

Abstract
The human endometrium is an essential component in human reproduction that has the unique characteristic of undergoing cyclic regeneration during each menstrual cycle. Vigorous regeneration after shedding may be sustained by stem/progenitor cells, for which molecular markers have not been fully identified. Although clonality analysis using X chromosome inactivation patterns has shown that normal human endometrial glands are composed of a monoclonal cell population, whether clonal expansion is derived from stem/progenitor cells remains unclear. Remarkable advances in next‐generation sequencing technology over the past decade have enabled somatic mutations to be detected in not only cancers, but also normal solid tissues. Unexpectedly frequent cancer‐associated mutations have been detected in a variety of normal tissues, and recent studies have clarified the mutational landscape of normal human endometrium. In epithelial glandular cells, representative cancer‐associated mutations are frequently observed in an age‐dependent manner, presumably leading to growth advantage. However, the extremely high mutation loads attributed to DNA mismatch repair deficiency and POLE mutations, as well as structural and copy number alterations, are specific to endometrial cancer, not to normal epithelial cells. The malignant conversion of normal epithelial cells requires these additional genetic hits, which are presumably accumulated during aging, and may therefore be a rare life event. These discoveries could be expected to shed light on the physiology and pathogenesis of the human endometrium and urge caution against the application of genetic screening for the early detection of endometrial cancer.
Keywords: endometrial cancer, endometriosis, mutation, next‐generation sequencing, normal endometrium
Human endometrial gland exhibits clonal growth in each menstrual cycle possibly via stem/progenitor cells, with harboring frequent somatic gene mutations in cancer‐associated genes. These mutations may plays role in endometrial regeneration and pathogenesis of endometriosis or endometrial cancer.
1. INTRODUCTION
The endometrium is a unique component of the uterus characterized by cyclic regeneration during menstrual cycles. After shedding during menstruation, the endometrium vigorously regenerates during a 14‐d proliferative phase, followed by a secretory change persisting until the next menstruation. Endometriosis is a disease experienced by many women of reproductive age in which the uterine endometrium grows outside the uterus. It is accompanied by a variety of complications, including dysmenorrhea, pelvic pain, and infertility. Sampson's retrograde menstruation theory, which states that normal endometrial cells flow back into the peritoneal cavity during menstruation, leading to the development of ectopic endometriotic lesions, is widely accepted. 1 Endometrial cancer also arises from epithelial cells in the human endometrium through atypical hyperplasia as precursors or de novo from those of the atrophic endometrium in postmenopausal women. Therefore, the vast majority of studies have used normal human endometrium as experimental controls for both diseases.
While the somatic mutational landscape of cancer in humans has been comprehensively characterized, 2 knowledge of somatic mutations in normal cells remains limited because of difficulties in accessing tissue, the small amounts of samples collected, the likely low frequency of mutations, and insufficient technical resolution. Recent advances in next‐generation sequencing (NGS) technology have helped to overcome these issues and uncover the mutational landscape of a variety of normal tissues, including the colon, 3 skin, 4 esophagus, 5 and endometrium. 6 , 7 , 8 This review introduces the clonal growth properties of endometrial glands, possibly via stem/progenitor cells, and summarizes recent findings regarding their mutational status, focusing on the roles of somatic gene mutations in the biology of endometrium and the pathogenesis of endometriosis and endometrial cancer.
2. NORMAL ENDOMETRIAL GLANDS ARE COMPOSED OF A MONOCLONAL CELL POPULATION
The way in which human endometrial glands regenerate after shedding during menstruation is not well known. While the stratum functionalis sheds during menstruation, the stratum basalis remains, providing proliferating glandular cells for the next cycle of regeneration. The clonality of human endometrial glands is crucial for understanding the mechanism of cyclic regeneration. Therefore, we investigated the clonality of normal human endometrial glands using X chromosome inactivation patterns as informative markers of tissue clonality. 9 During embryogenesis, DNA methylation randomly inactivates either the paternal or maternal X chromosome, and methylated alleles become resistant to DNA restriction enzymes. 10 Following treatment with restriction enzymes, polymerase chain reaction (PCR) can amplify intact alleles on the methylated, but not the unmethylated X chromosome, which generates 2 distinct PCR bands in electrophoresis, derived from the paternal and maternal alleles, respectively, because of allelic polymorphisms in polyclonal normal tissues. However, if the PCR generates the single band, the tissue is considered to comprise monoclonal cell population. DNA isolated from individual glands was treated with restriction enzymes, and the alleles of the androgen receptor gene on the X chromosome were amplified by PCR. The DNA recovered from almost all the glands of each patient produced a single band upon HhaI digestion (Figure 1A), suggesting that the glands were composed of a monoclonal cell population of either paternal or maternal origin. We further examined gland clonality in a mouse harboring a green fluorescent protein gene on either the maternal or paternal X chromosome. 11 The cells of this mouse harboring the transgene on the unmethylated active X chromosome exhibited fluorescence, whereas those with the transgene on the inactive X chromosome did not. 12 Therefore, this mouse genus, known as “green mice,” can be used to analyze the clonality of tissues of interest. Under fluorescence microscopy, histological sections of the uteri of this mouse revealed that the individual glands were composed of either fluorescent or nonfluorescent cells at similar ratios (Figure 1B), suggesting that the glands are composed of a monoclonal cell population.
FIGURE 1.
