

Abstract
Murine hematopoietic-acute radiation syndrome (H-ARS) survivors of total body radiation (TBI) have a significant loss of heart vessel endothelial cells, along with increased tissue iron, as early as 4 months post-TBI. The goal of the current study was to determine the possible role for excess tissue iron in the loss of coronary artery endothelial cells. Experiments utilized the H-ARS mouse model with gamma radiation exposure of 853 cGy (LD50/30) and time points from 1 to 12 weeks post-TBI. Serum iron was elevated at 1 week post-TBI, peaked at 2 weeks, and returned to non-irradiated control values by 4 weeks post-TBI. A similar trend was seen for transferrin saturation, and both results correlated inversely with red blood cell number. Perls’ Prussian Blue staining used to detect iron deposition in heart tissue sections showed myocardial iron was present as early as 2 weeks following irradiation. Pretreatment of mice with the iron chelator deferiprone decreased tissue iron, but not serum iron, at 2 weeks. Coronary artery endothelial cell density was significantly decreased as early as two weeks vs. non-irradiated controls (P<0.05), and the reduced density persisted to 12 weeks after irradiation. Deferiprone treatment of irradiated mice prevented the decrease in endothelial cell density at 2 and 4 weeks post-TBI compared to irradiated, non-treated mice (P<0.03). Taken together, the results suggest excess tissue iron contributes to endothelial cell loss early following TBI and may be a significant event impacting the development of delayed effects of acute radiation exposure.
Keywords: radiation dose, radiation damage, mice, whole body radiation
INTRODUCTION
Exposure to high levels of radiation, such as that from belligerent activity or accidents, results in both acute and long-term multi-organ damage. Short-term damage is reflected by development of the acute radiation syndromes (ARS) involving gastrointestinal and/or hematologic morbidity and mortality. Survivors of ARS will experience the delayed effects of acute radiation exposure (DEARE) which includes multiple chronic illnesses affecting multiple organ systems. There currently exists a gap in the understanding of the mechanisms that mediate the long-term effects of radiation, and this knowledge will be essential to develop novel therapies to both improve initial survival and to mitigate DEARE. It has been demonstrated that a mouse model of hematopoietic-ARS (H-ARS; <10 Gy) displays organ and vascular pathologies of DEARE similar to those of larger species, including humans (Unthank et al. 2015). Cardiac pathologies included pericardial thickening, loss of endothelial and smooth muscle cells in coronary vasculature, reduced ejection fraction, and extensive fibrosis. Vascular endothelium is considered to be one of the most radiation-sensitive non-hematopoietic tissues (Fajardo and Berthrong 1988) and to be related to multiple organ injury (Gaugler 2005, Satyamitra et al. 2016). Vascular injury may be a mechanism for delayed radiation-induced tissue injury (Fajardo and Berthrong 1988), but few studies have addressed DEARE at doses where survival from ARS is a possibility, i.e., less than 10 Gy. Recent results (Unthank et al. 2019) indicate that heart vessel endothelial cell loss occurs before 4 months post-total body irradiation (TBI) in the H-ARS mouse model, and is coincident with iron deposits (hemosiderin) known to be deleterious to organ function, possibly due to increased reactive oxygen species (Xie et al. 2016). Whole body irradiation has been shown to increase iron content in bone marrow and serum (Zhang et al. 2013, Xie et al. 2015), and disrupt iron metabolism as well as impair erythropoiesis, which contributes to hemosiderin formation in tissue. The current study was designed to define the time frame (≤3 months) of increased serum and tissue iron post-TBI and its contribution to mediating onset of cardiac endothelial cell loss in the H-ARS mouse model.
METHODS
Mice and irradiation
All studies followed the Public Health Service Policy on Humane Care and Use of Laboratory Animals, were compliant with animal welfare guidelines as reviewed by the Association for Assessment and Accreditation of Laboratory Animal Care, and were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee. Specific pathogen free C57BL/6 mice [50/50 male/female (male only for time course and ferrostatin experiments); Jackson Laboratory, Bar Harbor, ME, USA] were received at 10 wk of age and acclimated for 2 wk prior to irradiation. Mice were placed in single chambers of a Plexiglas® irradiation apparatus and were exposed to a single uniform total body dose of 8.53 Gy of gamma radiation (LD50/30) from a 137Cs radiation source (0.95–1.00 Gy min). Dosimetry was performed as previously described (Unthank et al. 2015). Mice were randomized into groups, identified by ear punch, and husbandry and health status monitoring were carried out as previously described (Plett et al. 2012). Survivors of the acute phase (30 days) were used in the experiments extending to 12 weeks post-TBI, and non-irradiated (NI) mice served as controls.
Agent Dosing
Deferiprone (3-Hydroxy-1,2-dimethyl-4(1H)-pyridone; Ferriprox) was administered in acidified drinking water ad libitum at a concentration of 2.5 g/liter (Cohen et al. 2009) for an estimated dose of 375 mg/kg/day for 14 days. Ferrostatin-1 (3-amino-4-(cyclohexylamino)-benzoic acid, ethyl ester) was administered i.p. once daily for 14 days in sterile PBS with 5% ethanol (vehicle) to achieve a dose of 2.5 umol/kg (Wang et al. 2017). Control mice received vehicle only, and both agents were started ~24 hours prior to irradiation. Deferiprone solutions were changed every 48 hours, and ferrostatin-1 doses were diluted before each use from a concentrated stock solution.
Tissue harvest, fixation, and histological staining
Mice were anesthetized via isoflurane inhalation and a maximum volume of blood was collected via a closed chest cardiac puncture. Serum was prepared and stored frozen at −80°C until use. The thoracic cavity was opened and mice were perfused via the left ventricle at 100–120 mm Hg with phosphate buffered saline (PBS) including vascular dilator (0.1 mM adenosine and 0.01 mM sodium nitroprusside). This was followed by 10% neutral buffered formalin (NBF). Following perfusion-fixation, the heart was placed in NBF for storage until processing. Formalin-fixed tissues were paraffin embedded, sectioned, and then stained with hematoxylin only (for endothelial cell density measurements), or Perls’ Prussian blue.
