

Ischemia-induced WRAP53 nuclear accumulation promotes DNA damage repair resulting in functional recovery after stroke.
Abstract
Failure of neurons to efficiently repair DNA double-strand breaks (DSBs) contributes to cerebral damage after stroke. However, the molecular machinery that regulates DNA repair in this neurological disorder is unknown. Here, we found that DSBs in oxygen/glucose-deprived (OGD) neurons spatiotemporally correlated with the up-regulation of WRAP53 (WD40-encoding p53-antisense RNA), which translocated to the nucleus to activate the DSB repair response. Mechanistically, OGD triggered a burst in reactive oxygen species that induced both DSBs and translocation of WRAP53 to the nucleus to promote DNA repair, a pathway that was confirmed in an in vivo mouse model of stroke. Noticeably, nuclear translocation of WRAP53 occurred faster in OGD neurons expressing the Wrap53 human nonsynonymous single-nucleotide polymorphism (SNP) rs2287499 (c.202C>G). Patients carrying this SNP showed less infarct volume and better functional outcome after stroke. These results indicate that WRAP53 fosters DNA repair and neuronal survival to promote functional recovery after stroke.
INTRODUCTION
Ischemic stroke is the leading cause of neurologic death and long-term adult disability worldwide, imposing a huge health and economic burden on society (1). Ischemia-induced oxidative stress (2) compromises genome integrity, resulting in DNA damage, neuronal death, and impairments in functional recovery after stroke (3, 4). Neuroprotective approaches are therefore necessary to ensure neuronal survival and better functional outcome of patients with stroke.
Effective repair mechanisms to maintain DNA integrity are essential for neuronal homeostasis (5, 6). Increased oxidative stress and damaged DNA occur during brain aging and neurological disorders, including neurodegenerative diseases and stroke (3, 4). Oxidative damage causes different DNA lesions, including base modifications, abasic sites, single-, and double-strand breaks (DSBs) in DNA (4). In contrast to DSB, neuronal single-strand break repair mechanisms have been extensively studied in the past decades due to their frequency, reversibility, and direct association with neurodegeneration and neurodevelopmental disorders (7–9). Recently, solid evidence demonstrates that the most severe DNA DSBs can be repaired in neurons both under physiological and pathological conditions (6, 10–12). Moreover, defects in the nonhomologous end joining (NHEJ) repair pathway in neurons progressively accumulate endogenous DSBs, leading to chronic DNA damage, cell dysfunction, and neuronal apoptosis (6, 13, 14), thus pointing out the relevance of an adequate neuronal DNA repair response to survive. However, the precise molecular mechanisms that activate DSB repair response in neurons are still poorly understood.
Wrap53 (WD40 encoding RNA antisense to p53) gene encodes both a regulatory RNA, a natural antisense transcript that stabilizes p53 mRNA (15) and a scaffold WD40 protein WRAP53 (also known as WRAP53, WDR79, and TCAB1), involved in trafficking of factors to Cajal bodies, telomeres, and DSBs in replicative cells, independently of p53 (16–18). Following DNA damage, several factors are directed to DSB sites to trigger a signaling cascade leading to DNA repair (19). In this context, WRAP53 is rapidly recruited to DSBs, in association with phosphorylated histone H2AX (γH2AX) and is required for the assembly of downstream repair proteins, including 53-binding protein 1 (53BP1), Breast cancer type 1 susceptibility protein (BRCA1), and DNA repair RAD51 protein, at DNA breaks, promoting DNA repair by both the homologous recombination and the NHEJ pathways (16, 20). Moreover, loss of WRAP53-mediated DNA repair contributes to carcinogenesis and progression of primary tumors (18). However, how WRAP53 works in neurons remain unexplored. Here, we aimed to examine the impact of WRAP53 on DSB repair and neuronal survival after ischemia and its potential clinical relevance in stroke.
We describe that experimental ischemia induced by oxygen and glucose deprivation (OGD) (21) promoted the nuclear accumulation of WRAP53, leading to the activation of DNA repair response. WRAP53 knockdown exacerbated DSBs, thus promoting neuronal susceptibility to ischemia-induced apoptosis; conversely, overexpression of WRAP53 induced DSB DNA repair and neuronal survival. We also show that patients carrying a highly efficient WRAP53 variant harboring the human nonsynonymous single-nucleotide polymorphism (SNP) rs2287499 (c.202C>G) show improved functional outcome after stroke. These results provide new molecular insights into the mechanism of DSB repair in neurons and unravel the role of WRAP53 to maintaining genome integrity and survival after stroke.
RESULTS
OGD induces WRAP53 accumulation and nuclear translocation to foster neuronal survival
We first evaluated DNA damage in neurons after the ischemic insult in vitro. Double staining of neuronal marker microtubule-associated protein 2 (MAP2) and DSB marker γH2AX (22, 23) revealed that OGD induced DNA DSBs in neurons (Fig. 1A). The WRAP53 protein regulates DNA repair by providing a scaffold for DNA repair factors at DSBs in replicative cells (16, 20). We found that OGD time dependently up-regulated Wrap53 mRNA (Fig. 1B) and protein (Fig. 1C and fig. S1A) expression. However, p53 mRNA remained unchanged after ischemia (fig. S1B) and then discarding a possible role of Wrap53 mRNA on p53 mRNA stabilization. Accordingly, WRAP53-activated DSB repair response is independent of p53 mRNA stabilization (15, 24). As shown in Fig. 1C, γH2AX expression increased after OGD in a time dependent manner. WRAP53 up-regulation paralleled γH2AX accumulation in ischemic neurons (Fig. 1C and fig. S1A), suggesting that WRAP53 might be involved in DNA damage response of neurons to ischemia.
Fig. 1. OGD increased DNA DSBs and WRAP53 expression, which translocates to the nucleus in neurons.
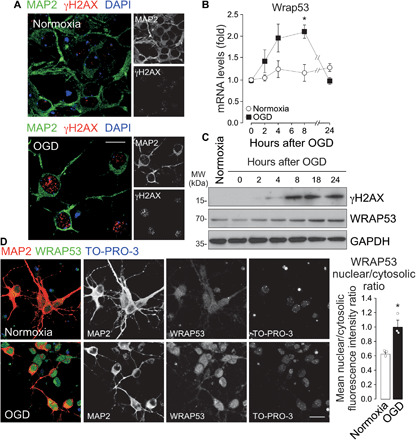
Neurons were subjected to normoxic (normoxia) or OGD conditions for 3 hours and were further incubated in culture medium for 8 hours or indicated time periods. (A) Representative image of cortical neurons stained with MAP2 (neuronal marker), γH2AX (DSB marker), and 4′,6-diamidino-2-phenylindole (DAPI; nuclear marker). Scale bar, 15 μm. (B) Time course of Wrap53 mRNA levels as quantified by real-time qPCR. The error bars are standard error deviation from three different neuronal cultures (*P < 0.05 versus normoxia; two-way ANOVA followed by the Bonferroni post hoc test). (C) Time course of WRAP53 and γH2AX expression levels as detected by Western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was probed as loading control. Representative blots are shown. Protein abundance quantification from three different neuronal cultures is shown in fig S1A. MW, molecular weight. (D) Representative image of cortical neurons stained with MAP2, WRAP53, and TO-PRO-3 (nuclear marker). Scale bar, 25 μm. The nuclear to cytosolic fluorescence intensity ratio of WRAP53 staining was quantified in around 145 neurons (45 to 50 neurons per culture). The error bars are standard error deviation from three different neuronal cultures (*P < 0.05 versus normoxia; Student’s t test).
Next, we studied subcellular localization of WRAP53 in neurons by immunofluorescence analysis and nucleus-cytosol fractionation. WRAP53 was present in both the nucleus and the cytosol in control (normoxia) neurons. However, OGD promoted WRAP53 translocation into the nucleus, particularly at 8 hours after the ischemic insult (Fig. 1D and fig. S1, C and D). Quantification of the blots (fig. S1D) and nuclear to cytosolic fluorescence ratios (Fig. 1D) revealed a 40% increase in WRAP53 nuclear localization, in relation to normoxia. Notably, the WRAP53 nuclear/cytosol fluorescence ratio after OGD was above the mean value found in normoxia in ~94% of the neurons. Thus, OGD up-regulates WRAP53 expression, which accumulates in neuronal nuclei after ischemia.
To evaluate the function of WRAP53 in neurons upon ischemia, we first down-regulated the expression of WRAP53 by using different small interfering RNA (siRNA) (siWrap53#1 and siWrap53#2). As shown in fig. S2, siWrap53#2 (hereinafter referred to as siWrap53) reduced mRNA (fig. S2A) and protein (fig. S2B) levels by 50%. WRAP53 knockdown (siWrap53) increased susceptibility to OGD-induced DNA damage, as shown by the increase in both γH2AX expression at earlier time points after ischemia (Fig. 2A and fig. S2C) and percentage of TUNEL+ (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling–positive) neurons at 4 and 8 hours after OGD (Fig. 2B), in comparison to control (siControl) neurons. Moreover, WRAP53 loss rendered neurons vulnerable to apoptosis following OGD, as revealed by both enhanced caspase-3 activation (Fig. 2C and fig. S2D) and increased percentage of apoptotic neurons (Fig. 2D), when compared with control neurons. Mitochondrial reactive oxygen species (ROS) production has an important role in DNA damage and neuronal apoptosis following ischemia (2, 4). Mitochondrial ROS generation and H2O2 production rapidly increased in neurons after OGD (fig. S2, E and F). However, WRAP53 knockdown did not affect OGD-induced mitochondrial ROS production in neurons (fig. S2E). Then, WRAP53 loss promoted DNA damage and neuronal apoptosis caused by ischemia, without interfering with mitochondrial ROS generation.
Fig. 2. WRAP53 knockdown enhanced DNA DSBs and neuronal susceptibility to OGD-induced apoptosis.
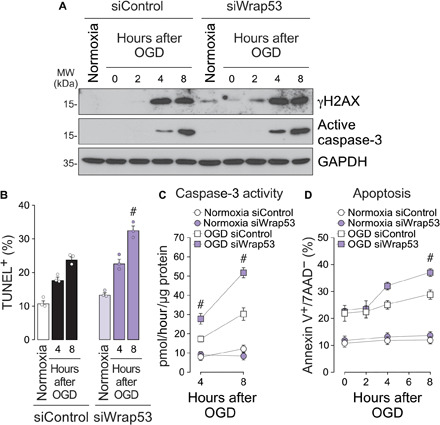
WRAP53 knockdown was performed by siRNA (siWrap53) transfection for 48 hours, and then neurons were subjected to OGD, as indicated in Fig. 1. (A) Time course of γH2AX and active caspase-3 expression levels detected by Western blotting. GAPDH was probed as loading control. Representative blots are shown. Protein abundance quantification from three different neuronal cultures is shown in fig. S2 (C and D). (B) The percentage of TUNEL-positive siControl and siWrap53-transfected cells was detected by flow cytometry at different time points after OGD. Data are means ± SEM from three different neuronal cultures (#P < 0.05 versus siControl OGD; two-way ANOVA followed by the Bonferroni post hoc test). (C and D) Analysis of caspase-3 activity and neuronal apoptosis in siControl and siWrap53-transfected neurons performed at different time periods after OGD. Data are means ± SEM from three different neuronal cultures (#P < 0.05 versus siControl OGD; two-way ANOVA followed by the Bonferroni post hoc test).
