

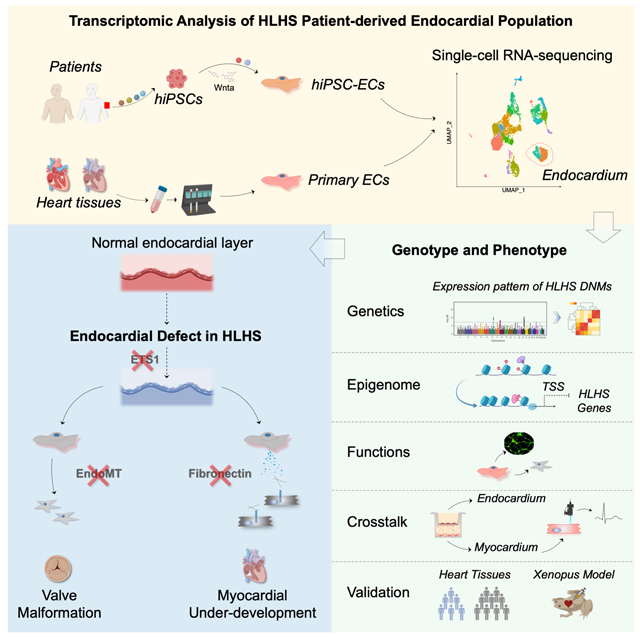
Summary
Hypoplastic left heart syndrome (HLHS) is a complex congenital heart disease characterized by abnormalities in the left ventricle, associated valves, and ascending aorta. Studies have shown intrinsic myocardial defects, but do not sufficiently explain developmental defects in the endocardial-derived cardiac valve, septum, and vasculature. Here we identify a developmentally-impaired endocardial population in HLHS, through single-cell RNA profiling of hiPSC-derived endocardium and human fetal heart tissue with an underdeveloped left ventricle. Intrinsic endocardial defects contribute to abnormal endothelial-to-mesenchymal transition, NOTCH signaling, and extracellular matrix organization, key factors in valve formation. Endocardial abnormalities cause reduced cardiomyocyte proliferation and maturation by disrupting fibronectin-integrin signaling, consistent with recently described de novo HLHS mutations associated with abnormal endocardial gene and fibronectin regulation. Together, these results reveal a critical role for endocardium in HLHS etiology and provide a rationale for considering endocardial function in regenerative strategies.
Keywords: Hypoplastic left heart syndrome, Single-cell RNA-seq, Induced pluripotent stem cells, Human heart tissue, Endocardium, Endothelial to mesenchymal transition, NOTCH, Fibronectin, De novo mutation, ETS1, CHD7
Graphical Abstract
eTOC Blurb
Gu and colleagues provide direct evidence that a developmentally impaired endocardium underlies the ventricular and valvular hypoplasia in hypoplastic left heart syndrome (HLHS), and relates recently discovered de novo mutations to the pathogenesis of this condition.
Introduction
Hypoplastic left heart syndrome (HLHS) is a congenital heart malformation characterized by severe underdevelopment of the left ventricle, mitral valve, aortic valve, and ascending aorta. Previous studies using human induced pluripotent stem cells (iPSCs) from HLHS patients as well as mouse forward genetics have shown an intrinsic defect in cardiomyocytes in the etiology of HLHS, with reduced cardiac differentiation efficiency, disorganized sarcomeres, abnormal mitochondria, and impaired NOTCH signaling (Hrstka et al., 2017; Liu et al., 2017; Yang et al., 2017). However, none of these features sufficiently explained the abnormal development of the cardiac valve, septum, and vasculature in HLHS.
