

Abstract
A previous randomized phase 2 study of hepatocellular carcinoma revealed that the c‐Met inhibitor tivantinib as second‐line treatment significantly prolonged progression‐free survival in a subpopulation whose tumor samples highly expressed c‐Met (MET‐high). Accordingly, this phase 3 study was conducted to evaluate the efficacy of tivantinib as a second‐line treatment for Japanese patients with MET‐high hepatocellular carcinoma. This randomized, double‐blind, placebo‐controlled study was conducted at 60 centers in Japan. Hepatocellular carcinoma patients with one prior sorafenib treatment and those with MET‐high tumor samples were eligible for inclusion. Registered patients were randomly assigned to either the tivantinib or placebo group at a 2:1 ratio and were treated with twice‐a‐day oral tivantinib (120 mg bid) or placebo until the discontinuation criteria were met. The primary endpoint was progression‐free survival while the secondary endpoints included overall survival and safety. Between January 2014 and June 2016, 386 patients provided consent, and 195 patients were randomized to the tivantinib (n = 134) or placebo (n = 61) group. Median progression‐free survival was 2.8 (95% confidence interval: 2.7‐2.9) and 2.3 (1.5‐2.8) mo in the tivantinib and placebo groups, respectively (hazard ratio = 0.74, 95% confidence interval: 0.52‐1.04, P = .082). Median overall survival was 10.3 (95% confidence interval: 8.1‐11.6) and 8.5 (6.2‐11.4) mo in the tivantinib and placebo group, respectively (hazard ratio = 0.82, 95% confidence interval: 0.58‐1.15). The most common tivantinib‐related grade ≥3 adverse events were neutropenia (31.6%), leukocytopenia (24.8%), and anemia (12.0%). This study did not confirm the significant efficacy of tivantinib as a second‐line treatment for Japanese patients with MET‐high hepatocellular carcinoma. (NCT02029157).
Keywords: biomarker, clinical study, c‐Met, hepatocellular carcinoma, tivantinib
Results of JET‐HCC.

Abbreviations
- AE
Adverse event
- CR
Complete response
- DCR
Disease control rate
- EM
Extensive metabolizer
- HBV
Hepatitis B virus
- HCC
Hepatocellular carcinoma
- HCV
Hepatitis C virus
- HGF
Hepatocyte growth factor
- IHC
Immunohistochemistry
- IRC
Independent review committee
- ITT
Intent‐to‐treat
- JET‐HCC
Japanese Evaluation of Tivantinib in Hepatocellular Carcinoma
- ORR
Overall response rate
- OS
Overall survival
- PFS
Progression‐free survival
- PM
Poor metabolizer
- PR
Partial response
- QOL
Quality of life
- RECIST
Response Evaluation Criteria in Solid Tumors
- SAE
Serious AE
- SD
Stable disease
1. INTRODUCTION
Hepatocellular carcinoma (HCC) is the seventh most common type of cancer, and the third leading cause of cancer‐related death worldwide. 1 According to the statistics in Japan in 2015, more than 30 000 individuals have died from HCC. 2 Systemic therapies are one of the options for the treatment of advanced or unresectable HCC. 3 Since 2017, many agents such as lenvatinib, regorafenib, cabozantinib, ramucirumab, nivolumab, and pembrolizumab have become newly available for the systemic treatment of HCC. 4 , 5 However, before the era of those new agents, sorafenib was the only systemic treatment, 6 , 7 and none of the second‐line agents had proven clinical efficacy for patients with HCC who had failed sorafenib therapy. 4 , 5
The receptor tyrosine kinase, c‐Met, is stimulated by its ligand, HGF, and activates intracellular signaling pathways such as MEK, AKT, and STAT. 8 The aberrant activation of c‐Met is involved in cancer metastasis and invasion, 9 and the elevated expression of c‐Met and HGF is associated with poor prognosis in many types of cancers. 10 , 11 Tivantinib is an orally available, small molecular inhibitor of c‐Met 12 that was clinically developed in many types of cancers, including non‐small‐cell lung cancer, gastric cancer, and HCC. 13 , 14 , 15 In humans, tivantinib is mainly metabolized by CYP2C19, which is well known for its polymorphism associated with PMs in some Asian populations, but is rare in the Western population. 15 , 16 , 17
The METIV‐HCC was a randomized, double‐blinded, placebo‐controlled phase 3 study that evaluated the efficacy and safety of tivantinib as a second‐line treatment for MET‐high HCC patients who had received one prior sorafenib‐containing regimen. 18 Patients in the METIV‐HCC were enrolled from centers in Australia, The Americas, Europe, and New Zealand from 2012‐2015, where no drugs, besides sorafenib, was approved for the systemic treatment of advanced HCC. Although the study rationale of the METIV‐HCC was clearly supported by a randomized phase 2 study (ARQ 197‐215 study), 19 however the METIV‐HCC study found that tivantinib did not improve OS relative to the placebo in the tested population. 18
The present phase 3 study, Japanese Evaluation of Tivantinib in Hepatocellular Carcinoma (JET‐HCC, NCT02029157 in ClinicalTrials.gov), was conducted independently of, and in parallel with, the METIV‐HCC. The primary objective of this study was to evaluate the efficacy of tivantinib as a second‐line treatment for Japanese patients with MET‐high HCC who had been previously treated with sorafenib. The study rationale for the JET‐HCC was provided by the ARQ 197‐215 study, which demonstrated that compared with placebo, oral tivantinib treatment could significantly improve PFS and OS in a subset of patients whose tumor samples highly expressed the c‐Met protein (MET‐high) but was not significant in all enrolled patients. 19 The tivantinib dose for Japanese patients with HCC was determined according to a previous phase 1 study in Japan. 15 Patients were screened by a biomarker test of c‐Met expression in tumor samples, and one of the inclusion criteria of the JET‐HCC was patients with MET‐high.