We next assessed the potential impact of WRAP53 on neuronal susceptibility to OGD damage. Ectopic expression of green fluorescent protein (GFP)–WRAP53 (fig. S3A) confirmed OGD-induced nuclear accumulation of WRAP53 (Fig. 3, A and B, and fig. S3B), as revealed by the increase in both nuclear/cytosolic fluorescence ratio (as in Fig. 3A) and nuclear fluorescence of GFP-WRAP53 (Fig. 3B and fig. S3B), in relation to normoxia. Thus, GFP-WRAP53 was mainly located in the cytosol under normoxia but accumulated in the nucleus after OGD, in contrast to the ubiquitous expression of GFP (Fig. 3, A and B, and fig. S3B). The GFP-WRAP53 nuclear/cytosol fluorescence ratio after OGD was above the mean value found in normoxia in ~90% of the neurons. Moreover, WRAP53 partially prevented OGD-induced neuronal apoptosis (Fig. 3C).
Fig. 3. Nuclear accumulation of WRAP53 fosters neuronal survival after OGD.
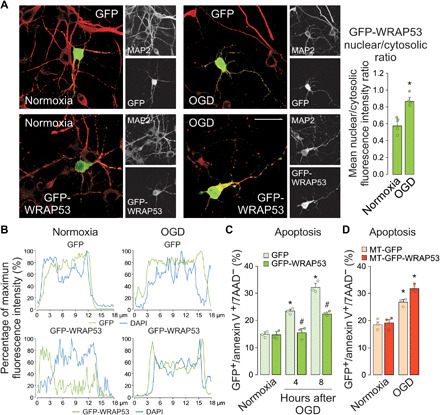
Neurons were transfected with GFP (empty vector) and GFP-WRAP53 for 24 hours and were subjected to OGD, as indicated in Fig. 1. (A) Representative image of cortical neurons stained with GFP and MAP2 (neuronal marker). Scale bar, 25 μm. The GFP-WRAP53 nuclear/cytosolic fluorescence intensity ratio was quantified in around 35 to 40 neurons (10 to 15 neurons per culture). The error bars are standard error deviation from three different neuronal cultures (*P < 0.05 versus normoxia; two-way ANOVA followed by the Bonferroni post hoc test). (B) Representative cross-sectional intensity profiles for GFP (green) and DAPI (blue) staining of GFP and GFP-WRAP53–transfected neurons. (C) Analysis of neuronal apoptosis in GFP and GFP-WRAP53–transfected neurons at 4 and 8 hours after OGD. The error bars are standard error deviation from three different neuronal cultures (*P < 0.05 versus normoxia; #P < 0.05 versus GFP; two-way ANOVA followed by the Bonferroni post hoc test). (D) Neurons were transfected with plasmids encoding GFP (MT-GFP) and GFP-WRAP53 (MT-GFP-WRAP53) fused to a mitochondrial-targeting (MT) sequence of human ornithine transcarbamylase for 24 hours and were subjected to a 3-hour OGD. Four hours after OGD, neuronal apoptosis in MT-GFP and MT-GFP-WRAP53–transfected neurons was analyzed. The error bars are standard error deviation from three different neuronal cultures (*P < 0.05 versus normoxia; two-way ANOVA followed by the Bonferroni post hoc test).
To study the potential role of nuclear accumulation of WRAP53 on neuronal survival, we directed newly synthesized GFP (MT-GFP) and GFP-WRAP53 (MT-GFP-WRAP53) away from the nucleus by using a mitochondrial-targeting (MT) sequence of human ornithine transcarbamylase (fig. S3C), consistent with the lack of identifiable nuclear localization signal in WRAP53. First, we confirmed the absence of nuclear protein accumulation by immunostaining and confocal microscopy (fig. S3, D to F), even after OGD. Cytosol sequestration of WRAP53 did not affect neuron viability under normoxic conditions while circumventing the neuroprotective role of WRAP53 observed in ischemic neurons (Fig. 3D). These findings support that nuclear accumulation of WRAP53 promotes neuronal survival after ischemia. WRAP53 may play a neuroprotective function against ischemic damage.
WRAP53 promotes DNA repair after OGD
Recent evidences support that WRAP53 activates DNA repair by providing a scaffold for DNA repair factors at DSB sites in replicative cells (16, 20). In this context, WRAP53 targets the ubiquitin E3 ligase RNF8 to DSBs and promotes the assembly of repair factor 53BP1 afterward (25). Once demonstrated that WRAP53 accumulates in the nucleus of neurons after an ischemic insult, we next analyzed the ability of WRAP53 to induce DNA repair in neurons after OGD. Foci formation of early DNA damage and repair markers, γH2AX and 53BP1, were quantified by immunostaining and confocal microscopy. As shown in Fig. 4A, a substantial increase in γH2AX and 53BP1 foci was observed in neurons after OGD, suggesting an activation of DNA repair response (25). However, WRAP53 knockdown (siWrap53) prevented DNA DSB repair, as shown by the decrease in the formation of 53BP1 foci and after OGD, in comparison with siControl neurons (Fig. 4A). The loss of WRAP53 increased the percentage of γH2AX foci–positive and 53BP1 foci–negative neurons after OGD, confirming that WRAP53 is necessary for the assembly of 53BP1 at DBS sites. Conversely, GFP-WRAP53 expression increased the number of 53BP1 foci and decreased the percentage of unrepaired DSBs (γH2AX foci–positive and 53BP1 foci–negative) neurons (Fig. 4B) at 8 hours after OGD. At a longer time (18 hours) following OGD, the percentage of 53BP1 foci decreased in neurons expressing GFP-WRAP53, indicating efficient DNA repair (fig. S4). Again, the expression of MT-GFP-WRAP53 that prevented the nuclear translocation of the protein and neuroprotection (fig. S3, D to F, and Fig. 3D) also failed to promote DNA repair after OGD (Fig. 4B and fig. S4). The deleterious effect of lacking 53BP1 foci was also confirmed in 53BP1 knockdown neurons (si53BP1) after OGD (fig. S5A). Then, nuclear accumulation of WRAP53 after OGD promotes DSB repair, leading to neuronal survival. These results highlight the neuroprotective function of WRAP53-induced DNA repair after ischemia.
Fig. 4. Nuclear accumulation of WRAP53 promoted DSB repair.
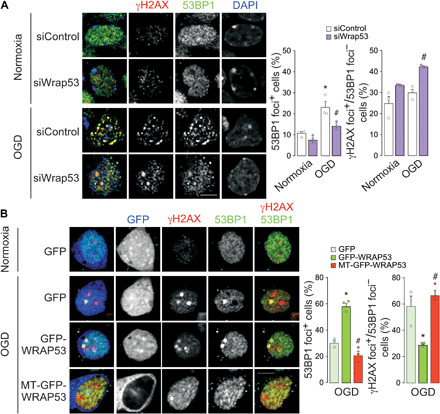
(A) WRAP53 knockdown was performed by siRNA (siWrap53) transfection for 48 hours and neurons were subjected to OGD, as indicated in Fig. 1. Neurons were immunostained for γH2AX and 53BP1 at 8 hours after OGD. Representative images are shown. DAPI, nuclear staining. Scale bar, 5 μm. Quantification of 53BP1 foci–positive neurons (percentage of neurons costaining γH2AX and 53BP1 foci) and percentage of γH2AX foci–positive and 53BP1 foci–negative neurons. The error bars are standard error deviation of 300 measurements from three different neuronal cultures (90 to 110 neurons per culture. *P < 0.05 versus normoxia; #P < 0.05 versus siControl OGD; two-way ANOVA followed by the Bonferroni post hoc test). (B) Neurons were transfected with plasmids encoding GFP, GFP-WRAP53, and MT-GFP-WRAP53 (MT sequence) for 24 hours and were subjected to OGD, as indicated in Fig. 1. Neurons were immunostained for GFP, γH2AX, and 53BP1 at 8 hours after OGD. Representative images are shown. Scale bar, 5 μm. Quantification of 53BP1 foci–positive neurons (percentage of neurons costaining γH2AX and 53BP1 foci) and percentage of γH2AX foci–positive and 53BP1 foci–negative neurons. The error bars are standard error deviation of 30 to 35 measurements from three different neuronal cultures (10 to 15 GFP+ neurons per culture. *P < 0.05 versus GFP; #P < 0.05 versus GFP-WRAP53; one-way ANOVA followed by the Bonferroni post hoc test).
Oxidative stress is necessary to induce WRAP53 accumulation and nuclear translocation after ischemia
To better understand the molecular mechanism of WRAP53-mediated DNA damage response in neurons, we next explored the stimulus responsible for WRAP53 nuclear translocation (Fig. 5, A and B). First, we focused on DNA damage as the endogenous trigger, and then we analyzed the effect of the topoisomerase I inhibitor camptothecin on WRAP53 expression (26). To avoid the induction of neuronal apoptosis observed at high doses of the drug (27), neurons were treated with 1 μM camptothecin for 8 hours. Under these conditions, cells suffered from DNA damage, as measured by the accumulation of γH2AX (Fig. 5A). In contrast to OGD (Fig. 1C and fig. S1A), camptothecin-induced DNA damage failed to up-regulate WRAP53 expression (Fig. 5A and fig. S5B) and to induce changes in subcellular compartmentalization of the protein (Fig. 5, C and D, and fig. S5C), which exclude DNA damage as the responsible stimulus.
Fig. 5. Oxidative stress is necessary to up-regulate WRAP53 expression and promote its traffic to the nucleus.