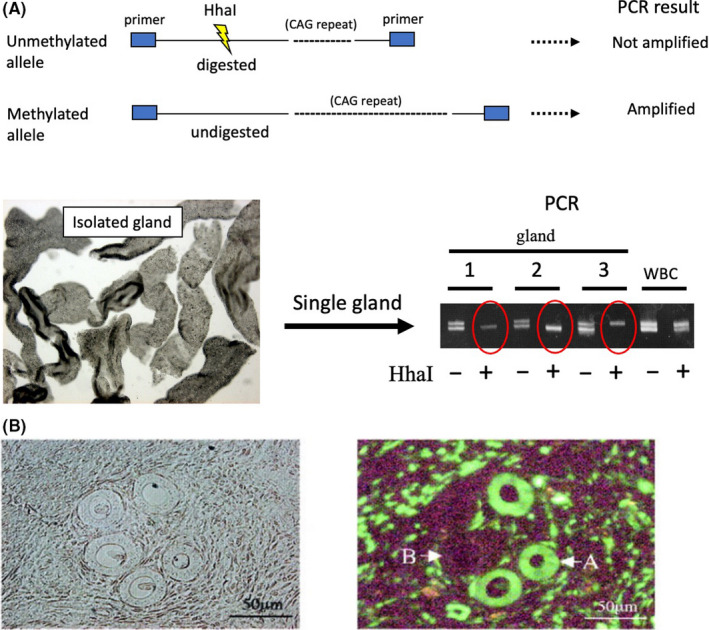
Clonality analysis of human endometrial glands. A, The principle of the clonality assay for androgen receptor (AR) gene is illustrated. Endometrial glands were individually isolated with microscopic manipulation as shown in the photograph. DNA from each gland was digested with HhaI, followed by PCR to amplify the CAG repeats in the AR gene. After digestion with HhaI, the DNA yielded single PCR bands (shown as red circles) derived from either paternal or maternal alleles, suggesting that glandular cells are monoclonal. DNA from each patient's white blood cells (WBC) were used as controls for the polyclonal population. B, Uterine sections of a female hemizygous transgenic mouse carrying an X‐linked green fluorescent protein transgene. Left: Histological view of endometrium. Right: Fluorescence microscopy shows endometrial glands composed of either fluorescent (A) or nonfluorescent (B) cells. (Adapted with alterations and permission from reference 9)
3. ARE THERE STEM CELLS IN HUMAN ENDOMETRIAL EPITHELIAL CELLS?
The monoclonal composition of endometrial glands reminds us of the presence of stem/progenitor cells, from which descendant cells compose a whole gland with clonal expansion; otherwise, the proliferative potential would become exhausted after several dozen menstrual cycles.
Endometrial epithelial stem cells are supposed to localize at the stratum basalis of the gland because they appear to be protected from shedding during menstruation. Extensive efforts have been made to identify endometrial epithelial stem cells. The side population (SP) phenotype is a universal marker of adult stem cells that is characterized as a low fluorescent phenotype by flow cytometry with Hoechst 33342 staining. 13 SP cells have been identified in cultured endometrial cells, 14 and endometrial SP cells sorted from short‐term cultures have been maintained in three‐dimensional (3D) culture for 3 mo, whereas non‐SP cells became senescent in shorter periods, which concurs with the longevity associated with adult stem cells.
Endometrial SP cells preferentially express several endothelial cell markers 15 and can differentiate into various phenotypes, including glandular epithelial, stromal, and endothelial cells in vitro. Furthermore, endometrial SP, but not non‐SP, cells can successfully reconstitute endometrium‐like tissues containing glandular components when transplanted into severely immunodeficient mice, but are mainly composed of endothelial tissues (46%), stromal tissues, (13%), and rarely, epithelial tissues (0.02‐8%), indicating their heterogeneity. 15
Several other candidate markers for endometrial epithelial stem cells have been identified. The embryonic stem cell‐surface marker, stage‐specific embryonic antigen 1 (SSEA‐1), marks human endometrial basal glandular cells. 16 Cultured SSEA‐1+ cells have increased telomerase activity and longer telomeres and generate larger gland‐like spheroids in 3D culture than do SSEA‐1– cells and N‐cadherin has been identified as a potential marker for endometrial epithelial progenitors. 17 Serial cloning assay has revealed that N‐cadherin+ epithelial cells exhibit greater self‐renewal capacity than do N‐cadherin– endometrial epithelial cells. N‐cadherin+ epithelial cells can differentiate into glandular structures lined with cytokeratin+ luminal cells in organotypic culture. Some other potential markers for endometrial epithelial cells have also been proposed and reviewed 18 , 19 , 20 , 21 , 22 , 23 , 24 (Table 1). Despite these extensive studies, no definitive marker specific to endometrial epithelial stem cells has been identified. One problem may lie in the controversy regarding the specificity of the antibodies used, as the reliability of some antibodies was not verified. Another concern is that a greater number of cells with candidate markers are present in the normal endometrium compared with the expected number for an adult stem cell population. Furthermore, the difficulty in establishing a reconstitution model in which the selected cell population, hopefully a single cell, with specific markers can reconstitute a whole endometrial gland, with or without other components, in 3D culture or a mouse model remains an urgent issue. Overcoming these issues could be expected to purify further glandular stem/progenitor cells, which is important for understanding the mechanisms of clonal expansion as well as whether they are mutation targets.
TABLE 1.
