit‐Robo2‐Ena/Vasp signaling activated by Eiger/TNF‐JNK signaling promotes extrusion of scrib cells by downregulating E‐Cadherin. 39 Eiger/TNF‐JNK signaling activated in wild‐type cells elevates Pvr‐Elmo‐Mbc signaling and thereby promotes engulfment of neighboring scrib cells. 40 Epithelial cells secrete a serine protease inhibitor Serpin 5, which inhibits Toll signaling and subsequently Yki‐mediated cell survival signaling in scrib cells. 42 Circulating blood insulin (Dilp) suppresses scrib cell elimination by insulin receptor (InR)‐mediated elevation of protein synthesis 45
While the elimination of scrib clones essentially depends on JNK signaling, JNK‐induced cell death does not fully account for the cell elimination as blocking cell death does not cause as drastic tumorigenesis as blocking JNK. 10 , 39 It was found through a genetic screen that JNK activation in scrib clones upregulates the evolutionarily conserved repulsive axon guidance ligand, receptor, and downstream target, namely Slit, Roundabout2 (Robo2), and Enabled (Ena)/VASP, respectively. This causes downregulation of E‐Cadherin and thus disruption of cell‐cell adhesion, which promotes extrusion of scrib cells from epithelium (Figure 3). 39
4. CELL ELIMINATION BY DIRECT CELL‐CELL INTERACTION
scrib mutant cells overproliferate in the absence of wild‐type neighbors, suggesting a non‐cell‐autonomous antitumor effect by juxtaposed wild‐type cells. Intriguingly, when scrib clones are induced in the eye imaginal discs, JNK activation is observed not only in scrib cells but also in surrounding wild‐type cells right next to the mutant cells. 27 , 40 JNK activation in surrounding wild‐type cells does not cause apoptosis, but instead, it upregulates the Drosophila platelet‐derived growth factor (PDGF)/vascular endothelial growth factor (VEGF) receptor (PVR), resulting in activation of ELMO (engulfment and cell motility, a Ced‐12 homolog)/Mbc (myoblast city, a Ced‐5/DOCK180 homolog)‐mediated engulfment pathway. As a consequence, wild‐type cells phagocytose nearby scrib cells, thereby promoting scrib cell elimination 40 (Figure 3). This mechanism first provided the molecular basis for the significance of neighboring wild‐type cells in the execution of tumor‐suppressive cell competition.
An ethyl methanesulfonate (EMS)‐based genetic screen in Drosophila identified cell‐surface ligand‐receptor proteins that regulate scrib cell elimination via cell‐cell interaction. The ligand Sas and its receptor PTP10D (a receptor tyrosine phosphatase) normally localize to the apical surface of epithelial cells, but at the interface between scrib and wild‐type cells they relocalize to the lateral membrane, where the ligand and receptor meet with each other in trans. This leads to activation of PTP10D signaling in scrib cells, resulting in suppression of epidermal growth factor receptor (EGFR) signaling and subsequent elimination of scrib cells 41 (Figure 3). In the absence of Sas‐PTP10D signaling, scrib clones elevate both EGFR and JNK signaling, which cooperate to activate the Hippo pathway effector Yorkie (Yki) and thus cause overgrowth. 41 It is likely that the lateral relocalization of Sas and PTP10D at the interface between scrib and wild‐type clones is triggered by lateral expansion of the apical domain in scrib cells. Thus, tumor‐suppressive cell competition seems to be triggered by an intrinsic cellular event that causes lateral relocalization of apical proteins in oncogenic polarity‐deficient cells, which would cause direct cell‐cell interaction with nearby cells.