2. PATIENTS AND METHODS
2.1. Study design
This randomized, double‐blind, placebo‐controlled Japanese phase 3 study was conducted at 60 centers in Japan. The eligibility criteria included: age ≥20 y; histologically, cytologically, or radiologically confirmed HCC diagnosed as ineligible for both surgery resection and locoregional therapy, such as transarterial chemoembolization; radiological disease progression or intolerance to prior sorafenib‐containing therapy; Eastern Cooperative Oncology Group performance status 0 or 1; Child‐Pugh Class A; having at least 1 measurable target lesion as defined by RECIST version 1.1 20 ; a life expectancy ≥12 wk; adequate organ function; and use of contraception for a designated period.
Another key eligible criterion was confirmed “MET‐high” in archival or biopsied tumor tissue samples. Patients could submit multiple tissue samples for the c‐Met expression test if several samples were collected on different dates and/or from different lesions. c‐Met expression in the tumor samples was examined by immunohistochemistry (IHC) using the SP‐44 anti‐c‐Met rabbit monoclonal antibody (Ventana, AZ, USA) in a central laboratory, LabCorp Clinical Trials (NC, USA). As carried out in the METIV study 18 and the ARQ 197‐215 study, 19 staining intensity (0, 1, 2, or 3) and percentage of cells stained were independently scored, and then tumor tissue sample was graded as “MET‐high” if more than 50% of the tumor cells were stained as moderate (2) or strong (3). Samples that were not graded as MET‐high were defined as “MET‐low.” Eligible patients had to have at least 1 tumor sample graded as “MET‐high.”
Patients were excluded if they had: received ≥2 prior systemic therapies; prior treatment with a c‐Met inhibitor, including antibodies; systemic anticancer therapy within 2 wk before randomization; locoregional therapy and/or major surgery within the 4 wk prior to randomization; a history of cardiac disease; active clinically serious infection (defined as grade ≥3 according to Common Terminology Criteria for Adverse Events [CTCAE] version 4.0 except for HCV and HBV, respectively, infection); a history of liver transplantation; or confirmed interstitial lung disease, pleural effusion, and/or clinically significant ascites.
Enrolled patients were randomly assigned at a 2:1 ratio to receive either the tivantinib (tivantinib group) or placebo (placebo group). The randomization was stratified by the absence or presence of vascular invasion, and ECOG PS 0 or 1. The starting dose of tivantinib was 120 mg twice daily, regardless of the CYP2C19 phenotype, which was recommended according to a previous phase 1 study with Japanese HCC patients. 15 Tivantinib and placebo tablets were supplied by the sponsor Kyowa Kirin Co., Ltd., were orally administered during or immediately after meals. Dose interruption or reduction was permitted in cases of drug‐related toxicity, per the guidance prespecified in the protocol. Patients continued the study treatments until they met the discontinuation criteria, which included disease progression or safety concerns. After treatment termination, OS was confirmed every 12 wk.
This study was sponsored by Kyowa Kirin Co., Ltd., and conducted in accordance with the institutional guidelines, Good Clinical Practice guidelines, and the Declaration of Helsinki. Documented approvals from the Institutional Review Boards were obtained at every study center. All patients provided written informed consent before any study‐related procedures were carried out. The sponsor consulted the Safety Review Committee, which could recommend continuation, temporary interruption, or termination of the study.
2.2. Assessments
The baseline evaluations included vital signs, body height/weight, ECOG PS, hepatitis virus markers, Child‐Pugh score, blood cell counts, blood biochemistry, blood coagulation, urine, QOL evaluation using FACT‐Hep version 4 and EQ‐5D‐3L (both were the Japanese editions), 12‐lead electrocardiograms, genotype of CYP2C19, and tumor imaging by CT or MRI. Based on the genotype, the phenotype of CYP2C19 was classified into 2 categories: EM or PM as described previously. 15 , 16 During treatment, vital signs, blood cell counts, and blood biochemistry were assessed every week for the first 4 wk and every 2 wk thereafter. ECOG PS, Child‐Pugh score, blood coagulation, urine, biomarkers, 12‐lead electrocardiography, QOL evaluation, and tumor imaging were performed every 6 wk. Tumor response was radiologically evaluated by an IRC according to the RECIST, version 1.1. 20 AEs were continuously assessed throughout the study and graded according to the CTCAE, version 4.0. For pharmacokinetic assessment, blood samples were collected on days 1, 22, 29, and 43.
2.3. Endpoints
The primary objective was to compare PFS between the 2 groups in the ITT population. PFS was defined as the time from the date of randomization to the date of either disease progression defined by the IRC or death from any causes. The secondary endpoints included OS, best overall response, and safety. OS was defined as the time from the date of randomization to the date of death from any causes. Best overall response was determined according to RESIST as CR, PR, SD, progressive disease (PD), or non‐CR/non‐PD. ORR and DCR were defined as the proportion of patients that achieved CR/PR and CR/PR/SD/(non‐CR/non‐PD), respectively. The exploratory endpoints were pharmacokinetics and QOL, however an analysis of those exploratory endpoints has not been completely performed. The results will thus be reported in a future publication.
2.4. Statistical analysis
Assuming a randomization ratio of 2:1 for the tivantinib group and the placebo group, median PFS of 8 wk for the placebo group 19 and 13 wk for the tivantinib group (ie, Hazard Ratio (HR) of 0.6), 19 an accrual period of 96 wk, a follow‐up period of 24 wk for the last patient, 80% power, and a two‐sided significance level of .05, 144 subjects were required to detect an HR of 0.60 by a SAS POWER Procedure. When an assumed dropout rate of 10% was used, the total required sample size was 160 (tivantinib: 107 patients; placebo: 53 patients).