Imaging
Digital images of heart sections were acquired with Leica Application Suite imaging software using a Leica DM 5000B microscope (Leica Microsystems, Inc., Buffalo Grove, IL, USA) with a Leica DMC 4500 digital camera. Multiple sections (4–5) from each mouse were evaluated for histopathological characteristics. Morphometric measurements for endothelial cell density were made with Image J as previously described (Unthank et al. 2019).
Serum assays
Serum obtained at euthanasia was analyzed for total iron and transferrin by The Center for Diabetes & Metabolic Diseases Translation Core, Indiana University School of Medicine. Red blood cell counts, hemoglobin, and hematocrit were obtained from complete blood counts (CBC) as previously described (Plett et al. 2012).
RESULTS
Serum iron experiments
Total serum iron, transferrin saturation, hematocrit, and red blood cells were quantified (Fig. 1) to determine short-term (1–12 weeks post-TBI) effects of radiation exposure on iron in serum. The results showed that serum iron was elevated at 1 week post-TBI, peaked at 2 weeks, and returned to non-irradiated control values by 4 weeks post-TBI. A similar trend was seen for transferrin saturation, and both results correlated approximately inversely with red blood cell number and hematocrit. Red blood cell hemoglobin concentrations also were determined and tracked with red blood cell number, as expected (data not shown).
Fig 1.
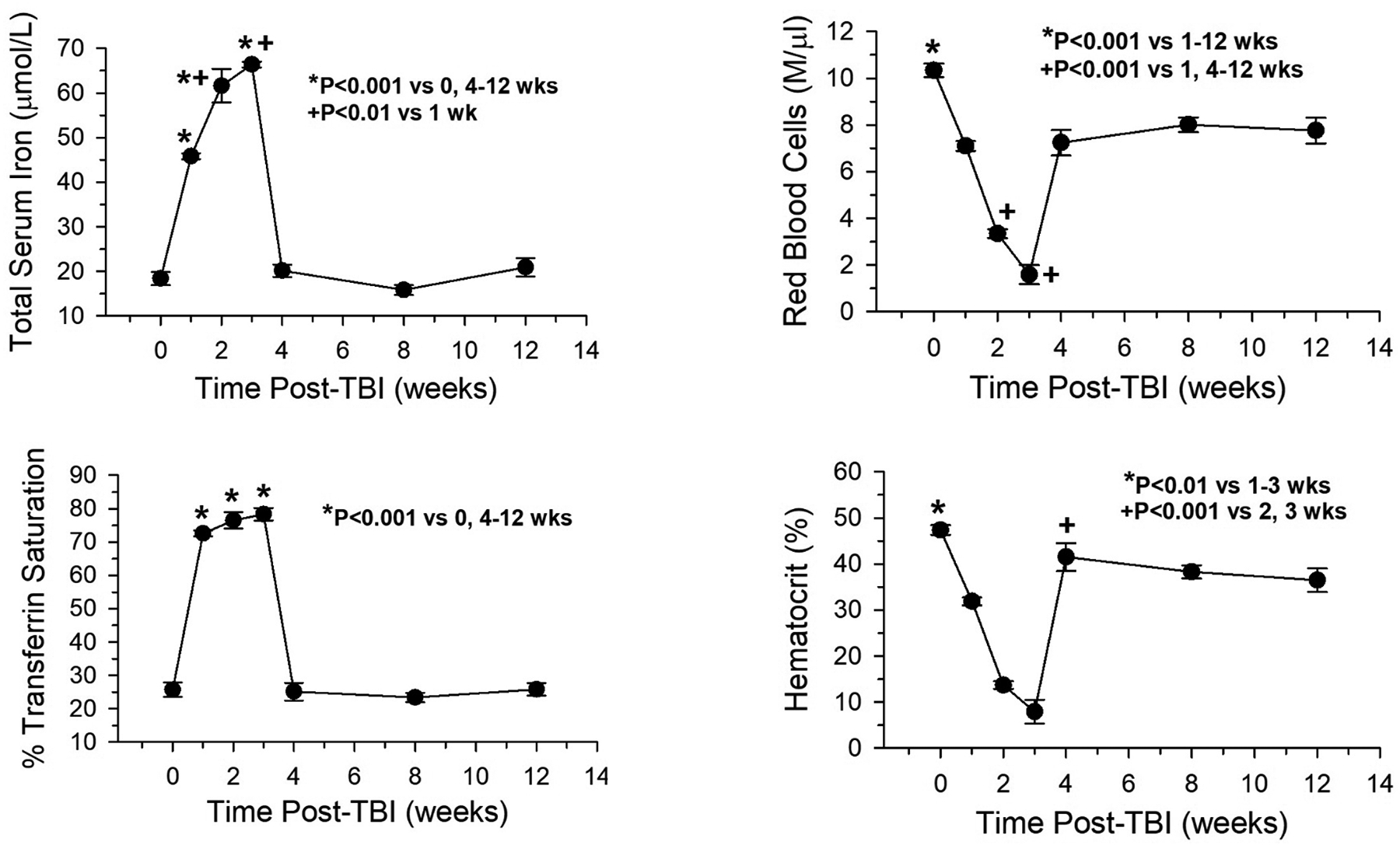
Total serum iron and transferrin saturation varied inversely with hematocrit and red blood cell (RBC) loss post-total body irradiation (TBI) (n=6, iron/transferrin; n=12, hematocrit, RBC). Zero weeks post-TBI = non-irradiated controls (n=8).
Detection of tissue Iron
Perls’ Prussian Blue staining was used to detect iron (hemosiderin) deposition in paraffin-embedded heart and spleen tissue cross sections at 1, 2, 4, 8, and 12 weeks post-TBI. Myocardial iron was present as early as 2 weeks post-TBI, and was still present at 12 weeks (Fig. 2). Hemosiderin content in spleen also was assessed as an index of serum iron clearance. Perls’ staining was very prominent at 1 week but almost absent at 4 weeks, consistent with the pattern of changes for serum iron and transferrin saturation at the same time points.