5. MICROENVIRONMENTAL REGULATION OF CELL ELIMINATION
While genetic studies in flies have clearly shown the critical role of cell‐cell interaction in driving tumor‐suppressive cell competition, it had been unclear whether cell competition is solely regulated by direct cell‐cell interaction. A genetic screen in Drosophila identified serpin 5 (spn5), which encodes a secreted serine protease inhibitor as a suppressor of tumor‐suppressive cell competition when mutated in surrounding wild‐type cells. 42 spn5 is one of the most abundantly expressed serpins in the imaginal discs, 42 whose protein product negatively regulates the Toll ligand, Spätzle (Spz). Therefore, downregulation of Spn5 in surrounding wild‐type cells leads to activation of Toll signaling in scrib cells. It has previously been shown that activation of Spz/Toll signaling causes elimination of loser cells in Minute or Myc‐induced cell competition. 43 , 44 Intriguingly, however, Toll activation in scrib cells does not promote their elimination but rather causes JNK activation and F‐actin accumulation, leading to activation of Yki and thus scrib overgrowth 42 (Figure 3). This suggests that restricting the basal level of Toll signaling in the epithelium is crucial for the induction of tumor‐suppressive cell competition and that Toll activation by infection may trigger tumorigenesis by abrogating cell competition. In this sense, Serpins act as microenvironmental “surveillance factors” that facilitate tumor‐suppressive cell competition.
6. SYSTEMIC REGULATION OF CELL ELIMINATION
Cancer development is comprehensively regulated by a variety of systemic factors within the human body. Significantly, a recent genetic study revealed that a systemic factor also critically regulates tumor‐suppressive cell competition in flies. A dominant modifier screen identified chico, which encodes an evolutionarily conserved insulin receptor substrate as a suppressor of tumor‐suppressive cell competition when deleted heterozygously in the animal. 45 Unexpectedly, Chico was not required in competing cells in the imaginal discs but was essential in insulin‐producing cells (IPCs) of the brain to execute cell competition remotely. Mechanistically, chico downregulation in IPCs causes hyperinsulinemia by upregulating a Drosophila insulin Dilp2, which activates insulin/target of rapamycin (TOR) signaling in scrib cells. Notably, scrib cells normally show decreased protein synthesis activity compared to wild‐type neighbors, but hyperinsulinemia‐induced insulin/TOR activation in scrib cells boosts protein synthesis and causes scrib overgrowth 45 (Figure 3). These observations provide an in vivo mechanistic explanation for why metabolic diseases such as type 2 diabetes are associated with increased cancer incidence in humans. This study also highlights an unexpected mechanistic link between tumor‐suppressive cell competition and classical Minute cell competition, both of which represent elimination of loser cells that have lower protein synthesis compared with neighboring winner cells.
7. ADDITIONAL ONCOGENIC ALTERATION REVERSES TUMOR‐SUPPRESSIVE CELL COMPETITION
Accumulation of oncogenic alterations is a hallmark of malignant progression of tumors. This suggests that additional oncogenic alterations induced in scrib cells could reverse cell competition and cause scrib tumorigenesis. A prominent example of such phenomena is the activation of Ras or Notch signaling. While the activation of Ras or Notch in the eye imaginal discs only causes moderate overgrowth of benign tumors, its activation in scrib clones results in drastic, neoplastic overgrowth of malignant tumors that aggressively invade adjacent organ ventral nerve cord 10 , 46 (Figure 4). The tumor malignancy of Ras‐activated scrib (RasV12 /scrib) cells is caused by JNK activation, E‐cadherin downregulation, and Yki activation. 10 , 27 , 46 , 47 In addition, RasV12 /scrib cells undergo endoreplication and thus become polyploid giant cells, which is essential for their malignant overgrowth. 48 It has also been reported that RasV12 /scrib cells in the eye discs induce nonautonomous autophagy (NAA) in their surrounding wild‐type cells and in distant tissues, which are essential to support aggressive growth and metastatic potential of RasV12 /scrib tumors, likely through nutrient supply. 49 A similar NAA is observed in losers of Minute cell competition; in this case, NAA causes cell death, 50 which may also promote the growth of neighboring winner cells like RasV12 /scrib‐induced NAA. Interestingly, while RasV12 /scrib cells aggressively overproliferate, they also undergo apoptosis at the boundary between RasV12 /scrib and neighboring wild‐type clones like losers of cell competition. 51
Figure 4.
