

Abstract
Aside from the induction of cell death, some anticancer chemotherapeutic drugs can modulate antitumor immune responses. In this study, we examined the anticancer effects of 5‐fluorouracil (5‐FU) and oxaliplatin (L‐OHP), which are standard chemotherapeutic drugs for colon cancer, combined with cyclophosphamide (CP) in two mouse colon cancer models (CT26 and MC38 colon adenocarcinoma models). In the CT26 model, two injections of 5‐FU/L‐OHP and CP significantly suppressed the growth of subcutaneously established CT26 tumors compared with either 5‐FU/L‐OHP or CP, without a significant loss of body weight. The anticancer effect was weakened in nude mice. Cured mice acquired protective immunity against CT26, and CT26‐specific cytotoxic T cells (CTLs) were induced from their spleen cells. Analysis of tumor‐infiltrating immune cells revealed that 5‐FU/L‐OHP treatment with or without CP increased the proportion of CD8+ T cells at tumor sites. The 5‐FU/L‐OHP treatment decreased the proportion of granulocytic myeloid‐derived suppressor cells (MDSCs) and increased monocytic MDSCs in tumor sites, whereas the addition of CP treatment reversed these changes. In the MC38 model, although significant anticancer effects of the triple combination therapy were seen, additional treatment with anti‐PD‐1 antibody increased the number of cured mice. These mice exhibited protective immunity against MC38, and MC38‐specific CTLs were generated from their spleen cells. Together, these results indicate that the antitumor effects of the combination of 5‐FU/L‐OHP and CP mainly depend on host T cells; moreover, the therapeutic efficacy can be effectively boosted by immune checkpoint blockade.
Keywords: anti‐PD‐1 antibody, chemotherapy, colon cancer, cytotoxic T lymphocytes, immunotherapy
Antitumor effects induced by the triple combination chemotherapy with 5‐FU/L‐OHP and cyclophosphamide.

1. INTRODUCTION
Numerous clinical studies on cancer immunotherapy have revealed that immune checkpoint blockade (ICB) therapy is effective against various types of cancers, including gastrointestinal cancer. 1 , 2 , 3 , 4 Programmed cell death 1 (PD‐1) blockade therapy has been approved for multiple malignancies, although its clinical effectiveness is limited; the overall response rate is only 20‐30%. 5 , 6 In addition, although colorectal cancer (CRC) with high microsatellite instability (MSI‐H) has been reported to be sensitive to PD‐1 blockade therapy, 7 the overall response rate for advanced colon cancer is only approximately 5‐6%. 8 Therefore, novel treatment modalities are needed to improve the therapeutic efficacy of ICB therapy for CRC patients. Given that the antitumor effects of PD‐1 blockade therapy depend on the presence of antitumor T cells at tumor sites, 9 methods to promote the infiltration of T cells into tumor sites are required.
In addition to direct cytotoxic effects on cancer cells, some anticancer chemotherapeutic drugs have the potential to modulate the immune system and augment antitumor responses in cancer‐bearing hosts when administered at low or moderate doses. 10 , 11 Recent studies have revealed the underlying mechanisms, namely regulation of the immunogenicity of tumor cells, 12 mitigation of regulatory T cell (Treg)–mediated immunosuppression, 13 reduction of myeloid‐derived suppressor cells (MDSCs), 14 induction of mature dendritic cells (DCs), 15 and induction of the homeostatic proliferation of T cells. 16 More specifically, cyclophosphamide (CP) decreases immunosuppression by Tregs when given at low/medium doses. 17 , 18 Some chemotherapeutic drugs, including doxorubicin and oxaliplatin (L‐OHP), can induce immunogenic cancer cell death. 11 , 19 Gemcitabine and 5‐fluorouracil (5‐FU) decrease MDSCs in cancer‐bearing hosts. 14 , 20 Theoretically, combining these chemotherapeutic drugs could mitigate immunosuppression by Tregs and MDSCs, and induce immunogenic cancer cell death, thereby promoting T cell infiltration into tumor sites.
5‐FU/L‐OHP are standard chemotherapeutic drugs for colon cancer. Recently, a study revealed that one injection of 5‐FU/L‐OHP resulted in tumor regression when combined with anti‐PD‐1 antibody in mouse colon cancer models. 21 On the other hand, we reported that CP is a useful immunomodulatory drug when used in combination with ICB antibodies. 22 , 23 In this study, we combined 5‐FU/L‐OHP with CP (triple combination chemotherapy). These drugs were administered twice at 8‐day intervals in two mouse colon cancer models. We demonstrated that the triple combination chemotherapy had superior antitumor effects to 5‐FU/L‐OHP therapy alone. We also found that the antitumor effects of the triple combination therapy were mainly attributable to host T cells, and that therapeutic efficacy could be boosted by ICB therapy.
2. MATERIALS AND METHODS
2.1. Mice, tumor cell lines, and reagents
Wild‐type BALB/c, BALB/c nu/nu, and C57BL/6 female mice (6‐7 weeks old) were purchased from CLEA Japan Inc and kept under specific pathogen‐free conditions. Experiments were carried out in accordance with the ethical guidelines for animal experiments of the Shimane University Faculty of Medicine (IZ30‐104, IZ31‐41, and IZ31‐66). CT26 and MC38 are colon carcinomas originating in BALB/c and C57BL/6 mice, respectively. MC38 was kindly provided by Dr Toshiyasu Ojima of the Department of Surgery, Wakayama Medical University. Both cell lines were maintained in RPMI 1640 media supplemented with 10% FCS and 20 μg/ml gentamicin (Sigma‐Aldrich) at a temperature of 37℃ in an atmosphere with 5% CO2. CP was purchased from Shionogi Co. Ltd. 5‐FU and L‐OHP were purchased from Nacalai Tesque, Inc and LCL Labo, respectively.
2.2. Treatment protocols
Wild‐type BALB/c mice were injected subcutaneously (s.c.) with 5.0 × 105 CT26 cells into the right flank. The mice were randomly divided into four groups 10 days after tumor inoculation. On days 10 and 18, the mice were injected intraperitoneally (i.p.) with either one or all of CP (50 mg/kg), 5‐FU (50 mg/kg), and L‐OHP (6 mg/kg). The doses of 5‐FU and L‐OHP were based on a previous report. 21 For BALB/c nude mice, the drugs were injected i.p. on days 8 and 16 because tumor growth is more rapid in nude mice compared to wild‐type BALB/c mice. The tumor volume (mm3) and body weight were measured every 4 days. The tumor volume was calculated as follows: tumor volume = (length × width2)/2. In the MC38 model, C57BL/6 mice were injected s.c. with 5.0 × 105 MC38 cells into the right flank. On days 10 and 18, the mice were injected i.p. with the indicated doses of chemotherapeutic drugs. On days 11 and 19, the mice were injected i.p. with anti‐PD‐1 monoclonal antibody (mAb; 200 μg/mouse) (clone RMP 1‐14; Bio X Cell Inc) at a volume of 200 μL. In some experiments, the same volume of rat IgG (200 μg/mouse) (Sigma‐Aldrich) was injected i.p. as a vehicle control. When tumors were not palpable at least 1 month after complete regression after therapy, the mice were judged as “cured.”