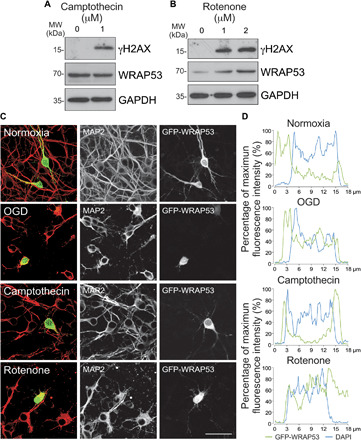
Neurons were treated with the topoisomerase inhibitor, camptothecin, and the mitochondrial inhibitor, rotenone, for 8 and 4 hours, respectively. (A and B) WRAP53 and γH2AX expression levels detected by Western blotting. GAPDH was probed as loading control. Representative blots are shown. Protein abundance quantification from three different neuronal cultures is shown in fig. S5B. (C and D) Neurons were transfected with WRAP53-GFP for 24 hours and were subjected to either OGD, as indicated in Fig. 1, or to camptothecin and rotenone treatments as described above. (C) Representative images of cortical neurons stained with GFP and MAP2 (neuronal marker). Scale bar, 25 μm. (D) Representative cross-sectional intensity profiles for GFP (green) and DAPI (blue) staining of GFP-WRAP53–transfected neurons.
Ischemia-induced oxidative stress compromises genome integrity, resulting in DNA damage and neuronal cell death (4). Taking into account that mitochondrial ROS generation and H2O2 production rapidly increased in neurons after OGD (fig. S2, E and F), we next analyzed whether oxidative stress was mediating nuclear migration of WRAP53. Neurons were treated with rotenone, which blocks the ubiquinone binding site of mitochondrial complex I to induce ROS generation in normoxia (28, 29). Rotenone treatment for 4 hours stimulated both the accumulation (Fig. 5B and fig. S5B) and nuclear migration of WRAP53 (Fig. 5, C and D, and fig. S5C). Thus, oxidative stress might be the trigger that promotes nuclear accumulation of WRAP53.
To confirm this observation, we pharmacologically prevented OGD-induced mitochondrial ROS production, and WRAP53 subcellular accumulation was analyzed in neurons. Reverse electron transport at complex I is the main source of ROS upon reperfusion of ischemic tissues (28). Mitochondrial-delivered nitric oxide using mitochondria-targeted S-nitrosothiol (MitoSNO) reversibly S-nitrosates—and prevents oxidation—of the Cys39 on the NADH dehydrogenase 3 (ND3) subunit of mitochondrial complex I (30). In this way, MitoSNO treatment during ischemia prevents the burst of ROS at complex I that takes place during the reperfusion phase, thus protecting against ischemia-reperfusion injury (30, 31). Therefore, we treated neurons with MitoSNO during OGD and DNA damage response after OGD was analyzed. As control, we used the nitric oxide donor SNAP (S-nitroso-N-acetyl penicillamine), which does not affect mitochondrial S-nitrosation (30). Accordingly, MitoSNO, but not SNAP, reduced ROS generation after OGD (Fig. 6A). Moreover, MitoSNO prevented DNA damage and WRAP53 accumulation (Fig. 6B and fig. S5D) and nuclear translocation after OGD (Fig. 6, C and D, and fig. S5E).
Fig. 6. Mitochondrial generation of ROS triggers WRAP53 nuclear accumulation after OGD.
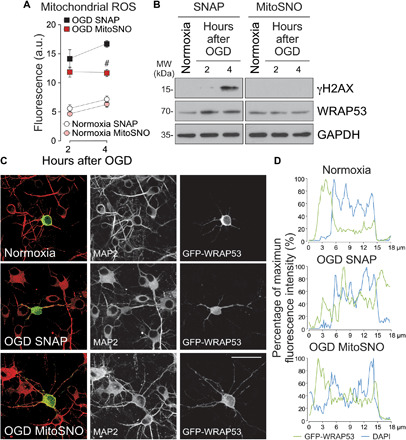
Neurons were subjected to OGD in the presence of the mitochondrial ROS production inhibitor, MitoSNO (mitochondria-selective S-nitrosating agent). As control, neurons were incubated with SNAP, which do not affect mitochondrial S-nitrosation and respiration. (A) Mitochondrial ROS generation after OGD. The error bars are standard error deviation from three different neuronal cultures (#P < 0.05 versus SNAP OGD; two-way ANOVA followed by the Bonferroni post hoc test). (B) Time course of WRAP53 and γH2AX expression levels as detected by Western blotting. GAPDH was probed as loading control. Representative blots are shown. Protein abundance quantification from three different neuronal cultures is shown in fig. S5D. (C and D) Neurons were transfected with GFP-WRAP53 for 24 hours and were treated with equal doses of SNAP or MitoSNO during OGD (3 hours), as indicated above. (C) Representative images of cortical neurons stained with GFP and MAP2 (neuronal marker). Scale bar, 25 μm. (D) Representative cross-sectional intensity profiles for GFP (green) and DAPI (blue) staining of representative GFP-WRAP53-transfected neurons. a.u., arbitrary units.
Ischemia-induced WRAP53 accumulation and nuclear translocation were confirmed in vivo [transient middle cerebral artery occlusion (tMCAO) model]. Figure 7 shows that ischemia enhanced WRAP53 expression (Fig. 7A) and nuclear accumulation in the damaged ipsilateral hemisphere, as compared with the contralateral hemisphere (Fig. 7, B and C). Moreover, these effects were not observed in mitochondrial catalase (mCAT) mice, which expresses the antioxidant enzyme catalase within the mitochondria (mCAT) to down-regulate endogenous mitochondrial ROS abundance (32). All these results demonstrate that mitochondrial ROS generation induces WRAP53 nuclear accumulation to maintain genome integrity and preserve neuronal survival after OGD.
Fig. 7. In vivo down-regulation of endogenous mitochondrial ROS prevents WRAP53 nuclear accumulation after ischemia.
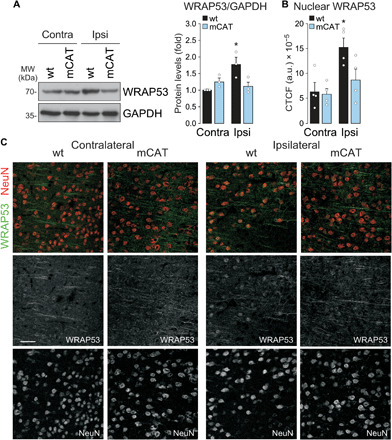
Wild-type (WT; +/+) and mCAT (mCAT/+; expressing the antioxidant enzyme catalase within the mitochondria to down-regulate endogenous mitochondrial ROS abundance) male mice were subjected to ischemic (tMCAO) stroke model and brains were analyzed 24 hours after surgery. (A) WRAP53 expression levels in the ipsilateral (Ipsi) hemisphere and contralateral (Contra) hemisphere as detected by Western blotting. GAPDH was probed as loading control. Representative blots and results of the protein abundance quantification were shown. The error bars are standard error deviation from three different animals (*P < 0.05 versus Contra hemisphere; two-way ANOVA followed by the Bonferroni post hoc test). (B and C) Brain sections were stained with WRAP53 and NeuN (neuronal nuclei marker) after 24 hours of tMCAO. (B) Quantification of nuclear WRAP53 using corrected total cell fluorescence (CTCF). The error bars are standard error deviation from four different animals (*P < 0.05 versus Contra hemisphere; two-way ANOVA followed by the Bonferroni post hoc test). (C) Representative image of brain sections stained with WRAP53 (green) and NeuN (red). Scale bar, 50 μm.
Wrap53 rs2287499 C>G SNP regulates protein nuclear translocation and functional outcome after stroke
Last, we evaluated the clinical relevance of our results. In this context, the common nonsynonymous SNP in codon 68 of the first coding exon of Wrap53 (rs2287499 c.202C>G) results in the amino acid change arginine (Arg68) to glycine (Gly68), which might affect protein function (33). In particular, the Wrap53 C>G SNP has been associated with primary tumor risk (33–37). However, the impact of this SNP on stroke remains unexplored. Taking into account that WRAP53 deficiency renders neurons vulnerable to apoptosis (Fig. 2, C and D), which conditions the functional outcome of stroke (38–40), we next examined the possible association between the Wrap53 C>G SNP and stroke prognosis. We found that stroke patients harboring the C/C genotype showed higher median modified Rankin scale (mRS) scores, indicating poor functional outcome at 3 months after stroke, than those with C/G and G/G genotypes. However, no differences were found between patients heterozygous and homozygous for the G allele (Fig. 8A). Moreover, the majority (100% G/G; 80.5% C/G) of patients harboring the G allele showed good functional outcome (mRS scores, ≤2) at 3 months after stroke. In contrast, a smaller proportion (53.7%) of patients harboring the C/C genotype showed good outcome, whereas 46.3% had poor functional outcome (mRS scores, >2) at 3 months after stroke (fig. S6A). Furthermore, the infarct volume at 4 to 7 days after stroke was higher in patients with the C/C genotype than those harboring the G allele (Fig. 8B). The logistic regression analysis revealed that the C/C genotype was an independent marker of poor functional outcome [odds ratio (OR), 2.95; 95% confidence interval (CI), 1.01 to 4.21; P = 0.001] at 3 months after stroke, after adjustment by age, neurological deficit evaluated by the National Institutes of Health Stroke Scale (NIHSS) score on admission, infarct volume, and early neurological deterioration (table S3). When patients were matched by infarct volume size, we found that, at infarct volume > 10 ml, the percentage of patients with poor functional outcome was higher in C/C patients, when compared with those with the G allele (table S4). All these results demonstrate that the Wrap53 C>G SNP (rs2287499) may be an independent biomarker of functional outcome after stroke.
Fig. 8. The Wrap53 C>G SNP (rs2287499) modulates protein nuclear translocation and functional outcome after stroke.
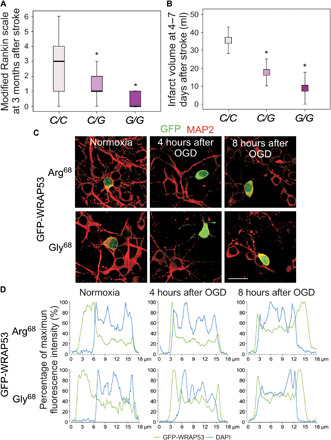
(A and B) The stroke cohort included 408 patients (rs2287499 C/C, 311; C/G, 87; G/G, 10). (A) mRS was used to evaluate the functional outcome of patients at 3 months after stroke. Boxplots show median values (horizontal line inside the box), quartiles (box boundaries), and the largest and smallest observed values (error bars) (*P < 0.0001 versus C/C patients; Mann-Whitney test). (B) Infarct volumes at 4 to 7 days after stroke were measured in patients with indicated Wrap53 genotypes. Data are expressed as means ± SD (*P < 0.0001 versus C/C patients; Student’s t test). (C and D) Neurons were transfected with the two polymorphic variants of WRAP53, Arg68 (ancestral, C allele) and Gly68 (mutant, G allele), fused to GFP (GFP-WRAP53) for 24 hours, and were subjected to OGD, as indicated in Fig. 1. (C) Representative images of cortical neurons stained with GFP and MAP2 (neuronal marker). Scale bar, 25 μm. (D) Representative cross-sectional intensity profiles for GFP (green) and DAPI (blue) staining of GFP-WRAP53 Arg68– and GFP-WRAP53 Gly68–transfected neurons.