PFS was compared between the treatment groups using stratified log‐rank tests, adjusting for ECOG PS and vascular invasion as stratification factors to obtain point estimates of the treatment HR and 95% confidence interval (95% CI). Patients with non‐PD until the cut‐off date or who were lost to follow‐up were censored at the last confirmed evaluation date, whichever occurred first. Median PFS was estimated by the Kaplan‐Meier method for each treatment group, and the corresponding 95% CIs were calculated. A stratified Cox proportional hazard regression model was also used for subgroup analyses.
3. RESULTS
3.1. Patients and treatment
From January 2014 to June 2016, 386 patients provided informed consent. Of those, 195 patients with MET‐high tumor sample were randomly assigned to either the tivantinib group or placebo group at a 2:1 ratio. With the exception of 1 patient that discontinued the study before treatment initiation, 194 of the 195 assigned patients received either tivantinib (n = 133) or placebo (n = 61). All treated patients discontinued the study by the data cut‐off for the final analysis, and most discontinued treatment due to disease progression (Figure 1). As shown in Table 1, the demographic and baseline characteristics of the randomized patients were generally well balanced between the groups.
FIGURE 1.
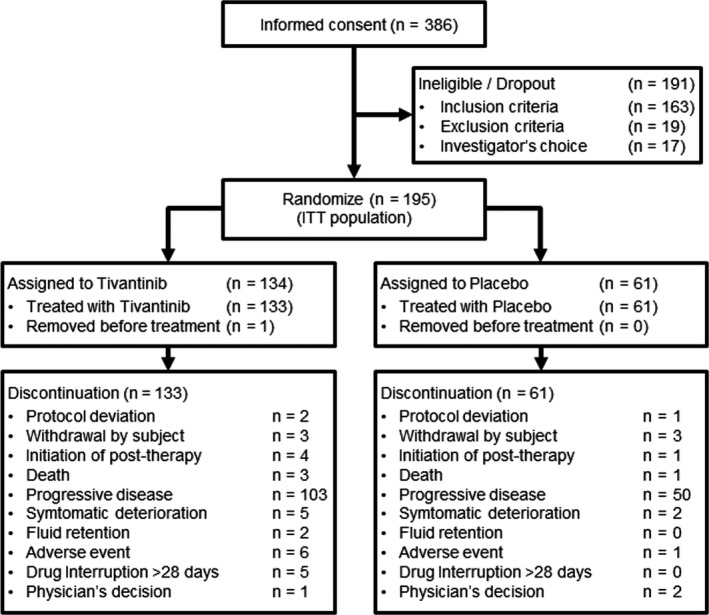
Summary of patient disposition. After providing consent, patients were screened via several tests, including a test of c‐Met expression in tumor samples. Eligible patients who had at least one tumor sample with MET‐high were randomly assigned to either the tivantinib group or the placebo group at a 2:1 ratio
TABLE 1.
Demographic and baseline characteristics of the randomized patients
| Characteristics | Number (%) | |
|---|---|---|
| Tivantinib (N = 134) | Placebo (N = 61) | |
| Age, years (median and range) | 70 (36‐86) | 72 (47‐83) |
| Sex | ||
| Male | 110 (82.1) | 52 (85.2) |
| Female | 24 (17.9) | 9 (14.8) |
| Height, cm (median and range) | 163.6 (136.2‐179) | 163.1 (136.9‐184.8) |
| Weight, kg (median and range) | 60.78 (33.4‐94.6) | 59.00 (33.0‐88.0) |
| CYP2C19 genotype | ||
| Extensive metabolizer (EM) | 119 (88.8) | 51 (83.6) |
| Poor metabolizer (PM) | 14 (10.4) | 10 (16.4) |
| ECOG performance status at randomized | ||
| 0 | 111 (82.8) | 51 (83.6) |
| 1 | 23 (17.2) | 10 (16.4) |
| Child‐Pugh classification | ||
| A | 130 (97.0) | 59 (96.7) |
| B | 4 (3.0) | 2 (3.3) |
| Cause of hepatocellular carcinoma (multiple selection) | ||
| HBV | 35 (26.1) | 16 (26.2) |
| HCV | 61 (45.5) | 31 (50.8) |
| Alcoholic | 27 (20.1) | 14 (23.0) |
| Non‐alcoholic steatohepatitis | 8 (6.0) | 3 (4.9) |
| Other | 14 (10.4) | 4 (6.6) |
| BCLC stage | ||
| 0 | 1 (0.7) | 0 |
| A | 6 (4.5) | 3 (4.9) |
| B | 38 (28.4) | 22 (36.1) |
| C | 89 (66.4) | 35 (57.4) |
| D | 0 | 1 (1.6) |
| Vascular invasion | ||
| No | 100 (74.6) | 44 (72.1) |
| Yes | 34 (25.4) | 17 (27.9) |
| Best overall response of prior sorafenib therapy | ||
| CR | 0 | 0 |
| PR | 7 (5.2) | 6 (9.8) |
| SD | 68 (50.7) | 24 (39.3) |
| Non‐CR/non‐PD | 1 (0.7) | 1 (1.6) |
| PD | 45 (33.6) | 17 (27.9) |
| NE | 13 (9.7) | 12 (19.7) |
| Reasons for discontinuation of prior sorafenib treatment | ||
| PD | 114 (85.1) | 47 (77.0) |
| Toxicity | 20 (14.9) | 14 (23.0) |
| History of locoregional therapy (multiple selection) | ||
| Surgical resection | 59 (44.0) | 26 (42.6) |
| Radiotherapy | 17 (12.7) | 6 (9.8) |
| Hepatic arterial embolization | 21 (15.7) | 6 (9.8) |
| Hepatic chemotherapeutic arterial embolization | 97 (72.4) | 45 (73.8) |
| Hepatic intra‐arterial chemotherapy | 45 (33.6) | 18 (29.5) |
| Radiofrequency ablation | 63 (47.0) | 23 (37.7) |
| Percutaneous ethanol injection therapy | 16 (11.9) | 6 (9.8) |
| Other | 9 (6.7) | 6 (9.8) |
The most frequent reason for a patient's ineligibility to participate in this study was the result of the c‐Met expression test; 127 of the 191 ineligible patients were ineligible because their samples were graded as MET‐low. In other words, c‐MET‐high rate per patients was approximately 67% in this study. In total, 539 tumor tissue samples were provided by the 386 patients who provided consent. Overall, 52.3% of the tissue samples were graded as MET‐high (Table 2). The rate of MET‐high was evidently higher in the samples which were obtained after prior sorafenib therapy (64.5%), compared with those obtained when the patient was sorafenib treatment‐naive (14.5%) (Table 2). Conversely, this study did not clarify the heterogeneity among metastases in different site at the time of the MET‐high result, because the time‐matched tumor sampling was not intended in this study.
