

Abstract
CD4+ T cells are critical to fighting pathogens, but a comprehensive analysis of human T cell specificities is hindered by the diversity of HLA alleles (>20,000) and the complexity of many pathogen genomes. We previously described GLIPH, an algorithm to cluster T cell receptors (TCRs) that recognize the same epitope and to predict their HLA restriction, but this method loses efficiency and accuracy when analyzing >10,000 TCRs. Here we describe an improved algorithm, GLIPH2, that can process millions of TCR sequences. We used GLIPH2 to analyze 19,044 unique TCRβsequences from 58 individuals latently infected with Mycobacterium tuberculosis (Mtb) and to group them according to their specificity. To identify the epitopes targeted by clusters of Mtb-specific T cells, we carried out a screen of 3,724 distinct proteins covering 95% of Mtb protein-coding genes using artificial antigen presenting cells (aAPC) and reporter T cells. We found that at least five PPE (Pro-Pro-Glu) proteins are targets for T cell recognition in Mtb.
CD4+ T cells perform many essential functions, including stimulating B cells to mature and secrete antibodies and supporting cytotoxic CD8+ T cells and phagocytes to mount a fast and effective protection against infections1. Depletion of CD4+ T cells in untreated HIV infection leads to opportunistic infections, such as Mycobacterium tuberculosis (Mtb), in areas where both are endemic2. Despite their importance, it has been difficult to comprehensively analyze CD4+ T cell responses in human beings. Tens of thousands of peptides predicted to bind HLA have been synthesized and screened for T cell recognition using ELISPOT analysis3, resulting in a very valuable collection of 300 Mtb antigenic peptides4. However, class II HLA binding motifs are only accurate for the best characterized molecules, particularly those common in cohorts of European origin. In addition, ELISPOT is limited in terms of its sensitivity and the requirement for a specific cytokine readout. Lastly, the Mtb genome is so large (4.4 Megabases) that using overlapping peptides for the entire genome is not a viable option.
Another strategy is the use of peptide-MHC tetramers5, where the development of combinatorial labeling and DNA barcode schemes allows hundreds to thousands of tetramers to be tested at once6–10. Although this approach has excellent sensitivity and is not dependent on the expression of any cytokine, it does depend on knowing the relevant HLA and epitopes, which in most cases cannot be predicted efficiently9, 11 and its throughput is likely to fall short when dealing with such large genomes as Mtb and Plasmodium falciparum (malaria). In addition, epitopes can be derived from sources other than translated genes, such as splicing variants, non-coding regions, and peptide fragments joined together during processing12–14. Furthermore, there are many examples of post-translationally modified peptides, such as deamidated, glycosylated, phosphorylated etc15, 16, for which simple T cell assays are not well developed. Whole pathogen lysates can be used to stimulate T cells, to enrich for antigen activated effector CD4+ T cells by CD154 upregulation and regulatory CD4+ T cells by CD137 upregulation17, 18, as well as to isolate Mtb-specific T cells19, 20. However, due to the complexity of pathogen lysates, it is difficult to define epitopes for these activated T cells or to know what might be missing versus an infection.
To overcome these challenges, we previously developed GLIPH, an algorithm to cluster TCRs that recognize the same epitope based on shared similarity in the complementarity-determining region 3 (CDR3), as well as to predict their HLA restriction19. It has been widely used for detection of antigen specific TCRs when the knowledge of the antigens is unavailable. But GLIPH loses efficiency and accuracy when analyzing >10,000 TCRs, where one starts to see artifactual clusters caused by “small-world” effect21. Meanwhile, other TCR clustering algorithms have become available, including TCRdist22, ALICE23 and iSMART24. However, most of these methods have not been systematically evaluated with a large and typically noisy dataset. And ALICE is designed to identify similar TCR variants from a single individual, but not to find shared specificities across multiple individuals. To identify the epitope for a TCR cluster, we previously introduced a T cell reporter system to efficiently map peptide epitopes19. However, the system cannot efficiently screen whole protein antigens due to its inefficient processing of exogenous proteins, and is thus unable to identify epitopes for TCR clusters only reactive to the Mtb lysate.
Here we broadly analyze the CD4+ T cell response to Mtb in T cells from latently infected individuals using GLIPH2, which can quickly analyze millions of TCR sequences with high clustering efficiency and accuracy after overcoming the “small-world” effect and algorithm optimization. To screen the Mtb genome for CD4+ T cell epitopes of interest, we expressed subpools of the 3,724 Mtb open reading frames (ORFs), endocytosed and processed by artificial APCs (aAPC) transfected with class II HLA genes, and used reporter T cells expressing TCR of interest to identify targeted epitopes. We show that this approach enables the identification of Mtb-specific TCRs that had not been previously described and represents a general strategy for analyzing human T cell responses.
Results
The CD4+ T cell response repertoire to Mtb
By combining the stimulation of specific T cells, TCR sequencing, repertoire analysis and antigen discovery we are able to agnostically analyze the TCR repertoire for Mtb at scale (Fig. 1a). We analyzed a dataset of 19,044 unique TCRβ sequences enriched with Mtb-specific variants from 58 latently infected individuals with GLIPH2, and grouped the TCRs into clusters on the basis of their specificities. A reporter system allowed us to screen 95% of Mtb proteins as potential sources of epitopes.
Figure 1. The workflow of Mtb-specific T cell repertoire and GLIPH2 analysis.
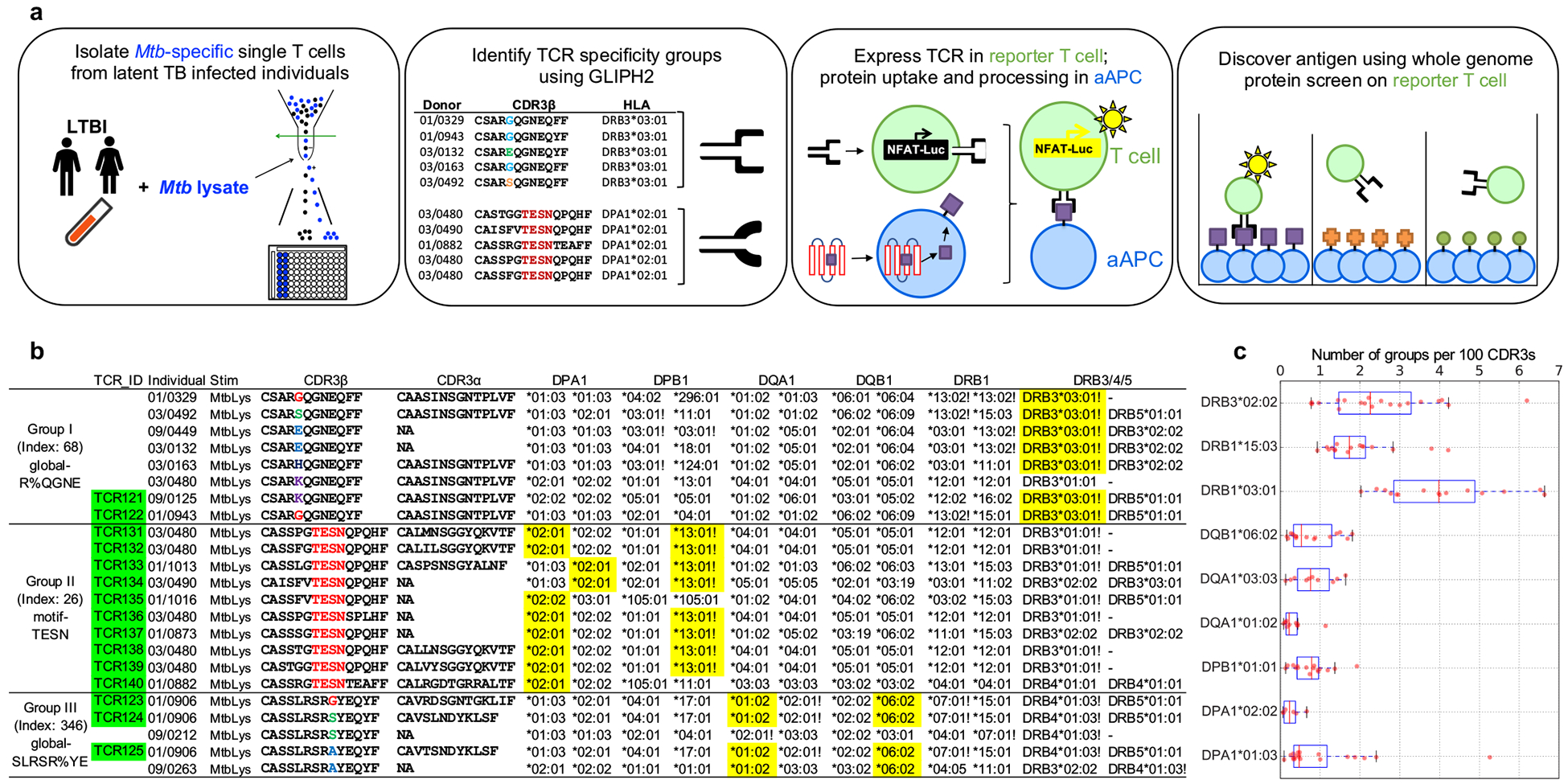
a, A schematic depicting the workflow. Briefly, PBMCs were isolated and stimulated with Mtb lysate for 12 hours. After stimulation, activated antigen-specific T cells were selected and single-cell sorted into 96-well plate for TCR sequencing. GLIPH2 analysis: TCRs with the same specificity contributed by different individuals sharing the same HLA allele were grouped together. Candidate TCRs were expressed in TCR-negative T cell line Jurkat 76; artificial antigen presenting cells (aAPC) were used to uptake protein antigen and present processed peptide. To identify antigens, reporter TCRs were screened against the whole proteome in microplate format. b, Representative TCR specificity groups and predicted HLA-restriction among Mtb-infected individuals (Group index from Supplementary Table 3). CDR3 α/β amino acid sequences from three GLIPH TCR specificity groups which only respond to Mtb lysate. Exclamation marks highlight the predicted common HLA class II alleles for each specificity group (combinatorial sampling probability Prob<0.1 DRB3*03 for group I, Prob<0.1 DPB1*13 for group II). Green colored boxes highlight the TCRs that have been validated in vitro. Yellow colored boxes indicate actual HLA as determined by reporter assay. c, The box plot shows the distribution of group numbers co-enriched with different HLA alleles among individuals (n=58). The y-axis indicates a specific HLA allele. The x-axis indicates the number of co-enriched specificity groups normalized to input CDR3 counts. Box-and-whisker plot shows 1 × interquartile ranges and 5–95th percentiles, centers indicate medians.