Fig 2.
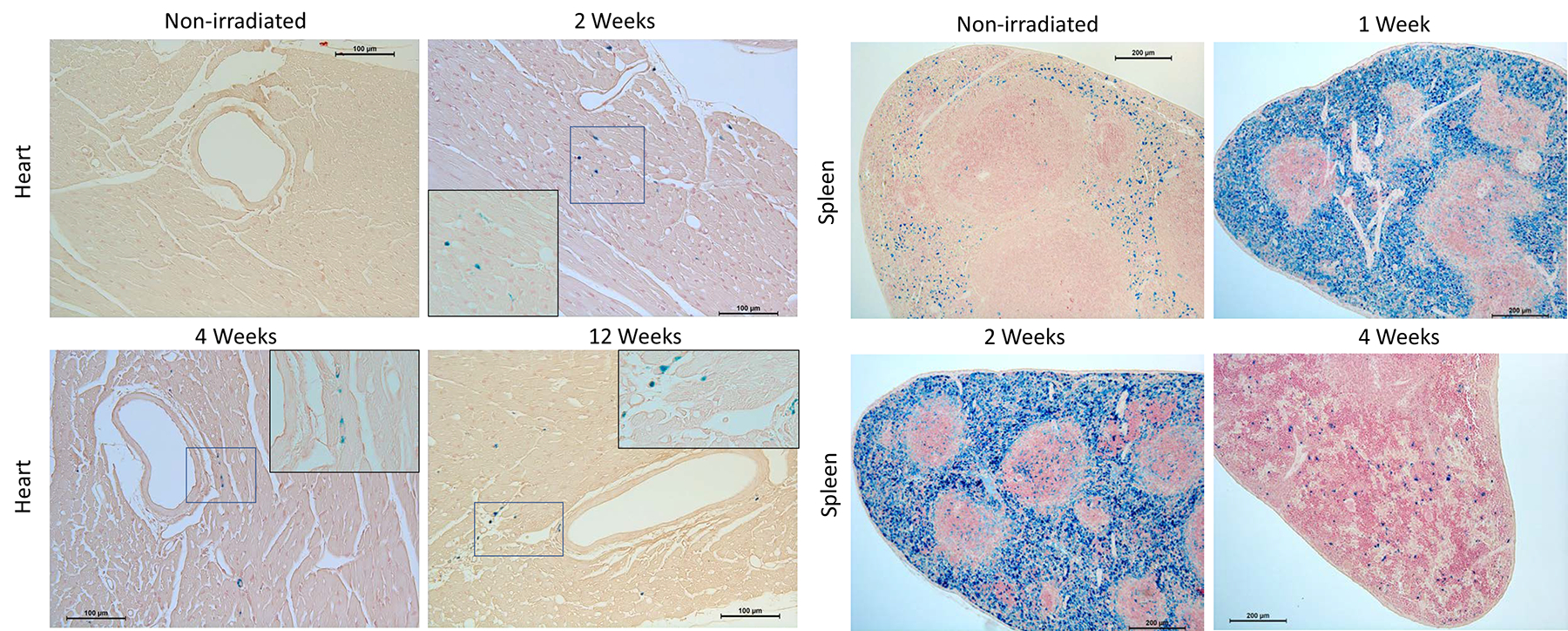
Representative images of tissue iron identified by Perls’ staining (blue). Iron in heart appeared at 2 weeks post-TBI and was still present at 12 weeks, whereas in spleen iron was present at an earlier time point and decreased to non-irradiated levels by 4 weeks post-TBI. Image inserts show higher magnification (cropped 400x image) of Perls’-positive areas indicated by the blue boxes.
Endothelial cell loss assessment
To determine potential effects of acute radiation exposure on coronary artery endothelial cells, a histological assessment of intimal nuclei density was performed, and intimal nuclei per unit length (endothelial cell density) was quantified. The results showed significantly decreased endothelial cells as early as two weeks post-TBI vs. non-irradiated controls, and the decrease persisted to 12 weeks post-TBI (Fig. 3a). It was noticed that in the majority of arteries the intimal cell loss was not uniform, with variable distances between endothelial cells in certain areas of the vessel lumen compared to non-irradiated controls (Fig. 3, b & c).
Fig 3.
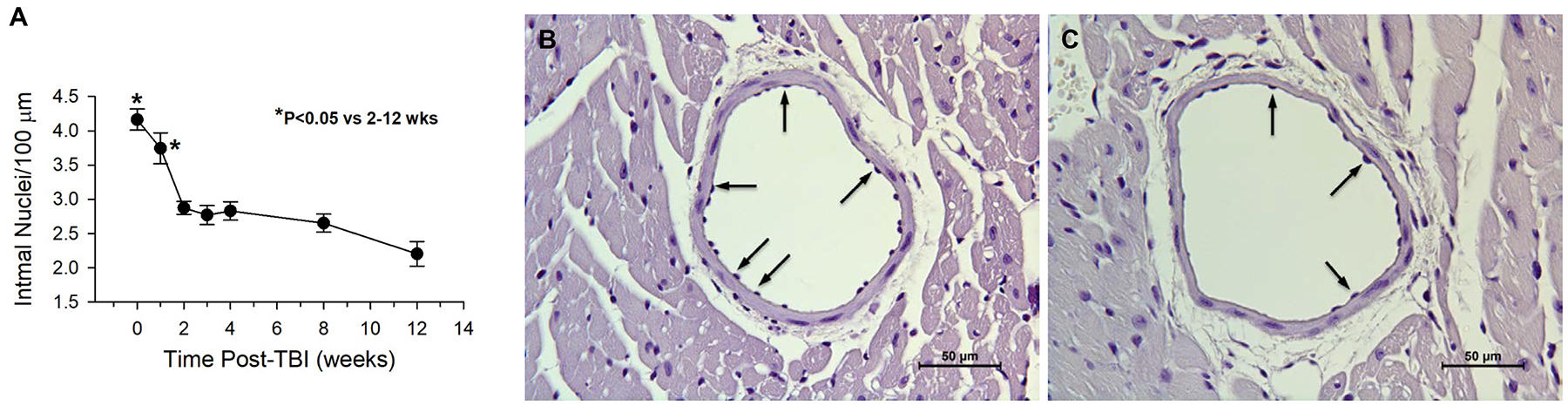
Endothelial cell density decreased after irradiation. (A) Intimal cell nuclear density in the largest coronary arteries is shown up to 12 weeks post-total body irradiation (TBI) compared to non-irradiated controls (zero weeks post-TBI) (n=4–6). Representative micrographs of hematoxylin only-stained heart tissue sections illustrate a decrease in endothelial cell density in coronary arteries of mice 4 weeks post-TBI (853 cGy) (C) compared to age matched non-irradiated control coronary arteries (B). Arrows indicate endothelial cell nuclei and variable density.