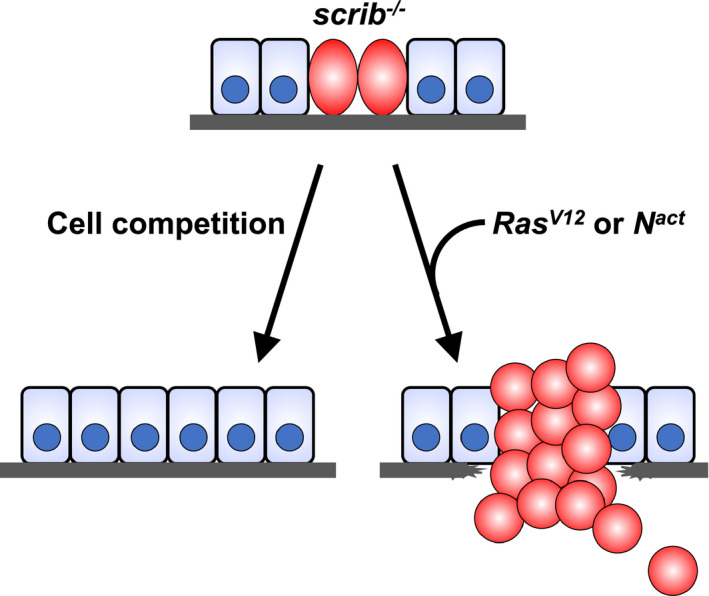
scrib cell competition is reversed when additional oncogenic mutations such as activating mutation in Ras (RasV12) or Notch (Nact) is introduced. RasV12/scrib or Nact/scrib tumors overgrow and invade surrounding tissues
The elimination of scrib clone is also reversed by overexpression of Myc, 52 which is consistent with the fact that increased level of Myc turns cells into supercompetitors. 53 , 54 In addition, it has been shown that clones defective in scrib or lgl are eliminated when located in the wing pouch (therefore this region is referred to as “tumor cold spots”), while they evade cell competition and overgrow when located at the hinge region, where endogenous JAK/STAT activity is elevated (therefore referred to as “tumor hot spots”). This suggests that tissue‐intrinsic local signaling activity or cytoarchitecture could regulate tumor‐suppressive cell competition. 55
8. CELL COMPETITION ELIMINATES SCRIB CELLS IN MAMMALS
The evolutionary conservation of the scrib gene 56 raises the question of whether elimination of scrib mutant cells by cell competition is conserved in mammals. This was studied in mammalian epithelial cell line Madin‐Darby canine kidney (MDCK) cells. While scrib knockdown MDCK cells are viable on their own, they undergo apoptosis and are extruded apically when cocultured with normal MDCK cells. 57 Apoptosis of scrib knockdown MDCK cells when surrounded by normal cells depends on cell‐autonomous activation of p38. 57 A subsequent study showed that scrib knockdown MDCK cells surrounded by normal cells are hypersensitive to mechanical compaction due to elevated p53 activity. 58 Compaction further upregulates p53 levels through Rho‐associated kinase (ROCK) and p38 in scrib knockdown cells, thereby inducing cell death. 58 This suggests that tumor‐suppressive cell competition is also regulated by mechanical insults. Thus, while the underlying mechanisms are different, tumor‐suppressive cell competition triggered by loss of scrib seems to be conserved in mammalian systems.
9. CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Genetic studies in Drosophila have established a concept that cell competition acts as an intrinsic tumor suppressor in the epithelium. 34 , 59 Although the basal expansion of the apical domain is currently the sole hallmark of losers of tumor‐suppressive cell competition, a recent study suggested that a reduction in protein synthesis may also contribute to establish the loser status. 45 The fact that the elimination of scrib cells is mediated not only by direct cell‐cell interaction but by microenvironmental and systemic factors 42 , 45 underscores that tumor‐suppressive cell competition is regulated by comprehensive mechanisms and can therefore be affected by a variety of cellular or environmental changes within the animal, just like human cancers.
An important outstanding question is what the initial trigger is for tumor‐suppressive cell competition. It would be important to clarify whether cellular changes other than loss of cell polarity can also trigger tumor‐suppressive cell competition. It would also be interesting to investigate whether the machinery of tumor‐suppressive cell competition is involved in physiological processes other than tumor suppressing. For instance, during normal development, there are a variety of cell‐cell interactions that couple with cell elimination and extrusion, 60 which may be regulated by the common machinery of tumor‐suppressive cell competition.