Proposed markers for endometrial epithelial stem cells
| SP | LRC | SSEA‐1 | SOX9 | Nuclear β‐catenin | LGR5 | N‐cadherin | |
|---|---|---|---|---|---|---|---|
| Localization in a gland | Not defined | Luminal | Basalis, Luminal | Basalis | Basalis | Luminal (but controversial) | Basalis |
| Glandular structure in 3D culture | + | − | + | + | − | − | + |
| Reconstitution of glandular structure in mice | + | − | − | − | − | + | − |
| Notes | Reconstruction in mice, but rarely with epithelial tissues | Co‐localizes with SSEA‐1 and nuclear β‐catenin | Weak endometrial reconstitution in mice | ||||
| Representative studies | 14, 15 | 21, 22 | 16, 18 | 16 | 16 | 18, 19, 20 | 17 |
Abbreviations: LGR5, leucine‐rich repeat‐containing G‐protein‐coupled receptor 5; LRC, label‐retaining cell; SOX9, sex‐determining region Y (SRY)‐box 9 protein; SP, side population; SSEA‐1, stage‐specific monoclonal antigen‐1
4. CANCER‐ASSOCIATED MUTATIONS IN ENDOMETRIOTIC LESIONS
In 2014, for the first time, Li et al 25 performed whole‐exome sequencing of endometriosis samples from 16 patients. This was the first study to detect unexpectedly numerous somatic mutations in endometriotic lesions and to find that the patients harbored various somatic mutations, including genes encoding histone methyltransferase, which is involved in histone H3 lysine 4 modification. In 2017, Anglesio et al reported somatic mutations in deeply infiltrating endometriotic (DIE) lesions. 26 Whole‐exome sequencing identified 80 nonsynonymous somatic mutations in 19 (79%) of 24 patients, with a mean of 3.3 mutations per lesion. Five patients (21%) had a known cancer driver mutation in either ARID1A, PIK3CA, KRAS, or PPP2R1A. Targeted sequencing and a droplet PCR assay identified additional KRAS mutations, and 10 (26%) of 39 DIE lesions had driver mutations, all of which were confined to the epithelial compartment of endometriotic lesions; these were not observed in stromal components, which raised a hypothesis regarding the differing origins of epithelial and stromal components. Eutopic endometria in 3 patients with DIE lesions harboring KRAS mutations were available, but were confirmed to have no detectable mutations. Overall, the frequency of cancer‐associated mutations detected in noncancerous DIE lesions surprised researchers, and was thought to explain the aggressive phenotype of this disease, which raises the question of whether superficial endometriosis has similar spectra of mutations.
The same research group further examined mutation status in incisional endometriosis (IE), a type of iatrogenic endometriosis resulting from surgical procedures such as a cesarean section. 27 Endometriosis tissue samples from patients with IE or DIE lesions were examined by target‐gene sequencing for 33 genes. Four (10%) of 40 patients with IE harbored somatic mutations in either KRAS, PIK3CA, or ERBB2. No somatic mutations were detected in any of the 4 corresponding eutopic endometrial specimens available. Eight patients (22.2%) with DIE lesions had somatic mutations in either KRAS or CTNNB1. Simultaneous immunohistochemical analyses for the loss of PTEN or ARID1A expression added potentially mutated cases, and the overall rates of somatic driver mutations in IE and DIE lesions were 27.5% and 36.1%, respectively. These findings demonstrated that both lesions had similar rates of somatic driver mutations. As IE arises from surgical implantation/endometrial seeding, whether these mutations are attributable to the eutopic endometrium prior to implantation/seeding remains a great concern, although no mutations were detected in eutopic endometria in the above study.
In 2018, Suda et al 6 performed whole‐exome sequencing of 13 ovarian endometriotic epithelial samples using laser microdissection techniques. Several cancer‐associated mutations, including KRAS, PIK3CA, FBXWJ, PPP2R1A, and PIK3R1, were recurrently detected, with KRAS and PIK3CA being the most frequent. These mutations were located within the hotspot, leading to gain‐of‐function mutations, which activated downstream signaling pathways that may lead to a growth advantage. Representative cancer‐associated mutations had high mutant allele frequencies (MAFs), being ranked in the top 5%, reaching nearly clonal status (MAF of 0.5). The authors examined the patterns of mutation sharing in multiple endometriotic regions collected from the same individual; KRAS and PIK3CA mutations were frequently shared across multiple regions of an individual subject, indicating the growth advantage of clonal populations harboring such mutations.
5. MUTATIONAL LANDSCAPE OF NORMAL HUMAN ENDOMETRIUM
Unexpectedly frequent somatic driver mutations in endometriosis prompted researchers to conduct an investigation in normal human endometrium. Suda et al 6 performed whole‐exome sequencing of 11 normal endometrial epithelial samples and found frequent somatic mutations in a variety of genes. Surprisingly, the number of somatic mutations per Mb did not significantly differ between endometriotic and normal endometrial epithelial samples; however the MAFs were significantly higher in the former than the latter. Again, representative cancer‐associated genes, such as KRAS, PIK3CA, FBXWJ, PPP2R1A, and PIK3R1, were recurrently mutated in normal endometrium. Information regarding the driver mutations in each individual is shown in Figure 2. In 5 patients for whom paired endometriotic and normal endometrial epithelial glands were available, the mutation profiles were discordant between both samples (Figure 2), which seems incompatible with retrograde menstruation theory. However, only selected clones with specific types of mutations may have the advantage of being able to evolve clonally and develop ectopic sites, which is not incompatible with retrograde menstruation theory.
FIGURE 2.