Effects of Iron chelation
In order to determine if increased serum and tissue iron was related to endothelial cell loss, mice were pretreated with the iron chelator deferiprone ~24 hours before irradiation and the dosage was continued for 2 weeks following irradiation. Tissue and serum analyses showed decreased myocardial iron deposition, but not a decrease in serum iron, at 2 weeks Post-TBI (Fig. 4, a & b). Analysis of coronary artery endothelial cell density demonstrated that deferiprone treatment prevented the decrease in density at both 2 and 4 weeks post-TBI compared to irradiated non-treated mice (Fig. 5).
Fig 4.
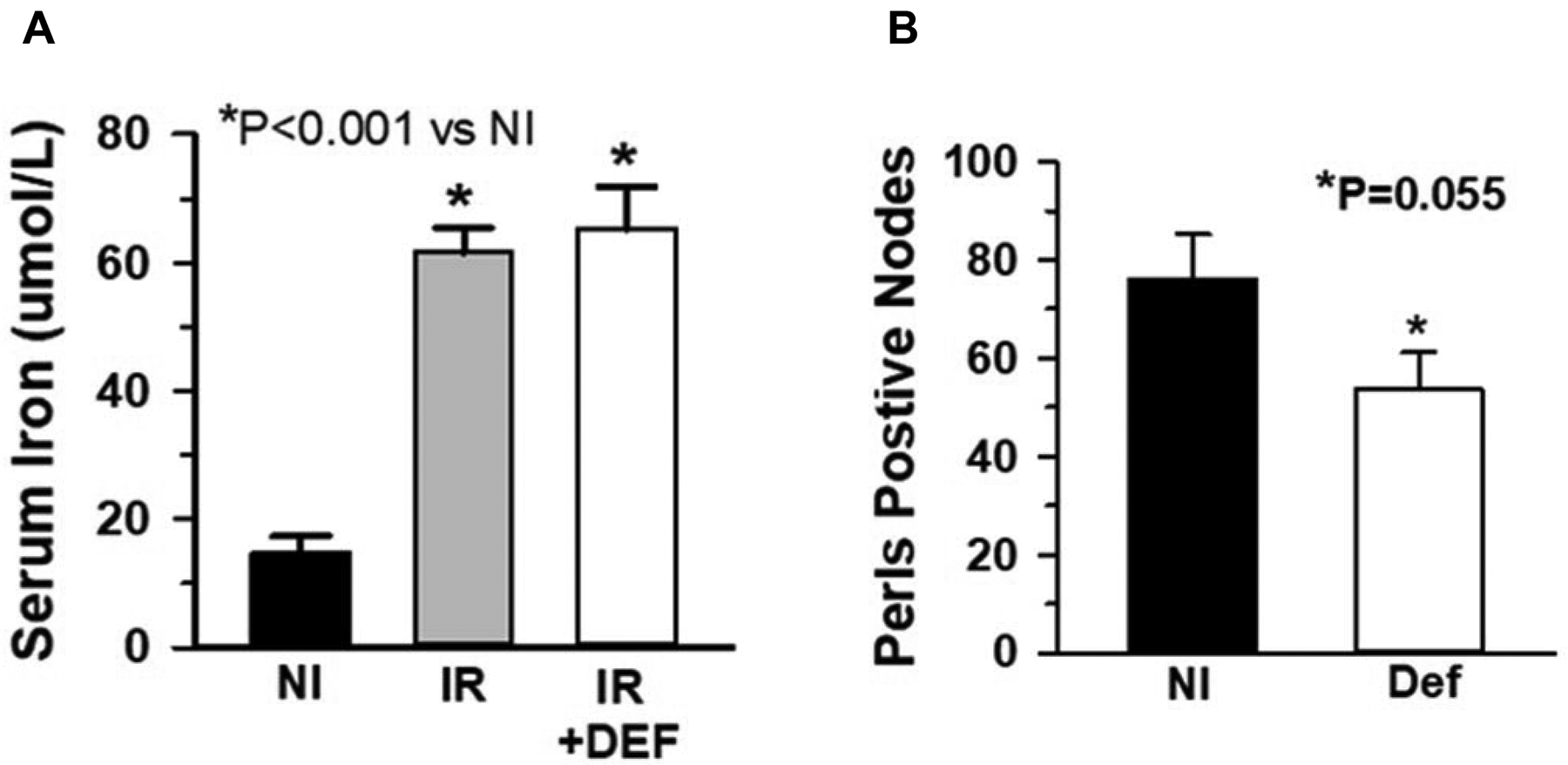
Deferiprone prevented tissue, but not serum iron increases. The iron chelator deferiprone (DEF) was administered in drinking water at time of irradiation (IR) to 2 weeks post-TBI. Iron in heart was determined by Perls’ Prussian Blue staining of tissue sections and compared to non-irradiated (NI) mice. While deferiprone treatment did not prevent the irradiation-mediated serum iron increase (n=6) (a), it inhibited accumulation of myocardial iron (n=4) (b).
Fig 5.