Although there is experimental evidence that mammalian epithelial cells can eliminate scrib cells via cell‐cell interaction, it would need further investigations to clarify whether similar machinery also exists in mammalian epithelial tissues. Further studies in mammalian systems could lead to the development of a novel anticancer strategy that potentiates tumor‐suppressive cell completion.
DISCLOSURE STATEMENT
A research funding was provided by Bayer.
ACKNOWLEDGMENTS
We apologize to those whose work we could not cite due to space constraints. The work in the Igaki laboratory was supported in part by grants from the MEXT/JSPS KAKENHI (Grant Number 26114002 and 19K22424) and Japan Agency for Medical Research and Development (Project for Elucidating and Controlling Mechanisms of Aging and Longevity, Grant Number 17938731).
Kanda H, Igaki T. Mechanism of tumor-suppressive cell competition in flies. Cancer Sci. 2020;111:3409–3415. 10.1111/cas.14575
REFERENCES
- 1. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307‐315. [DOI] [PubMed] [Google Scholar]
- 2. Evan GI, Littlewood TD. The role of c‐myc in cell growth. Curr Opin Genet Dev. 1993;3:44‐49. [DOI] [PubMed] [Google Scholar]
- 3. Zhu H, Blake S, Kusuma FK, Pearson RB, Kang J, Chan KT. Oncogene‐induced senescence: From biology to therapy. Mech Ageing Dev. 2020;187:111229. [DOI] [PubMed] [Google Scholar]
- 4. Ito T, Igaki T. Dissecting cellular senescence and SASP in Drosophila. Inflamm Regen. 2016;36:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dimri GP, Itahana K, Acosta M, Campisi J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol. 2000;20:273‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Damalas A, Kahan S, Shtutman M, Ben‐Ze'ev A, Oren M. Deregulated beta‐catenin induces a p53‐ and ARF‐dependent growth arrest and cooperates with Ras in transformation. EMBO J. 2001;20:4912‐4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593‐602. [DOI] [PubMed] [Google Scholar]
- 8. Morata G, Ripoll P. Minutes: Mutants of Drosophila autonomously affecting cell division rate. Dev Biol. 1975;42:211‐221. [DOI] [PubMed] [Google Scholar]
- 9. Marygold SJ, Roote J, Reuter G, et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 2007;8:R216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brumby AM, Richardson HE. Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22:5769‐5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kajita M, Fujita Y. EDAC: Epithelial defence against cancer‐cell competition between normal and transformed epithelial cells in mammals. J Biochem. 2015;158:15‐23. [DOI] [PubMed] [Google Scholar]
- 12. Bilder D, Perrimon N. Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature. 2000;403:676‐680. [DOI] [PubMed] [Google Scholar]
- 13. Fuja TJ, Lin F, Osann KE, Bryant PJ. Somatic mutations and altered expression of the candidate tumor suppressors CSNK1 epsilon, DLG1, and EDD/hHYD in mammary ductal carcinoma. Cancer Res. 2004;64:942‐951. [DOI] [PubMed] [Google Scholar]
- 14. Navarro C, Nola S, Audebert S, et al. Junctional recruitment of mammalian Scribble relies on E‐cadherin engagement. Oncogene. 2005;24:4330‐4339. [DOI] [PubMed] [Google Scholar]
- 15. Gardiol D, Zacchi A, Petrera F, Stanta G, Banks L. Human discs large and scrib are localized at the same regions in colon mucosa and changes in their expression patterns are correlated with loss of tissue architecture during malignant progression. Int J Cancer. 2006;119:1285‐1290. [DOI] [PubMed] [Google Scholar]
- 16. Kamei Y, Kito K, Takeuchi T, et al. Human scribble accumulates in colorectal neoplasia in association with an altered distribution of beta‐catenin. Hum Pathol. 2007;38:1273‐1281. [DOI] [PubMed] [Google Scholar]
- 17. Zhan L, Rosenberg A, Bergami KC, et al. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135:865‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lisovsky M, Dresser K, Baker S, et al. Cell polarity protein Lgl2 is lost or aberrantly localized in gastric dysplasia and adenocarcinoma: an immunohistochemical study. Mod Pathol. 2009;22:977‐984. [DOI] [PubMed] [Google Scholar]