To build an Mtb-specific TCR repertoire with broad coverage, we stimulated PBMCs from 24 latently infected individuals for 12 hours with Mtb lysate, which presumably represents the whole Mtb proteome. We selected activated CD4+ T cells based on increased surface expression of CD154 and CD69 (Supplementary Figs. 1a–c) and single cell sorted them into 96 well plates. Paired TCRα and TCRβ sequences were identified as previously described25 (Supplementary Table 1). To further increase the number of TCRβ sequences, we also performed bulk sequencing on all the Mtb lysate activated CD4+ T cells with a range of 1,000~5,000 cells sorted from each of 12 individuals26 (Supplementary Table 1). Combining these TCRs with our previous work19, we had 19,044 unique TCRβ sequences enriched with Mtb-specific variants from 58 individuals, to enable a much broader GLIPH analysis and potential for shared epitope discovery (Supplementary Table 1).
GLIPH2
In order to accommodate this much larger dataset, we have developed a new version of the GLIPH algorithm (Supplementary Code). In this new version, first, a TCR can be assigned to more than one cluster. Second, we solved the “small-world” effect by restricting TCR members to be the same length for a cluster based on global similarity and differ at the same position between exchangeable amino acids according to a BLOSUM matrix. For a cluster based on local similarity (motif-based), we restricted the position of the CDR3 motif to only vary within 3 amino acids. Third, we introduced Fisher’s exact test to assess the statistical significance of a given motif instead of the sampling method used in the initial algorithm. This removes the requirement for a large reference dataset and enables GLIPH2 to analyze much larger datasets. Fourth, motifs that include conserved N or P encoded amino acids were given extra weight, as they are more random than germline encoded sequences, as also used by TCRdist22. In all, GLIPH2 is designed to analyze large single cell and bulk TCR sequencing with less noise and higher efficiency, and now can routinely analyze over one million TCR sequences at a time (Supplementary Code).
We compared GLIPH2 against the three published TCR clustering tools, including GLIPH19, TCRdist22 and iSMART24. First, we collected 3,264 unique TCRβ sequences spanning 15 specificities from VDJdb27 and analyzed these with all four algorithms (Supplementary Table 2). We found that GLIPH2 correctly clustered a higher percentage of unique TCRs compared to the other three algorithms (Supplementary Fig. 2a). This difference becomes even more obvious when irrelevant TCRs were spiked in, to generate a noisier dataset—imitating real input data structure. As the amount of spike-in data increased, the clustering efficiency and accuracy dropped precipitously for GLIPH and TCRdist, but not for GLIPH2. Notably, more variance, caused by random artifactual clusters produced by the “small-world” effect, was observed for GLIPH and TCRdist, but not for GLIPH2 after adding the random TCRs. These results confirmed GLIPH2’s superiority of efficiently and accurately capturing TCR specificity groups even from noisy datasets. Second, GLIPH2, similar to iSMART, is about 1,000 times faster than GLIPH and TCRdist on the annotated dataset and is about 10,000 times faster on a larger dataset with a 5-fold spike-in (Supplementary Fig. 2b). Third, analyzing the expanded Mtb dataset discussed above, we found that GLIPH2 consistently clustered a higher percentage of TCRs than GLIPH as the amount of input dataset increased (Supplementary Fig. 2c). With the whole dataset, GLIPH2 clustered 36.2% of all TCRs into 4,185 clusters (Supplementary Table 3) in 25 seconds using the online server, while GLIPH took 56,139 seconds (almost one day) to cluster 30.5%. This also represents a marked improvement over the earlier study, which was only able to cluster 14–15% of the ~5,700 sequences, indicating that an increased number of sequences results in more efficient clustering.
In addition, an important feature of GLIPH was that it predicted the HLA restriction for the five most common specificity groups19. To see whether this feature is preserved in GLIPH2, we ran a permutation test to validate the co-enrichment between a certain HLA allele and TCR specificity groups. In the case of DRB1*0301, the number of co-enriched TCR clusters dropped substantially after randomly shuffling the HLA information among all the study participants (Supplementary Fig. 2d).
GLIPH2 analysis of the Mtb-specific TCR repertoire
Multiple parameters are available in the GLIPH2 output to help evaluate specificity groups, including Fisher’s score, number of individuals, number of unique CDR3, and various scores inherited from GLIPH. In general, filters with a higher stringency result in a smaller number of clusters that are more likely to be correct (Supplementary Table 3). To facilitate antigen discovery, we focused on the 354 specificity groups that contained at least 3 unique TCRs from 3 or more individuals and exhibited significant V-gene bias (P < 0.05). All the five validated specificity groups from our previous study19 were sustained and expanded with more TCR members, indicating its consistency (Supplementary Table 3). Among these specificity groups, 119 were reactive to the Mtb “megapool” peptide collection developed by the Sette group4 but 235 of them were only reactive to the lysate, suggesting that even very large peptide pools capture only a fraction of the possible antigens, and thus hundreds of new epitopes remain to be identified. To address the possibility that these represent bystander T cell activation events or non-canonical/modified peptides15, we selected three candidate groups only reactive to the Mtb lysate for antigen identification (Fig. 1b). Group I “global-R%QGNE” and II “motif-TESN”, which were the top-ranking ones according to the GLIPH2 final score, with group III “global-SLRSR%YE” identified in both the current and previous study19.
The large size of our Mtb TCR dataset and GLIPH2 specificity groups also enabled us to determine how diverse the T cell response to a specific pathogen is in a given individual. This diversity (or the lack thereof) may be a critical aspect of how resistant a person might be to a pathogen. Among the most abundant DP, DQ and DR alleles in our cohort, we calculated the number of co-enriched specificity groups for each allele and normalized it according to input sequences in order to remove sequencing depth bias (Fig. 1c). We found a large variance of co-enriched group numbers among individuals for most of the alleles. Particularly, we found the largest range of diversity from 1 to 6 per 100 CDR3s in DRB3*0202 and DRB1*0301.
A global screen of the Mtb proteome
To screen protein antigens, we adopted the strategy of previous studies on artificial APCs (aAPC), which showed that co-expressing HLA-DM and CD80 gives K562 cells the ability to process and present exogenous proteins28, 29. To test whether or not expressing both HLA-DM and CD80 in the K562 cell line could produce a robust aAPC (Fig. 2a and Supplementary Fig. 3), we selected two Mtb reactive TCRs—TCR004 restricted by DQA1*0102/DQB1*0602 and TCR052 restricted by DRB1*0301 from our previous report19. For both TCRs, only the aAPC with a correct HLA molecule could activate its corresponding TCR after adding the whole Mtb lysate (Fig. 2b, c). Notably, this approach showed a more than 100-fold induction of luciferase with peptide stimulation, compared with less than 10-fold induction with peptide stimulation previously (Fig. 2d)19.
Figure 2. A reporter system to efficiently screen protein antigen.