2.3. Assay of protective immunity
Control naïve or cured mice (after therapy) were injected s.c. with 5.0 × 105 CT26 or 2.5 × 105 MC38 cells into the flank. After tumor inoculation, tumor volume (mm3) was measured every 4 days.
2.4. Assay of cytotoxicity
To examine the cytotoxicity against cancer cells, the spleen cells of naïve or cured mice were cultured with the indicated tumor peptide and 20 U/mL interleukin (IL)‐2 for 4 days. H‐2Ld‐binding AH1 peptide (SPSYVYHQF) 24 and H‐2Kb‐binding p15E peptide (KSPWFTTL), 25 both derived from the envelope protein (gp70) of an endogenous murine leukemia virus, were used as tumor antigenic peptides for CT26 and MC38, respectively. All peptides were > 80% pure and were purchased from Invitrogen. Cytotoxicity was measured using a 5‐h 51Cr‐release assay.
2.5. Flow cytometry
To analyze tumor‐infiltrating immune cells, tumor tissues were resected from mice individually, 22 days after tumor inoculation (4 days after the second chemotherapy treatment). The tumors were minced, placed on glass slides, and passed through gauze mesh and nylon mesh immediately before flow cytometry. The following mAbs were used: APC‐conjugated anti‐CD45 mAb (BioLegend), FITC‐conjugated anti‐Gr‐1 mAb (BioLegend), PE‐conjugated anti‐CD11b mAb (BioLegend), FITC‐conjugated anti‐CD8 mAb (BioLegend), PE‐conjugated anti‐CD4 mAb (BioLegend), and PE/cy7‐conjugated anti‐Ly6C mAb (BioLegend). Similarly, spleen cells of tumor‐bearing and treated mice were analyzed. Analysis was performed using FACSCalibur (Becton‐Dickinson).
2.6. Statistical analysis
For the analysis of parametric and nonparametric data, Student's t test (two groups) and ANOVA with Tukey's post hoc test (more than two groups) were used. All tests were performed with JMP Pro14 software (SAS Institute Inc). P‐values < 0.05 were considered statistically significant.
3. RESULTS
3.1. T cell–dependent antitumor effects of the combination of 5‐FU/L‐OHP and CP in CT26‐bearing mice
We first examined the effect of combination therapy of a single administration of 5‐FU/L‐OHP and/or CP on day 10 after CT26 inoculation. 5‐FU/L‐OHP treatment with or without CP suppressed the tumor growth. However, the difference in tumor growth was not statistically significant compared with the untreated group (Fig. S1A–C). The tumor growth was quite variable in mice treated with CP alone.
Next, 5‐FU/L‐OHP and/or CP was administered twice, on days 10 and 18 after tumor inoculation. Although 5‐FU/L‐OHP treatment alone suppressed the tumor growth, the addition of CP to the regimen augmented the antitumor effects (Figure 1A and B). A significant difference was observed between the 5‐FU/L‐OHP plus CP triple combination therapy and either therapy alone (Figure 1C). The triple combination therapy cured two out of six mice. No body weight loss, which is considered a systemic adverse event, was observed in mice treated with any therapy (Figure 1D and E). In addition, two out of three cured mice, which were prepared by two independent experiments, rejected rechallenge with CT26, while no such rejection was observed in naïve mice (Figure 1F). Furthermore, higher levels of cytotoxicity against CT26 cells were generated in vitro from the spleen cells of cured mice compared with naïve mice after stimulation with the CT26 tumor antigenic peptide AH1 (Figure 1G). 24 Apparent cytotoxicity against CT26 was induced from the spleen cells of cured mice without in vitro stimulation of the AH1 peptide, which was almost half of that induced by the AH1 stimulation (Fig. S2A).
Figure 1.
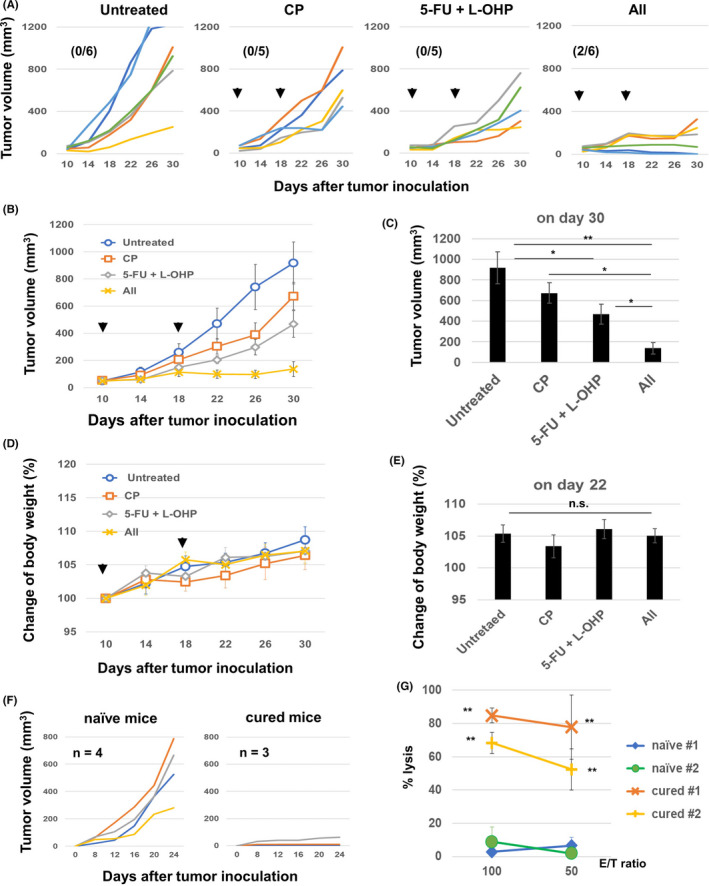
In vivo antitumor effects of the combination of chemotherapeutic drugs. A and B, BALB/c mice were injected subcutaneously (s.c.) with 5.0 × 105 CT26 cells into the right flank. On days 10 and 18, these mice were injected intraperitoneally with CP (50 mg/kg) and/or 5‐FU (50 mg/kg) and L‐OHP (6 mg/kg). The tumor volume was calculated as follows: (length × width2)/2. The arrows indicate the drug injection times. The data are presented as means ± SEM. The number in parentheses represents the ratio of cured mice to total mice. C, Tumor volume on day 30. *P < 0.05, **P < 0.01 (ANOVA). D and E, Similarly, the body weight was measured, and the tumor size data on day 22 are shown as means ± SEM. F, The cured BALB/c mice were inoculated s.c. with 5.0 × 105 CT26 cells 3 months after the last therapy (n = 3). Naïve mice were also inoculated with CT26 cells (n = 4). G, The spleen cells from cured and naïve mice were harvested and cultured with the AH1 peptide in the presence of IL‐2 (20 U/mL) for 4 days. The cytotoxicity of the cultured cells against CT26 cells was examined using a 5‐h 51Cr‐release assay. CP, cyclophosphamide; 5‐FU, 5‐fluorouracil; L‐OHP, oxaliplatin; n.s, not significant
We then determined whether the effects of combination chemotherapy with 5‐FU/L‐OHP and/or CP could occur in nude mice. 5‐FU/L‐OHP therapy, with or without CP, marginally but significantly suppressed CT26 tumor growth (Figure 2A–C). Given that the antitumor effects in nude mice were apparently weaker compared with those in wild‐type mice (Figure 1B and C), the triple combination chemotherapy with 5‐FU/L‐OHP and CP was suggested to be mainly dependent on host T cells.