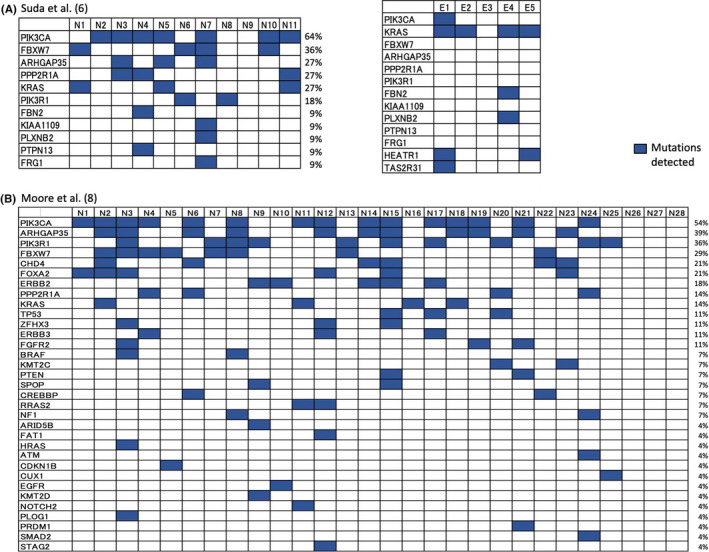
Oncoplot of driver mutations and distribution across individual patients by whole‐exome sequencing. The results of the references 6 (A) and 8 (B) are summarized, with some alterations. The listed genes (shown in the vertical columns) differed between the 2 studies; patient numbers are shown in the horizontal rows. N and E indicate normal endometrial and endometriosis samples, respectively. Paired samples were analyzed in 5 patients (patients 1 to 5) (A). The frequency (%) of patients with mutations are shown beside the plot for each gene. (Adapted from alterations and permission from references 6 and 8 )
The authors further performed target‐gene sequencing of single whole endometrial glands, in which fresh normal endometrial samples were minced and single individual glands were manually isolated under microscopy and subjected to sequencing. PIK3CA was the most frequently mutated across the single glands; less frequent mutations were observed in KRAS and ARID1A. The MAFs of these mutations were near 0.5, suggesting clonal expansion within a gland, consistent with our previous study. 9 Of particular interest is that the mutation in each gland was distinct with a unique amino acid substitution, suggesting gland‐to‐gland genomic variation.
In 2019, Lac et al examined the presence of somatic driver mutations in histologically normal endometrium in 25 hysterectomy and 85 curettage or biopsy specimens. 7 The authors found that 54% of the women had somatic mutations in KRAS, PIK3CA, FGFR2, NRAS, AKT1, and ERBB2 (variant allele frequencies [VAFs] ranged from 0.12 to 0.8). The most common mutations were hotspot KRAS and PIK3CA mutations, and none was observed in ARID1A. The prevalence of such oncogenic mutations increased with patient age; the likelihood of a woman having a somatic mutation increased by 5% per year. Compared with patient data in 33 genes from The Cancer Genome Atlas, 28 patients with endometrial cancer had many more somatic mutations at a given age. Gene‐specific analysis revealed that mutation frequencies, except for KRAS mutations, were higher in cancer specimens; a greater proportion of normal endometria showed KRAS mutations (28%) compared with endometrial cancer (19%). In regard to KRAS mutations, the rate of mutation in normal samples increased with age, while the opposite trend was observed in cancer samples.
In 2020, Moore et al performed whole genome sequencing of 292 normal human endometrial glands of 28 women. 8 The somatic mutation burdens ranged from 209 to 2833 base substitutions (median: 1521) and from 1 to 358 indels (median: 180), and were associated with patient age, in that a linear accumulation of 29 bp substitutions per gland per year was observed. Somatic copy number alterations and genome rearrangements were found in 36 (14%) of 257 normal endometrial glands, the majority of which exhibited a single change.
Most glands exhibited VAFs ranging from 0.3 to 0.5 and showed a monoclonal composition. Many glands were found to have driver mutations but, unexpectedly, the median VAFs were similar between glands with or without driver mutations, indicating that driver mutations are not likely to be a determinant of clonality.
In total, 25 (89%) of 28 women had potential driver mutations in 33 genes, among whom, the youngest carrier was a 24‐y‐old woman with a KRAS G12D mutation. Approximately 60% of endometrial glands carried at least one driver mutation, and 20% at least 2. Information regarding the different driver mutations in individual patients is shown in Figure 2. Four (14%) of 28 women carried driver mutations in all glands examined. The mostly frequently mutated gene was PIK3CA, for which over 50% the women had at least 1 mutation. Phylogenetically closely related glands were likely to be located in close proximity to one another, ranging a distance of several millimeters, which indicated gland colonization with the same clonality. The mutational status of normal endometrial epithelial cells in the abovementioned studies is summarized in Table 2.
TABLE 2.