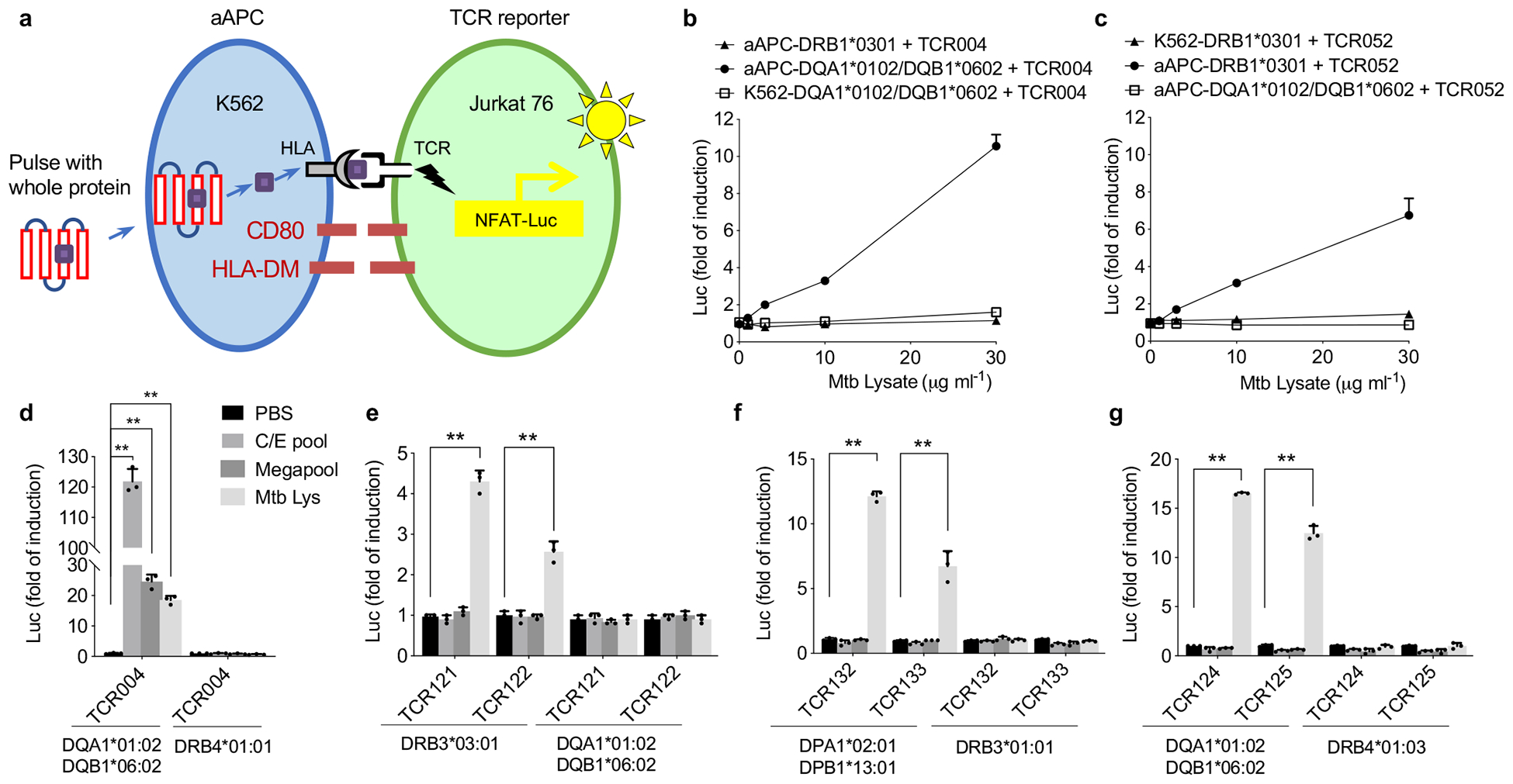
a, A schematic overview of the reporter system. Briefly, aAPC was built on K562 cell line with stable expression of HLA-DM, CD80 molecules and a candidate HLA allele. TCR reporter was built on TCR negative cell line Jurkat 76 with stable expression of Luciferase gene under NFAT response element. Exogeneous protein was endocytosed, processed and presented by aAPC, the resulted peptide MHC was recognized by its corresponding TCR. b, Dose-dependent response of TCR004 to Mtb lysate, paired with three different formats of APC: aAPC with a mismatched allele DRB1*0301, aAPC with a correct HLA DQA1*0102 and original K562 with a correct HLA DQA1*0102. c, Dose-dependent response of TCR052 to Mtb lysate, paired with three different formats of APC. Mean ± s.d. (n=3, biological replicates) shown. d, TCR004 was tested against its restricted HLA-DQA1*0102/DQB1*0602 allele and a mismatched DRB4*0101 allele using C/E pool, megapool peptides and Mtb lysate. Negative controls, PBS. e-g, Group I (e), group II (f) and group III (g) TCRs were tested against candidate HLA alleles as described in (d). Negative controls, PBS. Mean ± s.d. (n=3, biological replicates) shown. **P < 0.005 two-tailed Student’s t-tests.
To screen the whole Mtb proteome and discover novel antigens, we sought to produce a protein library de novo. We obtained a large set of Mtb cDNA clones from BEI Resources, which includes 3,294 ORFs from strain H37Rv and 430 from CDC1551, for a total of 3,724 distinct proteins covering 95% of the Mtb genome (Supplementary Table 4). To validate this collection, we first tested two TCRs of known specificity—TCR004, which recognizes a CFP10 epitope and TCR052, which recognizes a Rv3804c epitope. Using an in vitro cell-free protein expression system, we produced enough of these proteins to stimulate each TCR target (Supplementary Fig. 4a, b). Even after a thousand-fold dilution of the protein product, the activation signal remained significantly higher than the baseline. Of note, TCR004 only responded to its target antigen CFP10 but not Rv3804c and TCR052 only responded to Rv3804c but not CFP10. This demonstrates that the expression strategy can be used for T cell epitope screening with a robust signal to noise ratio. Therefore, we expressed pools of 12 clones each to cover all 3,724 proteins to speed proteome production and screening, resulting in 321 subpools in four 96-well plates (Supplementary Fig. 5).
Antigen discovery for lysate reactive TCR specificity groups
All 58 of the latently infected individuals discussed here were comprehensively HLA-typed by sequencing to facilitate GLIPH2 analysis and antigen discovery30. To determine whether or not the predicted HLA allele is correct, we chose two TCRs from different individuals from the three specificity groups (I—III) (Fig. 1b). Using the modified reporter system and Mtb lysate as the antigen source, we found that, as predicted, group I (TCR121 and 122) responded to DRB3*0301, group II (TCR132 and 133) responded to DPA1*0201/DPB1*1301 and group III (TCR124 and 125) responded to DQA1*0102/DQB1*0602 (Fig. 2e–g). In accordance with our selection criteria, none of these TCRs responded to the CFP-10/ESAT-6 (C/E) pool or Megapool stimulation. As a positive control, TCR004, which is known to respond to both C/E pool and Megapool stimulation, showed robust activation signals with all three conditions (Fig. 2d).
We then screened our Mtb proteome collection for each specificity group. For group II, we found one subpool (32F) was positive for TCR132 and subsequently identified espA (Rv3616c) as the specific antigen (Fig. 3a, b). EspA has been reported to be critical for mycobacterial survival and virulence. It is required for the secretion of EsxA (ESAT-6) and EsxB (CFP-10), as well as for the maintenance of mycobacterial cell surface integrity31, 32. To identify the specific epitope, we tested the top five peptide candidates predicted by NetMHCIIpan, and screened 77 overlapping peptides spanning the entire protein. We found the peptide “STRQALRPRADGPVG” to be the epitope for TCR132 (Fig. 3c). This epitope was ranked 335th among the 378 predicted peptides from espA by NetMHCIIpan, underscoring the fact that MHC binding predictions are only accurate for extensively characterized alleles33. In addition to TCR132, we took nine more TCR pairs from specificity group II, and found that eight out of ten can recognize the same peptide-MHC ligand (Fig. 3d, e). We also performed a glycine mutagenesis scan of TCR131, which confirmed that the GLIPH2 predicted contact motif “TESN” was indeed critical, with even a single amino acid change in this motif being sufficient to abolish specificity, but not in residues flanking either side (Fig. 3f).
Figure 3. Antigen discovery using proteome screening.