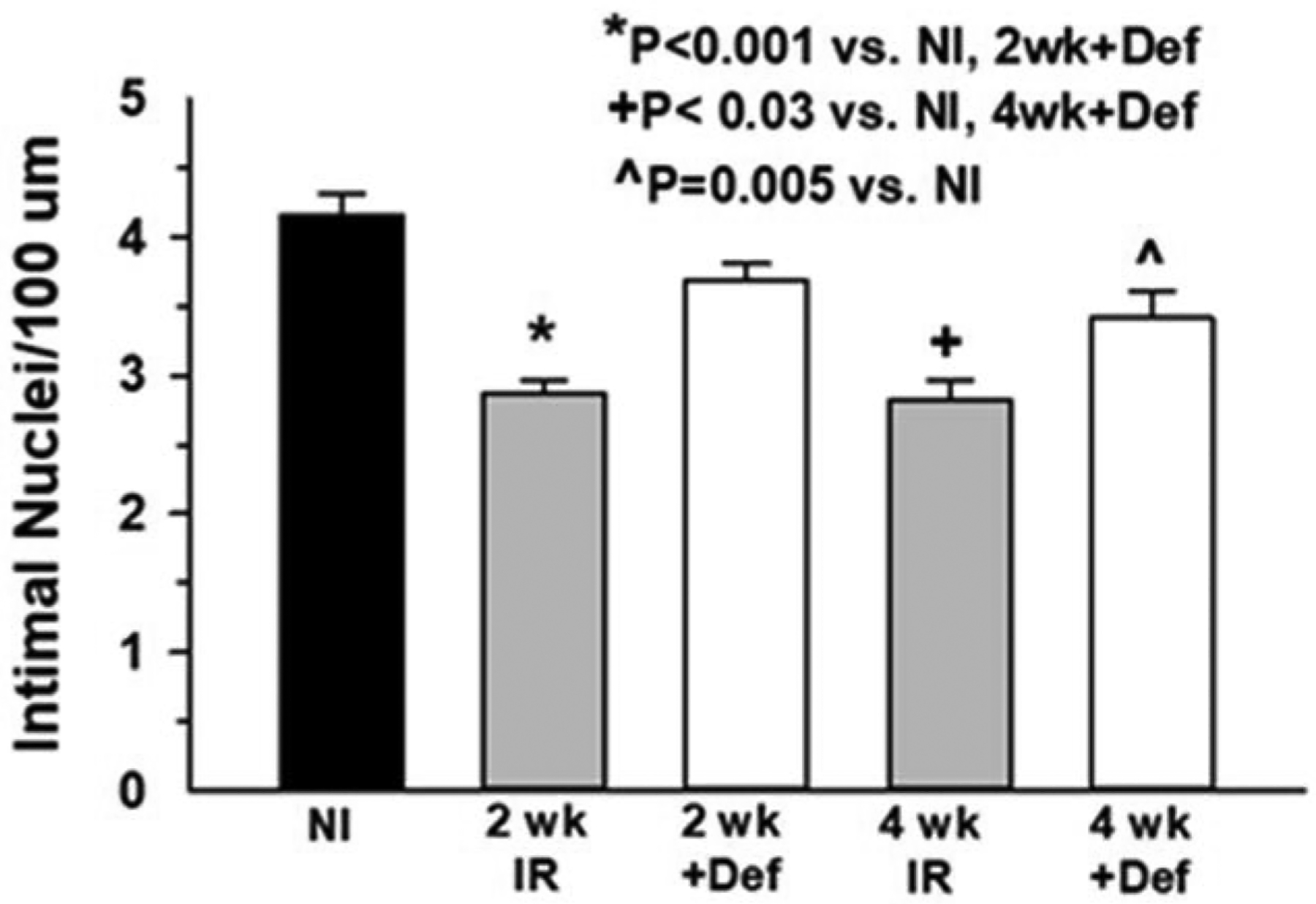
Deferiprone prevented cardiac endothelial cell loss. Degree of endothelial cell loss in treated and untreated mouse heart vessels was determined by quantifying intimal nuclei density at 2 and 4 weeks post-TBI and comparing to non-irradiated (NI) mice (n=4–6).
Ferroptosis inhibition and endothelial cell density
The ferroptosis inhibitor ferrostatin-1 was administered to determine if the increase in iron could be associated with ferroptosis. Administration of ferrostatin-1 for 2 weeks resulted in a partial protective effect compared to non-irradiated controls, however, the change in coronary artery EC density was not significantly different compared to that of vehicle alone (Fig. 6). In addition, vehicle administration alone was able to completely protect from endothelial cell loss, thus any potential effect of ferrostatin was inconclusive.
Fig 6.