Summary results of studies reporting somatic mutations in normal endometrial epithelium
| Study | Sample size | Patient age (y) | Detection method | Sample collection | Mutation burden | Mutation frequency (MAF or VAF) | Driver mutations or representative mutations detected | Percentage of patients with driver mutations | Notes |
|---|---|---|---|---|---|---|---|---|---|
| Suda et al (2018) | N = 11 (discovery cohort) | 40‐57 (median: 47) | Whole‐exome sequencing | Laser‐captured microdissection | NA (72‐536 base substitutions [median: 128]) in individuals of 13 endometriotic and 11 normal endometrial patients | Roughly 0.1‐0.4 | KRAS, PIC3CA, TTN, FBXW7, ARGAP35, MUC6, PLXNB2, PPP2R1A, CSMD3, FBN2, HEATR1, KIAA1109, PIK3R1, TAS2R31, PTPN13, FRG1 | 10/11 (91%) | The mutation profiles were discordant between paired endometriotic and endometrial samples. The number of mutations was similar, but the MAFs were much higher in the endometriotic samples |
| N = 29 (validation cohort) | 38‐52 (median: 45) | Target‐gene sequencing (covering 84 genes) | Laser‐captured microdissection | NA | Roughly 0‐0.5 | PIK3CA, KRAS, HGAP35, PIK3R1, TAF1, FAT1, FBXW7, FGFR2, KMT2C, PTEN, ZFHX3, POLE, CTNNB1, PP2R1A | 49 of 71 (69%) glands examined | The MAFs of driver mutations were near 0.5, suggesting clonal expansion within a gland | |
| N = 109 (glands from 3 patients: 51, 39, 19 in each patient) | 38, 47, 49 | Target‐gene sequencing (covering 84 genes) | Manual collection under microscope | 82 mutations and indels in 109 glands | Close to 0.5 | PIK3CA, ARHGAP35, FBXW7, PPP2R1A, PIK3R1, KRAS, ARID1A | 65 of 109 glands (26 of 51, 29/39, 10/19, in each patient) | Single gland analysis revealed gland‐to‐gland genomic variation | |
| Lac et al (2019) | N = 110 | 21‐61 (median: 37) | Target‐gene sequencing (covering 33 genes) and immunohistochemistry for PTEN and ARID1A | Macrodissection | NA | 0.008‐0.119 | KRAS, PICK3CA, FGFR2, NRAS, AKT1, ERBB2, PTEN. | 59/110 (54%) (loss of PTEN expression was not correlated with sequencing results, but judged as having mutation) | The mutations increased with aging by 5% per year |
| Moore et al (2020) | N = 28 | 19‐81 (median: 39) | Whole‐exome sequencing | Laser‐captured microdissection | 225‐2890 base substitutions (mean: 1324) and 3‐243 indels (mean: 85) in individuals | 0‐1.0 (with a peak between 0.3 and 0.5) | PIK3CA, PIK3R1, ARHGAP35, FBXW7, ZFHX3, FOXA2, ERBB2, CHD4, KRAS, SPOP, PPP2R1A, REBB3,CREBBP, FGFR2, ARID5B, RRAS2, FAT1, HRAS, NF1, TP53, BRAF, PTEN, HRAS, ATM, CDKN1B, CUX1, EGFR, KMT2 D, NOTCH2, PLOG1, PRDM1, SMAD2, STAG2 | 25/28 (89%) | A linear accumulation of 29 base substitutions per gland per year. The median VAFs were similar between glands with or without driver mutations. Overall, 14% of women carried driver mutations in all glands examined. Phylogenetically closely related glands were likely to be located in close proximity |
Abbreviations: NA, not applicable; MAF, mutant allele frequency; VAF, variant allele frequency.
The Pan Cancer Analysis of the Whole Genomes data set 2 showed that endometrial cancers have mutation loads for base substitutions that are 5 times higher than those for normal endometria. Most importantly, extremely high mutation loads attributed to DNA mismatch repair deficiency and mutations of catalytic subunit of the DNA polymerase epsilon (POLE) are specific to endometrial cancer, and not observed in normal endometrium. Structural and copy number alterations were observed in endometrial cancer much more frequently than in normal endometrium (medians of 23 vs. 0, respectively). The repertoire of cancer genes also differed between endometrial cancer and normal endometrium; the former had frequent mutations in PTEN, CTCF, CTNNB1, and ARID1A in endometrioid subtype and in p53 in serous carcinoma. Biallelic mutations of these genes are required for conferring a growth advantage and conversion to malignancy; these were not observed in normal endometrium. The mutational status of potential precursors, endometrial intraepithelial neoplasia (EIN) and atypical endometrial hyperplasia (AEH), were also analyzed using NGS by recent studies, 29 and the paired EIN/AEH and endometrial cancer had identical somatic mutations, while private mutations were observed in each lesion. These findings are consistent with that EIN/AEH are precursors of endometrial cancer, but the step toward cancer is not simply a linear accumulation of genetic hits.
Overall, driver mutations detected in normal endometrium may confer a growth advantage to the glands and clonal colonization. However, the acquisition of additional hits, such as DNA mismatch repair deficiency, POLE mutations, copy number alterations, and biallelic mutations, in specific tumor suppressor genes is required for malignant conversion; these were seldom observed in normal cells. Considering the fact that normal endometrium is extensively colonized by clones with driver mutations, even in early childhood, but that the overall lifetime risk of endometrial cancer is only 3%, such colonization does not appear to be directly associated with malignant progression.
6. MUTATIONAL STATUS OF STROMAL COMPARTMENTS IN NORMAL ENDOMETRIUM AND ENDOMETRIOTIC LESIONS
While somatic mutations in epithelial cells have been extensively analyzed, little information has been available for those in stromal cells. Suda et al performed targeted‐gene sequencing of laser‐microdissected stromal samples from endometriotic lesions and normal endometria. 30 In total, 18 and 16 mutations were detected in endometriotic stromal and normal endometrial stromal samples, respectively, but without any mutations shared with paired epithelial samples. Furthermore, the MAFs of mutations in stromal samples were extremely low compared with those of the paired epithelial samples. The completely different mutation profiles between paired samples suggested their independent origin and biology.
In relation, Noë et al 31 analyzed 19 somatic passenger mutations in 24 patients with DIE lesions and found that these mutations were predominantly detected in the epithelial compartment, whereas very few mutations were observed in the stromal ones.
7. WHAT HAVE WE LEARNED FROM MOUSE MODELS?
Mouse studies have provided useful information on the effect of driver mutations in endometrium. As PTEN homozygous null mice are embryonic lethal, PTEN heterozygous mice are widely used for cancer studies in earlier periods. 32 PTEN heterozygous females have been reported to show AEH. 33 The development of the Cre‐loxP system has enabled conditional knockout in endometrium, and some studies with PTEN homozygous null mice have demonstrated the development of carcinoma, 34 whereas others have reported the development of precursor lesions, such as atypical hyperplasia. 35 , 36 The additional introduction of PIK3CA mutation to PTEN‐null mice efficiently created cancer in endometrium. 35
Cheng et al developed a mouse model of endometriosis in which the endometrium of transgenic mice with conditional knock‐in of KRAS G12V/+ was subcutaneously transplanted into immunocompetent recipient mice. 37 The implanted endometrium developed endometriosis‐like lesions with intact glandular structures, while that of the control mice with the wild‐type allele formed smaller lesions with many dying cells. This model suggests that oncogenic KRAS mutation in eutopic endometrium plays an essential role in the development of endometriotic lesions via implantation.