- 19. Lisovsky M, Ogawa F, Dresser K, Woda B, Lauwers GY. Loss of cell polarity protein Lgl2 in foveolar‐type gastric dysplasia: correlation with expression of the apical marker aPKC‐zeta. Virchows Arch. 2010;457:635‐642. [DOI] [PubMed] [Google Scholar]
- 20. Ouyang Z, Zhan W, Dan L. hScrib, a human homolog of Drosophila neoplastic tumor suppressor, is involved in the progress of endometrial cancer. Oncol Res. 2010;18:593‐599. [DOI] [PubMed] [Google Scholar]
- 21. Pearson HB, Perez‐Mancera PA, Dow LE, et al. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J Clin Invest. 2011;121:4257‐4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113‐116. [DOI] [PubMed] [Google Scholar]
- 23. Ji T, Zhang L, Deng M, et al. Dynamic MAPK signaling activity underlies a transition from growth arrest to proliferation in Drosophila scribble mutant tumors. Dis Model Mech. 2019;12:dmm040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223‐1237. [DOI] [PubMed] [Google Scholar]
- 25. de Vreede G, Schoenfeld JD, Windler SL, Morrison H, Lu H, Bilder D. The Scribble module regulates retromer‐dependent endocytic trafficking during epithelial polarization. Development. 2014;141:2796‐2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woods DF, Bryant PJ. The discs‐large tumor suppressor gene of Drosophila encodes a guanylate kinase homolog localized at septate junctions. Cell. 1991;66:451‐464. [DOI] [PubMed] [Google Scholar]
- 27. Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol. 2006;16:1139‐1146. [DOI] [PubMed] [Google Scholar]
- 28. Lu H, Bilder D. Endocytic control of epithelial polarity and proliferation in Drosophila. Nat Cell Biol. 2005;7:1232‐1239. [DOI] [PubMed] [Google Scholar]
- 29. Herz HM, Woodfield SE, Chen Z, Bolduc C, Bergmann A. Common and distinct genetic properties of ESCRT‐II components in Drosophila. PLoS One. 2009;4:e4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson BJ, Mathieu J, Sung HH, Loeser E, Rorth P, Cohen SM. Tumor suppressor properties of the ESCRT‐II complex component Vps25 in Drosophila. Dev Cell. 2005;9:711‐720. [DOI] [PubMed] [Google Scholar]
- 31. Ballesteros‐Arias L, Saavedra V, Morata G. Cell competition may function either as tumour‐suppressing or as tumour‐stimulating factor in Drosophila. Oncogene. 2014;33:4377‐4384. [DOI] [PubMed] [Google Scholar]
- 32. Woodfield SE, Graves HK, Hernandez JA, Bergmann A. De‐regulation of JNK and JAK/STAT signaling in ESCRT‐II mutant tissues cooperatively contributes to neoplastic tumorigenesis. PLoS One. 2013;8:e56021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abdelilah‐Seyfried S, Cox DN, Jan YN. Bazooka is a permissive factor for the invasive behavior of discs large tumor cells in Drosophila ovarian follicular epithelia. Development. 2003;130:1927‐1935. [DOI] [PubMed] [Google Scholar]
- 34. Igaki T, Pastor‐Pareja JC, Aonuma H, Miura M, Xu T. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev Cell. 2009;16:458‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Igaki T, Kanda H, Yamamoto‐Goto Y, et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002;21:3009‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moreno E, Yan M, Basler K. Evolution of TNF Signaling Mechanisms. Curr Biol. 2002;12:1263‐1268. [DOI] [PubMed] [Google Scholar]
- 37. Andersen DS, Colombani J, Palmerini V, et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature. 2015;522:482‐486. [DOI] [PubMed] [Google Scholar]
- 38. Cordero JB, Macagno JP, Stefanatos RK, Strathdee KE, Cagan RL, Vidal M. Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev Cell. 2010;18:999‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vaughen J, Igaki T. Slit‐Robo repulsive signaling extrudes tumorigenic cells from Epithelia. Dev Cell. 2016;39:683‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohsawa S, Sugimura K, Takino K, Xu T, Miyawaki A, Igaki T. Elimination of oncogenic neighbors by JNK‐mediated engulfment in Drosophila. Dev Cell. 2011;20:315‐328. [DOI] [PubMed] [Google Scholar]