On the basis of results showing that nuclear accumulation of WRAP53 promoted neuronal survival after ischemia (Figs. 2 and 3), one determining factor of functional outcome of stroke patients (38–40), we examined whether the SNP affects WRAP53 nuclear localization after an ischemic insult. Neurons were then transfected with plasmids expressing the two polymorphic variants of WRAP53, Arg68 and Gly68, fused to GFP. Immunofluorescence images revealed that OGD-induced nuclear translocation was faster (4 hours) in neurons expressing the Gly68 variant than those with the Arg68 variant (Fig. 8, C and D, and fig. S6, B and C), which would promote DNA repair and consequently neuronal survival, all leading to good functional outcome of patients harboring the G allele (Fig. 8, A and B). However, a delay in the nuclear translocation of the Arg68 variant after OGD (Fig. 8, C and D, and fig. S6, B and C) would be detrimental for functional recovery of C/C stroke patients (Fig. 8, A and B). Thus, the Wrap53 C>G SNP (rs2287499) conditions WRAP53 nuclear accumulation after ischemia, which would affect functional outcome after stroke.
DISCUSSION
Our results show that ROS generated during brain ischemia activates the translocation of WRAP53 to the nucleus, where it promotes 53BP1 assembly to DNA DSBs to facilitate DNA repair and neuronal protection. We also show that patients harboring the Gly68 polymorphic variant of WRAP53, which is efficiently recruited to the nucleus, dictate good functional prognosis after stroke.
DNA damage is a detrimental consequence of increased redox stress after stroke (3) and determines the balance between neuronal death and survival (4). The activation of the DNA repair response is therefore a critical step in the endogenous activation of the brain repair machinery during stroke recovery (4). Only the neurons displaying early DNA repair and hence keeping the genomic integrity survive at long term in humans (3). We found that ischemia promotes DNA DSB repair by a mechanism involving ROS-induced up-regulation and nuclear accumulation of WRAP53, leading to neuronal survival after OGD. We previously found that neuronal susceptibility to ischemia dictates brain damage, which finally determines functional recovery after stroke (38, 39). Our results provide new insights into DNA repair regulation in neurons and poses WRAP53 as an attractive target for the development of new neuroprotective strategies in ischemic stroke. Accumulated DSBs after ischemia compromises DNA integrity not only in neurons but also in cells forming the neurovascular unit, including endothelial cells, astrocytes, and pericytes. In particular, oxidative DNA damage and repair in pericytes are poorly understood, and pericyte loss is as an important contributor for blood-brain barrier disruption and neurovascular unit dysfunction in ischemic stroke (41). Further studies are needed to unravel the potential function of WRAP53 in preserving the neurovascular unit function after stroke.
We show that the Wrap53 rs2287499 C>G SNP determines functional recovery of stroke patients. Mechanistically, nuclear translocation of the polymorphic variant Gly68—encoded by the Wrap53 G allele—occurred faster than that of the Arg68—encoded by the Wrap53 C allele—upon ischemia. Cytosolic sequestration of WRAP53 was ineffective at preventing DNA repair and ischemic damage, which is in line with previous observations indicating that low levels of nuclear—but not cytosolic— WRAP53 are associated with deficient DNA repair response in tumor cells and poor prognosis of patients with cancer (24, 42). However, and in contrast to neurons, nuclear down-regulation of WRAP53 enhances radioresistance and survival of tumor cells (42). Therefore, other mechanisms, including WRAP53 loss–induced telomere dysfunction, might be considered in proliferative cells. As postmitotic cells, telomere length in neurons remains stable throughout life (43). However, telomere shortening enhances tumor risk and resistance to radiotherapy (44). Moreover, no correlation was found between the Wrap53 rs2287499 SNP and WRAP53 nuclear localization in tumor cells (24, 42), which is in apparent paradox with our data found in ischemic neurons. Therefore, the different mechanisms underlying WRAP53 function in neurons and cancer cells are unknown and deserve further investigation.
Our data also contributes to a better understanding of the mechanism responsible for DSB resolution in neurons. Specifically, we demonstrate that redox stress is an important component of the cytonuclear signaling mechanism that induces the traffic of WRAP53 to the nucleus. Mitochondrial complex I is a major endogenous generator of ROS (28, 29) that is amenable to pharmacological modulation. Rotenone, which blocks complex I ubiquinone binding site to activate complex I–derived ROS, induced both protein expression and nuclear accumulation of WRAP53 in the absence of OGD. Conversely, the presence of MitoSNO during OGD, by S-nitrosating the complex I ND3 subunit Cys39 (31), prevented complex I–mediated ROS formation after OGD and fully rescued protein expression and nuclear accumulation of WRAP53. Together, these results indicate that mitochondrial ROS, but not DNA damage itself, accounts for WRAP53 nuclear accumulation to stimulate 53BP1 recruitment on DNA DSBs to enhance DSBs repair.
In conclusion, here, we describe a previously unknown pathway involving ROS-mediated WRAP53 activation to maintain genome integrity and to preserve neuronal survival against stroke. Thus, ischemia-induced mitochondrial ROS generation triggers WRAP53 localization at nuclear DNA lesions to interact with γH2AX and to promote 53BP1 recruitment, DNA repair, and neuronal survival. In cycling cells, 53BP1 has been shown to favor the NHEJ pathway over the homologous recombination to repair DSBs (45). Neurons defective of the NHEJ pathway show progressive accumulation of endogenous DSBs leading to neuronal apoptosis (6, 13). Our data showing the 53BP1 involvement in neuronal DSB repair thus support the notion that the NHEJ pathway would be a preferred DNA damage protective mechanism after stroke. We identify the Wrap53 rs2287499 C>G SNP as a novel genetic biomarker of functional recovery of stroke patients. These results may be important not only in stroke but also in other neurological disorders associated with oxidative DNA damage. In this context, neurons are metabolically highly active cells and therefore are subjected to frequent oxidative DNA lesions during their long life (46). Neuronal activity–induced DSBs occur in early-response gene promoters leading to gene induction (11), and most early-response genes encode transcription factors with roles in cognitive function (47). Given that, according to our data, ROS triggers WRAP53 nuclear recruitment to repair DSBs in neurons, it is tempting to propose whether this mechanism would be important to delay the cognitive decline associated with aging. Moreover, increased DNA breaks and fragmentation reminiscent of inefficient DNA repair are likely involved in the cognitive decline associated with Alzheimer’s disease (12). WRAP53 has been described to accumulate in persistent DNA damage foci in sensory ganglion neurons (48). Furthermore, loss in WRAP53-containing Cajal bodies (49) occurs in motor neuron diseases (50), and WRAP53 depletion in worms and flies causes locomotion impairment and motor neuron death (51). Recently, Bergstrand et al. (52) described that heterozygous mutations in WRAP53 cause Hoyeraal-Hreidarsson syndrome, which is associated with impaired brain development and neurological decline (53). Whether a defect in the WRAP53-mediated DNA repair pathway herein described contributes to these and/or other neurological diseases is an interesting possibility deserving further research.
MATERIALS AND METHODS
Study design
In the current study, we examined the function of WRAP53 on DSB repair and neuronal survival after ischemia and the underlying mechanism, both in vitro and in vivo. In vitro experiments were performed on primary cultures of cortical neurons subjected to OGD, a widely used model of in vitro stroke (21). WRAP53 levels were modulated in neurons by plasmid expressing GFP-fused WRAP53 and siRNA transfections. The study design in vitro mainly included quantitative immunocytochemistry, flow cytometry, and Western blot analysis. Cerebral ischemia in vivo by tMCAO was performed in both wild-type (WT) and mCAT mice, which express catalase in the mitochondria to knocking down endogenous mitochondrial ROS accumulation (32). The study design included the tMCAO model, quantitative immunohistochemistry, and Western blot analysis on mice brain tissue. Last, we used a hospital-based cohort of ischemic stroke patients (408 patients; male, 59.3%; mean age, 72.8 ± 12.1 years) to study the association between the human nonsynonymous Wrap53 SNP (rs2287499, c.202C>G) that results in the amino acid change arginine (Arg68) to glycine (Gly68) and functional outcome of patients at 3 months after stroke, as measured by the mRS. An mRS score of >2 was considered poor functional outcome, as previously done (38, 40). Genotyping of the Wrap53 rs2287499 SNP was performed by authors blinded to the clinical status of patients by sequencing techniques. The mechanism was studied by immunocytochemistry in primary neurons expressing the WRAP53 SNP variants, Arg68 and Gly68, fused to GFP.
Animals
Animals were maintained in specific pathogen–free facilities at the University of Salamanca. All animal procedures we performed according to the European Union Directive 86/609/EEC and Recommendation 2007/526/EC, enforced in Spanish legislation under the directive RD1201/2005, following the Guide for the Care and Use of Laboratory Animals. All protocols were approved by the Bioethics Committee of the Institute of Biomedical Research of Salamanca. All efforts were made to minimize the numbers of animals used and ensure minimal suffering.
Neuronal cultures
Primary neuronal cultures were prepared from C57BL/6 J (The Jackson Laboratories, Sacramento, CA, USA) mouse embryo (E14.5) cortices. Neurons were seeded at 1.8 × 105 cells/cm2 in complete Neurobasal medium (Invitrogen, Madrid, Spain), supplemented with 2% B-27 (Invitrogen) and 2 mM glutamine (Invitrogen) and incubated at 37°C in a humidified 5% CO2-containing atmosphere. Half of the culture medium was replaced with fresh medium every 3 days. Neurons were used for the experiments on days 9 to 10 in vitro (54). All experiments were replicated in the number (3–4) of neuronal cultures indicated in the figure legends, which were performed from different pregnant females.
OGD protocol
After 9 to 10 days in culture, neurons were subjected to OGD by incubating cells at 37°C in an incubator equipped with an air lock and continuously gassed with 95% N2/5% CO2 for 3 hours. The incubation medium (Neurobasal medium without glucose) was previously gassed with 95% N2/5% CO2 for 5 min. In parallel, neurons were incubated in complete Neurobasal medium (normoxia condition) at 37°C in a humidified atmosphere of 95% air/5% CO2. After OGD, neurons were further incubated in Neurobasal medium at 37°C in a humidified atmosphere of 95% air/5% CO2 (reoxygenation or time after OGD) (21).