TABLE 2.
Results of c‐Met IHC tests for patient screening
| Number (%) | ||
|---|---|---|
| MET‐high | MET‐low | |
| Overall samples of tumor tissue (N = 539) | 282 (52.3) | 257 (47.7) |
| Samples obtained at | ||
| Before sorafenib therapy (N = 131) | 19 (14.5) | 112 (85.5) |
| After sorafenib therapy (N = 408) | 263 (64.5) | 145 (35.5) |
3.2. Efficacy
The primary objective of this study was the IRC‐defined PFS in the ITT population, with 134 in the tivantinib group and 61 in the placebo group. Median PFS was 2.8 mo (95% CI, 2.7‐2.9) in the tivantinib group and 2.3 mo (95% CI, 1.5‐2.8) in the placebo group (HR, 0.74; 95% CI, 0.52‐1.04; P = .082, Figure 2A). Median OS in the ITT population was 10.3 mo (95% CI, 8.1‐11.6) in the tivantinib group and 8.5 mo (95% CI, 6.2‐11.4) in the placebo group (HR, 0.82; 95% CI, 0.58‐1.15, Figure 2B). Although the median value of both PFS was numerically greater in the tivantinib group, this study did not meet the prespecified significance level of .05.
FIGURE 2.
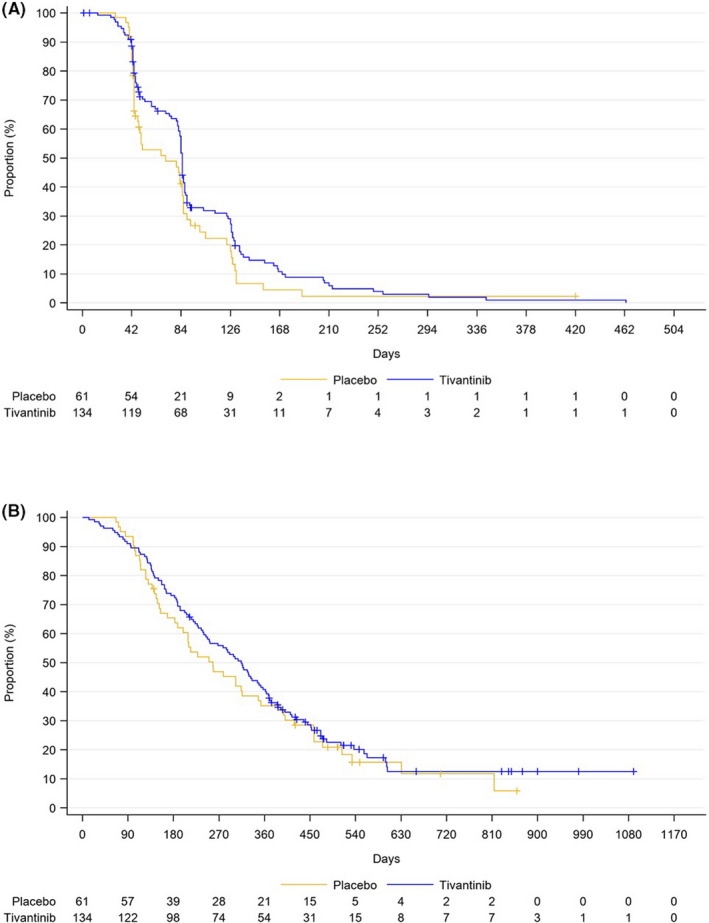
Kaplan‐Meier curves of the intent‐to‐treat (ITT) population in the tivantinib group (blue line) vs the placebo group (yellow line). A, Progression‐free survival (PFS) based on independent radiological committee. B, Overall survival (OS)
The subpopulation analyses of PFS and OS are depicted in a forest plot (Figure 3). The HR for nearly all markers indicated that the tivantinib group was favorable. PFS analysis revealed that tivantinib appeared particularly favorable in the subpopulation with early‐stage tumor, low alpha‐fetoprotein level, or absence of extrahepatic metastasis (Figure 3A), but this result was not supported by the OS analysis (Figure 3B). There was no obvious difference between CYP2C19 EM and PM.
FIGURE 3.