Endocardial cells are specialized endothelial cells (EC) that form the innermost layer of the heart wall. The endocardium plays several pivotal roles in heart development and disease (Sharma et al., 2018). It serves as the source of mesenchymal cells in endocardial cushions that give rise to the structural elements of the atrioventicular valves as well as the atrial and membranous ventricular septa (Harris and Black, 2010). The “no flow, no grow” theory suggested that the abnormal cardiac valve could limit blood flow, leading to the ventricular hypoplasia (Boselli et al., 2015). Endocardial cells also contributed to a small amount of coronary vascular ECs in the developing heart (Tian et al., 2015). Moreover, the reciprocal endocardial-myocardial interaction constitutes an important signaling center that mediates the trabeculation and maturation of the heart chamber (Bressan et al., 2014; de la Pompa and Epstein, 2012). Given that the majority of congenital heart defects are represented by valvular abnormalities, septal defects, and/or ventricular underdevelopment (Botto et al., 2001; Hoffman and Kaplan, 2002), it is important to understand the role of the endocardium in the pathogenesis of these conditions. This will also inform the development of therapeutic strategies aimed at ventricular and valvular regeneration.
Previously, it has been reported that HLHS patient hearts had lower capillary density compared with normal hearts (Gaber et al., 2013). HLHS fetal heart ECs also exhibited the greatest susceptibility to genotoxic injury compared with other cardiac cell types (Gaber et al., 2013). Several genetic variants (e.g., in NOTCH1, HAND1) that are robust determinants of the generic ECs function (Luxan et al., 2016; Theodoris et al., 2015), are also implicated in HLHS (Hrstka et al., 2017; Kobayashi et al., 2014; Yang et al., 2017). However, the role of the intrinsic endocardial abnormality in HLHS remains controversial. In the commonest variant of HLHS, endocardial fibroelastosis (EFE) is present as a result of de novo deposition of subendocardial fibrous tissue layers, and has been considered the hallmark of endocardial injury (Grossfeld et al., 2019). However, a recent study using a lineage tracing mouse model reported that the excessive fibroblasts in the EFE-like tissues mainly originated from the epicardium-derived mesenchymal cells, not endocardial cells (Zhang et al., 2017). Other than the EFE related studies, the nature of endocardial insufficiency as an underlying mechanism in causing HLHS during heart development remains poorly understood.
To further elucidate the transcriptomic and functional defects in the HLHS endocardium in human, we carried out single-cell RNA sequencing (scRNA-seq) in both HLHS iPSC-ECs and fetal heart tissue with an underdeveloped left ventricle. We identified a developmentally impaired endocardial population in HLHS. Subsequent functional assays using purified iPSC-derived endocardial cells revealed that the endocardial defect in HLHS could lead to impaired endocardial to mesenchymal transition and angiogenesis. Additionally, endocardial abnormalities in HLHS could contribute to the reduced proliferation and maturation of cardiomyocytes (CMs), mediated by a disrupted fibronectin-integrin α5β1 interaction. These features were mediated through reduced expression of the transcription factor ETS proto-oncogene 1 (ETS1) and other epigenetic regulators such as CHD7, which have been previously reported to have a de novo mutation associated with HLHS (Jin et al., 2017). Taken together, our findings have uncovered a mechanism whereby a developmentally impaired endocardium could underlie the ventricular and valvular hypoplasia in HLHS.
Results
HLHS patient de novo mutations were highly enriched in the endocardial and endothelial populations in a developing human heart.
It is well appreciated that the genetics of HLHS is complex. Previously, a set of congenital heart disease (CHD) related de novo mutations (DNMs) were reported (Jin et al., 2017), some of which were strongly associated with HLHS. To further understand the role of these HLHS-associated DNMs in the development of human heart, we first examined their expression levels at single cell resolution. A normal human fetal heart at a gestational age of 83 days was micro-dissected into the left- and right-side of the heart. Each side was enzymatically dispersed as single cells, enriched with endocardial/endothelial cells using a CD144 antibody and processed for single-cell RNA-sequencing (scRNA-seq) (Figure 1A). The majority of the cell types expected to be present in the heart were successfully retrieved by scRNA-seq analysis as shown in the UMAP projection, including a certain amount of CMs (TNNI3+) in addition to pan-ECs (CDH5+) (Figure 1B & C). Of note, CDH5+ populations clustered into three subgroups: endocardium (NPR3+), endothelium (APLN+) (unless specified, endothelium refers to the vascular endothelium), and lymphatic endothelium (LYVE1+) (Figure 1C).