Small interfering RNA
Specific depletion of WRAP53 was achieved by using siRNA designed specifically to target the coding sequence of the mouse Wrap53 mRNA. The following siRNA sequences were used: siWrap53 #1, 5′-GCAUCUACUUUGAUCUGGA-3′ (accession number NM_144824, nucleotides 1471 to 1489); siWrap53(#2), 5′-GGACCUACAGCUUCUCGCA-3′ (nucleotides 661 to 679). To achieve the silencing of 53BP1, the commercial probe s77592 was used. As negative control, we used Silencer Select Negative Control No. 1 siRNA (siControl). All siRNAs were purchased from Ambion (Thermo Fisher Scientific, Offenbach, Germany). mRNA depletion and protein knockdown were confirmed by quantitative real time reverse transcription polymerase chain reaction (RT-qPCR) and Western blot, respectively.
Plasmid constructions
Plasmid vector expressing human Wrap53 full-length complementary DNA (cDNA) fused with the GFP cDNA (pWrap53) was obtained from Addgene (pEGFP-WRAP53beta, Addgene plasmid no. 64676) (16). Mitochondrial translocation of GFP-WRAP53 was achieved by inserting a mitochondrial targeting (MT) sequence of human ornithine transcarbamylase (accession number NM_000531) (55), at the N terminus of WRAP53. Briefly, cDNA fragment encoding the first 32 amino acids of human ornithine transcarbamylase was cloned into Bgl II and Eco RI digested pEGFP-WRAP53beta (GFP-WRAP53) plasmid to generate the new MT-GFP-WRAP53 plasmid. The forward and reverse oligonucleotides including the MT sequence of ornithine transcarbamylase were, respectively, 5′-GATCTATGCTGTTTAATCTGAGGATCCTGTTAAACAATGCAGCTTTTAGAAATGGTCACAACTTCATGGTTCGAAATTTTCGGTGTGGACAACCACTACAAGCG-3′ and 5′-AATTCGCTTGTAGTGGTTGTCCACACCGAAAATTTCGAACCATGAAGTTGTGACCATTTCTAAAAGCTGCATTGTTTAACAGGATCCTCAGATTAAACAGCATA-3′ (Bgl II and Eco RI sites underlined, Thermo Fisher Scientific). Oligos were annealed by heating to 95°C for 4 min and then slowly cooling to 22°C. Ligation was performed by the T4 DNA ligase (Thermo Fisher Scientific) enzyme for 10 min at 22°C, followed by heat inactivation for 10 min at 65°C. The PCR product was purified with ExoSAP-IT Express PCR Cleanup Reagent (Thermo Fisher Scientific) and sequence-verified. Where appropriate, GFP or MT-GFP plasmids were used as controls. GFP-WRAP53 was subjected to site-directed mutagenesis on Arg68, which were replaced by Gly to obtain GFP-WRAP53 Gly68, using a QuikChange XL kit (200524; Stratagene, CA, USA), followed by Dpn I digestion. As control, GFP-WRAP53 (corresponding to the ancestral variant GFP-WRAP53 Arg68) was used (GenBank accession number NM_018081.2). The forward and reverse oligonucleotides designed were 5′-GTGTCCCAGGAGCTAGGGGAGGGG-3′ and 5′-CCCCTCCCCTAGCTCCTGGGACAC-3′, respectively. GFP-transfected cells were identified by fluorescence microscopy and flow cytometry.
Cell transfections and treatments
For silencing experiments, neurons were transfected with siRNA (10 nM) using Lipofectamine RNAiMAX (Invitrogen), following the manufacturer’s instructions. Neurons were further incubated in Neurobasal medium for 48 hours before their use. Plasmid transfection was performed using Lipofectamine LTX (Invitrogen), according to the manufacturer’s instructions. Neurons were transfected with plasmid vectors (1.5 μg/μl) and used 24 hours later. When indicated, the OGD protocol was performed in the presence of the mitochondrial-selective S-nitrosating agent MitoSNO (1 μM), to decrease ROS production, or the control probe SNAP (1 μM); both compounds were donated by M. Murphy, Cambridge University (56). In some experiments, neurons were treated with the topoisomerase I inhibitor camptothecin (1 μM; Sigma-Aldrich, MO, USA) for 8 hours or the electron transport complex I inhibitor rotenone (1 to 2 μM; Sigma-Aldrich) for 4 hours.
Flow cytometry detection of apoptotic neuronal death
Neurons were carefully detached from the plates using 1 mM EDTA tetrasodium salt in phosphate-buffered saline [PBS; 136 mM NaCl, 2.7 mM KCl, 7.8 mM Na2HPO4, and 1.7 mM KH2PO4 (pH 7.4)] at room temperature. Neurons were stained with annexin V–DY634 (Immunostep, Salamanca, Spain) and 7-aminoactinomycin D (7-AAD; Becton Dickinson Biosciences, NJ, USA) in binding buffer (100 mM Hepes, 140 mM NaCl, and 2.5 mM CaCl2), and 5 × 104 (siRNA-transfected neurons) or 1.5 × 104 (plasmid-transfected neurons, identified as GFP+ cells) cells were analyzed, in three replicates per condition, on a FACScalibur flow cytometer (15-mW argon ion laser tuned at 488 nm; CellQuest software, Becton Dickinson Biosciences) to quantitatively determine the percentage of apoptotic neurons. The annexin V–DY634–stained neurons that were 7-AAD negative were considered to be apoptotic (38). Data were obtained from three different neuronal cultures and were expressed as percentages.
TUNEL assay
TUNEL staining was performed in cortical neurons by using the APO-DIRECT kit (BD Pharmingen, BD Biosciences) and following the manufacturer’s protocol. Briefly, neurons were carefully detached using 1 mM EDTA tetrasodium salt in PBS and suspended at a concentration of 1 to 2 × 106 cells/ml in 1% (w/v) (in PBS) paraformaldehyde for 45 min on ice. Cells were resuspended in cold 70% (v/v) ethanol and stored overnight at −20°C. Neurons were incubated in the DNA labeling solution, containing terminal deoxynucleotidyl transferase enzyme and fluorescein isothiocyanate–conjugated dUTPs, for 60 min at 37°C, and 7.5 × 104 siRNA-transfected neurons were analyzed, in three replicates per condition, on a FACScalibur flow cytometer (15-mW argon ion laser tuned at 488 nm; CellQuest software) to quantitatively determine the percentage of TUNEL+ cells. Data were obtained from three different neuronal cultures and were expressed as percentages.
Caspase-3 activity determination
A fluorimetric caspase-3 assay kit (Sigma-Aldrich) was used following the manufacturer’s protocol. Cells were lysed with 50 mM Hepes, 5 mM CHAPS, and 5 mM dithiothreitol (DTT) (pH 7.4) for 20 min on ice, and the assay buffer containing the Ac-DEVD-AMC (acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin) substrate [20 mM Hepes, 2 mM EDTA, 0.1% CHAPS, 5 mM DTT, and 16 μM Ac-DEVD-AMC, (pH 7.4)] was added. Aliquots of 200 μl were transferred to a 96-well plate, and the fluorescence was recorded at 5-min intervals for 30 min at 37°C using a Fluoroskan Ascent FL (Thermo Fisher Scientific) fluorimeter (excitation, 360 nm; emission, 460 nm). Caspase-3 activity was determined as AMC release rate extrapolating the slopes to those obtained from the AMC standard curve. Results were expressed as picomol per hour per microgram protein.
Determination of ROS generation
Mitochondrial ROS were detected using the fluorescent MitoSox probe (Invitrogen). Cells were incubated in buffered Hanks’ solution [134.2 mM NaCl, 5.26 mM KCl, 0.43 mM KH2PO4, 4.09 mM NaHCO3, 0.33 mM Na2HPO4, 5.44 mM glucose, 20 mM Hepes, and 4 mM CaCl2 (pH 7.4)] with 2 μM MitoSox Red reagent for 30 min at 37°C in a 5% CO2 atmosphere. Neurons were then washed with PBS and trypsinized. MitoSox fluorescence (580 nm) was analyzed on a FACScalibur flow cytometry. H2O2 production was measured by the Amplex Red assay (Invitrogen). Briefly, 1.8 × 104 suspended cells were incubated in Krebs-Ringer phosphate buffer [145 mM NaCl, 5.7 mM Na2PO4, 4.86 mM KCl, 0.54 mM CaCl2, 1.22 mM MgSO4, and 5.5 mM glucose (pH 7.4)], containing 100 μM Amplex Red reagent and horseradish peroxidase (0.1 U/ml). The luminescence was recorded at 20-min intervals for 1 hour using a Fluoroskan Ascent FL (Thermo Fisher Scientific) fluorimeter (excitation, 538 nm; emission, 604 nm), and the slopes were used for calculations (55). Results obtained as nanomol per hour per microgram protein were expressed as the fold change between normoxia and OGD.
RT-qPCR analysis
Total RNA samples were purified from cells using a commercially available kit (Sigma-Aldrich), and RT-qPCR was performed with Power SYBR Green RNA-to-CT TM 1-Step kit (Applied Biosystems, CA, USA). RT was carried out at 48°C for 30 min, and PCR conditions were 10 min at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C using the Prime PCR Assay Trp53 (Mmu #10025636, Bio-Rad Laboratories, CA, USA) and appropriate forward and reverse primers (0.3 μM), respectively (Thermo Fisher Scientific): 5′-AGCCCGAAAGGATGCTGAAC-3′ and 5′-ACCGCTCGGGTCCAGATCAAA-3′ (Wrap53 Mmu); 5′-TCAGCAATGCCTCCTGCACCA-3′ and 5′-GCATGGACTGTGGTCATGAG-3′ (Gapdh Mmu). The mRNA levels of each transcript were normalized to the Gapdh mRNA abundance obtained from the same sample. The relative mRNA levels were calculated using the ΔΔCt method and were expressed as the fold change between sample and calibrator (40).
Subcellular fractionation
To fractionate cytosol from nucleus, cells were washed with cold PBS containing 1 mM MgCl2 and harvested with cytosolic buffer [10 mM Hepes, 1.5 mM MgCl2, 10 mM KCl, 1 mM EDTA, NP-40 0.1% (v/v), and 1.5 M sucrose] supplemented with phosphatase inhibitors [1 mM Na3VO4 and 50 mM sodium fluoride (NaF)] and protease inhibitors [100 μM phenylmethylsulfonyl fluoride, anti-papain (50 μg/ml), pepstatin (50 μg/ml), amastatin (50 μg/ml), leupeptin (50 μg/ml), bestatin (50 μg/ml), and soybean (50 μg/ml)]. To promote cell lysis, samples were resuspended with a micropipette, stored on ice for 30 min, and vortexed. After checking cell lysis under a light microscope, extracts were centrifuged at 830g for 10 min, and the cytosolic fraction (supernatant) was removed and boiled for 5 min. Lysis of the nuclei was performed by repeatedly resuspending with a micropipette the nuclear pellet in nuclear buffer [50 mM Hepes, 1.5 mM MgCl2, 10 mM KCl mM, 0.5 mM NaCl, 1 mM EDTA, and NP-40 1% (v/v) (pH 7.9)] including protease and phosphatase inhibitor cocktail. Subsequently, nucleus extracts were stored on ice for 2 hours, then vortexed, boiled for 5 min, and sonicated for another 5 min (29).