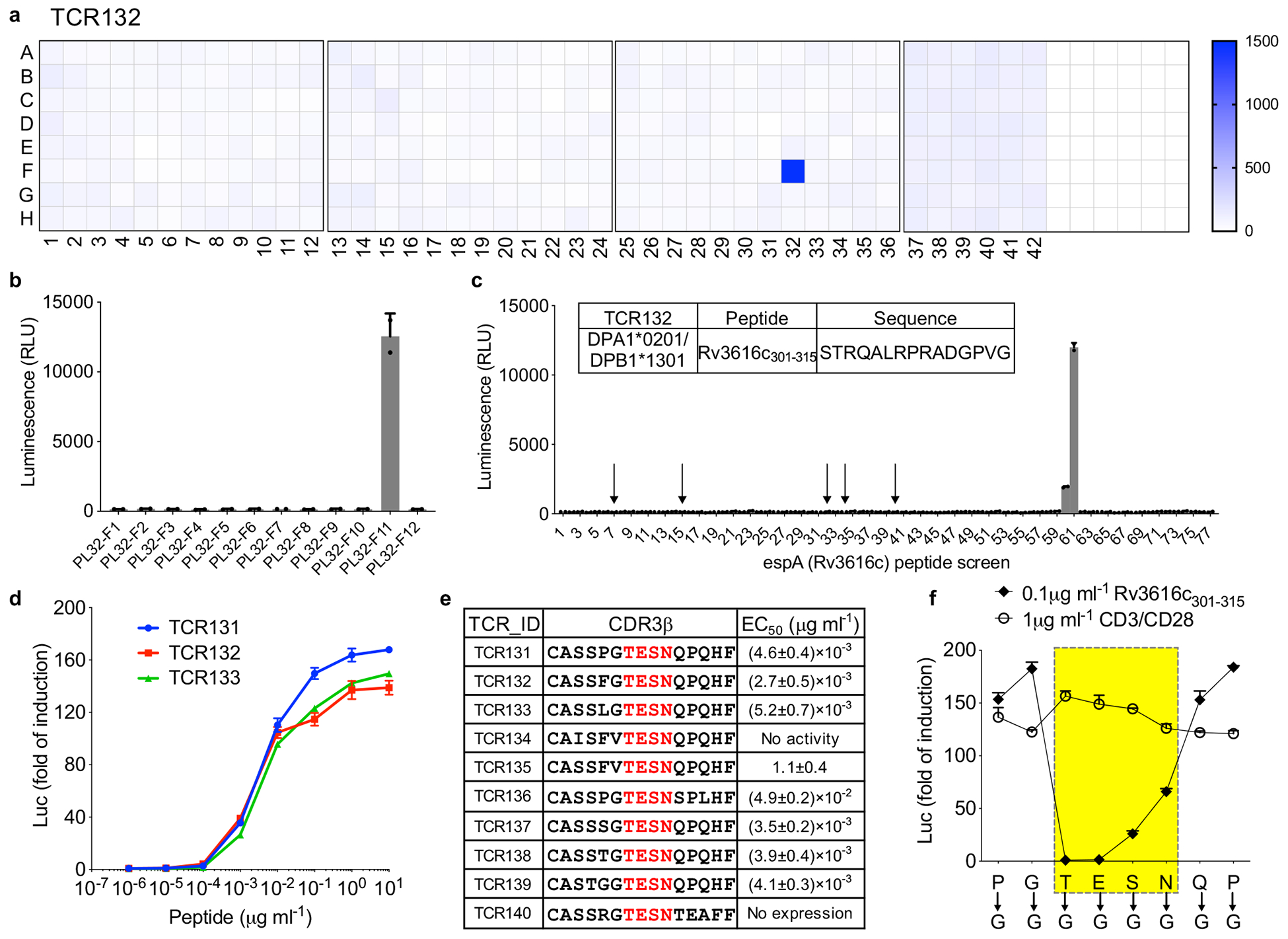
a, Screening of the whole Mtb proteome (321 subpools displayed in 4 plates) for TCR132. Color scale indicates the luminescence signal after stimulation. Representative of two independent experiments. b, Individual protein from the positive subpool (PL32F) was expressed separately and screened against TCR132. PL32-F11 (Rv3616c) showed positive activation. c, Overlapping peptides spanning protein espA (Rv3616c) were screened against TCR132. The top five candidate peptides predicted by NetMHCIIpan are labeled with an arrow. Insert table lists identified peptide antigen. Mean ± s.d. (n=2, biological replicates) shown. d, Dose-dependent response of Group II TCRs to its peptide antigen. TCR131-TCR133 were shown as examples. Mean ± s.d. (n=3, biological replicates) shown. e, EC50 values for TCR131–140 were determined from dose-response curves obtained by fitting the data from (d) to a nonlinear variable slope model. The average EC50 value and S.D. for each ligand were calculated from three different experiments. f, Glycine scan of CDR3β of TCR131. Each mutant was stimulated by DPA1*0201/DPB1*1301-restricted Rv3616c301–315, as well as a CD3/CD28-positive control. Mean ± s.d. (n=3, biological replicates) shown.
Similarly, we identified the antigen mIHF (Rv1388) and epitope “LTDEQRAAALEKAAA” for TCR123, TCR124 and TCR125 from group III (Fig. 4a–d). mIHF is one of the four mycobacterial nucleoid associated proteins (NAPs) that impact the expression of hundreds of genes by shaping chromatin architecture, and thus directly and indirectly controlling genes required for pathogenesis and for housekeeping functions34. The same peptide was tested in a previous report using ELISPOT analysis but did not yield a positive readout according to IEDB (http://www.iedb.org). Its identification as a TCR target in this study further illustrates the superior sensitivity of this system to identify T cell epitopes.
Figure 4. Antigen discovery for TCR specificity group III.
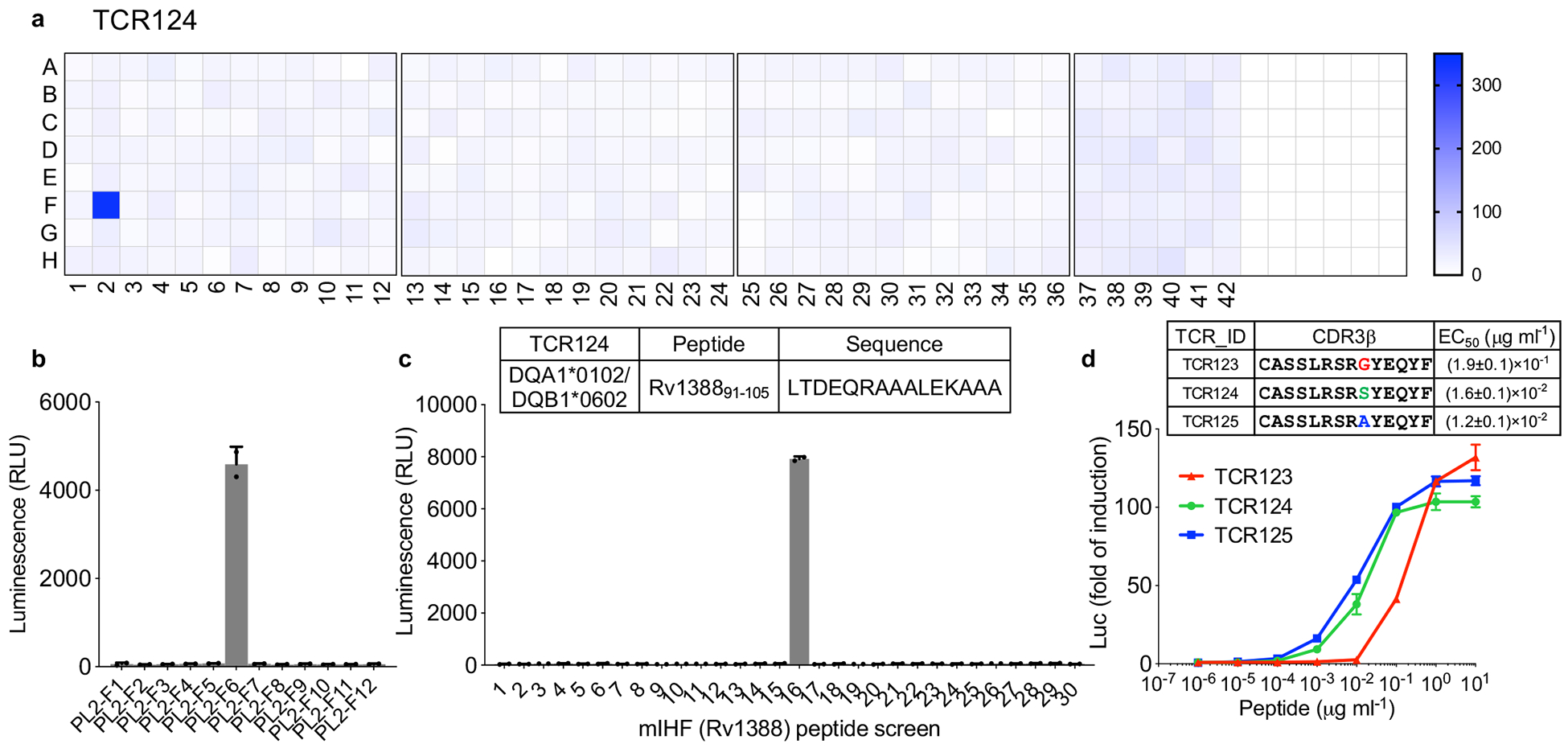
a, Antigen screen for TCR124 as described in figure 3. b, Individual protein from the positive subpool (PL2F) were expressed separately and screened against TCR124. PL2-F6 (Rv1388) showed positive activation. c, Overlapping peptides spanning protein mIHF (Rv1388) were screened against TCR124. Insert table lists identified peptide antigen. Mean ± s.d. (n=2, biological replicates) shown. d, Dose-dependent response of Group III TCR123-TCR125 to its peptide antigen. Mean ± s.d. (n=3, biological replicates) shown. EC50 values were determined as described in figure 3.
A highly conserved epitope from the PPE family of proteins
In the case of the group I TCRs, we found that four different subpools (6A, 32G, 34B and 35H) all robustly stimulated TCR121 (Fig. 5a) and subsequently identified a total of five proteins (Fig. 5b). All of them belong to the PPE family of proteins (Fig. 5c), characterized by the presence of a highly conserved N-terminal domain. Similar to ESAT6 and CFP10, PPE proteins are among the most immunogenic Mtb proteins, but there are 69 of them, which makes it difficult to determine which of these antigens or their epitopes are dominant and should be included in a vaccine35. To determine the epitope, we applied NetMHCIIpan to predict DRB3*0301 binding peptides for all the five PPE proteins and identified a consistent “AANR” binding motif among the top ranked peptides, indicating that this could be a conserved TCR epitope (Fig. 5c). Using synthesized peptides, we found that all of the five peptides could stimulate TCR121 and TCR122 with varying potency (Fig. 5d, e). While TCR121 responded robustly to all the five peptides, TCR122 only responded robustly to the high potency peptides PPE33107–121 and PPE29109–120, indicating a single amino acid difference in the CDR3β can introduce varying recognition pattern. Quantitatively, we calculated the half maximal effective concentration (EC50) for each peptide, with 25.9 pg/ml for PPE33107–121 activating TCR121 as the lowest and 21.4ng/ml for PPE65108–122 activating TCR122 as the highest (Fig. 5f).