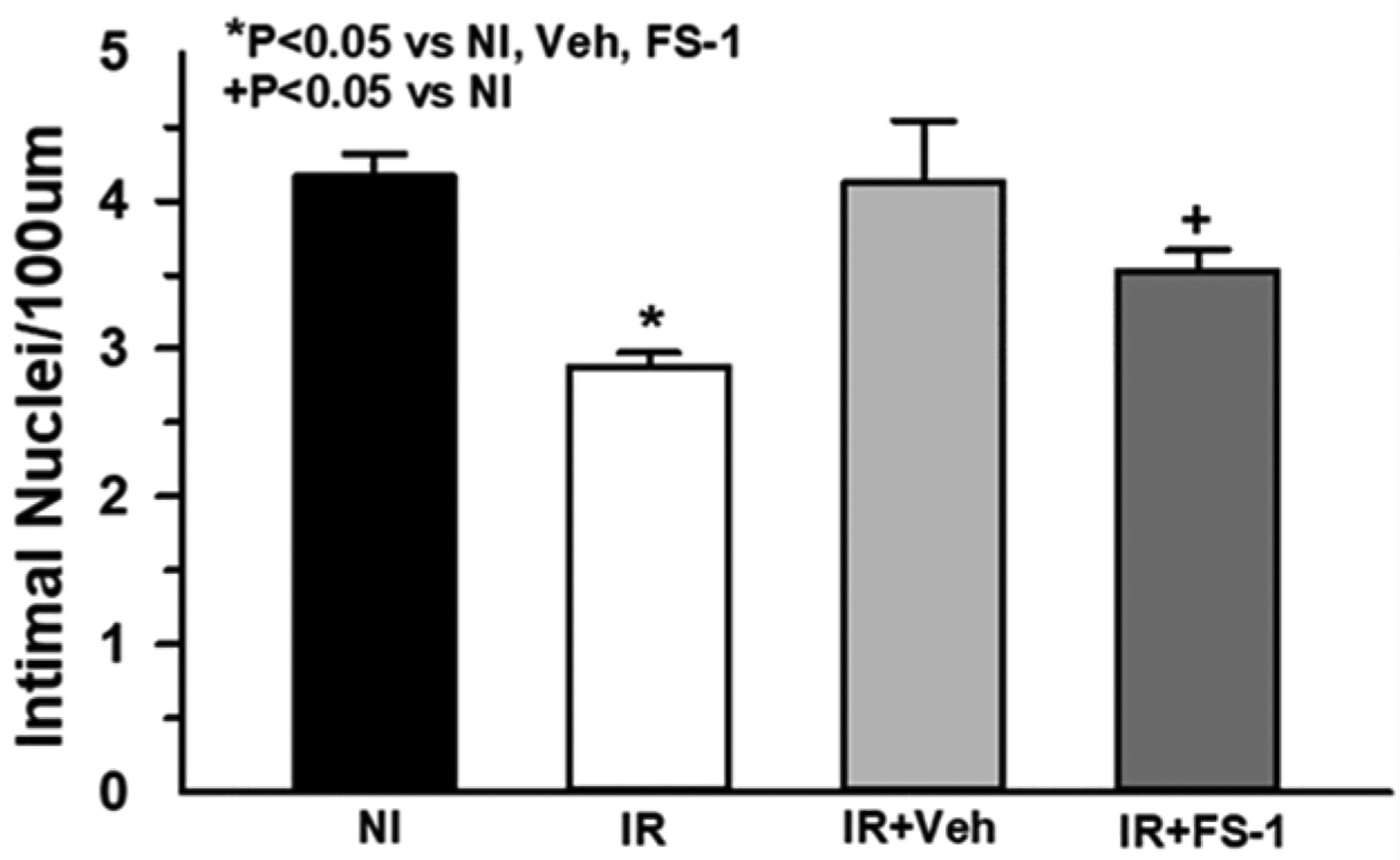
Ferrostatin-1 (FS-1), a ferroptosis inhibitor, or vehicle alone (Veh) was administered i.p. for 2 weeks post-irradiation (IR). Coronary artery endothelial cell density was assessed by counting intimal nuclei and normalizing to 100 um luminal perimeter (n=7). While FS-1 treatment appeared to partially rescue EC loss compared to non-irradiated (NI) controls, vehicle-treated mice were not significantly different from FS-1 treated mice (P=0.23).
DISCUSSION
A previous study has shown Perls’ positive staining in heart myocardium and coronary arteries, along with endothelial cell loss, as early as 4 months post-TBI (Unthank et al. 2019). In this study we show that iron in heart tissue sections was detectable as early as 2 weeks post-TBI, and also was associated with endothelial cell loss. The observation that iron chelation started prior to irradiation prevented the decrease in endothelial cell density suggests a causative role for elevated iron in vascular damage, and thus may be a causative factor in the development of DEARE-related pathologies.
Vascular endothelium is known to be linked to radiation damage of several major organs and is recognized as an important component of strategies for radiation mitigation (Satyamitra et al. 2016). The acute phase of damage occurs within hours to weeks, and is characterized by edema, increased permeability due to denudation, lymphocyte adhesion/infiltration, and apoptosis among others (Paris et al. 2001). These early endothelial events precede later tissue damage such as fibrosis, but the molecules and pathways involved are unclear. Results from the current study showed reduced density of intimal cell nuclei as early as 1 week post-TBI, and this loss of endothelial cells was statistically significant by 2 weeks. This observation of early cell loss is consistent with other studies, and it has been demonstrated that once endothelial cell loss occurs in heart it does not recover within 18 months post-TBI (Unthank et al. 2015). However, endothelial cells are heterogeneous in nature and other vascular beds and organs may differ in the response to radiation exposure (Nolan et al. 2013). Kidney does not appear to have a reduction in endothelial cell density following irradiation (Unthank et al. 2019), whereas intestine is very sensitive to endothelial cell damage (Wang et al. 2007). The current data support a potential role for endothelial cell damage in the progression of cardiac DEARE-related pathology and dysfunction, although the initiating mechanism is unclear. Accordingly, restoring or preventing loss of endothelial cells may serve to prevent DEARE. Although potential effects on DEARE development are unclear, transplantation of vascular endothelial cells has been shown to improve survival and stimulate hematopoietic recovery of irradiated mice (Chute et al. 2007).
Few studies have examined a role for iron in radiation-mediated tissue damage, but a recent report suggests iron-mediated damage of cells in bone marrow (Zhang et al. 2020). Excess iron causes a disruption in iron metabolism, with a concomitant increase in serum iron. Serum iron levels have been shown to be acutely radiation dose-dependent and reach high concentrations (Zhang et al. 2013). These reports are consistent with the current data showing maximally elevated serum iron and transferrin saturation at 2 days post-TBI at the LD50/30 for C57BL/6.