Western blot analysis
Neurons were lysed in buffer containing 1% (v/v) nonidet NP-40, 5 mM EDTA di-K+, 20 mM tris-HCl (pH 8.0), 137 mM NaCl, and 10% (v/v) glicerol supplemented with phosphatase and protease inhibitor cocktail, stored on ice for 30 min, and centrifuged at 13000g for 5 min. Supernatants were collected and kept at −80°C until its use. Protein concentrations were determined with the bicinchoninic acid method, using bovine serum albumin as a standard (Pierce BCA Protein Assay kit, Thermo Fisher Scientific). Neuronal extracts were subjected to SDS–polyacrylamide gel electrophoresis on acrylamide gels (MiniProtean; Bio-Rad Laboratories). The resolved proteins were transferred electrophoretically to nitrocellulose membranes (Amershan, Little Chalfont, UK). Membranes were blocked with 5% (w/v) low-fat milk in 20 mM tris, 150 NaCl, and 0.1% (v/v) Tween 20 (pH 7.5) for 1 hour. After blocking, membranes were immunoblotted with primary antibodies overnight at 4°C. The antibodies used were anti-WRAP53 (1:500; Orb2180, Biorbyt, Cambridge, UK), anti-cleaved caspase-3 (1:2000; Asp175, 9661, Cell Signaling, MA, USA), anti-γH2AX (1:500; 05-636, Millipore, Darmstadt, Germany), anti-GFP (1:2000; ab290, Abcam, Cambridge, UK), anti-Lamin B (1:100; Sc-374015, Santa Cruz Biotechnology, Heidelberg, Germany), and anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:10,000; Ambion). GAPDH was used as loading control. After incubation with horseradish peroxidase–conjugated goat anti-rabbit immunoglobulin G (IgG) (Pierce, Thermo Fisher Scientific) or goat anti-mouse IgG (Bio-Rad) for 1 hour at room temperature, membranes were incubated with the enhanced chemiluminescence SuperSignal West Dura (Thermo Fisher Scientific) or Pierce ECL Plus Western Blotting Substrate (Thermo Fisher Scientific) for 5 min, before exposure to Kodak XAR-5 film, and the autoradiograms were scanned. Band intensities were quantified using ImageJ1.48v software.
Immunocytochemistry and image analysis
Neurons grown on glass coverslips were fixed with 4% (w/v) (in PBS) paraformaldehyde for 30 min and immunostained with rabbit anti-WRAP53 (1:200; Orb2180, Biorbyt), mouse anti-MAP2 (1:300; M#1406, Sigma-Aldrich), mouse anti-γH2AX (1:500; 05-636, Millipore), rabbit anti-HSP60 (1:500; ab46798, Abcam), rabbit anti-GFP (1:2000; ab290, Abcam), and rabbit anti-53BP1 (1:1000; NB-100-304, NovusBiological, CO, USA). Immunolabeling was detected using IgG-Cy2 (1:500), IgG-Cy3 (1:500), or DyLight 405 (1:500) secondary antibodies (Jackson ImmunoResearch. Cambridge, UK). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; D9542, Sigma-Aldrich) or with TO-PRO-3 iodide (T3, T3605, Molecular Probes, OR, USA). Coverslips were washed, mounted with SlowFace light antifade reagent (Invitrogen), and examined under an Olympus IX81 Spinning disk confocal microscope (Olympus, Tokyo, Japan) or a spectral laser confocal microscope (TSC-SL; Leica Microsystems, Mannheim, Germany) with three lasers: multiline argon (488 nm), helium-neon (543 nm), and helium-neon (633 nm) and equipped with 40×, 63× (1.4) HCX PL Apo oil immersion objectives for high-resolution imaging. All microscope settings were set to collect fluorescent images below saturation and were kept constant for all images taken in the experiment. Images were analyzed with the ImageJ 1.48v software (National Institutes of Health).
The nucleocytoplasmic distribution of WRAP53 was calculated as the ratio of the nuclear mean fluorescence to the cytoplasmic mean fluorescence of endogenous WRAP53 staining, measured in 145 neurons (45 to 50 neurons per culture; three different neuronal cultures). In the case of GFP-WRAP53–transfected neurons, the nuclear/cytosolic mean fluorescence intensity ratio was quantified in around 35 to 40 neurons (10 to 15 neurons per culture; three different neuronal cultures). Nuclei were identified by TO-PRO staining. Nuclear accumulation of WRAP53 after OGD was also quantified by measuring the mean fluorescence intensity of GFP, GFP-WRAP53, MT-GFP, and MT-GFP-WRAP53 within the nucleus. Once the background fluorescence was subtracted, cross-sectional intensity profiles were achieved for GFP+ cells. Values were normalized as the percent maximum intensity for both, GFP-WRAP53 (green) and nuclear (blue; DAPI staining) intensity profiles. Mean values were obtained from 30 to 50 transfected neurons (10 to 15 GFP+ neurons per culture) from three to four different neuronal cultures.
Neuronal DNA repair foci were assessed by quantifying cells with nuclear 53BP1 foci colocalizing with γH2AX protein (53BP1 foci–positive cells) (25). Results were expressed as the percentage of 53BP1 foci–positive cells in relation to total neurons, identified by DAPI staining. The percentage of γH2AX foci–positive cells that were 53BP1 foci–negative was measured and expressed as percentage related to total neurons, identified by DAPI staining. In siRNA-transfected neurons, approximately 300 neurons, from three different neuronal cultures (90 to 110 neurons per culture), were analyzed. In neurons transfected with plasmid vectors, analysis was performed in 30 to 35 GFP+ neurons from three different neuronal cultures (10 to 15 GFP+ cells per culture).
Transient middle cerebral artery occlusion
A knock-in mouse model that expresses mitochondrial catalase (mCAT) constitutively was used to down-modulate endogenous mitochondrial ROS in vivo (32). The transgenic mCAT mouse harboring the full-length cDNA encoding catalase fused to the cytochrome c oxidase subunit VIII–mitochondrial leading sequence was generated by homologous recombination in the Rosa26 locus under a C57BL/6 background, as detailed previously (32). Surgical endovascular insertion of a silicon-coated monofilament (Doccol Corporation, MA, USA, reference number: 602012PK10) was performed to induce tMCAO for 30 min of ischemia followed by filament removal to allow reperfusion, as previously described (40) Briefly, 16-week-old WT (+/+) and mCAT (mCAT/+) male mice (n = 3 to 4 animals per condition) were anesthetized with sevoflurane (4% for induction, 3% for maintenance) in a mixture of O2/N2O (30/70%). After surgical exposure of the right carotid artery tree, the filament was inserted through the external carotid artery and advanced through the internal carotid artery until it reached the middle cerebral artery. The regional cerebral blood flow was monitored during surgery with a laser Doppler probe (Moor Instruments, Devon, UK). After 30 min of ischemia, the filament was removed to allow reperfusion. Body temperature was maintained at 37° ± 0.5°C using a heating pad connected to a rectal probe (BAT-12 thermometer; Physitemp, NJ, USA). The animals were then sutured and allowed to recover for 24 hours. Brain tissue from ipsilateral and contralateral hemispheres was next collected for protein determination by Western blot analysis (three mice per condition), or animals (four mice per condition) were perfused for further immunohistochemical analysis.
Mouse perfusion and immunohistochemistry
The WT (+/+) and mCAT (mCAT/+) mice were anesthetized 24 hours after tMCAO by intraperitoneal injection of a mixture (1:4) of xilacine hydrochloride (Rompun; Bayer, Germany) and ketamine hydrochloride/chlorbutol (Imalgene; Merial, France), using 1 ml of the mixture per kilogram of body weight and then perfused intra-aortically with 0.9% NaCl, followed by 5 ml/g per body weight of Somogy’s fixative [4% (w/v) paraformaldehide and 0.2% (w/v) picric acid in 0.1 M phosphate buffer (pH 7.4)]. After perfusion, brains were dissected out coronally in three parts and postfixed, using the Somogy’s fixative, overnight at 4°C. Brain blocks were rinsed with 0.1 M phosphate buffer (PB) and sequentially immersed in 10, 20, and 30% (w/v) sucrose in PB until they sank. After cryoprotection, 20-μm-thick coronal sections were obtained with a freezing-sliding cryostate (Leica CM 1950 AgProtect, Leica). Coronal sections were rinsed in 0.1 M PBS three times each for 5 min and then incubated in (i) 1:200 anti-WRAP53 (Orb2180, Biorbyt), 1:1000 anti-NeuN (MAB377, Millipore), in 0.2% Triton X-100 (Sigma-Aldrich) and 5% goat serum (Jackson ImmunoResearch) for 48 hours at 4°C in 0.1 M PB; (ii) fluorophore-conjugated secondary antibodies (Jackson ImmunoResearch) in 0.05% Triton X-100 and 2% goat serum in 0.1 M phosphate buffer, for 2 hours at room temperature. Nuclei were stained either with DAPI (Sigma-Aldrich D9542) or TO-PRO-3 for 10 min. After rinsing with PBS, sections were mounted with Fluoromount (Sigma-Aldrich) aqueous mounting medium and examined under a spectral laser confocal microscope (Leica TSC-SL, Leica Microsystems) (39). Maximum z-projections of confocal image stacks (2 μm) were analyzed using ImageJ 1.48v software as described previously (57), whereby regions of interest containing NeuN-positive cells were traced. The corrected total cell fluorescence (CTCF) intensities corresponding to nuclear WRAP53 were determined by subtracting the area of selected NeuN-positive cells multiplied by the mean fluorescence of background readings from the integrated density of cells. CTCF = Integrated density − (area of selected cell × mean fluorescence of background readings). The CTCF mean value was calculated from 9 to 10 images of ipsilateral and contralateral cortical areas per animal (four animals per condition) and was expressed in arbitrary units.