Figure 2.
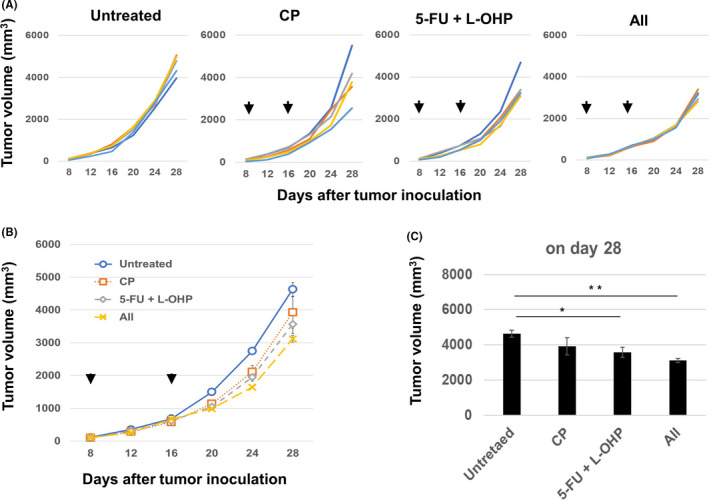
Antitumor effects of triple combination chemotherapy in nude mice. A, BALB/c nu/nu mice were injected subcutaneously with 5.0 × 105 CT26 cells. On days 8 and 16, the mice were injected intraperitoneally with CP (50 mg/kg) and/or 5‐FU (50 mg/kg) and L‐OHP (6 mg/kg). The arrows indicate the drug injection times. The tumor volume was calculated as follows: (length × width2)/2. B, Means ± SEM of five mice. C, Tumor size data on day 28 as means ± SEM. * P < 0.05, ** P < 0.01 (ANOVA). CP, cyclophosphamide; 5‐FU, 5‐fluorouracil; L‐OHP, oxaliplatin
3.2. T cells in CT26‐bearing mice treated with combined chemotherapy
Next, we examined T cells in treated mice with CT26 tumors. Although there was no difference in the proportion of CD45+ immune cells among tumor sites (Figure 3A), treatment with 5‐FU/L‐OHP significantly increased the proportions of CD4+ T cells and CD8+ T cells (Figure 3B). The increase in the proportion of CD8+ T cells was particularly drastic. The additional CP treatment decreased the proportion of CD4+ T cells but showed no change in that of CD8+ T cells. Representative flow cytometric results are shown in Figure 3C. We also examined T cells in spleen and found that treatment with CP or 5‐FU/L‐OHP or both significantly decreased the numbers of spleen cells (Figure 3D). In addition, treatment with CP or 5‐FU/L‐OHP or both slightly increased the proportions of CD8+ T cells (Figure 3E). Representative flow cytometry results are shown in Figure 3F.
Figure 3.
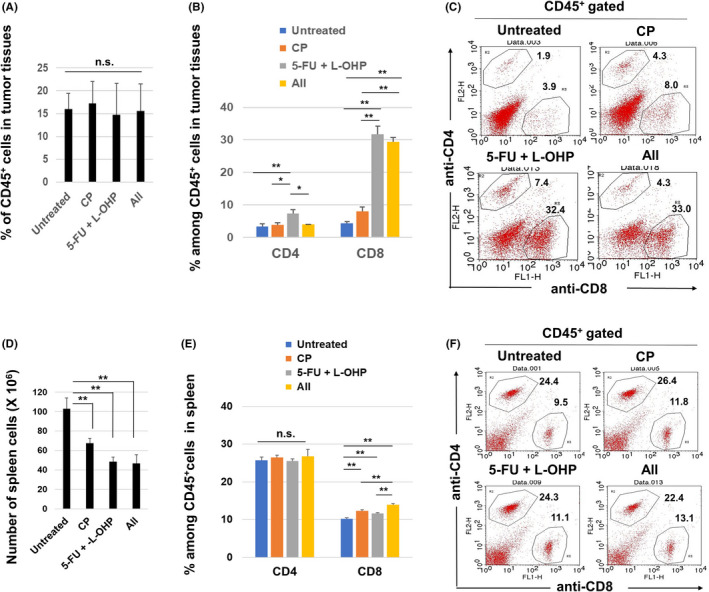
Flow cytometric analysis of T cells in CT26‐bearing mice. A, At 22 days after CT26 inoculation (4 days after the second treatment), tumor tissues were harvested, and tumor‐infiltrating CD45+ immune cells were examined. B, Percentages of CD4+ and CD8+ cells in tumor tissues. The mean ± SEM data of five mice are shown. C, Representative results; numbers are percentages. D, Similarly, spleens were harvested and their cell numbers were counted. E, Percentages of CD4+ and CD8+ cells of spleen cells. The means ± SEM of four mice are shown. F, Representative results; numbers are percentages. * P < 0.05, ** P < 0.01 (ANOVA). CP, cyclophosphamide; 5‐FU, 5‐fluorouracil; L‐OHP, oxaliplatin; n.s., not significant
3.3. MDSCs in CT26‐bearing mice treated with combined chemotherapy
Next, we examined MDSCs in treated mice with CT26 tumors. Treatment with 5‐FU/L‐OHP with or without CP decreased the proportion of CD11b+ cells among CD45+ cells at tumor sites (Figure 4A). Figure 4B shows the staining strategy used to discriminate the following MDSC subsets: CD11b+ Gr‐1high Ly6C med granulocytic MDSCs (G‐MDSCs), CD11b+ Gr‐1med Ly6C high monocytic MDSCs (M‐MDSCs), and CD11b+ Gr‐1med Ly6C med “others.” The proportion of G‐MDSCs was decreased following 5‐FU/L‐OHP treatment, while the addition of CP treatment significantly increased this population (Figure 4C). In contrast, the proportion of M‐MDSCs increased following treatment with 5‐FU/L‐OHP, while the addition of CP treatment significantly decreased this population. There was no obvious change in the “others” population, probably tumor‐associated macrophages. Representative flow cytometry results are shown in Figure 4D. We also examined MDSCs in spleen. Triple combination chemotherapy slightly decreased the proportions of CD11b+ cells among CD45+ cells in spleen (Figure 4E). In terms of MDSCs, treatment with 5‐FU/L‐OHP showed the tendency to increase the proportion of G‐MDSCs but decrease that of M‐MDSCs in spleen, and the additional CP treatment reversed these changes (Figure 4F). Representative flow cytometry results are shown in Figure 4G. Interestingly, changes of the proportions of G‐MDSCs and M‐MDSCs in tumor sites and spleen of CT26‐bearing mice were contrasting.