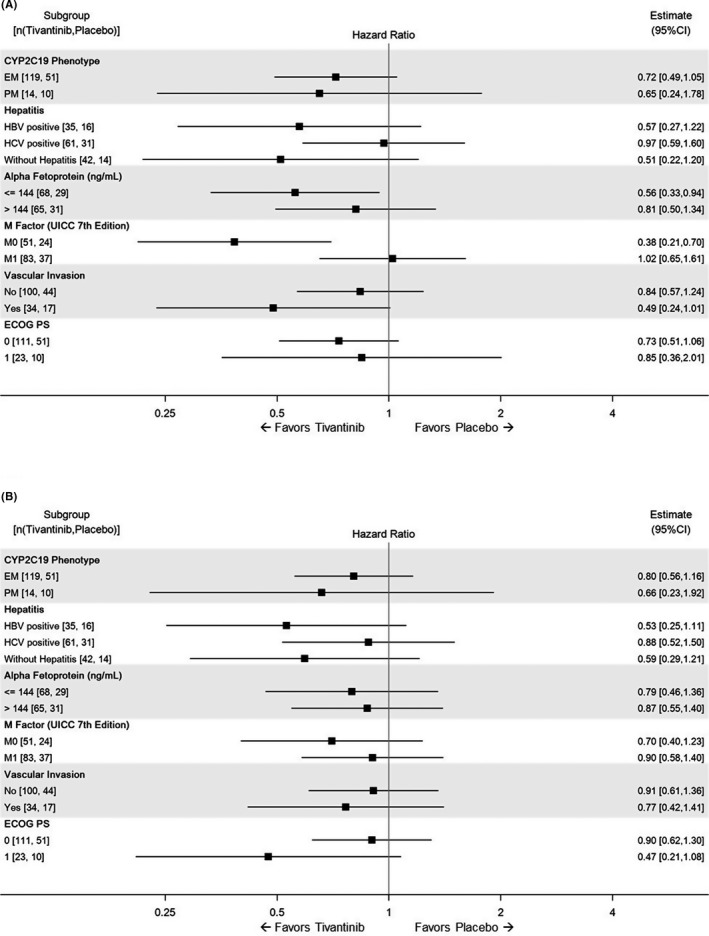
Subpopulation analysis of progression‐free survival (A) and overall survival (B) in the intent‐to‐treat population. Hazard ratios of the comparisons of the tivantinib and placebo groups are shown for the subgroups stratified by the indicated factors
The best objective response in the ITT population is summarized in Table 3. Generally, there were no significant differences between the groups, however the number of patients with PD as the best response was relatively lower in the tivantinib group. The ORR and DCR (including non‐CR/non‐PD) were generally similar between both groups: 0.7% (95% CI, 0.0‐4.1) and 67.2% (95% CI, 58.5‐75.0) in the tivantinib group, and 1.6% (95% CI, 0.0‐8.8) and 55.7% (95%CI, 42.4‐68.5) in the placebo group.
TABLE 3.
Best objective response in the ITT population
| Best overall response | Number (%) | |
|---|---|---|
| Tivantinib (N = 134) | Placebo (N = 61) | |
| CR | 0 | 0 |
| PR | 1 (0.7) | 1 (1.6) |
| SD | 82 (61.2) | 33 (54.1) |
| Non‐CR/non‐PD | 7 (5.2) | 0 |
| PD | 38 (28.4) | 27 (44.3) |
| NE | 6 (4.8) | 0 |
3.3. Safety
A safety analysis was conducted with patients in the safety analysis set who were administered at least 1 tivantinib treatment (n = 133) or placebo (n = 61). The number of patients who had at least one AE was 92.5% and 88.5% in the tivantinib group and placebo group, respectively. Serious AEs (SAEs) were reported in 30.1% of patients in the tivantinib group, with neutropenia (6.0%) and febrile neutropenia (3.8%) as the most commonly reported SAEs. The common (>5%) drug‐related AEs of any grade or grade ≥3 are shown in Table 4. Neutropenia, leukocytopenia, and anemia were the most common drug‐related AEs in the tivantinib group, however such frequent and severe hematological toxicities were not found in the placebo group. The frequency of such hematological AEs was similar between CYP2C19 EM and PM patients. Neutropenia, leukopenia, and anemia were respectively reported in 41.2%, 37.0%, and 33.6% of the tivantinib‐treated CYP2C19 EM patients (n = 119), and 42.9%, 42.9%, and 35.7% of the tivantinib‐treated CYP2C19 PM patients (n = 14). The drug‐related AEs that led to treatment interruption, dose reduction, or discontinuation were reported in 45.1% and 9.8% of patients in the tivantinib group and placebo group, respectively. The most frequent dose‐limiting toxicities in the tivantinib group were neutropenia (33.8%), leukopenia (18.8%), and anemia (8.3%). A study drug‐related death was reported in 1 patient in the tivantinib group. This patient developed febrile neutropenia and died of sepsis.
TABLE 4.