Current and previous data (Plett et al. 2012) show a maximal decrease in red blood cell numbers by approximately 21 days following total-body irradiation, returning to near normal by 4 weeks, reflecting a decrease and recovery in erythropoiesis. The pattern of iron detected in spleen is consistent with the clearance of damaged erythrocytes, although the liver also has been shown to be an important organ for storing excess iron and recycling it (Kohgo et al. 2008, Theurl et al. 2016). Evidence to date in several species suggests hemolysis is not a major factor in acute radiation damage; thus, dysfunctional erythropoiesis with the resulting lack of iron incorporation into hemoglobin in addition to abnormal iron metabolism may explain, at least in part, the observation of tissue iron deposits in heart early after radiation exposure. Cardiac iron deposition occurs during iron overload, and several pathways regulating iron metabolism are involved. Hepcidin is an important regulator of iron metabolism, and controls expression of the iron transport protein ferroportin and thus subsequent tissue iron release and transport from tissues, especially the liver and duodenum. Control of hepcidin expression is complex, related to multiple pathways, and is not completely understood. Increased expression of hepcidin causes inhibition of ferroportin expression and resultant inhibition of iron release from tissue macrophages, thus contributing to hemosiderin formation (Kremastinos and Farmakis 2011, Ganz and Nemeth 2012). Hepcidin expression is known to be regulated by radiation (Christiansen et al. 2007, Iizuka et al. 2016), and thus may be a potential factor contributing to iron overload in heart.
The mechanism of iron-mediated endothelial cell damage may be related to the fact that hemosiderin present in tissues releases iron under conditions of hypoxia or inflammation, leading to production of iron-catalyzed hydroxyl radicals via the Fenton reaction (Ozaki et al. 1988). Redox-active iron may promote vascular injury, including denudation, via hemoglobin oxidation (Woollard et al. 2009). Contrary to our results using deferiprone in heart, experiments in kidney show no effects of iron chelation on radiation-induced nephropathy (Cohen et al. 2009), consistent with a limited role for reactive oxygen species-related damage in that organ. The effects of iron chelation in preventing loss of coronary artery endothelial cells, however, is consistent with results showing elevated oxidant stress in heart, at least at 4 months post-TBI (Unthank et al. 2019). Although only total hemosiderin deposits were quantified, previous results (Unthank et al. 2015) show hemosiderin may be present both in the myocardium and in arterioles, and the arteriolar-associated hemosiderin is more persistent. Presumably it is the presence of vascular iron which is associated with the loss of endothelium. Although the quantity of Perls’ staining around arterioles appears to be relatively low, the actual amount of iron may be underestimated, since this is a relatively insensitive histological method of detection. Future studies will utilize atomic absorption to accurately quantitate total heart iron levels.
Additional evidence for an oxidant stress-mediated mechanism is related to haptoglobin (Hp) and hemopexin (Hpx), acute phase proteins produced by the liver with high binding affinity for hemoglobin and heme, respectively, that limit their oxidative activity (Schaer et al. 2013). Haptoglobin is used clinically in Japan (Schaer et al. 2013), and studies have shown that Hpx may function to attenuate endothelial cell activation and renal damage (Tolosano et al. 1999, Vinchi et al. 2008). Low Hp and high hemoglobin have been associated with carbonylation and nitrotyrosine levels in sickle cell anemia where iron overload occurs (Kupesiz et al. 2012), and elevated nitrotyrosine has been detected in preliminary experiments with kidney and heart (unpublished data) from irradiated mice at 4 and 18 months post-TBI, which correlates with the presence of hemosiderin.
Other mechanisms, possibly ferroptosis, may be responsible, at least in part, for the radiation effects on cardiac tissue iron and endothelial cell loss. Thus, the ferroptosis inhibitor ferrostatin-1 was tested for its ability to block or decrease the early loss of endothelial cells following irradiation. The results were inconclusive due to the significant vehicle effect, which we speculate was caused by the daily supportive fluid administration. However, further investigation using alternative inhibitors is warranted before ferroptosis is eliminated as a potential mechanism. A recent study by Zhang et al. (Zhang et al. 2020) showed that radiation exposure induced ferroptosis in granulocyte-macrophage hematopoietic progenitor cells in mouse bone marrow, and Thermozier et al. demonstrated that ferroptosis inhibition enhanced the mitigation effect of total-body irradiation when combined with agents that inhibited apoptosis and necroptosis (Thermozier et al. 2020).
Development of novel, effective agents that offer protection and/or mitigation from radiation effects is a high priority of government agencies. The current study showed that deferiprone administration prior to radiation exposure decreased acute tissue iron deposition and prevented loss of endothelial cells and suggests a role for excess iron in the progression of cardiac DEARE. Thus, additional experiments assessing effects of iron chelation on development of DEARE will be important to determine if abnormal iron regulation represents a new target for development of radiation countermeasures. In addition to lethal radiation, the data also have possible relevance for cancer treatment-related radiation exposure, because there is evidence for deposition of tissue iron at doses as low as 100 cGy. Although iron chelation and inhibitors of ferroptosis may be effective for preventing iron-mediated damage, administration of haptoglobin or hemopexin also may be useful agents to prevent acute elevation of iron following irradiation and thus reduce development of DEARE.
ACKNOWLEDGEMENTS
We wish to thank Jennifer Stashevsky for her excellent histology work, and Abigail Russell (supported by an American Physiological Society STRIDE Fellowship) and Sameen Siddiqui (a participant in the Indiana University SEED/STEM Program) for their help with the project.
Serum analyses were performed at The Center for Diabetes & Metabolic Diseases Translation Core (P30DK097512).
Support: This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, NIH 1U01AI107340-01 (C. Orschell), Indiana University Purdue University Indianapolis (IUPUI) Research Support Funds Grant (S. Miller).