A recent knockout/knock‐in mouse study indicated the role of ARID1A and PIK3CA mutations in normal endometrium. 38 Neither conditional knockout of ARID1A nor knock‐in of PIK3CA mutant allele in normal endometrial epithelia had any gross phenotypes, however the combination of both conferred phenotypic change as atypical hyperplasia.
Thereafter, in a mouse model, the above authors challenged the mice to mimic the spread of endometrium into the peritoneal cavity. 39 Conditional knockout mice displaying a loss of ARID1A expression concomitant with oncogenic PIK3CA H1047R expression in endometrium were prepared and subjected to a surgical procedure to facilitate endometrial spread into the peritoneal cavity, in which salpingectomy was performed to remove the barrier of the uterotubal junction, facilitating mutant endometrial cells to invade outside the uterus. Following salpingectomy, endometriotic lesions were formed in the mice on various sites in the pelvic space, whereas no specific lesions were formed in control mice with the wild‐type allele. These findings support the hypothesis that mutated phenotypes of eutopic endometrium are essential for the development of ectopic sites.
Overall, these mouse studies demonstrated that a single hit of driver mutations in endometrial epithelial cells is insufficient for the acquisition of complete cancer phenotypes; additional hits are required. They also demonstrated that KRAS mutations in eutopic endometrium may play pivotal roles in the development of endometriosis.
8. PITFALLS OF MOLECULAR‐BASED SCREENING AND EARLY DETECTION OF ENDOMETRIAL CANCER
Due to the lack of cellular atypia in endometrial cells, cytological screening of endometrial cancer is not easy compared with that of cervical cancer. Several studies have proposed the utility of genetic analysis for cancer screening using cytological samples or washing fluid in the endometrial cavity. The use of Sanger sequencing targeting representative cancer‐associated genes has frequently revealed somatic mutations in 80%‐90% of endometrial cancer specimens, but rarely in normal endometrial samples. 40 , 41 A targeted NGS panel for 8 genes (ARID1A, CTNNB1, KRAS, MTOR, PIK3CA, POLE, PTEN and TP53) detected somatic mutations in over 90% of endometrial cancer specimens, while only 6% of women without cancer exhibited PIK3CA mutations. 42
Collectively, these studies demonstrated the potential utility of genetic analysis for the early detection or screening of endometrial cancer, based on the assumption that normal endometrium has an extremely low frequency of cancer‐associated mutations. However, Nair et al reported the results of NGS for 12 genes using uterine lavage fluids. 43 Of 95 women without cancer, 51 were identified as having somatic mutations; the most frequent driver mutations were KRAS G12C (8 patients), KRAS G12S (10 patients), and PIK3CA H1047R (8 patients) with relatively high VAFs (1%‐30.4%). These findings suggest that NGS is likely to detect unexpectedly frequent somatic mutations in normal endometrial samples, and that additional or alternative strategies are needed to distinguish cancerous from noncancerous mutations for screening or early detection of endometrial cancer.
9. CONCLUSION AND FUTURE DIRECTIONS
Recent NGS technologies have detected unexpectedly greater numbers of somatic mutations in endometrial epithelial cells compared with in stromal cells in an age‐dependent manner, with substantial rates of glands having cancer‐associated mutations. The biological significance of such mutations remains unclear, but may confer a growth advantage to glands, thereby contributing to efficient regeneration in a menstrual cycle; this should be confirmed in functional studies.
Somatic mutations in endometriosis show a similar set of driver mutations, but with higher MAFs. Endometrial epithelial cells with driver mutations, especially KRAS mutation, may therefore evolve clonally to develop ectopic sites, which supports retrograde menstruation theory (Figure 3). It is, however, unclear when and where additional genetic hits occur within eutopic or ectopic sites during the development of endometriosis.
FIGURE 3.
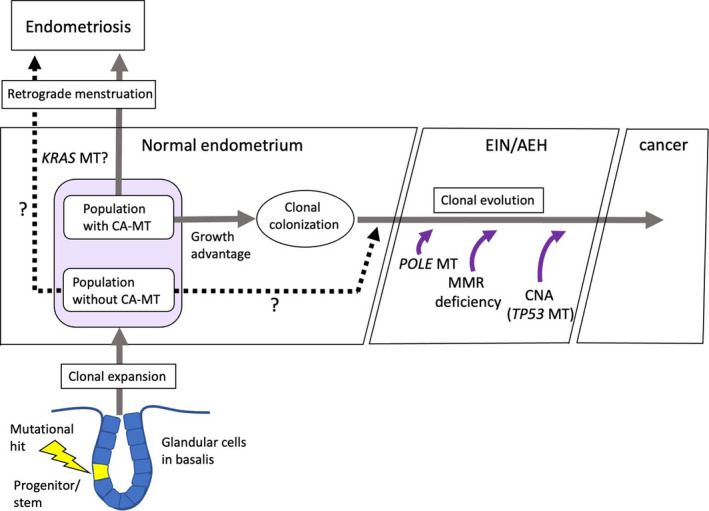
Proposed consequence of somatic mutations in normal endometrial glands. Stem/progenitor cells in normal endometrial glands, possibly located in the stratum basalis, may clonally expand with or without somatic mutations in cancer‐ or noncancer‐associated genes. Both populations, probably the former in favor, may have a growth advantage, forming clonal colonization. Additional genetic hits, including POLE mutation, mismatch repair (MMR) deficiency, and/or copy number alterations (CNAs), facilitate malignant conversion. Similarly, endometriosis may arise via clonal evolution, possibly in the population with KRAS mutation. AEH, Atypical endometrial hyperplasia; CA, cancer‐associated mutation; EIN, Endometrial intraepithelial lesion; MT, mutation
Endometrial cancer also has a similar set of drive mutations, but with a much higher frequency. The very high mutation loads attributed to DNA mismatch repair deficiency and POLE mutations, as well as structural variants and copy number changes, were specific to endometrial cancer. Malignant conversion of normal epithelial cells requires these additional hits accumulated in an age‐dependent manner, and may therefore be a rare lifetime event (Figure 3). Knockout/knock‐in mouse studies also support multiple hits of key driver mutations required for the development of cancer.