Drug‐related adverse events occurring at >5% in either of the treatment groups
| Drug‐related adverse events | Tivantinib group (N = 133) | Placebo group (N = 61) | ||||||
|---|---|---|---|---|---|---|---|---|
| Any Grade | Grade ≥ 3 | Any Grade | Grade ≥ 3 | |||||
| n % n % n % n % | ||||||||
| Neutropenia | 58 | 43.6 | 42 | 31.6 | 3 | 4.9 | 1 | 1.6 |
| Leukocytopenia | 49 | 36.8 | 33 | 24.8 | 0 | 0.0 | 0 | 0.0 |
| Anemia | 38 | 28.6 | 16 | 12.0 | 4 | 6.6 | 0 | 0.0 |
| Alopecia | 23 | 17.3 | 0 | 0.0 | 2 | 3.3 | 0 | 0.0 |
| Decreased appetite | 18 | 13.5 | 1 | 0.8 | 7 | 11.5 | 1 | 1.6 |
| Lymphocytopenia | 17 | 12.8 | 9 | 6.8 | 0 | 0.0 | 0 | 0.0 |
| Malaise | 14 | 10.5 | 0 | 0.0 | 4 | 6.6 | 0 | 0.0 |
| Sinus bradycardia | 12 | 9.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| Thrombocytopenia | 11 | 8.3 | 2 | 1.5 | 0 | 0.0 | 0 | 0.0 |
| Bradycardia | 10 | 7.5 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| Pyrexia | 9 | 6.8 | 0 | 0.0 | 2 | 3.3 | 0 | 0.0 |
| Edema peripheral | 9 | 6.8 | 0 | 0.0 | 1 | 1.6 | 0 | 0.0 |
| Diarrhea | 8 | 6.0 | 1 | 0.8 | 5 | 8.2 | 1 | 1.6 |
| Febrile neutropenia | 8 | 6.0 | 8 | 6.0 | 0 | 0.0 | 0 | 0.0 |
| Rash | 7 | 5.3 | 0 | 0.0 | 2 | 3.3 | 0 | 0.0 |
4. DISCUSSION
To date, 3 randomized studies have been conducted to compare tivantinib to placebo in HCC patients who have received a prior sorafenib therapy. The first of these 3 studies was a phase 2 study, ARQ 197‐215, in which HCC patients in Western countries were enrolled. 19 The subpopulation analysis in this phase 2 study demonstrated that tivantinib significantly prolonged PFS and OS in the subpopulation patients with MET‐high tumor (ie, this did not occur in all enrolled patients including those with MET‐low tumor). 19 Therefore, this phase 2 study provided the rationale for the 2 subsequent phase 3 studies, the METIV‐HCC in the Western population 18 and the JET‐HCC study in the Japanese population. The JET‐HCC study demonstrated that oral daily tivantinib did not significantly prolong PFS in Japanese patients with MET‐high HCC who had relapsed or were refractory or intolerant to a prior treatment with sorafenib. Consequently, not only the METIV‐HCC, 18 but also the JET‐HCC did not meet their primary endpoints (OS and PFS, respectively), and neither could confirm the hypothesis generated by ARQ 197‐215. 19
The potential reasons for the inconsistency between the phase 2 and phase 3 studies were variously provided have been discussed extensively in the previous report of the METIV‐HCC. 18 Of those, in our discussion, we would like to emphasize the difference in the enrolled population between the phase 2 and phase 3 studies (ie, the difference derived from the biomarker‐driven patient selection). The ARQ 197‐215 study enrolled HCC patients regardless of c‐Met expression, and the IHC‐based assessment to grade MET‐high or MET‐low was retrospectively conducted after the enrollment. Conversely, in the phase 3 studies, patients who provided consent had to confirm the c‐Met expression levels in their tumor during the screening test for eligibility. Thereafter, they were randomized for treatment initiation if their tumor sample was graded as MET‐high. Figure 4 illustrates the procedure in each study from consent to randomization. It is notable that the IHC‐based assessment of c‐Met was similar among the phase 2 and phase 3 studies. As shown in Table 2, most MET‐high results were observed in the tissue samples obtained from patients who had a history of a prior sorafenib treatment. This finding indicated that those samples were newly obtained by biopsy after a patient consented to participate in the phase 3 studies and which was implementing a biomarker‐driven patient selection. However, the patients in the phase 3 studies had to wait to obtain their results from the c‐Met expression test, which consisted of many steps: consenting, biopsy collection, sample shipment to the US laboratory, IHC staining, pathological reviewing, and reporting. Such procedures required a longer time from informed consent to randomization of patients in this study than the ARQ197‐215 study. It is assumed that the duration of the biomarker test might cause a bias as patients with early progress during the screening period may have dropped out of the study. Conversely, JET‐HCC and METIV‐HCC comprised many patients who could wait for the c‐Met expression result. As described previously, MET‐high is a poor prognostic factor for HCC. 11 Thus, it is assumed that several early progressions may occur after consent to the phase 3 studies. This assumption might be supported by the result that survival data of the placebo group with MET‐high was the shortest in the ARQ 197‐215 study. Median PFS were 1.4, 2.0, and 2.3 mo, and median OS were 3.8, 9.1, and 8.5 mo in ARQ 197‐215, METIV‐HCC, and JET‐HCC, respectively. 18 , 19 Based on the discussion above, a point is suggested to improve the planning and designing of a biomarker‐driven clinical study in the future. The duration of biomarker test for patient selection may sometimes be long enough to affect the study population consisting of patients with poor prognosis. When planning or designing a phase 3 study based on a phase 2 study, differences in study design should be taken into account, as shown in Figure 4, as these could change the study population and lead to different results between phase 2 and phase 3 studies.
FIGURE 4.