Figure 1. HLHS de novo mutations (DNMs) were enriched in the endocardium and endothelium based on scRNA-seq analysis of human fetal heart.

(A) Schematic of the workflow for micro-dissection of normal human fetal heart and scRNA-seq. Endocardial/endothelial populations were enriched with a CD144 antibody and Magnetic-Activated Cell Sorting. PA: pulmonary artery; RA: right atrium; LA: left atrium; RV: right ventricle; LV: left ventricle. (B) UMAP projection of various cell types from day 83 normal human fetal heart. SMC: smooth muscle cell; RBC: red blood cell; CM: cardiomyocyte; EC: endothelial cell. (C) UMAP projection of represented genes for various cell types colored by represented genes’ expression level (purple indicates high expression level) in human fetal heart. (D) Cell type-specific RNA expression of HLHS DNM genes based on scRNA-seq from day 83 normal fetal heart. Row Z-score indicates RNA level. Red denotes high expression, blue minimal expression. See also Figure S1.
An in-depth analysis of CD144+ populations from the fetal heart further defined several endocardial and endothelial subpopulations (Figure S1A). The homologues of several well-defined genes that label mouse endocardium (Zhang et al., 2016), such as NPR3, CDH11, HAPLN1, and ADGRG6 were also enriched in the human heart endocardium (Figure S1B). A small cluster of cells that were in close proximity to the endocardium highly expressed valve markers (FST and PROX1) (Figure S1C) (Hulin et al., 2019; Su et al., 2018). Lymphatic EC was defined by high expression of LYVE1 (Figure S1D). Within the vascular endothelial populations, there were distinct clusters expressing artery, vein, and capillary markers (Su et al., 2018; Wolf et al., 2019) as well as intermediate clusters that were positive for both arterial and venous/capillary genes (Figure S1E & F).
After generating the human heart atlas at single cell resolution, we next examined the expression levels of the previously reported HLHS-associated DNMs in different cell types (Jin et al., 2017). Intriguingly, the majority of these genes were highly expressed in the endocardium, endothelium, and lymphatic ECs (Figure 1D, red dashed box), as opposed to the cardiomyocytes (Figure 1D, black dashed box). These observations reinforce the pivotal role for endocardial/endothelial dysfunctions in the pathogenesis of HLHS.
scRNA-seq of HLHS iPSC-ECs uncovered a developmentally impaired endocardial population.
To further investigate the role of EC dysfunction in causing HLHS, iPSCs from four controls and three HLHS patients were differentiated into heterogenous ECs including an endocardial population using a previously established protocol (Gu, 2018) (Figure 2A). iPSC lines from HLHS cases that developed early-onset right heart failure within the first decade of life, as defined in Table S1 were used in this study. After purification with CD144 antibody, day 8 iPSC-derived ECs (iPSC-ECs) expressed both endocardial and endothelial/capillary markers as compared to day 0 undifferentiated iPSCs (Figure S2A). scRNA-seq of iPSC-ECs from a control (control 1) and a HLHS patient (patient 3) revealed several EC sub-clusters (Figure 2B & C). These iPSC-ECs expressed both endocardial marker genes (Figure 2D & E) and endothelial/capillary marker genes (Figure S2B & C, control: cluster 3 and 5; HLHS: cluster 1 and 4). Hierarchal clustering analysis showed that HLHS cluster 6 grouped together with control clusters 2 and 0 (endocardial clusters), while HLHS clusters 3 and 5 grouped together with control clusters 4 and 1 (endothelial clusters, Figure 2F). Control iPSC-ECs were highly enriched with endocardial genes (clusters 2 and 0); whereas HLHS iPSC-ECs showed significantly reduced endocardial gene expression within cluster 6 (Figure 2D, E). To further validate this finding, we enriched for the endocardial population from the heterogenous iPSC-ECs by fluorescence-activated cell sorting (FACS) with an antibody against the endocardial cell surface marker NPR3. 20.2% of differentiated control iPSC-ECs were positive for NPR3 (Figure S2D). NPR3+ iPSC-ECs expressed a high level of endocardial marker genes (NPR3 and HAPLN1) when compared to NPR3- iPSC-ECs, indicating that the enrichment was successful (Figure S2E). Next, we examined the differentiation efficiency of iPSC-derived endocardial endothelial cells (iEECs) from each control and HLHS patient. As revealed by FACS analysis, the variation amongst the different samples were comparable between control and HLHS lines (Figure S2F, F test, p value=0.4519, ns), and the percentage of HLHS iEECs was lower compared with control iEECs. However, this did not reach statistical significance (Figure S2F, p=0.1865, ns). Although the change in the endocardial population in HLHS was not significant, endocardial gene expression levels were significantly decreased in three HLHS iEECs when compared with four control iEECs, consistent with the scRNA-seq results (Figure 2G).