Patients
An observational prospective study was performed on a cohort of consecutive patients with ischemic stroke admitted to the University Hospital of Santiago de Compostela (Galicia, Spain). Inclusion criteria were patients admitted within the first 12 hours after the onset of symptoms (or from the start of sleep in those with symptoms upon awakening) who were previously independent for daily living activities. Patients receiving tissue reperfusion therapies (65), those included in clinical trials (33), and those showing severe systemic disease (28) were excluded. In addition, some patients (24) refused to participate in the study, and others (36) were lost during the follow-up. Thus, the final cohort included 408 patients with ischemic stroke (male, 59.3%; mean age, 72.8 ± 12.1 years; C/C, 311; G/C, 87; C/C, 10 patients). Demographic and clinical features of stroke patients are included in table S1. In all cases, the research was carried out in accordance with the Declaration of Helsinki of the World Medical Association (2013) and approved by the Ethics Committee of Santiago de Compostela (2019/616). Informed consent was obtained from each patient or their relatives, after full explanation of the procedures and before taking part of the study.
Clinical variables
Patients were admitted to the Acute Stroke Unit (University Hospital of Santiago de Compostela, Spain) and treated according to the Guidelines of the Cerebrovascular Disease Study Group of the Spanish Society of Neurology (58). Medical history, previous treatments, potential vascular risk factors, blood and coagulation tests, 12-lead electrocardiogram, chest x-ray, and carotid ultrasonography were recorded upon admission. Stroke subtypes were classified according to the TOAST (Trial of Org 10172 in Acute Stroke Treatment) criteria (59). Stroke severity was assessed by a certified neurologist using the NIHSS upon admission and after 24 and 48 hours (table S1). Functional outcome was evaluated at 3 months using the mRS. An mRS score of >2 was considered poor functional outcome, as previously done (38, 40).
Neuroimaging studies
Patients were subjected to computed tomography (CT) scans upon admission and at days 4 to 7. The infarct volume was calculated in the second CT scan using an automated planimetric method. All neuroimaging evaluations were performed by neuroradiologists blinded to the patients’ clinical and laboratory results.
Outcome variables
The main outcome measure for all patients was functional outcome evaluated by mRS at 3 months ± 15 days. The infarct volume was considered as the secondary variable.
Human SNP analysis
Genotyping of Wrap53 SNP was performed by authors blinded to the clinical status of patients by sequencing techniques. The nonsynonymous SNP in codon 68 of Wrap53 (rs2287499 c.202C>G) was detected by amplifying genomic DNA with the forward primer 5′-CAACCGTTAGCTCCGGACTGCTG-3′ and the reverse primer 5′-GTGGAGTCTGGGGAGATGAA-3′. The 742-bp DNA fragment amplified was purified with the commercially available kit USB PCR Product Pre-Sequencing Kit (Affymetrix, CA, USA) following the manufacturer instructions. DNA fragments were sent to the sequencing service of the University of Salamanca (Spain). Analysis of sequenced data was performed by Geneious software (Geneious Biologics). The distribution of genotype frequencies between the stroke patients was within the Hardy-Weinberg equilibrium (P > 0.1 in all cases).
Statistical analysis
Clinical results are expressed as percentages for categorical variables. Results from continuous variables are expressed as mean (SD) or median (25th and 75th percentiles), depending on their normal distribution or not, respectively. The Student’s t test (normal data) or the Mann-Whitney test (non-normal data) was used to compare continuous variables between the groups. Proportions were compared using the χ2 test. To exclude a nonrandom mating population, the allele frequencies for Hardy-Weinberg equilibrium were tested with a goodness-of-fit χ2. The influence of the SNP on functional outcome was assessed by logistic regression analysis, after adjusting for the main baseline variables related to each main variable in the univariate analysis (enter approach and probability of entry P < 0.05). The results are expressed as adjusted ORs with the corresponding 95% CIs.
Experimental results are expressed as means ± SEM. A one-way or two-way analysis of variance (ANOVA) with a least significant difference post hoc test was used to compare values between multiple groups, and a two-tailed, unpaired Student’s t test was used for two-group comparisons. In all instances, P < 0.05 was considered significant. Statistical analyses were performed using SPSS Statistics 24.0 for Macintosh (IBM). In the Supplementary Materials, the raw data for experimental results (data S1) and statistical analysis (data S2) are reported.
Supplementary Material
Acknowledgments
We acknowledge the technical assistance of L. Martin, E. Prieto, M. Carabias-Carrasco, and M. Resch. Funding: This work was funded by the Instituto de Salud Carlos III (PI18/00265 and RD16/0019/0018 to A.A., RD16/0019/0001 to J.C., and CPII17/00027 to T.S.), European Regional Development Fund (FEDER) to Ministerio de Ciencia en Innovación (SAF2016-78114-R and RED2018-102576-T to J.P.B. and SAF2017-90794-REDT to A.A.), CIBERFES (CB16/10/00282 to J.P.B.), European Union’s Horizon 2020 Research and Innovation Programme (grant agreement 686009 to A.A.), Junta de Castilla y León (IES007P17 to A.A. and Escalera de Excelencia CLU-2017-03 Cofinanciado por el P.O. FEDER de Castilla y León to A.A. and J.P.B.), Fundación Ramón Areces (to A.A. and J.P.B.), and Ayudas Equipos Investigación Biomedicina 2017 Fundación BBVA (to J.P.B.). Author contributions: A.A. conceived the idea. I.S.-M., C.R., and A.A. designed the study. I.S.-M., C.R., J.P.B., and A.A. analyzed and interpreted the data and contributed to manuscript writing. I.S.-M., C.R., R.L., and J.A. performed the experiments. T.S. and J.C. contributed to the characterization of patient cohorts and clinical statistical studies. All authors discussed the results and critically revised and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/41/eabc5702/DC1
REFERENCES AND NOTES
- 1.Benjamin E. J., Virani S. S., Callaway C. W., Chamberlain A. M., Chang A. R., Cheng S., Chiuve S. E., Cushman M., Delling F. N., Deo R., de Ferranti S. D., Ferguson J. F., Fornage M., Gillespie C., Isasi C. R., Jimenez M. C., Jordan L. C., Judd S. E., Lackland D., Lichtman J. H., Lisabeth L., Liu S., Longenecker C. T., Lutsey P. L., Mackey J. S., Matchar D. B., Matsushita K., Mussolino M. E., Nasir K., O’Flaherty M., Palaniappan L. P., Pandey A., Pandey D. K., Reeves M. J., Ritchey M. D., Rodriguez C. J., Roth G. A., Rosamond W. D., Sampson U. K. A., Satou G. M., Shah S. H., Spartano N. L., Tirschwell D. L., Tsao C. W., Voeks J. H., Willey J. Z., Wilkins J. T., Wu J. H., Alger H. M., Wong S. S., Muntner P.; American Heart Association Council on Epidemiology and Prevention Statistics Commitee and Stroke Statistics Subcommittee , Heart disease and stroke statistics—2018 update: A Report From the American Heart Association. Circulation 137, e67–e492 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Bolaños J. P., Moro M. A., Lizasoain I., Almeida A., Mitochondria and reactive oxygen and nitrogen species in neurological disorders and stroke: Therapeutic implications. Adv. Drug Deliv. Rev. 61, 1299–1315 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Huttner H. B., Bergmann O., Salehpour M., Rácz A., Tatarishvili J., Lindgren E., Csonka T., Csiba L., Hortobagyi T., Méhes G., Englund E., Solnestam B. W., Zdunek S., Scharenberg C., Strom L., Stahl P., Sigurgeirsson B., Dahl A., Schwab S., Possnert G., Bernard S., Kokaia Z., Lindvall O., Lundeberg J., Frisen J., The age and genomic integrity of neurons after cortical stroke in humans. Nat. Neurosci. 17, 801–803 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Li P., Stetler R. A., Leak R. K., Shi Y., Li Y., Yu W., Bennett M. V. L., Chen J., Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology 134, 208–217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKinnon P. J., Maintaining genome stability in the nervous system. Nat. Neurosci. 16, 1523–1529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madabhushi R., Pan L., Tsai L.-H., DNA damage and its links to neurodegeneration. Neuron 83, 266–282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Canugovi C., Misiak M., Ferrarelli L. K., Croteau D. L., Bohr V. A., The role of DNA repair in brain related disease pathology. DNA Repair 12, 578–587 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKinnon P. J., DNA repair deficiency and neurological disease. Nat. Rev. Neurosci. 10, 100–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caldecott K. W., Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Suberbielle E., Sanchez P. E., Kravitz A. V., Wang X., Ho K., Eilertson K., Devidze N., Kreitzer A. C., Mucke L., Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat. Neurosci. 16, 613–621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madabhushi R., Gao F., Pfenning A. R., Pan L., Yamakawa S., Seo J., Rueda R., Phan T. X., Yamakawa H., Pao P.-C., Stott R. T., Gjoneska E., Nott A., Cho S., Kellis M., Tsai L.-H., Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161, 1592–1605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suberbielle E., Djukic B., Evans M., Kim D. H., Taneja P., Wang X., Finucane M., Knox J., Ho K., Devidze N., Masliah E., Mucke L., DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 6, 8897 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kannan A., Bhatia K., Branzei D., Gangwani L., Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 46, 8326–8346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karanjawala Z. E., Murphy N., Hinton D. R., Hsieh C. L., Lieber M. R., Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr. Biol. 12, 397–402 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Mahmoudi S., Henriksson S., Corcoran M., Mendez-Vidal C., Wiman K. G., Farnebo M., Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol. Cell 33, 462–471 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Henriksson S., Rassoolzadeh H., Hedstrom E., Coucoravas C., Julner A., Goldstein M., Imreh G., Zhivotovsky B., Kastan M. B., Helleday T., Farnebo M., The scaffold protein WRAP53β orchestrates the ubiquitin response critical for DNA double-strand break repair. Genes Dev. 28, 2726–2738 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henriksson S., Farnebo M., On the road with WRAP53β: Guardian of Cajal bodies and genome integrity. Front. Genet. 6, 91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergstrand S., O’Brien E. M., Farnebo M., The Cajal body protein WRAP53β prepares the scene for repair of DNA double-strand breaks by regulating local ubiquitination. Front. Mol. Biosci. 6, 51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polo S. E., Jackson S. P., Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 25, 409–433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coucoravas C., Dhanjal S., Henriksson S., Bohm S., Farnebo M., Phosphorylation of the Cajal body protein WRAP53β by ATM promotes its involvement in the DNA damage response. RNA Biol. 14, 804–813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Almeida A., Delgado-Esteban M., Bolaños J. P., Medina J. M., Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J. Neurochem. 