Figure 4.
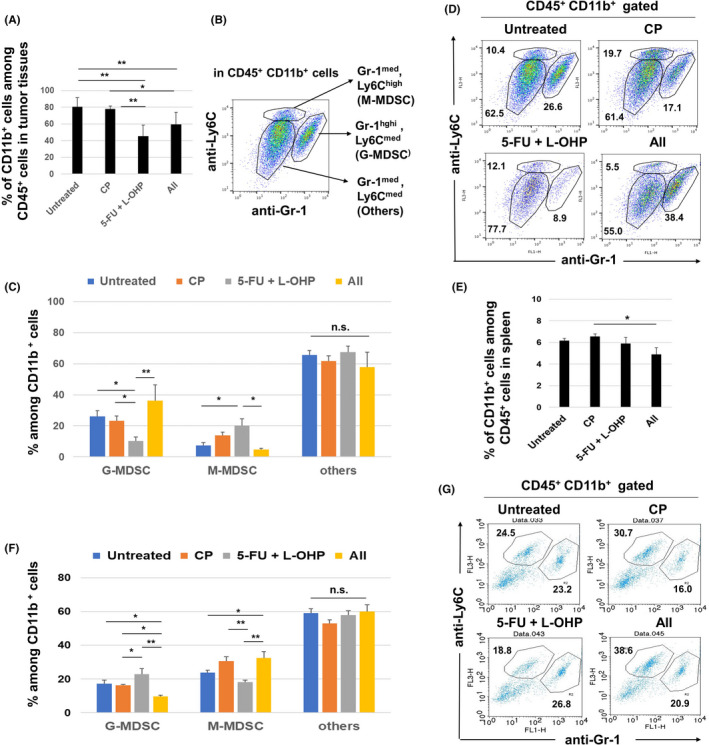
Flow cytometric analysis of MDSCs in CT26‐bearing mice. At 22 days after tumor inoculation (4 days after the second treatment), tumor tissues were harvested and analyzed by flow cytometry. A, Proportion of CD11b+ cells. B, The staining strategy used to discriminate MDSC subsets. C, Percentages of M‐MDSCs, G‐MDSCs, and “others.” The mean ± SEM data of five mice are shown. D, Representative results; numbers are percentages. E, Similarly, the proportion of CD11b+ cells in spleen is shown. F, Percentages of M‐MDSCs, G‐MDSCs, and “others.”. The means ± SEM of four mice are shown. G, Representative results; numbers are percentages. * P < 0.05, ** P < 0.01 (ANOVA). CP, cyclophosphamide; 5‐FU, 5‐fluorouracil; L‐OHP, oxaliplatin; MDSC, myeloid‐derived suppressor cell; n.s., not significant
3.4. Antitumor effects of the triple combination chemotherapy in MC38‐bearing mice
We further tested if triple combination chemotherapy was effective in MC38 colon carcinoma–bearing C57BL/6 mice. Chemotherapy with either 5‐FU/L‐OHP or CP decreased the tumor volume significantly (Figure 5A–C). The triple combination chemotherapy decreased the tumor volume to almost half that of tumors treated with either 5‐FU/L‐OHP or CP. However, no mice were cured after receiving triple combination chemotherapy and there was no obvious change in body weight (Figure 5D).
Figure 5.
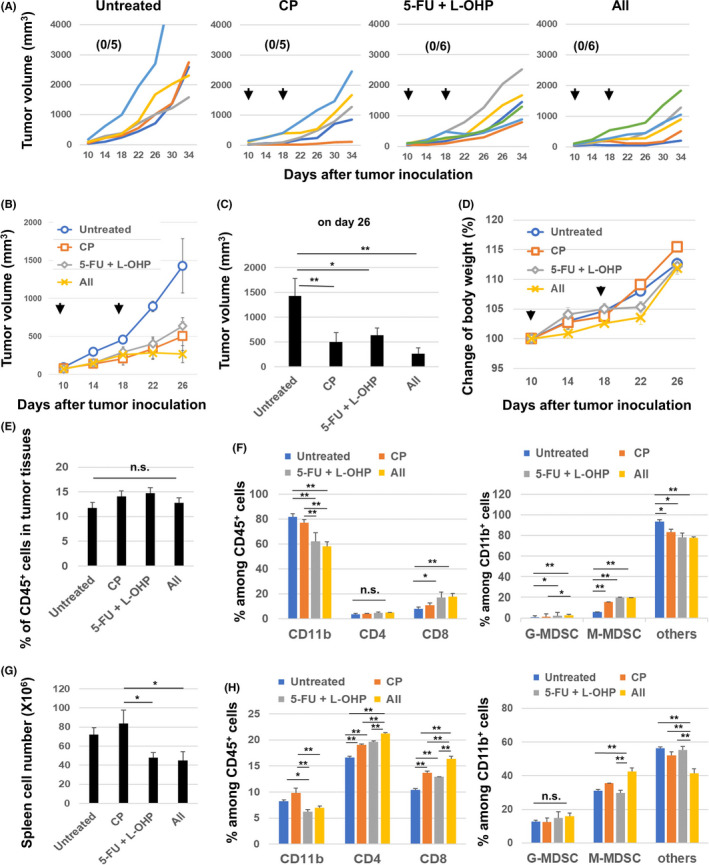
In vivo antitumor effects of the combination of chemotherapeutic drugs in the MC38 model. C57BL/6 mice were injected subcutaneously with 5.0 × 105 MC38 cells into the right flank. On days 10 and 18, the mice were injected intraperitoneally with CP (50 mg/kg) and/or 5‐FU (50 mg/kg) and L‐OHP (6 mg/kg). The tumor volume was calculated as follows: (length × width2)/2. The arrows indicate the drug injection times. The number in parentheses is the cured mice to total mice ratio. B, The data are presented as means ± SEM. C, Tumor volume data on day 26. D, Similarly, the body weight was measured every 4 days. E, At 22 days after MC38 inoculation (4 days after the second treatment), tumor tissues were harvested and tumor‐infiltrating CD45+ immune cells were examined. F, Percentages of CD11b+, CD4+, CD8+ cells, G‐MDSCs, and M‐MDSCs in tumor tissues. The means ± SEM of four mice are shown. G, Similarly, spleens were harvested, and their cell number was counted. H, Percentages of CD11b+, CD4+, CD8+ cells, G‐MDSCs, and M‐MDSCs in spleens. The means ± SEM of four mice are shown. * P < 0.05, ** P < 0.01 (ANOVA). CP: cyclophosphamide; 5‐FU: 5‐fluorouracil; L‐OHP: oxaliplatin; n.s., not significant
3.5. T cells and MDSCs in MC38‐bearing mice treated with combined chemotherapy
Treatment with CP or 5‐FU/L‐OHP or both showed no change of the proportions of CD45+ cells in tumor sites (Figure 5E). Among CD45+ cells in tumor sites, 5‐FU/L‐OH treatment decreased the proportions of CD11b+ cells but increased those of CD8+ T cells (Figure 5F). Among CD11b+ cells in tumor sites, CP or 5‐FU/L‐OHP or both increased the proportions of M‐MDSCs. Although changes of the proportions of G‐MDSC were observed, their levels were relatively low. In terms of spleen, 5‐FU/L‐OHP treatment decreased the numbers of spleen cells in MC38‐bearing mice (Figure 5G). 5‐FU/L‐OHP treatment decreased the proportions of CD11b+ cells. Treatment with CP or 5‐FU/L‐OHP or both increased the proportions of CD4+ T cells and CD8+ T cells (Figure 5H), and the triple combination increased those of M‐MDSCs among CD11b+ cells. Representative flow cytometry results are shown in Figure S3.