Figure 2. scRNA-seq of iPSC-ECs unraveled a developmentally impaired endocardial population in HLHS patients.

(A) Illustration of 10X Genomics scRNA-seq of iPSC-ECs from one healthy control and one HLHS patient. (B) UMAP projection of iPSC-ECs from both the control and HLHS patient. Each color defines one subpopulation based on the transcriptomic phenotype. (C) UMAP projection colored by cell origin. (D) UMAP plots of iPSC-ECs colored by expression levels of endocardial markers. Purple denotes high expression, white minimal expression. (E) Violin plot visualization of endocardial gene expression distribution across different subpopulations and normalized by cell number. False discover rate (FDR) indicated the significance of difference between control (cluster 2, 0, 5, and 3) vs HLHS (cluster 6, 4, and 1). (F) Hierarchal clustering analysis of EC clusters from scRNA-seq. Red lines: endocardial cluster branch. (G) Confirmation of endocardial genes expression changes between control (n=4) and HLHS (n=3) iEECs by quantitative PCR (qPCR). (H) Immunostaining of pan-EC (CD31) and endocardial (CDH11) proteins in fetal hearts from control (n=6) and HLHS (n=12) patients. In (G) and (H), data shown as the mean ± SEM. **p<0.01, control vs HLHS. See also Figure S2 and Table S1, S2, and S4.
The abnormality in endocardial gene expression was also evident in HLHS patient heart tissues, where a partial reduction in the endocardial marker CDH11 expression was observed (Figure 2H, Figure S2G & H). A CD31 antibody clearly labeled the endocardium, coronary endothelium, and capillary endothelium (Figure S2G), while CDH11 was restricted to the endocardium, but not the coronary endothelium (Figure S2H). In control samples, the majority of the endocardial layer was positive for both CD31 and CDH11. However, CDH11 positive staining in the HLHS endocardium was significantly reduced (Figure 2H).
HLHS iEECs exerted functional abnormalities compared with controls.
Based on the scRNA-seq results from iPSC-ECs, gene ontology (GO) enrichment analysis on endocardial clusters (clusters 2 and 0 in control; cluster 6 in HLHS) revealed suppression of several signaling pathways strongly relevant to endocardial functions as well as valvular structural remodeling and development, such as extracellular matrix (ECM) deposition, immune system, VEGF signaling and NOTCH signaling (Figure 3A) (Combs and Yutzey, 2009; Hulin et al., 2019; MacGrogan et al., 2010; Schroeder et al., 2003). ECM deposition is closely related to the endocardial to mesenchymal transition (EndoMT) (Brickner et al., 2000; Eisenberg and Markwald, 1995), which is a critical process for the development of valvular structures. Thus, we stimulated EndoMT in control and HLHS iEECs using TGFβ2, and found that the protein level of the mesenchymal cell marker α-smooth muscle actin (αSMA) (Figure 3B) as well as RNA levels of EndoMT-related genes (Figure 3C) were significantly reduced in HLHS iEECs compared with control cells.
Figure 3. iPSC-ECs and iEECs revealed functional defects in the HLHS endocardium.