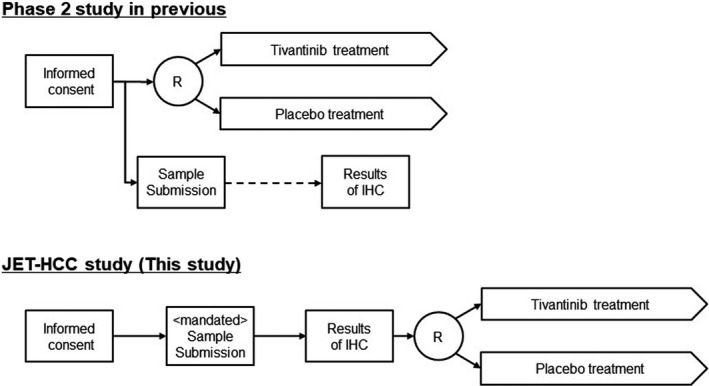
The differences in the procedure between the previous phase 2 and the JET‐HCC study (phase 3). Each text box indicates each step in the studies and the time at which these steps were taken is indicated (from left to right). The circled R indicates randomization. The scheme of the phase 2 study is summarized from reference 18
Another issue of the biomarker‐driven patient selection study may involve the control of the number of enrollments. In the JET‐HCC study, a new consent was closed when enrollment had reached the established number of 160, which was the planned sample size of the JET‐HCC. Thus, patients who gave consent before the closure could be registered, with an allowance of 3 mo. JET‐HCC was a last‐line study for HCC, and no standard treatment was established at the time of the JET‐HCC enrollment. During the extended time of 3 mo, patients who had already provided consent could proceed and wait for the c‐Met expression test. Consequently, additional patients were registered and 195 patients were enrolled.
No new findings were observed for the safety profile of tivantinib in Japanese patients with HCC. As indicated from the previous Japanese phase 1 study with HCC patients, 15 hematological toxicities were the most common AE in the tivantinib group of the JET‐HCC study. For example, neutropenia at any grade and that at grades ≥3 were 43.6% and 31.6% in the JET‐HCC study (n = 133, tivantinib‐treated patients), and 46.4% and 28.6% in the previous phase 1 study with HCC patients (n = 28). 15 Additionally, both Japanese tivantinib studies revealed that the frequency and severity of tivantinib‐induced hematotoxicity were generally similar between CYP2C19 EM and PM. 15 The agreement between these Japanese studies may suggest that the safety profile of tivantinib was well evaluated in both studies of Japanese patients with HCC.
Compared with the METIV‐HCC study, the JET‐HCC study provided 2 notably different results: a higher rate and severity of neutropenia and a larger split in the Kaplan‐Meier curve for PFS. In the METIV‐HCC study, neutropenia at any grade and that at grades ≥3 were 8% and ~4%, respectively. These values were nearly one‐fifth to one‐eighth lower than those of JET‐HCC. 18 Based on a prior early phase study, the plasma concentration of tivantinib would be well associated with neutropenia in frequency and severity. 16 Therefore, although both studies administered a tivantinib dose of 120 mg twice daily, the plasma concentration of tivantinib would be higher in Japanese patients in the JET‐HCC study than in the Western population in the METIV‐HCC study. 18 Generally, the body weight of members of the Asian population is less than that of individuals in the Western population and, therefore, a higher plasma concentration in Asian patients may occur if the same dose level is administered. Additionally, in general, the higher plasma concentration of an anticancer agent may have more opportunity for attainment of efficacy. The HRs from the PFS curves generated to compare the tivantinib group and placebo group were 0.74 and 0.96 in JET‐HCC and METIV‐HCC, respectively. 18 Collectively, a PK analysis is important to compare these 2 studies, and a further evaluation is warranted to explain the cause of the difference. Altogether, such findings may enable us to evaluate more precisely the therapeutic window of tivantinib in patients with HCC.
In conclusion, the present JET‐HCC study did not confirm the significant efficacy of tivantinib as a second‐line treatment for Japanese patients with MET‐high HCC. In fact, the JET‐HCC was negative in accordance with the Western phase 3 METIV‐HCC study that evaluated the effect of tivantinib in a similar population. Although JET‐HCC was a negative study, it has provided important implications for future studies employing a biomarker‐driven patient selection.
DISCLOSURE
The study was designed under the responsibility of Kyowa Kirin Co., Ltd. The study was funded by Kyowa Kirin Co., Ltd. Study drug was provided by Kyowa Kirin Co., Ltd; Kyowa Kirin Co., Ltd collected and analyzed the data and contributed to the interpretation of the study. All authors had full access to all of the data in the study and had final responsibility for the decision to submit for publication. Masatoshi Kudo has received honoraria, research funds and/or grants from Abbvie, Bayer, Bristol‐Myers Squibb, EA Pharma, Eisai, Eli Lilly, Gilead, MSD, Ono Pharmaceutical, Otsuka Pharmaceutical, Roche, Sumitomo Dainippon Pharma, Taiho Pharmaceutical, and Takeda Pharmaceutical. Manabu Morimoto has received research funds from MSD, Ono Pharmaceutical, Boehringer Ingelheim, Eli Lilly, AstraZeneca, and Takeda Pharmaceutical. Namiki Izumi and Michihisa Moriguchi have received honoraria from Bayer and Eisai. Tetsuji Takayama has received grants from Abbvie, Eisai, Taiho Pharmaceutical, Daiichi Sankyo, Eli Lilly, Bayer, Teijin Pharma, and Zeria Pharmaceutical. Keisuke Hino has received honoraria and/or grants from MSD, Abbvie, Gilead, Sumitomo Dainippon Pharma, and Otsuka Pharmaceutical. Hitoshi Yoshiji, Tetsuhiro Chiba, Junko Kato and Kentaro Yasuchika have no conflicts of interest to declare. Kenta Motomura has received honoraria from Eisai. Akio Ido has received honoraria, research funds and/or grants from Gilead, Abbvie, Bristol‐Myers Squibb, EA Pharma, Fujifilm, Otsuka Pharmaceutical, Eisai, and SNBL, and belongs to an endowed chair funded by Eisai. Takashi Sato and Daisuke Nakashima have been employees of Kyowa Kirin. Kazuomi Ueshima has received honoraria from Eisai, Bayer, and Eli Lilly. Masafumi Ikeda has received honoraria and/or research funds from Bayer, Eisai, Eli Lilly, Novartis, Taiho Pharmaceutical; Bristol‐Myers Squibb, AstraZeneca, Chugai Pharmaceutical, MSD, Ono Pharmaceuticals, Yakult Honsha, Aslan Pharmaceutical, Merck Serono, and J‐Pharma. Takuji Okusaka has received research funds from AstraZeneca, Chugai Pharmaceutical, Eisai, Novartis, and Bristol‐Myers Squibb. Kazuo Tamura has received remuneration from AC Medical. Junji Furuse has received honoraria and/or research funds from Bayer, Ono Pharmaceutical, Eisai, Taiho Pharmaceutical, Fujifilm, Chugai Pharmaceutical, Astellas Pharma, Novartis, Yakult Honsha, Teijin Pharma, MSD, Sumitomo Dainippon Pharma, J‐Pharma, and AstraZeneca.