3.6. Antitumor effects of triple combination chemotherapy and anti‐PD‐1 antibody in MC38‐bearing mice
To improve the therapeutic efficacy of the triple combination chemotherapy, we combined it with anti‐PD‐1 antibody treatment; this did not further decrease tumor volume (Figure 6A and B). However, only one mouse was cured by the triple chemotherapy alone, whereas three out of six mice were cured after receiving the combined chemotherapy and ICB treatment. Combined chemotherapy significantly decreased the body weight of mice with MC38 tumors on day 14 after MC38 inoculation. However, the body weight loss seemed to be slightly attenuated on day 22. Triple chemotherapy and anti‐PD‐1 antibody combination therapy accelerated body weight loss up to day 22, 4 days after the second injection with anti‐PD‐1 antibody (Figure 6C and D).
Figure 6.
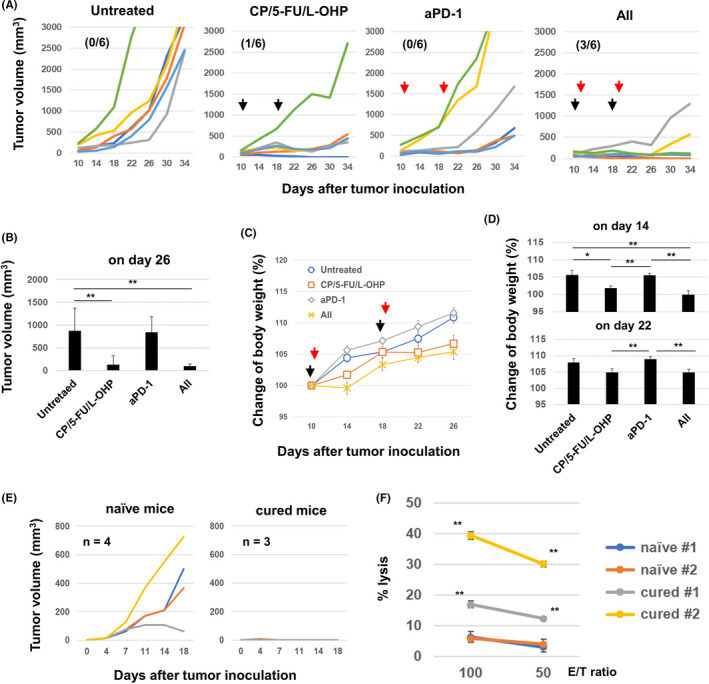
In vivo antitumor effects of the combination of chemotherapeutic drugs and anti‐PD‐1 antibody therapy. A, C57BL/6 mice were injected subcutaneously (s.c.) with 5.0 × 105 MC38 cells into the right flank. On days 10 and 18, the mice were injected intraperitoneally (i.p.) with CP (50 mg/kg), 5‐FU (50 mg/kg), and L‐OHP (6 mg/kg). On days 11 and 19, anti‐PD‐1 antibody (200 μg/mouse) was injected i.p. The tumor volume was calculated as follows: (length × width2)/2. The black and red arrows indicate the injection times of chemotherapeutic drugs and antibody, respectively. The number in parentheses represents the ratio of cured mice to total mice. B, Tumor volume data on day 26. The data are presented as means ± SEM. **P < 0.01 (ANOVA). C, D, Similarly, the body weight was measured every 4 days, and the body weight data on days 14 and 22 are shown as means ± SEM. E, The cured C57BL/6 mice were inoculated s.c. with 2.5 × 105 MC38 cells (n = 3). Naïve mice were also inoculated s.c. with 2.5 × 105 MC38 cells (n = 4). F, The spleen cells from cured and naïve mice were harvested and cultured with the p15E peptide in the presence of IL‐2 (20 U/mL) for 4 days. The cytotoxicity of the cultured cells against MC38 cells was examined using a 5‐h 51Cr‐release assay. CP, cyclophosphamide; 5‐FU, 5‐fluorouracil; L‐OHP, oxaliplatin
Next, acquired protective immunity in cured mice was investigated. Three cured mice, one mouse from the chemotherapy group and two from the group that received ICB and chemotherapy, were rechallenged with MC38. Naïve mice showed MC38 tumor growth, while cured mice rejected rechallenge with MC38 (Figure 6E). The generation of anti‐MC38 cytotoxic T cells (CTLs) in cured mice was also investigated (Figure 6F). Spleen cells from two cured mice were used. The first cured mouse (#1) was investigated 6 weeks after MC38 regression, and the second (#2) was investigated 3 weeks after rechallenge with MC38. As a result, higher levels of cytotoxicity against MC38 cells were generated from their spleen cells after in vitro stimulation with the MC38 tumor antigenic peptide p15E 25 compared with naïve mice. In particular, a high level of cytotoxicity against MC38 was induced from the spleen cells of cured mouse #2. Apparent cytotoxicity against MC38 was induced from the spleen cells of cured mice without in vitro stimulation of the p15E peptide, which was almost the same as that induced by the p15E stimulation (Fig. S2B).