(A) Functional enrichment in endocardial clusters from control (cluster 2 & 0) and HLHS (cluster 6) iPSC-EC scRNA-seq. −log10FDR indicates the significance of enrichment. GO: gene ontology. α smooth muscle actin (αSMA) staining (B) and EndoMT related gene expression (C) in iEECs from control and HLHS patients after 7 days treatment with TGFβ2 (10 ng/μl). DAPI: nucleus. (D) Adhesion assay of iEECs with different ECMs to coat the culture dish. UMAP (E) and violin plots (F) of the represented genes of the NOTCH pathway from scRNA-seq. FDR indicated the significance of difference between control (cluster 2, 0, 5, and 3) vs HLHS (cluster 6, 4, and 1). (G) Expression levels of NOTCH pathway related genes were measured by qPCR in control and HLHS patient iEECs. (H) Tube formation of control and HLHS iEECs after seeding for 6 hr. iEECs were stained with Calcein AM (green) before seeding. Pictures are under 10X magnification. In (C), (D), (G), (H), data shown as the mean ± SEM. *p<0.05, **p<0.01, control (n=4) vs HLHS (n=3).
Through binding to integrin receptors, different ECMs act as extracellular factors to initiate intracellular signaling cascades related to EndoMT, cell growth, apoptosis, and migration (Carey, 1991; Post et al., 2019; Schroeder et al., 2003; Short et al., 1998). Reduced integrin signaling predicted by scRNA-seq in iPSC-ECs was confirmed by an adhesion assay. Acute adhesion to several ECM components, such as collagen IV, gelatin, and laminin were profoundly suppressed in HLHS iEECs (Figure 3D).
NOTCH signaling plays a pivotal role during cardiac and valvular development (Luxan et al., 2016; MacGrogan et al., 2010). The mutation or haploinsufficiency of NOTCH are strongly implicated in the incidence of CHD (Hrstka et al., 2017; Kobayashi et al., 2014; Theodoris et al., 2015; Yang et al., 2017). Based on our scRNA-seq and qPCR analyses, we demonstrated that NOTCH signaling related genes were significantly down-regulated in HLHS iEECs compared with controls (Figure 3E-G). The imbalanced homeostasis of this process could lead to abnormal valve formation.
Reduced vascular endothelial growth factor (VEGF) expression has been described in the left ventricle from severe HLHS patients (Gaber et al., 2013). In accordance with the GO prediction shown in Figure 3A, tube formation assays demonstrated a significant suppression of angiogenesis in HLHS iEECs compared with controls (Figure 3H).
HLHS iEECs impaired cardiomyocyte functions.
We hypothesized that other than suppressing normal valve development, a defective endocardium in HLHS could also impair cardiomyocyte proliferation and contribute to the ventricular underdevelopment. To recapitulate the crosstalk between the endocardium and myocardium in human heart, we used a transwell co-culture system (Figure 4A) to culture day 15 normal iPSC-derived cardiomyocytes (iPSC-CMs) at its upmost dynamic stage with normal or HLHS iPSC-ECs. RNA-seq analysis revealed distinct gene expression in cell cycle, cardiac conduction, cardiac muscle contraction, and sarcomere pathways when comparing iPSC-CMs co-cultured with control vs. HLHS iPSC-ECs (Figure 4B & C, Figure S3A). Similarly, KEGG pathway analysis also indicated repression of cardiac muscle contraction signaling (Figure S3B). Based on our current knowledge on iPSC-CMs differentiation (Ahmed et al., 2020; Sharma et al., 2018), these cells are highly proliferative at early stage, and then exit active cell cycle to fully enter the maturation stage at later time point. Thus, we co-cultured purified iEECs with iPSC-CMs at either day 15 early stage to test proliferation, or day 30 late stage for maturation assays. The HLHS iEECs-induced dysfunctional iPSC-CMs were evident by reduced proliferation with fewer Ki67+ iPSC-CMs (Figure 4D), as well as reduced expression levels of proliferation genes (Figure 4E). Decreased velocity of contraction and relaxation with a comparable underlying beating rate were also observed in iPSC-CMs co-cultured with HLHS iEECs (Figure 4F). Sarcomere structure, another indicator of the cell maturation, was impaired in these iPSC-CMs based on computational alignment analysis (Figure 4G). In line with these assays, the gene expression levels of maturation markers were reduced in iPSC-CMs co-cultured with HLHS iEECs (Figure 4H). Several studies have indicated that the endocardium could alter myocardial growth through direct signal transduction, such as Notch ligand-receptor crosstalk (D'Amato et al., 2016; Papoutsi et al., 2018; Wang et al., 2013; Zhao et al., 2019). Therefore, we performed a direct-contact co-culture assay mixing iPSC-CMs with either control or HLHS iEECs in the same culture dish (Figure S3C). Consistent with our previous findings, iPSC-CMs co-cultured with HLHS iEECs showed reduced expression of cell proliferation- and maturation-related genes compared with the those co-cultured with control iEECs, indicating that the endocardium could promote cardiomyocyte development in both indirect (paracrine) and direct manner (Figure S3D & E). These data indicated that the HLHS endocardium could hamper myocardial development by impairing valve formation, as well as cardiomyocyte growth and maturation.