LITERATURE CITED
- Christiansen H, Saile B, Hermann RM, Rave-Frank M, Hille A, Schmidberger H, Hess CF, Ramadori G. Increase of hepcidin plasma and urine levels is associated with acute proctitis and changes in hemoglobin levels in primary radiotherapy for prostate cancer. J Cancer Res Clin Oncol 133:297–304; 2007. [DOI] [PubMed] [Google Scholar]
- Chute JP, Muramoto GG, Salter AB, Meadows SK, Rickman DW, Chen B, Himburg HA, Chao NJ. Transplantation of vascular endothelial cells mediates the hematopoietic recovery and survival of lethally irradiated mice. Blood 109:2365–2372; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen EP, Fish BL, Irving AA, Rajapurkar MM, Shah SV, Moulder JE. Radiation nephropathy is not mitigated by antagonists of oxidative stress. Radiat Res 172:260–264; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo LF, Berthrong M. Vascular lesions following radiation. Pathol Annu 23 Pt 1:297–330; 1988. [PubMed] [Google Scholar]
- Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta 1823:1434–1443; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler MH. A unifying system: Does the vascular endothelium have a role to play in multi-organ failure following radiation exposure? Brit J Radiat supplement / BIR 27:100–105; 2005. [Google Scholar]
- Iizuka D, Yoshioka S, Kawai H, Okazaki E, Kiriyama K, Izumi S, Nishimura M, Shimada Y, Kamiya K, Suzuki F. Hepcidin-2 in mouse urine as a candidate radiation-responsive molecule. J Radiat Res 57:142–149; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J. Body iron metabolism and pathophysiology of iron overload. Int J Hematol 88:7–15; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremastinos DT, Farmakis D. Iron overload cardiomyopathy in clinical practice. Circulation 124:2253–2263; 2011. [DOI] [PubMed] [Google Scholar]
- Kupesiz A, Celmeli G, Dogan S, Antmen B, Aslan M. The effect of hemolysis on plasma oxidation and nitration in patients with sickle cell disease. Free Radic Res 46:883–890; 2012. [DOI] [PubMed] [Google Scholar]
- Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, Ding BS, Schachterle W, Liu Y, Rosenwaks Z, Butler JM, Xiang J, Rafii A, Shido K, Rabbany SY, Elemento O, Rafii S. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell 26:204–219; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki M, Kawabata T, Awai M. Iron release from haemosiderin and production of iron-catalysed hydroxyl radicals in vitro. Biochem J 250:589–595; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, Haimovitz-Friedman A, Cordon-Cardo C, Kolesnick R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 293:293–297; 2001. [DOI] [PubMed] [Google Scholar]
- Plett PA, Sampson CH, Chua HL, Joshi M, Booth C, Gough A, Johnson CS, Katz BP, Farese AM, Parker J, MacVittie TJ, Orschell CM. Establishing a murine model of the hematopoietic syndrome of the acute radiation syndrome. Health Phys 103:343–355; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyamitra MM, DiCarlo AL, Taliaferro L. Understanding the pathophysiology and challenges of development of medical countermeasures for radiation-induced vascular/endothelial cell injuries: Report of a NIAID workshop, August 20, 2015. Radiat Res 186:99–111; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121:1276–1284; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermozier S, Hou W, Zhang X, Shields D, Fisher R, Bayir H, Kagan V, Yu J, Liu B, Bahar I, Epperly MW, Wipf P, Wang H, Huq MS, Greenberger JS. Anti-ferroptosis drug enhances total-body irradiation mitigation by drugs that block apoptosis and necroptosis. Radiat Res DOI: 10.1667/RR15486.1; 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, He S, Gerhardt LM, Holderried TA, Seifert M, Sopper S, Fenn AM, Anzai A, Rattik S, McAlpine C, Theurl M, Wieghofer P, Iwamoto Y, Weber GF, Harder NK, Chousterman BG, Arvedson TL, McKee M, Wang F, Lutz OM, Rezoagli E, Babitt JL, Berra L, Prinz M, Nahrendorf M, Weiss G, Weissleder R, Lin HY, Swirski FK. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med 22:945–951; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosano E, Hirsch E, Patrucco E, Camaschella C, Navone R, Silengo L, Altruda F. Defective recovery and severe renal damage after acute hemolysis in hemopexin-deficient mice. Blood 94:3906–3914; 1999. [PubMed] [Google Scholar]
- Unthank JL, Miller SJ, Quickery AK, Ferguson EL, Wang M, Sampson CH, Chua HL, DiStasi MR, Feng H, Fisher A, Katz BP, Plett PA, Sandusky GE, Sellamuthu R, Vemula S, Cohen EP, MacVittie TJ, Orschell CM. Delayed effects of acute radiation exposure in a murine model of the h-ars: Multiple-organ injury consequent to <10 gy total body irradiation. Health Phys 109:511–521; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unthank JL, Ortiz M, Trivedi H, Pelus LM, Sampson CH, Sellamuthu R, Fisher A, Chua HL, Plett A, Orschell CM, Cohen EP, Miller SJ. Cardiac and renal delayed effects of acute radiation exposure: Organ differences in vasculopathy, inflammation, senescence and oxidative balance. Radiat Res 191:383–397; 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinchi F, Gastaldi S, Silengo L, Altruda F, Tolosano E. Hemopexin prevents endothelial damage and liver congestion in a mouse model of heme overload. Am J Pathol 173:289–299; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, An P, Xie E, Wu Q, Fang X, Gao H, Zhang Z, Li Y, Wang X, Zhang J, Li G, Yang L, Liu W, Min J, Wang F. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66:449–465; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Boerma M, Fu Q, Hauer-Jensen M. Significance of endothelial dysfunction in the pathogenesis of early and delayed radiation enteropathy. World J Gastroenterol 13:3047–3055; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woollard KJ, Sturgeon S, Chin-Dusting JPF, Salem HH, Jackson SP. Erythrocyte hemolysis and hemoglobin oxidation promote ferric chloride-induced vascular injury. The Journal of Biological Chemistry 284:13110–13118; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie LH, Zhang XH, Hu XD, Min XY, Zhou QF, Zhang HQ. Mechanisms of an increased level of serum iron in gamma-irradiated mice. Radiat Environ Biophys; 55: 81–88; 2016. [DOI] [PubMed] [Google Scholar]
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: Process and function. Cell Death Differ 23:369–379; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-h, Lou Z-c, Wang A-l, Hu X-d, Zhang H-q. Development of serum iron as a biological dosimeter in mice. Radiat Res 179:684–689; 2013. [DOI] [PubMed] [Google Scholar]
- Zhang X, Xing X, Liu H, Feng J, Tian M, Chang S, Liu P, Zhang H. Ionizing radiation induces ferroptosis in granulocyte-macrophage hematopoietic progenitor cells of murine bone marrow. Int J Radiat Biol 96:584–595; 2020. [DOI] [PubMed] [Google Scholar]