3.7. Comparison of antitumor effects induced by double and triple combination chemotherapy with or without anti‐PD‐1 antibody
Finally, we compared the antitumor effects induced by double combination (5‐FU/L‐OHP) or triple combination (5‐FU/L‐OHP/CP) with anti‐PD‐1 antibody therapy (Figure 7A and B). Although the antitumor effects of 5‐FU/L‐OHP were enhanced by the additional treatment with CP, this change was not statistically significant. However, compared with the antitumor effects by anti‐PD‐1 therapy alone, additional combination with triple chemotherapy significantly enhanced the antitumor effects, whereas that with double chemotherapy was not significant (Figure 7B). These results suggest that triple combination chemotherapy is more effective than double combination chemotherapy when combined with anti‐PD‐1 therapy. On the other hand, triple combination chemotherapy induced body weight loss 14 days after MC38 inoculation, whereas the additional anti‐PD‐1 antibody therapy mitigated the body weight loss on days 22 and 26 (Figure 7C and D).
Figure 7.
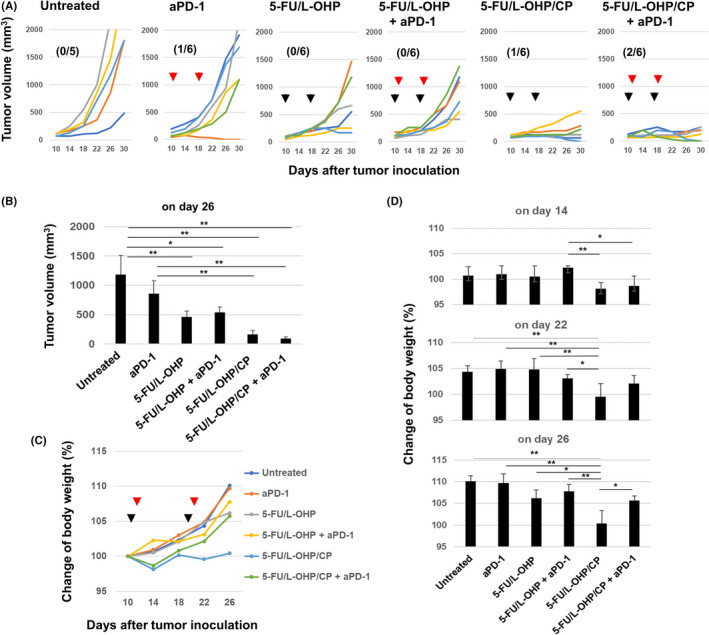
In vivo antitumor effects of double or triple combination of chemotherapeutic drugs with anti‐PD‐1 antibody therapy. A, C57BL/6 mice were injected subcutaneously (s.c.) with 5.0 × 105 MC38 cells into the right flank. On days 10 and 18, the mice were injected intraperitoneally (i.p.) with CP (50 mg/kg) or 5‐FU (50 mg/kg) and L‐OHP (6 mg/kg). On days 11 and 19, anti‐PD‐1 antibody (200 μg/mouse) was injected i.p. The tumor volume was calculated as follows: (length × width2)/2. The black and red arrows indicate the injection times of chemotherapeutic drugs and antibody, respectively. The number in parentheses represents the ratio of cured mice to total mice. B, Tumor volume data on day 26. The data are presented as means ± SEM. C, D, The body weight data on days 14, 22, and 26 are shown as means ± SEM. * P < 0.05, ** P < 0.01 (ANOVA). CP, cyclophosphamide; 5‐FU, 5‐fluorouracil; L‐OHP, oxaliplatin
4. DISCUSSION
ICB therapy has been used for treating various types of malignancies. However, to improve the therapeutic efficacy of ICB, its use in combination with other anticancer therapies is needed. When an ICB is considered in combination with anticancer immunotherapy for CRC treatment, chemotherapeutic drugs are preferred. To this end, 5‐FU and L‐OHP were used in this study.
5‐FU is an antimetabolite in the pyrimidine analog family that blocks the action of thymidylate synthase and prevents the production of DNA. In addition, Vincent et al reported that 5‐FU decreased tumor‐associated MDSCs in a mouse model of thymoma. 14 L‐OHP is a platinum‐based anticancer drug that blocks DNA duplication and causes immunogenic cell death by inducing calreticulin expression and the release of high mobility group box 1 (HMGB1). 19 Currently, 5‐FU and L‐OHP are widely used in the first‐line regimen called FOLFOX, as adjuvant chemotherapy for colon cancer. 26 Interestingly, Dosset et al revealed that a single injection of 5‐FU/L‐OHP resulted in tumor regression when combined with anti‐PD‐1 antibody in mouse colon cancer models. 21 In this study, CP was added to this combination because it is a useful immunomodulatory drug for use in combination with an ICB antibody. 22 , 23 Although CP has been used for treating hematological malignancy, as well as an immunosuppressant for autoimmune diseases and organ transplants, this drug is also known to decrease Tregs in murine models and cancer patients. 17 , 18 We compared antitumor effects induced by triple combination (5‐FU/L‐OHP/CP) and double combination (5‐FU/L‐OHP) using the MC38 model. The results were that the triple combination chemotherapy was superior to double combination chemotherapy to elicit in vivo antitumor effects when combined with or without anti‐PD‐1 antibody therapy (Figure 7A and B). CP may be a useful combination drug for FOLFOX. The triple combination chemotherapy of 5‐FU/L‐OHP and CP is expected to target two major immunosuppressive cell types, MDSCs and Tregs, while eliciting immunogenic, ie, immune response–inducing, cancer cell death.
For successful anticancer immunotherapy, the presence of immune cells in tumor microenvironments is key. 27 , 28 An increase in tumor‐infiltrating Tregs has been reported as a poor prognostic factor. 29 Similarly, it has been reported that the greater the increase in MDSCs in peripheral blood, and the greater the number of tumor‐infiltrating MDSCs, the poorer the prognosis. 30 Therefore, we examined tumor‐infiltrating immune cells in tumor tissues after the combined chemotherapy. We analyzed tumor‐infiltrating immune cells 4 days after the second therapy using the CT26 and MC38 models. As a result, 5‐FU/L‐OHP therapy with or without CP increased the proportion of CD8+ T cells in tumor sites (Figure 3B; Figure 5F). Although a similar tendency was observed in terms of spleen cells (Figure 3E; Figure 5H), the increase in the proportions of CD8+ T cells was more distinguishable in tumor sites.
We also examined MDSCs at tumor sites. The subset of MDSCs that infiltrate the tumor site differs by cancer type. 31 The 5‐FU/L‐OHP treatment significantly decreased the proportion of G‐MDSCs, and increased the proportion of M‐MDSCs, at the tumor sites of CT26‐bearing mice (Figure 4C). However, additional CP treatment reversed these effects. We recently reported that a single CP treatment increased the proportion of M‐MDSCs. 22 , 23 As shown in Figure 4C, 5‐FU/L‐OHP therapy decreased the proportion of G‐MDSCs, and increased that of M‐MDSCs, whereas additional CP treatment reversed these changes. We also examined MDSCs and T cells in spleen of CT26 and MC38 models. Changes of CD4+ T cells and CD8+ T cells showed a similar tendency to those of tumor sites, whereas those of MDSCs were different between the two models (Figure 4F and G; Figure 5G and H).