Figure 5. Antigen discovery for TCR specificity group I.
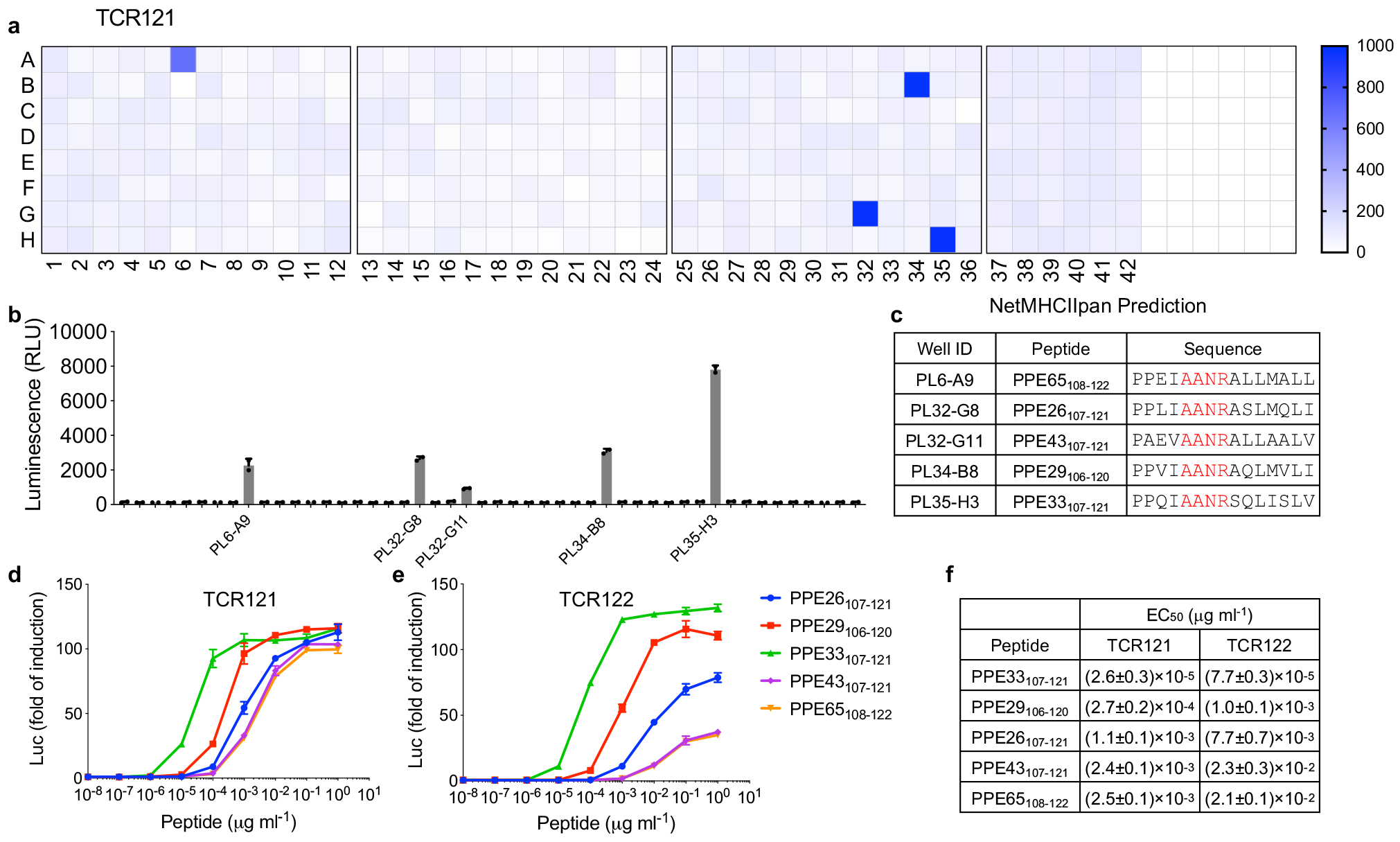
a, Antigen screen for TCR121 as described in figure 3. b, Individual protein from the four positive subpools (a) were expressed separately and screened against TCR121. Mean ± s.d. (n=2, biological replicates) shown. c, The five PPE proteins identified from (b) were analyzed by NetMHCIIpan. Top ranked peptides with high binding affinity to DRB3*0301 were listed. d-e, Dose-dependent response of Group I TCR121 (d) and TCR122 (e) to the top ranked peptides. Mean ± s.d. (n=3, biological replicates) shown. f, EC50 values were determined as described in figure 3.
To determine whether additional PPE family members could be recognized by the group I TCR, we collected a total of 68 PPE proteins from mycobacterial strain H37Rv using the UniProt database. After alignment, we found 64 of them contain the “AANR” motif (Supplementary Fig. 6) and summarized the sequence conservation using Weblogo (Fig. 6b)36. Using fold induction as the readout, 31 of the 64 peptides lead to a more than 2-fold induction at a high concentration (1 μg/ml) and 13 of them at a much lower concentration (1 ng/ml) (Fig. 6a). To summarize the correlation between peptide sequence and stimulation potency, we converted the data into a Weblogo plot using the fold induction as weight for each peptide sequence (Fig. 6c).
Figure 6. The discrepancy between peptide and protein stimulation.
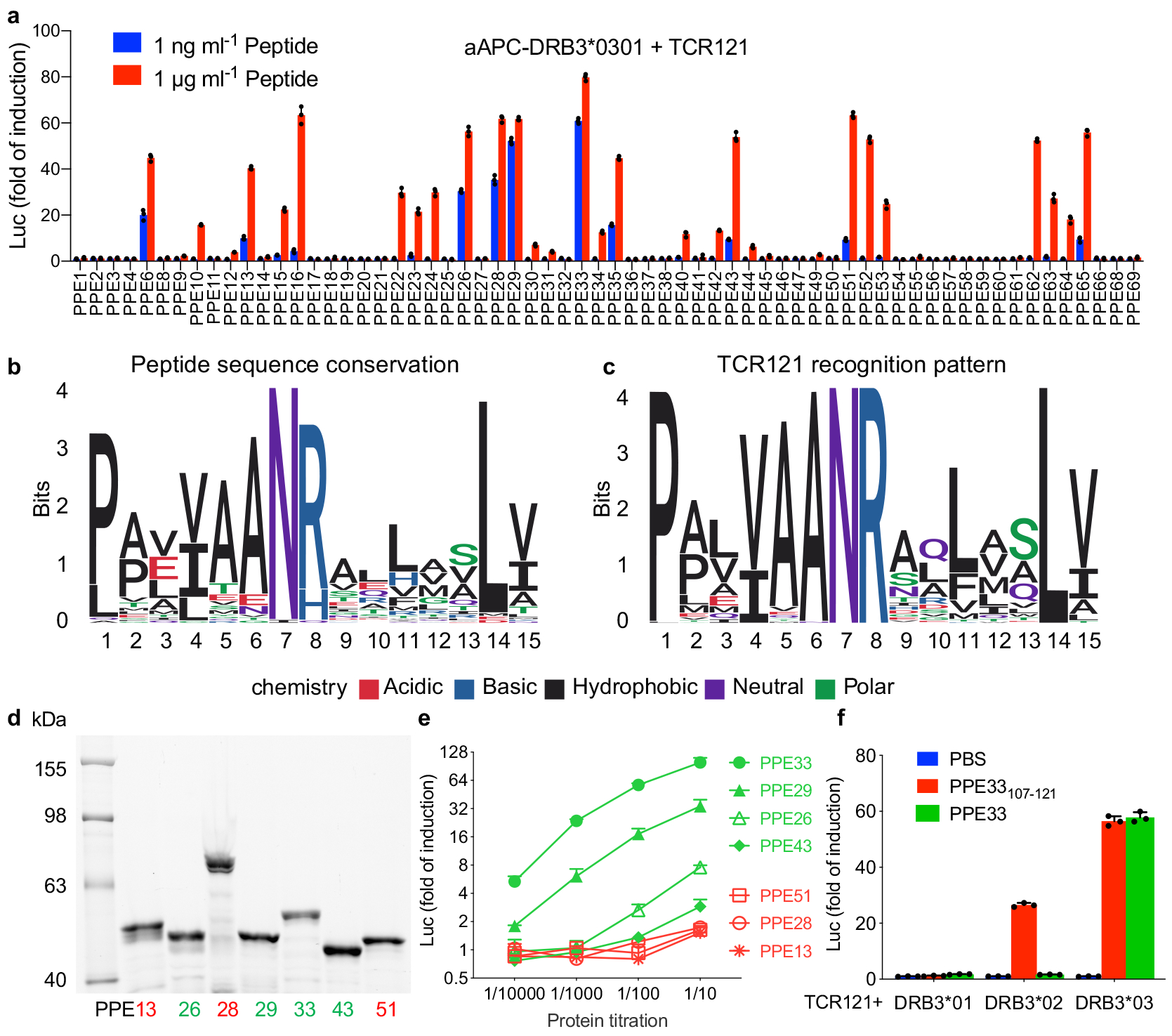
a, Dose response of TCR121 to all the available PPE protein-derived peptides containing the “AANR” region and restricted to HLA-DRB3*0301. The color indicates peptide concentration. Mean ± s.d. (n=3, biological replicates) shown. b, All peptides containing the “AANR” region derived from PPE family of proteins were aligned and visualized as a sequence logo based on sequence conservation. c, Recognition pattern of TCR121 to PPE protein-derived peptides, visualized as a sequence logo based on the activation data from (a). d, SDS-PAGE gel stained with Lumio green detection kit shows the expression level of each PPE protein. Marker in first lane and labeled on the left. PPE proteins in second to eighth lane, labeled on bottom and colored in accordance with (e). Representative of two independent experiments. e, Activation of TCR121 with different PPE proteins. Green indicating the four positive PPE proteins with a known potency and red indicating the three proteins with an unknown potency. Mean ± s.d. (n=3, biological replicates) shown. f, The response of TCR121 to both peptide PPE33107–121 and the whole PPE33 protein, restricted to three HLA-DRB3 homologous alleles. Mean ± s.d. (n=3, biological replicates) shown.