- 41. Yamamoto M, Ohsawa S, Kunimasa K, Igaki T. The ligand Sas and its receptor PTP10D drive tumour‐suppressive cell competition. Nature. 2017;542:246‐250. [DOI] [PubMed] [Google Scholar]
- 42. Katsukawa M, Ohsawa S, Zhang L, Yan Y, Igaki T. Serpin facilitates tumor‐suppressive cell competition by blocking toll‐mediated Yki activation in Drosophila. Curr Biol. 2018;28(1756–1767):e1756. [DOI] [PubMed] [Google Scholar]
- 43. Meyer SN, Amoyel M, Bergantinos C, et al. An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science. 2014;346:1258236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alpar L, Bergantinos C, Johnston LA. Spatially restricted regulation of spatzle/toll signaling during cell competition. Dev Cell. 2018;46(706–719):e705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sanaki Y, Nagata R, Kizawa D, Léopold P, Igaki T. Hyperinsulinemia Drives Epithelial Tumorigenesis by Abrogating Cell Competition. Dev Cell. 2020;53 (4):379–389. e5. [DOI] [PubMed] [Google Scholar]
- 46. Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science. 2003;302:1227‐1231. [DOI] [PubMed] [Google Scholar]
- 47. Doggett K, Grusche FA, Richardson HE, Brumby AM. Loss of the Drosophila cell polarity regulator Scribbled promotes epithelial tissue overgrowth and cooperation with oncogenic Ras‐Raf through impaired Hippo pathway signaling. BMC Dev Biol. 2011;11:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cong B, Ohsawa S, Igaki T. JNK and Yorkie drive tumor progression by generating polyploid giant cells in Drosophila. Oncogene. 2018;37:3088‐3097. [DOI] [PubMed] [Google Scholar]
- 49. Katheder NS, Khezri R, O'Farrell F, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017;541:417‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nagata R, Nakamura M, Sanaki Y, Igaki T. Cell competition is driven by autophagy. Dev Cell. 2019;51(99–112):e114. [DOI] [PubMed] [Google Scholar]
- 51. Menendez J, Perez‐Garijo A, Calleja M, Morata G. A tumor‐suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc Natl Acad Sci USA. 2010;107:14651‐14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen CL, Schroeder MC, Kango‐Singh M, Tao C, Halder G. Tumor suppression by cell competition through regulation of the Hippo pathway. Proc Natl Acad Sci USA. 2012;109:484‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moreno E, Basler K. dMyc transforms cells into super‐competitors. Cell. 2004;117:117‐129. [DOI] [PubMed] [Google Scholar]
- 54. de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila Myc regulates organ size by inducing cell competition. Cell. 2004;117:107‐116. [DOI] [PubMed] [Google Scholar]
- 55. Tamori Y, Suzuki E, Deng WM. Epithelial tumors originate in tumor hotspots, a tissue‐intrinsic microenvironment. PLoS Biol. 2016;14:e1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bilder D. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev. 2004;18:1909‐1925. [DOI] [PubMed] [Google Scholar]
- 57. Norman M, Wisniewska KA, Lawrenson K, et al. Loss of Scribble causes cell competition in mammalian cells. J Cell Sci. 2012;125:59‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wagstaff L, Goschorska M, Kozyrska K, et al. Mechanical cell competition kills cells via induction of lethal p53 levels. Nat Commun. 2016;7:11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nagata R, Igaki T. Cell competition: Emerging mechanisms to eliminate neighbors. Dev Growth Differ. 2018;60:522‐530. [DOI] [PubMed] [Google Scholar]
- 60. Ohsawa S, Vaughen J, Igaki T. Cell extrusion: a stress‐responsive force for good or evil in Epithelial homeostasis. Dev Cell. 2018;44:284‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]