In terms of underlying mechanisms of antitumor effects induced by the triple combination chemotherapy, T cells, especially CD8+ T cells, must be main effectors because the antitumor effects were apparently attenuated in nude mice (Figure 2). However, M‐MDSCs and Tregs could affect the antitumor effects by the triple combination chemotherapy. In the CT26 model, the proportion of CD11b+ cells among CD45+ cells in tumor sites was decreased after the triple chemotherapy in CT26‐bearing mice (Figure 4A). In addition, the triple chemotherapy decreased the proportions of M‐MDSCs and CD4+ T cells in tumor sites (Figure 3B; Figure 4G). Given that M‐MDSCs have a greater suppressive effect than G‐MDSCs on antigen‐stimulated CD8+ T cells 32 and that Tregs are CD4+ T cells, the decrease in proportions of M‐MDSCs and/or CD4+ T cells at tumor sites could participate in the therapeutic efficacy of the triple combination therapy in CT26‐bearing mice. On the other hand, different results were obtained in the MC38 model; the triple chemotherapy increased the proportions of CD8+ T cells and M‐MDSCs but showed no effect on that of CD4+ T cells in tumor sites (Figure 5F). An increase in the proportions of CD8+ T cells was more apparent in CT26 tissues than in MC38 tissues (Figure 3B, Figure 5F). This difference may account why the MC38 model needed additional anti‐PD‐1 antibody therapy.
Either a partial response or complete cure is critical for a good prognosis in cancer‐bearing hosts. In the CT26 model, triple combination chemotherapy was completely curative in some mice. The cured mice showed protective immunity and generated CT26‐specific CTLs from their spleen cells (Figure 1F and G). It should be noted that additional treatment with anti‐PD‐1 antibody was completely curative in half or one third of the MC38‐bearing mice (Figure 6A and Figure 7A). As for the mechanism why the additional anti‐PD‐1 antibody therapy increased the numbers of cured mice, we propose the following: The triple combination chemotherapy promoted the infiltration of MC38‐specific CTLs into tumor sites, as a result of the attenuation of Treg/MDSC‐mediated immunosuppression and the induction of immunogenic cancer cell death. However, such tumor‐infiltrating T cells increased the PD‐L1 expression on MC38 cells, so‐called adaptive immune resistance, as previously reported. 21 Anti‐PD‐1 antibody therapy must overcome this resistance. On the other hand, transient weight loss, as an immune‐related adverse event, was observed in MC38‐bearing mice after triple combination therapy, and the additional anti‐PD‐1 antibody treatment exacerbated this to some degree (Figure 6D). However, the additional anti‐PD‐1 antibody treatment unexpectedly mitigated the body weight loss by the triple combination chemotherapy (Figure 7C and D). Although we have no clear answer on these inconsistent results, these observations may call attention to combine chemotherapy with ICB therapy.
In conclusion, we demonstrated that a combination of 5‐FU/L‐OHP and CP is a promising immunogenic chemotherapy. The antitumor effects of the triple combination therapy were mainly due to host T cells. Also, the therapeutic efficacy of triple combination therapy was boosted by ICB therapy. These results could contribute to the development of more effective chemotherapy plus ICB combination therapy for CRC patients.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Fig S1
{kind=link}
Fig S2
{kind=link}
Fig S3
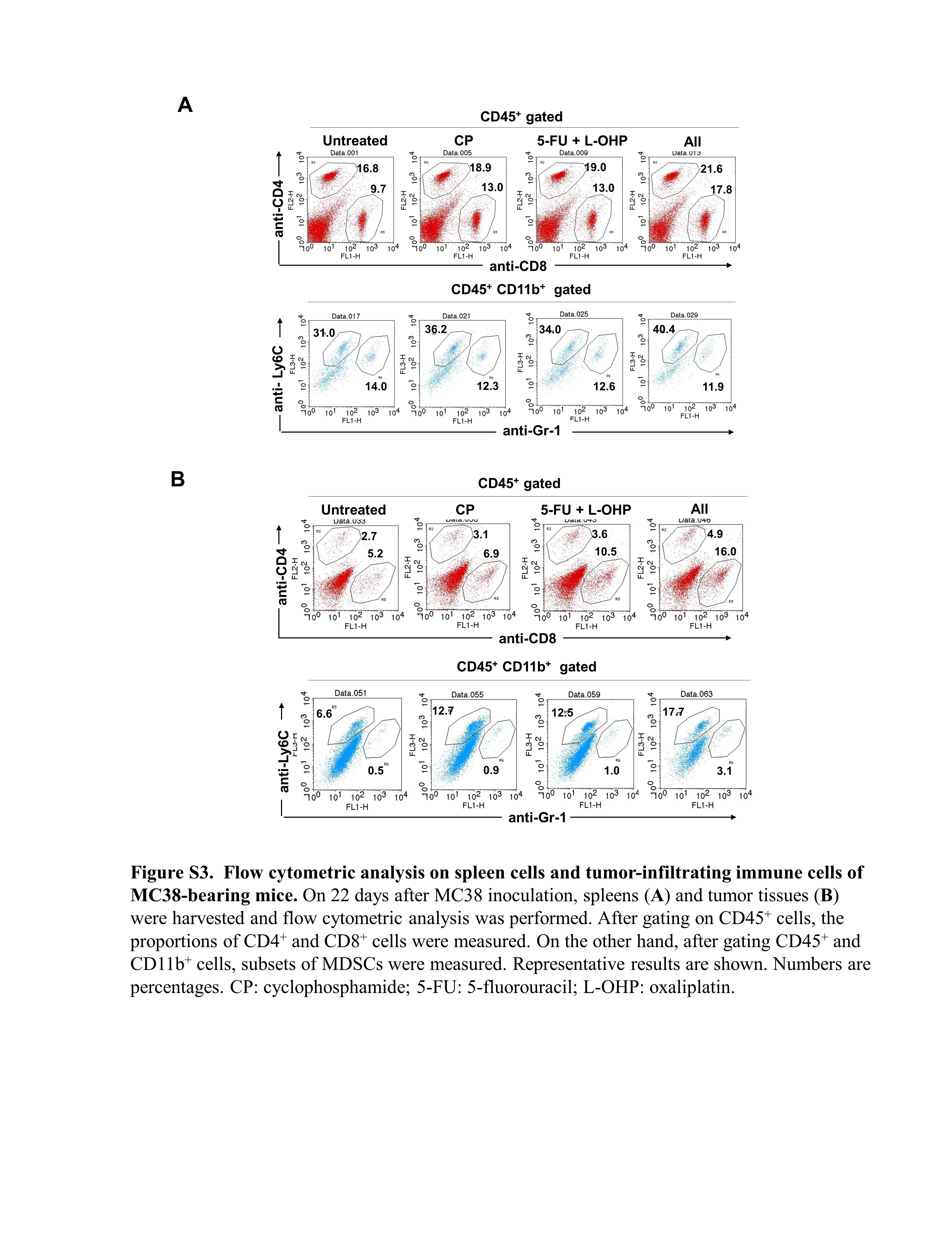{kind=link}
ACKNOWLEDGEMENT
This study was supported in part by the JSPS KAKENHI Grant (no. 17K07217 to M. Harada), the Shimane University “SUIGANN” Project, and the Trust Fund of Advanced Medical Service of Shimane University Hospital (A7A00522 to M. Harada).