Discordant TCR activation by peptide and whole protein
Among the positive peptide hits, PPE28109–123 showed strong stimulation and a similar EC50 as peptide PPE29106–120. PPE13110–124 and PPE51107–121 showed modest stimulation and a similar EC50 as peptide PPE43107–121 (Supplementary Fig. 7a, b). Searching within our Mtb protein library, we found these three proteins (PPE28, 13 and 51) were included but not identified as positive subpools during the screening. This was unexpected and we reasoned that either we missed these subpools due to a low sensitivity of our reporter system or that these proteins behave differently than their derived peptides. To investigate this further, we expressed seven PPE proteins individually including the four positive proteins (PPE26, 29, 33 and 43) and three unknowns (PPE13, 28 and 51) at similar levels (Fig. 6d), and measured their stimulation potency using TCR121 activation as the readout. At the highest concentration, all four of the positive proteins showed strong to modest stimulation. The three unknowns showed only a modest activation at the highest concentration (Fig. 6e). In particular, the EC50 of peptide PPE28109–123 ranked third among the seven peptides (Supplementary Fig. 7b), whereas protein PPE28 showed little activation. To ensure the aAPC system does not lead to false-negatives, we used monocyte-derived dendritic cells as APC to activate TCR121 and obtained the same results (Supplementary Fig. 7c). This shows that all seven peptides can activate this TCR robustly, but the seven proteins are not equally processed and presented, possibly due to different protein structures or variant flanking sequences.
In another experiment, we asked whether an epitope and the protein it is derived from could be presented by different HLA alleles. We chose the most potent peptide PPE33107–121 and its protein PPE33 and tested with three closely related alleles DRB3*01, DRB3*02 and DRB3*03. At an optimal concentration, with their original HLA restriction DRB3*03, both the peptide and protein could activate TCR121 to similar levels (Fig. 6f). But with allele DRB3*01, neither peptide nor protein PPE33 could activate TCR121. With allele DRB3*02, while the peptide retained partial activation potency, the protein PPE33 lost its stimulatory ability completely. Similar results were obtained from monocyte-derived dendritic cells as APCs (Supplementary Fig. 7d). This confirms that stimulation potency can be very different between a peptide and its original protein, and further supports the idea that multiple factors can contribute to the process of epitope selection by HLA molecules37, 38.
Discussion
Here we describe a general strategy for analyzing the diverse T cell responses to a major infectious disease. By first analyzing the CD4+ “TCRome” using GLIPH2, we are able to reduce the inherent redundancy of TCR sequences, where there can be hundreds or thousands of variant sequences coding for the same specificity39, into shared specificity groups linked to particular HLA alleles. We then identified antigens for CD4+ T cells across the entire (and very large, at 4.4 Megabases) Mtb genome. We used this system to analyze Mtb-specific CD4+ T cells from latently infected individuals and successfully identified multiple antigens not previously described, and that may be useful in the development of more effective vaccines.
The initial part of this two-part strategy is to collect large numbers of TCR sequences from T cells highly enriched for pathogen specificity. We used a mixture of bulk TCRβ sequencing together with single cell paired sequences25, 26. While the former is excellent for rapidly populating a database, the latter is necessary to reconstruct an entire TCR heterodimer and thus determine its specificity. With the sequence data we are able to create a “TCRome” that is, a database of the most common CD4+ T cell responses one is likely to encounter in this population, parsed into likely specificity groups using GLIPH2. This parsing could be considered a dimensionality reduction tool for αβ TCRs, since the amount of sequence diversity is vast40, whereas the number of specificities is much smaller and is generally the more relevant parameter41. A more diverse response is likely to be a more protective one (Fig. 1c), but this parameter has been difficult to measure in human immune responses, especially for very large pathogens such as Mtb. Thus this type of analysis could be a valuable new metric for evaluating candidate vaccine responses, or as a prognostic indicator of infectious disease outcomes.
It is also important to note that GLIPH analysis does not capture all of the input TCRs, with the first version able to put only ~15% of the input TCRs into specificity groups, with the limited data then available19. With many more sequences to work with here and the superiority of GLIPH2, we now find that 36.2% of the enriched Mtb-specific TCRs could be clustered. The TCRs that don’t cluster could be because they are restricted by rare HLA alleles (of which there are many in this South Africa cohort) or they use rare TCR sequence modalities. Both of these issues should improve with an increased number of sequences.
The second part of our strategy, we targeted the most prominent GLIPH2 defined groups that had not been analyzed previously for antigen discovery. Traditionally, MHC binding algorithms have been used to predict peptides derived from a specific pathogen genome33. From a different perspective, we start with disease-relevant T cells and TCR repertoire analysis, then apply high-throughput protein screening to agnostically identify epitopes for TCRs of interest. Using this strategy, we identified several new Mtb T cell epitopes, in particular, a series of conserved peptides derived from the PPE family of proteins. All of these peptides, plus the five groups from our previous study, are from regularly translated Mtb proteins, which shows firstly that the TCR specificity groups we have identified are indeed Mtb specific rather than from bystander activation, and secondly, that while there are non-canonical peptide epitopes, such as spliced or post translationally modified peptides12, 15, these do not seem to be a major component of the CD4+ T cell response to Mtb in our studies.
Taken together, we have established a strategy that combines pathogen specific CD4+ T cell repertoire analysis and antigen discovery. Besides Mtb, the platform could be easily adapted to study other pathogens and CD4+ T cells that recognize them.
Method
Mtb-infected study participants
36 adolescent participants, aged 12 to 18 years, were randomly selected from a subset of the Adolescent Cohort Study, which was enrolled in the town of Worcester, approximately 100 km from Cape Town, South Africa, between 2005 and 200742. This study was approved by the Faculty of Health Sciences Human Research Ethics Committee of the University of Cape Town and Human Research Protection Program (HRPP) at Stanford University. Written informed consent was obtained from the parents or legal guardians of adolescents and assent was obtained from each adolescent. Latent Mtb infection was diagnosed by QuantiFERON® TB Gold In-tube (Qiagen) (QFT)42. Venous blood was collected for PBMC isolation, obtained by density gradient centrifugation using Ficoll and cryopreserved using freezing medium containing 90% fetal bovine serum and 10% DMSO. All samples used in this study were from asymptomatic QFT-positive adolescents. All the participants were HLA typed at Sirona Genomics (now Immucor inc.), under the supervision of Dr. Michael Mindrinos, as described in Thorstenson et al Hum Imm 201830.
Antigen-specific T cell capture
The capture of activated CD4+ T cells was performed as previously reported19. Briefly, PBMCs were thawed in complete RPMI 1640 medium at 2 × 106 cells/ml and recovered 12 hours before stimulation. PBMCs were stimulated with an Mtb lysate (10 μg/ml) for 12 hours in the presence of 1 μg/ml purified anti-CD49d antibody and anti-CD154-BV421. After stimulation, cells were harvested and stained with antibodies to various cell surface markers, including anti-CD3-Alexa700, anti-TCR α/β-PE/Cy7, anti-CD4-PerCP/Cy5.5, anti-CD8-BV605, anti-CD69-APC/Cy7 abs from BioLegend; purified anti-CD49d and anti-CD154-BV421 abs from BD Biosciences. Dead cells were stained using LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit from Thermo Fisher scientific. Activated CD4+ T cells were either single-cell sorted into 96-well plate for single cell TCRα/β sequencing or bulk sorted into an individual tube for TCRβ library preparation.