81, 207–217 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Rogakou E. P., Pilch D. R., Orr A. H., Ivanova V. S., Bonner W. M., DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Crowe S. L., Tsukerman S., Gale K., Jorgensen T. J., Kondratyev A. D., Phosphorylation of histone H2A.X as an early marker of neuronal endangerment following seizures in the adult rat brain. J. Neurosci. 31, 7648–7656 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedström E., Pederiva C., Farnebo J., Nodin B., Jirstrom K., Brennan D. J., Farnebo M., Downregulation of the cancer susceptibility protein WRAP53β in epithelial ovarian cancer leads to defective DNA repair and poor clinical outcome. Cell Death Dis. 6, e1892 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pederiva C., Bohm S., Julner A., Farnebo M., Splicing controls the ubiquitin response during DNA double-strand break repair. Cell Death Differ. 23, 1648–1657 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pommier Y., Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 6, 789–802 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Morris E. J., Geller H. M., Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: Evidence for cell cycle-independent toxicity. J. Cell Biol. 134, 757–770 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chouchani E. T., Pell V. R., James A. M., Work L. M., Saeb-Parsy K., Frezza C., Krieg T., Murphy M. P., A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Veas-Pérez de Tudela M., Delgado-Esteban M., Maestre C., Bobo-Jiménez V., Jimenez-Blasco D., Vecino R., Bolaños J. P., Almeida A., Regulation of Bcl-xL–ATP synthase interaction by mitochondrial cyclin B1–cyclin-dependent kinase-1 determines neuronal survival. J. Neurosci. 35, 9287–9301 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prime T. A., Blaikie F. H., Evans C., Nadtochiy S. M., James A. M., Dahm C. C., Vitturi D. A., Patel R. P., Hiley C. R., Abakumova I., Requejo R., Chouchani E. T., Hurd T. R., Garvey J. F., Taylor C. T., Brookes P. S., Smith R. A. J., Murphy M. P., A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 106, 10764–10769 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chouchani E. T., Pell V. R., Gaude E., Aksentijevic D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N. J., Smith A. C., Eyassu F., Shirley R., Hu C. H., Dare A. J., James A. M., Rogatti S., Hartley R. C., Eaton S., Costa A. S. H., Brookes P. S., Davidson S. M., Duchen M. R., Saeb-Parsy K., Shattock M. J., Robinson A. J., Work L. M., Frezza C., Krieg T., Murphy M. P., Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vicente-Gutierrez C., Bonora N., Bobo-Jimenez V., Jimenez-Blasco D., Lopez-Fabuel I., Fernandez E., Josephine C., Bonvento G., Enriquez J. A., Almeida A., Bolaños J. P., Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 1, 201–211 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Cao H. Y., Wang S., Zhang Z. Y., Lou J. Y., Association between the WRAP53 gene rs2287499 C>G polymorphism and cancer risk: A meta-analysis. Genet. Mol. Res. 15, 10.4238/gmr.15037976, (2016). [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Closas M., Kristensen V., Langerød A., Qi Y., Yeager M., Burdett L., Welch R., Lissowska J., Peplonska B., Brinton L., Gerhard D. S., Gram I. T., Perou C. M., Børresen-Dale A.-L., Chanock S., Common genetic variation in TP53 and its flanking genes, WDR79 and ATP1B2, and susceptibility to breast cancer. Int. J. Cancer 121, 2532–2538 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Schildkraut J. M., Goode E. L., Clyde M. A., Iversen E. S., Moorman P. G., Berchuck A., Marks J. R., Lissowska J., Brinton L., Peplonska B., Cunningham J. M., Vierkant R. A., Rider D. N., Chenevix-Trench G., Webb P. M., Beesley J., Chen X., Phelan C., Sutphen R., Sellers T. A., Pearce L., Wu A. H., Van Den Berg D., Conti D., Elund C. K., Anderson R., Goodman M. T., Lurie G., Carney M. E., Thompson P. J., Gayther S. A., Ramus S. J., Jacobs I., Kjaer S. K., Hogdall E., Blaakaer J., Hogdall C., Easton D. F., Song H., Pharoah P. D., Whittemore A. S., McGuire V., Quaye L., Anton-Culver H., Ziogas A., Terry K. L., Cramer D. W., Hankinson S. E., Tworoger S. S., Calingaert B., Chanock S., Sherman M., Garcia-Closas M.; Australian Ovarian Cancer Study Group , Single-nucleotide polymorphisms in the TP53 region and susceptibility to invasive epithelial ovarian cancer. Cancer Res. 69, 2349–2357 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahmoudi S., Henriksson S., Farnebo L., Roberg K., Farnebo M., WRAP53 promotes cancer cell survival and is a potential target for cancer therapy. Cell Death Dis. 2, e114 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pouladi N., Abdolahi S., Farajzadeh D., Hosseinpour Feizi M. A., Haplotype and linkage disequilibrium of TP53-WRAP53 locus in Iranian-Azeri women with breast cancer. PLOS ONE 14, e0220727 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gomez-Sanchez J. C., Delgado-Esteban M., Rodriguez-Hernandez I., Sobrino T., Perez de la Ossa N., Reverte S., Bolaños J. P., Gonzalez-Sarmiento R., Castillo J., Almeida A., The human Tp53 Arg72Pro polymorphism explains different functional prognosis in stroke. J. Exp. Med. 208, 429–437 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodríguez C., Sobrino T., Agulla J., Bobo-Jiménez V., Ramos-Araque M. E., Duarte J. J., Gómez-Sánchez J. C., Bolaños J. P., Castillo J., Almeida Á., Neovascularization and functional recovery after intracerebral hemorrhage is conditioned by the Tp53 Arg72Pro single-nucleotide polymorphism. Cell Death Differ. 24, 144–154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodriguez C., Ramos-Araque M. E., Domínguez-Martínez M., Sobrino T., Sánchez-Morán I., Agulla J., Delgado-Esteban M., Gómez-Sánchez J. C., Bolaños J. P., Castillo J., Almeida A., Single-nucleotide polymorphism 309T>G in the MDM2 promoter determines functional outcome after stroke. Stroke 49, 2437–2444 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernández-Klett F., Potas J. R., Hilpert D., Blazej K., Radke J., Huck J., Engel O., Stenzel W., Genové G., Priller J., Early loss of pericytes and perivascular stromal cell-induced scar formation after stroke. J. Cereb. Blood Flow Metab. 33, 428–439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garvin S., Tiefenböck K., Farnebo L., Thunell L. K., Farnebo M., Roberg K., Nuclear expression of WRAP53β is associated with a positive response to radiotherapy and improved overall survival in patients with head and neck squamous cell carcinoma. Oral Oncol. 51, 24–30 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Assani G., Xiong Y., Zhou F., Zhou Y., Effect of therapies-mediated modulation of telomere and/or telomerase on cancer cells radiosensitivity. Oncotarget 9, 35008–35025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeggo P. A., Pearl L. H., Carr A. M., DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 16, 35–42 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Daley J. M., Sung P., 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 34, 1380–1388 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKinnon P. J., Genome integrity and disease prevention in the nervous system. Genes Dev. 31, 1180–1194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cholewa-Waclaw J., Bird A., von Schimmelmann M., Schaefer A., Yu H., Song H., Madabhushi R., Tsai L. H., The role of epigenetic mechanisms in the regulation of gene expression in the nervous system. J. Neurosci. 36, 11427–11434 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mata-Garrido J., Casafont I., Tapia O., Berciano M. T., Lafarga M., Neuronal accumulation of unrepaired DNA in a novel specific chromatin domain: Structural, molecular and transcriptional characterization. Acta Neuropathol. Commun. 4, 41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venteicher A. S., Abreu E. B., Meng Z., McCann K. E., Terns R. M., Veenstra T. D., Terns M. P., Artandi S. E., A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644–648 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lafarga M., Tapia O., Romero A. M., Berciano M. T., Cajal bodies in neurons. RNA Biol. 14, 712–725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Giorgio M. L., Esposito A., Maccallini P., Micheli E., Bavasso F., Gallotta I., Verni F., Feiguin F., Cacchione S., McCabe B. D., Di Schiavi E., Raffa G. D., WDR79/TCAB1 plays a conserved role in the control of locomotion and ameliorates phenotypic defects in SMA models. Neurobiol. Dis. 105, 42–50 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Bergstrand S., Böhm S., Malmgren H., Norberg A., Sundin M., Nordgren A., Farnebo M., Biallelic mutations in WRAP53 result in dysfunctional telomeres, Cajal bodies and DNA repair, thereby causing Hoyeraal-Hreidarsson syndrome. Cell Death Dis. 11, 238 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glousker G., Touzot F., Revy P., Tzfati Y., Savage S. A., Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br. J. Haematol. 170, 457–471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delgado-Esteban M., García-Higuera I., Maestre C., Moreno S., Almeida A., APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nat. Commun. 4, 2879 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Quintana-Cabrera R., Fernandez-Fernandez S., Bobo-Jimenez V., Escobar J., Sastre J., Almeida A., Bolaños J. P., γ-Glutamylcysteine detoxifies reactive oxygen species by acting as glutathione peroxidase-1 cofactor. Nat. Commun. 3, 718 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chouchani E. T., Methner C., Nadtochiy S. M., Logan A., Pell V. R., Ding S., James A. M., Cochemé H. M., Reinhold J., Lilley K. S., Partridge L., Fearnley I. M., Robinson A. J., Hartley R. C., Smith R. A., Krieg T., Brookes P. S., Murphy M. P., Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 19, 753–759 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hawkins S. J., Crompton L. A., Sood A., Saunders M., Boyle N. T., Buckley A., Minogue A. M., McComish S. F., Jiménez-Moreno N., Cordero-Llana O., Stathakos P., Gilmore C. E., Kelly S., Lane J. D., Case C. P., Caldwell M. A., Nanoparticle-induced neuronal toxicity across placental barriers is mediated by autophagy and dependent on astrocytes. Nat. Nanotechnol. 13, 427–433 (2018). [DOI] [PubMed] [Google Scholar]
- 58.de Lecinana M. A., Egido J. A., Casado I., Ribo M., Davalos A., Masjuan J., Caniego J. L., Martinez Vila E., Diez Tejedor E.; ad hoc committee of the SEN Study Group for Cerebrovascular Diseases, Fuentes B., Álvarez-Sabin J., Arenillas J., Calleja S., Castellanos M., Castillo J., Díaz-Otero F., López-Fernández J. C., Freijo M., Gállego J., García-Pastor A., Gil-Núñez A., Gilo F., Irimia P., Lago A., Maestre J., Martí-Fábregas J., Martínez-Sánchez P., Molina C., Morales A., Nombela F., Purroy F., Rodríguez-Yañez M., Roquer J., Rubio F., Segura T., Serena J., Simal P., Tejada J., Vivancos J.; Spanish Neurological Society , Guidelines for the treatment of acute ischaemic stroke. Neurologia 29, 102–122 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Adams H. P. Jr., Bendixen B. H., Kappelle L. J., Biller J., Love B. B., Gordon D. L., Marsh E. E. III, Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 24, 35–41 (1993). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/41/eabc5702/DC1