Taniura T, Iida Y, Kotani H, Ishitobi K, Tajima Y, Harada M. Immunogenic chemotherapy in two mouse colon cancer models. Cancer Sci. 2020;111:3527–3539. 10.1111/cas.14624
REFERENCES
- 1. Ohigashi Y, Sho M, Yamada Y, et al. Clinical significance of programmed death‐1 ligand‐1 and programmed death‐1 ligand‐2 expression in human esophageal cancer. Clinical Cancer Res. 2005;11:2947‐2953. [DOI] [PubMed] [Google Scholar]
- 2. Muro K, Chung HC, Shankaran V, et al. Pembrolizumab for patients with PD‐L1‐positive advanced gastric cancer (KEYNOTE‐012): a multicentre, open‐label, phase 1b trial. Lancet Oncol. 2016;17:717‐726. [DOI] [PubMed] [Google Scholar]
- 3. Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ott PA, Hodi FS, Robert C. CTLA‐4 and PD‐1/PD‐L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19:5300‐5309. [DOI] [PubMed] [Google Scholar]
- 6. Anagnostou VK, Brahmer JR. Cancer immunotherapy: a future paradigm shift in the treatment of non‐small cell lung cancer. Clin Cancer Res. 2015;21:976‐984. [DOI] [PubMed] [Google Scholar]
- 7. Le DT, Uram JN, Wang H, et al. PD‐1 Blockade in tumors with mismatch‐repair deficiency. New Engl J Med. 2015;372:2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiao Y, Freeman GJ. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015;5:16‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature. 2017;541:321‐330. [DOI] [PubMed] [Google Scholar]
- 10. Apetoh L, Mignot G, Panaretakis T, Kroemer G, Zitvogel L. Immunogenicity of anthracyclines: moving towards more personalized medicine. Trends Mol Med. 2008;14:141‐151. [DOI] [PubMed] [Google Scholar]
- 11. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59‐73. [DOI] [PubMed] [Google Scholar]
- 12. Ramakrishnan R, Assudani D, Nagaraj S, et al. Chemotherapy enhances tumor cell susceptibility to CTL‐mediated killing during cancer immunotherapy in mice. J Clin Invest. 2010;120:1111‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen CA, Ho CM, Chang MC, et al. Metronomic chemotherapy enhances antitumor effects of cancer vaccine by depleting regulatory T lymphocytes and inhibiting tumor angiogenesis. Mol Ther. 2010;18:1233‐1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vincent J, Mignot G, Chalmin F, et al. 5‐Fluorouracil selectively kills tumor‐associated myeloid‐derived suppressor cells resulting in enhanced T cell‐dependent antitumor immunity. Cancer Res. 2010;70:3052‐3061. [DOI] [PubMed] [Google Scholar]
- 15. Salem ML, Diaz‐Montero CM, Al‐Khami AA, et al. Recovery from cyclophosphamide‐induced lymphopenia results in expansion of immature dendritic cells which can mediate enhanced prime‐boost vaccination antitumor responses in vivo when stimulated with the TLR3 agonist poly(I:C). J Immunol. 2009;182:2030‐2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gameiro SR, Caballero JA, Higgins JP, Apelian D, Hodge JW. Exploitation of differential homeostatic proliferation of T‐cell subsets following chemotherapy to enhance the efficacy of vaccine‐mediated antitumor responses. Cancer immunol, immunother. 2011;60:1227‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sistigu A, Viaud S, Chaput N, et al. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Seminars Immunopath. 2011;33:369‐383. [DOI] [PubMed] [Google Scholar]
- 18. Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336‐344. [DOI] [PubMed] [Google Scholar]
- 19. Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482‐491. [DOI] [PubMed] [Google Scholar]
- 20. Suzuki E, Kapoor V, Jassar AS, et al. Gemcitabine selectively eliminate splenic Gr‐1+/CD11b+ myeloid suppressor cells in tumor‐bearing animals and enhance antitumor immune activity. Clin Cancer Res. 2005;11:6713‐6721. [DOI] [PubMed] [Google Scholar]
- 21. Dosset M, Vargas TR, Lagrange A, et al. PD‐1/PD‐L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy on colorectal cancer. Oncoimmunol. 2018;7:e1433981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iida Y, Harashima N, Motoshima T, et al. Contrasting effects of cyclophosphamide on anti‐CTLA‐4 blockade therapy in two tumor mouse models. Cancer Sci. 2017;108:1974‐1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Satoh Y, Kotani H, Iida Y, et al. Supplementation of L‐arginine augments the therapeutic efficacy of anti‐cancer chemoimmunotherapy. Cancer Sci. 2020;111:2248‐2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang AY, Gulden PH, Woods AS, et al. The immunodominant major histocompatibility complex class I‐restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci U S A. 1996;93:9730‐9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zeh HJ III, Donna PL, Dudley ME, et al. High avidity CTLs for two self‐antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. 1999;162:989‐994. [PubMed] [Google Scholar]
- 26. Andre T, Boni C, Mounedji‐Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. New Engl J Med. 2004;350:2343‐2351. [DOI] [PubMed] [Google Scholar]
- 27. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014‐1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Current Opin Immunol. 2016;39:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759‐767. [DOI] [PubMed] [Google Scholar]
- 30. Gonda K, Shibata M, Ohtake T, et al. Myeloid‐derived suppressor cells are increased and correlated with type 2 immune responses, malnutrition, inflammation, and poor prognosis in patients with breast cancer. Oncol Lett. 2017;14:1766‐1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Becht E, Giraldo NA, Germain C, et al. Immune contexture, immunoscore, and malignant cell molecular subgroups for prognostic and theranostic classifications of cancers. Adv Immunol. 2016;130:95‐190. [DOI] [PubMed] [Google Scholar]
- 32. Dolcetti L, Peranzoni E, Ugel S, et al. Hierarchy of immunosuppressive strength among myeloid‐derived suppressor cell subsets is determined by GM‐CSF. Eur J Immunol. 2010;40:22‐35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3