ACKNOWLEDGMENT
We thank the patients, their families, caregivers, and all personnel who contributed to patient care and data collection in the present study of tivantinib. We also thank the members of the Safety Review Committee (Drs. Kiyohiko Hatake, Akihiko Gemma, and Satoshi Mochida). This research was sponsored by Kyowa Kirin Co., Ltd.
Kudo M, Morimoto M, Moriguchi M, et al. A randomized, double‐blind, placebo‐controlled, phase 3 study of tivantinib in Japanese patients with MET‐high hepatocellular carcinoma. Cancer Sci. 2020;111:3759–3769. 10.1111/cas.14582
REFERENCES
- 1. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941‐1953. [DOI] [PubMed] [Google Scholar]
- 2. Kudo M. Surveillance, diagnosis, treatment, and outcome of liver cancer in Japan. Liver Cancer. 2015;4:39‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kudo M. Clinical practice guidelines for hepatocellular carcinoma differ between Japan, United States, and Europe. Liver Cancer. 2015;4:85‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kudo M. A new era of systemic therapy for hepatocellular carcinoma with regorafenib and lenvatinib. Liver Cancer. 2017;6:177‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Faivre S, Rimassa L, Finn RS. Molecular therapies for HCC: Looking outside the box. J Hepatol. 2020;72:342‐352. [DOI] [PubMed] [Google Scholar]
- 6. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378‐390. [DOI] [PubMed] [Google Scholar]
- 7. Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia‐Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double‐blind, placebo‐controlled trial. Lancet Oncol. 2009;10:25‐34. [DOI] [PubMed] [Google Scholar]
- 8. Peters S, Adjei AA. MET: a promising anticancer therapeutic target. Nat Rev Clin Oncol. 2012;9:314‐326. [DOI] [PubMed] [Google Scholar]
- 9. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834‐848. [DOI] [PubMed] [Google Scholar]
- 10. Qiao H, Hung W, Tremblay E, et al. Constitutive activation of met kinase in non‐small‐cell lung carcinomas correlates with anchorage‐independent cell survival. J Cell Biochem. 2002;86:665‐677. [DOI] [PubMed] [Google Scholar]
- 11. Wang ZL, Liang P, Dong BW, et al. Prognostic factors and recurrence of small hepatocellular carcinoma after hepatic resection or microwave ablation: a retrospective study. J Gastrointest Surg. 2008;12:327‐337. [DOI] [PubMed] [Google Scholar]
- 12. Munshi N, Jeay S, Li Y, et al. ARQ 197, a novel and selective inhibitor of the human c‐Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9:1544‐1553. [DOI] [PubMed] [Google Scholar]
- 13. Yoshioka H, Azuma K, Yamamoto N, et al. A randomized, double‐blind, placebo‐controlled, phase III trial of erlotinib with or without a c‐Met inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall‐cell lung cancer harboring wild‐type epidermal growth factor receptor (ATTENTION study). Ann Oncol. 2015;26:2066‐2072. [DOI] [PubMed] [Google Scholar]
- 14. Kang YK, Muro K, Ryu MH, et al. A phase II trial of a selective c‐Met inhibitor tivantinib (ARQ 197) monotherapy as a second‐ or third‐line therapy in the patients with metastatic gastric cancer. Invest New Drugs. 2014;32:355‐361. [DOI] [PubMed] [Google Scholar]
- 15. Okusaka T, Aramaki T, Inaba Y, et al. Phase I study of tivantinib in Japanese patients with advanced hepatocellular carcinoma: Distinctive pharmacokinetic profiles from other solid tumors. Cancer Sci. 2015;106:611‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamamoto N, Murakami H, Nishina T, et al. The effect of CYP2C19 polymorphism on the safety, tolerability, and pharmacokinetics of tivantinib (ARQ 197): results from a phase I trial in advanced solid tumors. Ann Oncol. 2013;24:1653‐1659. [DOI] [PubMed] [Google Scholar]
- 17. Kubota T, Chiba K, Iga T. Frequency distribution of CYP2C19, CYP2D6, and CYP2C9 mutant‐alleles in several different populations. Xenobio Metabol Dispos. 2001;16:69‐74. [Google Scholar]
- 18. Rimassa L, Assenat E, Peck‐Radosavljevic M, et al. Tivantinib for second‐line treatment of MET‐high, advanced hepatocellular carcinoma (METIV‐HCC): a final analysis of a phase 3, randomised, placebo‐controlled study. Lancet Oncol. 2018;19:682‐693. [DOI] [PubMed] [Google Scholar]
- 19. Santoro A, Rimassa L, Borbath I, et al. Tivantinib for second‐line treatment of advanced hepatocellular carcinoma: a randomised, placebo‐controlled phase 2 study. Lancet Oncol. 2013;14:55‐63. [DOI] [PubMed] [Google Scholar]
- 20. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228‐247. [DOI] [PubMed] [Google Scholar]