Antigens
Mtb whole cell lysate (strain H37Rv) and Mycobacterium tuberculosis Gateway® Clone Set (Plates 1–42) were kindly provided by Bei Resources. Mtb CFP10/ESAT-6 (C/E) peptide pool: 22 peptides spanning the length of the CFP10 molecule and 21 peptides spanning the length of the ESAT-6 molecule were purchased from Elim Biopharm. Each peptide was 15 amino acids long and overlapped with its adjacent peptide by 11 residues. Peptides were dissolved in DMSO at 100 μg/ml and then mixed together to make the C/E peptide pool. Megapool peptides, containing 300 epitopes from 90 Mtb proteins were kindly provided by Dr. Alessandro Sette (La Jolla Institute for Allergy & Immunology). For protein Rv1388 and Rv3616, overlapping peptide libraries were purchased from Elim Biopharm.
Single cell sequencing and analysis of TCRs
For the single cell TCR sequencing of antigen-specific T cells, we used our previously published method25. Briefly, single cells were sorted into 96-well plates containing 12μl of oneSTEP RT reaction buffer. The cells were then amplified for TCRβ and TCRα sequencing, using multiplex primers, a DNA-nesting and multiplex process as previously described25. During the PCR priming, DNA multiplex barcodes were attached to all the amplicons in a given well such that all wells could be combined to produce a single MiSeq 2×300bp sequencing run. For single-cell samples, the total population of reads is analyzed within each given well, identifying a single cell only if empirically determined boundary cutoffs of dominance for a single TCRβ and TCRα clone are encountered, as previously reported25. The resulting full sequence for the TCRα chain(s) and TCRβ chain is then combined with any index FACS phenotypic markers specific to these single cells.
Bulk sequencing and analysis of TCRs
For the bulk sequencing of sorted antigen-specific T cells, we used our previously published protocol for repertoire sequencing with purified mRNA as the template with some modifications26. In short, the method is based on the template switch mechanism in the reverse transcriptase which adds an anchor sequence at the 3’ end of the new synthesized ss cDNA (Clontech’s SMARTScribe TM reverse transcriptase). The template switch oligo also includes random nucleotides for unique molecular barcoding (isoC-GCGTCAGATGTGTATAAGAGACAGNNNNNNNNNNrGrGrG). The whole transcriptome was amplified with Clontech SeqAmp Polymerase. Based on the obtained ds cDNA as template TCR specific amplification was succeeded through a specific TCR constant primer (TRB: TGCTTCTGATGGCTCAAACACAGCG) using the NEB Q5 polymerase. Beckmann SPRIselect purification was used to clean the PCR product before sequencing on Illumina MiSeq. Filtering and merging of reads were done as previously described and VDJ and CDR3 annotations were assigned using IMGT/HighV-Quest (IMGT®).
Calculating TCR convergence in GLIPH2
For each cluster, a contingency table is constructed with the number of CDR3s that would fit the cluster in the query set, the number of unique CDR3s in the query set, the number of CDR3s that would fit this cluster in the reference set, the number of unique CDR3s in the reference set. A Fisher’s exact test is then carried out to compute the p-value for this contingency table. For each member CDR3 of a cluster based on local similarity, if the local motif of the member CDR3 is partially coded by non-template sequence, the Fisher’s exact p-value of the cluster is divided by 2 to boost the significance of convergence.
In the GLIPH2 algorithm, samples from an individual at different conditions, different time points, different subsets, or different tissue types are treated as different samples even though the HLA information for those samples is identical. To accommodate this situation, each sample is labeled as subject:condition.
Calculating TCR local convergence in GLIPH2
Within any set of T-cell receptors, a collection of all continuous 2mers, 3mers, 4mers and 5mers, can be extracted and evaluated for their frequency within the set. Instead of using sampling method in the previous GLIPH algorithm to check whether positive selection is observed for a particular motif, a Fisher’s exact test is carried out on a contingency table with number of motifs found in query set, number of unique CDR3s in query set, number of motifs found in reference set, and number of unique CDR3s in reference set. The change does not affect the results, but eliminates the requirement that the reference set needs to be much larger than the query set. In addition, by using Fisher’s exact test, instead of sampling, the GLIPH2 algorithm is able to run faster.
Performance comparison of GLIPH2, GLIPH, iSMART and TCRdist
To evaluate the performance among GLIPH2, GLIPH, iSMART and TCRdist, 3,264 unique CDR3β sequences for 15 epitopes, together with V and J information, were compiled from VDJdb, similarly as the authors did for iSMART24. For TCRdist, a customized code was used to convert amino acids CDR3 sequences back to nucleotide sequences with the original V and J genes. To mimic the real situation where the epitope is unknown for input CDR3s, epitopes were masked and labeled as “Unknown” during clustering. In addition, 3264 (1x), 6528(2x), 9792 (3x), 13056(4x) and 16320(5x) CDR3β sequences from irrelevant CD4+ T cells were spiked into the original dataset to mimic real noise in the data. Since TCRdist does not output clusters, its code was adjusted to generate TCR clusters in order to compare the output with the other programs. All four tools were run with default parameters. As each CDR3 is uniquely linked to one epitope in the benchmark dataset, we defined cluster purity (p) as the number of the most abundant antigen divided by the number of CDR3s in a cluster. We used the percent of accurate (p>90%) clusters as a measure for specificity.
Cell culture and cell lines
The Jurkat 76 T-cell line, deficient for both TCRα and TCRβ chains, was kindly provided by Dr. Shao-An Xue (Department of Immunology, University College London). The NFAT reporter cell line (J76-NFATRE-luc) was constructed using lentiviral transfer of pNL[NlucP/NFAT-RE/Hygro] (Promega) into the Jurkat 76 cell line. Single cell clones with the best fold induction were selected based on the induction of luciferase after PMA/Ionomycin stimulation. K562 cell line was obtained from the ATCC and cultured under standard conditions. Artificial antigen presenting cells (aAPC) were constructed using lentiviral transduction of CD80, HLA-DM molecules and different HLA alleles (gBlock ordered from IDT) into K562 cells. The surface expression of CD80 and HLA-DM on K562 was confirmed using anti-CD80-BV421 and anti-HLA-DM-PE antibodies from BioLegend.
Generation of dendritic cells (DCs)
Dendritic cells were generated as antigen-presenting cells according to a well-established protocol43. Briefly, HLA typed PBMCs carrying the required HLA alleles were plated in 6-well plate at 10×106 cells per well in 3ml of RPMI 1640 medium supplemented with 10% AB serum, 10mM HEPES and Penicillin-Streptomycin. After a 2-hour incubation, cells were washed twice gently by pipetting the medium up and down several times so that only adherent monocytes were left in the plate. After the wash, cells were incubated with 50ng/ml IL-4 and 50ng/ml GM-CSF (PeproTech) at 37°C in 5% CO2 for 5 days. The culture medium and cytokines were renewed every other day. On day 5, DCs were harvested and resuspended at 5×105/ml and split into 96-well plate at 50 μl per well. Individual peptide or protein antigens were pre-loaded for 24 hours, then 50 μl TCR transduced J76-NFATRE-luc cells (106/ml) were added to each well and co-cultured with antigen loaded DCs for 4 hours before measuring luciferase activity.
Lentiviral TCR transduction
Lentiviral transduction was performed as previously described19. Briefly, TCRα chain, P2A linker and β chain fusion gene fragments were ordered from IDT and cloned into MCS of N103 vector (nLV Dual Promoter EF-1a-MCS-PGK-Puro). HEK-293T cells were plated on 10-cm dishes at 5×106 cells/plate 24 h prior to transfection. The culture medium was changed prior to transfection. Lentiviral supernatants were prepared by co-transfection of 293T cells, using 10 μg of transfer vector, 7.5 μg of envelope vector (pMD2.G), 2.5 μg of packaging vector (psPAX2) and 75 μl PEI (Sigma). The culture medium was replaced 16 hours after transfection and the viral supernatant was collected 48 hours later. The viral supernatants were filtered through a 0.45 μm SFCA syringe filter (Corning) and concentrated by centrifugation with a 100K Amicon Ultra-15 filter (Millipore). Concentrated viruses were used for J76-NFATRE-luc cell transduction using spinoculation for 2 hours in the presence of 6 μg/ml polybrene (Sigma). Forty-eight hours after transduction, TCR expression was analyzed by flow cytometry and TCR positive cells were sorted for epitope screening.
Protein expression and whole proteome production
Escherichia coli (E. coli) DH10B-T1 cells containing individual ORF vector pDONR 221 were uniquely picked and cultured overnight in LB medium, 1 μl of which was used as template for PCR amplification of target ORF in a 50 μl PCR mixture. PCR was done using HotStart ReadyMix (Kapa Biosystems) with a pair of primers containing T7 promoter (T7 Forward: 5’-GATCTCGATCCCGCGAAATTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTTTGTTTAACTTTAAGAAGGAGATATACATATGACTTTGTACAAAAAAGTTGCCCATATG-3’) and T7 terminator (T7 Reverse: 5’- TCCTTTCAGCAAAAAACCCCTCAAGACCCGTTTAGAGGCCCCAAGGGGTTATGCTAGTGCTAGTTAAACTTTGTACAAGAAAGTTGCAAGCTT-3’). PCR product was purified using AMPure XP beads (Beckman Coulter). 500 ng of the purified PCR product was used as template for protein expression using Expressway™ Cell-Free Expression System (Thermo Fisher scientific) in a single tube according to the instruction. The protein product was diluted accordingly and used to activate its corresponding TCR without further purification.