How somatic mutations are consistently retained after complete shedding of the functionalis during menstruation remains unclear. This is a characteristic feature of the endometrium, unlike other regenerative tissues, such as the colon. “Stem cell hit theory” may explain this consistency clearly; descendants will take them over with clonal expansion during each menstrual cycle. The rate and spectra of mutation status in endometrial stem cells are crucial to gaining a better understanding of the biological significance of somatic mutations in normal endometrial glands and the pathogenesis of both endometriosis and endometrial cancer. Although specific markers for endometrial epithelial stem cells have not been clearly identified, organoid cultures derived from single cells under selected culture conditions may be available to resolve such issues.
Finally, caution should be urged when detecting cancer‐associated mutations for cancer screening and diagnosis. Molecular‐based screening or diagnostic strategies require further improvements to distinguish cancerous from noncancerous mutations.
CONFLICT OF INTEREST
None of the authors has any conflicts of interest to disclose.
ACKNOWLEDGMENT
We greatly appreciate the technical assistance provided by Ms Razia Sultana and Shanta Kamrunnahar, and the fruitful discussions with all members of the Department of Obstetrics and Gynecology. This review was supported in part by the Japan Society for the Promotion of Science (JSPS) (KAKENHI grant No. JP18H02946).
Kyo S, Sato S, Nakayama K. Cancer-associated mutations in normal human endometrium: Surprise or expected? Cancer Sci. 2020;111:3458–3467. 10.1111/cas.14571
REFERENCES
- 1. Sampson JA. Metastatic or embolic endometriosis, due to the menstrual dissemination of endometrial tissue into the venous circulation. Am J Pathol. 1927;3:93‐110. [PMC free article] [PubMed] [Google Scholar]
- 2. Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee‐Six H, Olafsson S, Ellis P, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019;574:532‐537. [DOI] [PubMed] [Google Scholar]
- 4. Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martincorena I, Fowler JC, Wabik A, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362:911‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suda K, Nakaoka H, Yoshihara K, et al. Clonal expansion and diversification of cancer‐associated mutations in endometriosis and normal endometrium. Cell Rep. 2018;24:1777‐1789. [DOI] [PubMed] [Google Scholar]
- 7. Lac V, Nazeran TM, Tessier‐Cloutier B, et al. Oncogenic mutations in histologically normal endometrium: the new normal? J Pathol. 2019;249:173‐181. [DOI] [PubMed] [Google Scholar]
- 8. Moore L, Leongamornlert D, Coorens THH, et al. The mutational landscape of normal human endometrial epithelium. Nature. 2020;580:640‐646. [DOI] [PubMed] [Google Scholar]
- 9. Tanaka M, Kyo S, Kanaya T, et al. Evidence of the monoclonal composition of human endometrial epithelial glands and mosaic pattern of clonal distribution in luminal epithelium. Am J Pathol. 2003;163:295‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mashal RD, Lester SC, Sklar J. Clonal analysis by study of X chromosome inactivation in formalin‐fixed paraffin‐embedded tissue. Cancer Res. 1993;53:4676‐4679. [PubMed] [Google Scholar]
- 11. Okabe M, Ikawa M, Kominami K, et al. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313‐319. [DOI] [PubMed] [Google Scholar]
- 12. Hadjantonakis AK, Gertsenstein M, Ikawa M, et al. Non‐invasive sexing of preimplantation stage mammalian embryos. Nat Genet. 1998;19:220‐222. [DOI] [PubMed] [Google Scholar]
- 13. Goodell MA, Brose K, Paradis G, et al. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kato K, Yoshimoto M, Kato K, et al. Characterization of side‐population cells in human normal endometrium. Hum Reprod. 2007;22:1214‐1223. [DOI] [PubMed] [Google Scholar]
- 15. Masuda H, Matsuzaki Y, Hiratsu E, et al. Stem cell‐like properties of the endometrial side population: implication in endometrial regeneration. PLoS One. 2010;5:e10387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Valentijn AJ, Palial K, Al‐Lamee H, et al. SSEA‐1 isolates human endometrial basal glandular epithelial cells: phenotypic and functional characterization and implications in the pathogenesis of endometriosis. Hum Reprod. 2013;28:2695‐2708. [DOI] [PubMed] [Google Scholar]
- 17. Nguyen HPT, Xiao L, Deane JA, et al. N‐cadherin identifies human endometrial epithelial progenitor cells by in vitro stem cell assays. Hum Reprod. 2017;32:2254‐2268. [DOI] [PubMed] [Google Scholar]
- 18. Tempest N, Baker AM, Wright NA, et al. Does human endometrial LGR5 gene expression suggest the existence of another hormonally regulated epithelial stem cell niche? Hum Reprod. 2018;33:1052‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cervelló I, Gil‐Sanchis C, Santamaría X, et al. Leucine‐rich repeat‐containing G‐protein‐coupled receptor 5‐positive cells in the endometrial stem cell niche. Fertil Steril. 2017;107:510‐519. [DOI] [PubMed] [Google Scholar]
- 20. Gil‐Sanchis C, Cervelló I, Mas A, et al. Leucine‐rich repeat‐containing G‐protein‐coupled receptor 5 (Lgr5) as a putative human endometrial stem cell marker. Mol Hum Reprod. 2013;19:407‐414. [DOI] [PubMed] [Google Scholar]
- 21. Chan RW, Gargett CE. Identification of label‐retaining cells in mouse endometrium. Stem Cells. 2006;24:1529‐1538. [DOI] [PubMed] [Google Scholar]
- 22. Chan RW, Kaitu'u‐Lino T, Gargett CE. Role of label‐retaining cells in estrogen‐induced endometrial regeneration. Reprod Sci. 2012;19:102‐114. [DOI] [PubMed] [Google Scholar]
- 23. Tempest N, Maclean A, Hapangama DK. Endometrial stem cell markers: current concepts and unresolved questions. Int J Mol Sci. 2018;19:e3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gargett CE, Schwab KE, Deane JA. Endometrial stem/progenitor cells: the first 10 years. Hum Reprod Update. 2016;22:137‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li X, Zhang Y, Zhao L, et al. Whole‐exome sequencing of endometriosis identifies frequent alterations in genes involved in cell adhesion and chromatin‐remodeling complexes. Hum Mol Genet. 2014;23:6008‐6021. [DOI] [PubMed] [Google Scholar]
- 26. Anglesio MS, Papadopoulos N, Ayhan A, et al. Cancer‐associated mutations in endometriosis without cancer. N Engl J Med. 2017;376:1835‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lac V, Verhoef L, Aguirre‐Hernandez R, et al. Iatrogenic endometriosis harbors somatic cancer‐driver mutations. Hum Reprod. 2019;34:69‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Russo M, Broach J, Sheldon K, et al. Clonal evolution in paired endometrial intraepithelial neoplasia/atypical hyperplasia and endometrioid adenocarcinoma. Hum Pathol. 2017;67:69‐77. [DOI] [PubMed] [Google Scholar]
- 30. Suda K, Nakaoka H, Yoshihara K, et al. Different mutation profiles between epithelium and stroma in endometriosis and normal endometrium. Hum Reprod. 2019;34:1899‐1905. [DOI] [PubMed] [Google Scholar]
- 31. Noë M, Ayhan A, Wang TL, et al. Independent development of endometrial epithelium and stroma within the same endometriosis. J Pathol. 2018;245:265‐269. [DOI] [PubMed] [Google Scholar]
- 32. Knobbe CB, Lapin V, Suzuki A, et al. The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue‐by‐tissue survey. Oncogene. 2008;27:5398‐5415. [DOI] [PubMed] [Google Scholar]
- 33. Vilgelm A, Lian Z, Wang H, et al. Akt‐mediated phosphorylation and activation of estrogen receptor alpha is required for endometrial neoplastic transformation in Pten+/– mice. Cancer Res. 2006;66:3375‐3380. [DOI] [PubMed] [Google Scholar]
- 34. Daikoku T, Hirota Y, Tranguch S, et al. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 2008;68:5619‐5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liang X, Daikoku T, Terakawa J, et al. The uterine epithelial loss of Pten is inefficient to induce endometrial cancer with intact stromal Pten. PLoS Genet. 2018;14:e1007630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Joshi A, Miller C Jr, Baker SJ, Ellenson LH. Activated mutant p110α causes endometrial carcinoma in the setting of biallelic Pten deletion. Am J Pathol. 2015;185:1104‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng CW, Licence D, Cook E, et al. Activation of mutated K‐ras in donor endometrial epithelium and stroma promotes lesion growth in an intact immunocompetent murine model of endometriosis. J Pathol. 2011;224:261‐269. [DOI] [PubMed] [Google Scholar]
- 38. Wilson MR, Reske JJ, Holladay J, et al. ARID1A and PI3‐kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat Commun. 2019;10:3554 10.1038/s41467-019-11403-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilson MR, Holladay J, Chandler RL. A mouse model of endometriosis mimicking the natural spread of invasive endometrium. Hum Reprod. 2020;35:58‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y, Li L, Douville C, et al. Evaluation of liquid from the Papanicolaou test and other liquid biopsies for the detection of endometrial and ovarian cancers. Sci Transl Med. 2018;10:eaap8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mota A, Colás E, García‐Sanz P, et al. Genetic analysis of uterine aspirates improves the diagnostic value and captures the intra‐tumor heterogeneity of endometrial cancers. Mod Pathol. 2017;30:134‐145. 10.1038/modpathol.2016.143 [DOI] [PubMed] [Google Scholar]
- 42. Reijnen C, van der Putten LJM, Bulten J, et al. Mutational analysis of cervical cytology improves diagnosis of endometrial cancer: a prospective multicentre cohort study. Int J Cancer. 2020;146:2628‐2635. [DOI] [PubMed] [Google Scholar]
- 43. Nair N, Camacho‐Vanegas O, Rykunov D, et al. Genomic analysis of uterine lavage fluid detects early endometrial cancers and reveals a prevalent landscape of driver mutations in women without histopathologic evidence of cancer: a prospective cross‐sectional study. PLoS Medicine. 2016;13:e1002206. [DOI] [PMC free article] [PubMed] [Google Scholar]