For the whole proteome production, every 12 ORF clones from each plate row were pooled together as a subpool and cultured overnight. This minimized the whole proteome to four plates of subpools (Supplementary Fig. 5). The pooled bacteria were used as template and proceeded to protein production as described above.
Alternatively, for PPE protein production, the selected ORFs were transferred from pDONR 221 vector to pEXP3-DEST vector with Lumio™-tag using LR recombination (Thermo Fisher scientific). The resulted vectors were used as template for cell-free protein production as described above.
SDS-PAGE gel detection of PPE proteins
SDS-PAGE gel detection of Lumio™-tagged PPE proteins was done according to the manufacturer’s protocol. Briefly, 10 μl of PPE protein products from the cell-free expression system were precipitated with cold acetone and resuspend with 20 μl of 1X Lumio™ Gel Sample Buffer. After adding Lumio™ Green Detection Reagent and In-Gel Detection Enhancer, 20 μl of each sample was loaded into 8% SDS-PAGE gel and run until the dye front ran off the bottom of the gel. Protein bands were visualized using a UV transilluminator.
Antigen screen
For protein stimulation, 50 μl aAPC (106/ml) were pre-loaded with the Expressway product mixture, and individual proteins at a range of 10—10000 dilutions or protein subpools at 10-fold dilution for 3 hours at 37 °C in the standard cell culture medium. 50 μl of TCR transduced J76-NFATRE-luc cells (106/ml) were added and co-cultured with aAPC for 8 hours. Then cells were harvested, and Luciferase activity was measured using Nano-Glo® Luciferase Assay (Promega). Fold induction of luciferase activity was calculated referring to unstimulated samples. For peptide stimulation, 50 μl of TCR transduced J76-NFATRE-luc cells (106/ml) were co-cultured with 50 μl HLA transduced K562 cells (106/ml) in a 96-well plate. Peptide pool or individual peptide was added to the well at 2 μg/ml. After 8 hours incubation, cells were harvested, and Luciferase activity was measured.
Generation of sequence logos
The sequence logos were generated in R using ggseqlogo36. For PPE peptide conservation, all the 64 peptide sequences were loaded equally as input to generate a sequence logo. For PPE peptide stimulation potential, the fold induction from each peptide stimulation were log2 transformed and used as weight value for each sequence. The weighted sequences are loaded as input to generate the sequence logo.
Statistical analysis
Statistical analysis was performed using two-tailed unpaired t-tests. All statistical analysis was performed in GraphPad Prism v.8.1.0. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information files.
Code availability
Two compiled standalone versions of GLIPH2 (Executable for MacOS >= 10.14.14 and Linux server Centos 7) are provided as Supplementary Code. Also, a web tool for GLIPH2 analysis is available at http://50.255.35.37:8080/.
Supplementary Material
Mtb specific TCR sequences and summary
TCR sequences from VDJdb
Gene list of the whole Mtb ORF clone set
Two compiled standalone versions of GLIPH2
TCR specificity groups from GLIPH2 analysis
Acknowledgements
We would like to thank the Stanford Human Immune Monitoring Center for their high-throughput sequencing support for this project, M. Mindrinos and co-workers at Sirona Genomics for the HLA typing, S. Xue (Department of Immunology, University College London) for providing Jurkat 76 T-cell line, J. Li for providing HLA typed PBMCs, L. Chen and S. Chiou for valuable discussions regarding GLIPH2 optimization, H. Mahomed, W. Hanekom and members of the Adolescent Cohort Study (ACS) group for enrolment and follow-up of the Mtb-infected adolescents, R. DiFazio for help making schematic overview and Y. Chien for constructive criticism of the manuscript, J. Pavlovitch-Bedzyk for proofreading. This work was supported by the Bill and Melinda Gates Foundation OPP1113682 and the Howard Hughes Medical Institute.
Footnotes
Competing interests
The authors declare no competing financial interests.
References
- 1.Zhu J & Paul WE CD4 T cells: fates, functions, and faults. Blood 112, 1557–1569 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corbett EL et al. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch Intern Med 163, 1009–1021 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Lindestam Arlehamn CS & Sette A Definition of CD4 Immunosignatures Associated with MTB. Front Immunol 5, 124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindestam Arlehamn CS et al. A Quantitative Analysis of Complexity of Human Pathogen-Specific CD4 T Cell Responses in Healthy M. tuberculosis Infected South Africans. PLoS Pathog 12, e1005760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altman JD et al. Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94–96 (1996). [PubMed] [Google Scholar]
- 6.Bentzen AK et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat Biotechnol 34, 1037–1045 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Hadrup SR et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods 6, 520–526 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Newell EW, Klein LO, Yu W & Davis MM Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nat Methods 6, 497–499 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newell EW et al. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat Biotechnol 31, 623–629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang SQ et al. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat Biotechnol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simoni Y et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Mishto M & Liepe J Post-Translational Peptide Splicing and T Cell Responses. Trends Immunol 38, 904–915 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Laumont CM et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med 10 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Kahles A et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 34, 211–224 e216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engelhard VH, Altrich-Vanlith M, Ostankovitch M & Zarling AL Post-translational modifications of naturally processed MHC-binding epitopes. Curr Opin Immunol 18, 92–97 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Cobbold M et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med 5, 203ra125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacher P et al. Human Anti-fungal Th17 Immunity and Pathology Rely on Cross-Reactivity against Candida albicans. Cell 176, 1340–1355 e1315 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Bacher P et al. Regulatory T Cell Specificity Directs Tolerance versus Allergy against Aeroantigens in Humans. Cell 167, 1067–1078 e1016 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Glanville J et al. Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang H et al. Select sequencing of clonally expanded CD8(+) T cells reveals limits to clonal expansion. Proc Natl Acad Sci U S A 116, 8995–9001 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pool I.d.S. & Kochen M Contacts and influence. Social Networks 1, 5–51 (1978). [Google Scholar]
- 22.Dash P et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pogorelyy MV et al. Detecting T cell receptors involved in immune responses from single repertoire snapshots. PLoS Biol 17, e3000314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B et al. Investigation of antigen-specific T cell receptor clusters in human cancers. Clin Cancer Res (2019). [DOI] [PubMed] [Google Scholar]
- 25.Han A, Glanville J, Hansmann L & Davis MM Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat Biotechnol 32, 684–692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubelt F et al. Individual heritable differences result in unique cell lymphocyte receptor repertoires of naive and antigen-experienced cells. Nat Commun 7, 11112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagaev DV et al. VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res 48, D1057–D1062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butler MO et al. A panel of human cell-based artificial APC enables the expansion of long-lived antigen-specific CD4+ T cells restricted by prevalent HLA-DR alleles. Int Immunol 22, 863–873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosskopf S et al. Creation of an engineered APC system to explore and optimize the presentation of immunodominant peptides of major allergens. Sci Rep 6, 31580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thorstenson YR et al. Allelic resolution NGS HLA typing of Class I and Class II loci and haplotypes in Cape Town, South Africa. Hum Immunol 79, 839–847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fortune SM et al. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci U S A 102, 10676–10681 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garces A et al. EspA acts as a critical mediator of ESX1-dependent virulence in Mycobacterium tuberculosis by affecting bacterial cell wall integrity. PLoS Pathog 6, e1000957 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karosiene E et al. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 65, 711–724 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Odermatt NT, Sala C, Benjak A & Cole ST Essential Nucleoid Associated Protein mIHF (Rv1388) Controls Virulence and Housekeeping Genes in Mycobacterium tuberculosis. Sci Rep 8, 14214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brennan MJ The Enigmatic PE/PPE Multigene Family of Mycobacteria and Tuberculosis Vaccination. Infect Immun 85 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagih O ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 33, 3645–3647 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Blum JS, Wearsch PA & Cresswell P Pathways of antigen processing. Annu Rev Immunol 31, 443–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boucau J & Le Gall S Antigen processing and presentation in HIV infection. Mol Immunol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song I et al. Broad TCR repertoire and diverse structural solutions for recognition of an immunodominant CD8+ T cell epitope. Nature structural & molecular biology 24, 395–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robins HS et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 114, 4099–4107 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis MM & Boyd SD Recent progress in the analysis of alphabetaT cell and B cell receptor repertoires. Curr Opin Immunol 59, 109–114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahomed H et al. Predictive factors for latent tuberculosis infection among adolescents in a high-burden area in South Africa. The international journal of tuberculosis and lung disease : the official journal of the International Union against Tuberculosis and Lung Disease 15, 331–336 (2011). [PubMed] [Google Scholar]
- 43.O’Neill DW & Bhardwaj N Differentiation of peripheral blood monocytes into dendritic cells. Curr Protoc Immunol Chapter 22, Unit 22F 24 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mtb specific TCR sequences and summary
TCR sequences from VDJdb
Gene list of the whole Mtb ORF clone set
Two compiled standalone versions of GLIPH2
TCR specificity groups from GLIPH2 analysis
Data Availability Statement
The data supporting the findings of this study are available within the paper and its Supplementary Information files.