Figure 4. HLHS iEECs impeded cardiomyocyte proliferation and maturation.

(A) Normal iPSC-CMs were co-cultured with iPSC-ECs from control or HLHS patients for 48 hr. (B) Transcriptomic profiling and functional enrichment analysis were performed on iPSC-CMs from (A) by bulk RNA-seq (n=3). −log10P indicates the significance of enrichment. Z-score defines the changing trend of enriched functions between HLHS vs control; z-score<0 means down-regulation in iPSC-CMs co-cocultured with HLHS iPSC-ECs. (C) Heatmap visualization of DEGs from GO terms in (B). (D) Immunostaining of Ki67 and TNNT2 in D15 iPSC-CMs co-cultured with control or HLHS iEECs for 48 hr. DAPI: nucleus; Green: Ki67 positive nucleus; Red: TNNT2 positive cardiomyocytes. (E) Gene expression related to proliferation in iPSC-CMs from (D). Contraction velocity (F), sarcomere organization (G), and maturation related gene expressions (H) in D30 iPSC-CMs co-cultured with control or HLHS iEECs. In (D-H), data shown as the mean ± SEM. *p<0.05, **p<0.01. See also Figure S3.
Endocardial homeostatic functions and endocardium-myocardium crosstalk were dependent on fibronectin 1, which was absent in HLHS.
We next compared the differentially expressed genes (DEGs) in a normal left ventricle (LV) versus an underdeveloped LV (ULV) from human fetal hearts (Figure S4A). The underdeveloped human fetal heart was micro-dissected, dissociated, and sequenced at single cell level. scRNA-seq analysis captured the major repertoire of the cardiac cell types (Figure S4B & C), including endocardium (NPR3+), coronary endothelium/capillary (APLN+), and CMs (TNNI3+). Further analysis of CD144+ cells identified various EC subpopulations (Figure S4D-G). Integration of scRNA-seq datasets from normal heart (day 83) and ULV (day 84) ECs showed similar endocardial and endothelial populations on the UMAP projection (Figure S4H-K). GO analysis of the DEGs between ULV versus control fetal heart showed dysregulated pathways in the ULV endocardial cell population (Figure S4L), similar to that observed in the HLHS iPSC-ECs scRNA-seq analysis (Figure 3A). These aberrant pathways include impaired focal adhesions, proliferation and increased cell death. DEGs from the CM population in ULV also showed similar functional defects as revealed by bulk RNA-seq from normal iPSC-CMs co-cultured with control or HLHS iEECs (Figure S4M-O). 17 genes associated with myocardial proliferation and maturation were shared by iPSC-CMs co-cultured with HLHS iEECs and CMs from fetal heart with ULV (Figure S4N & O).
Genes altered in both HLHS iPSC-ECs and endocardial cells from ULV compared with controls were selected for further analysis (Figure 5A). In total, 259 DEGs (117 down-regulated and 142 up-regulated) were identified between ULV vs. normal human fetal left heart endocardium, while 216 DEGs (190 down-regulated and 26 up-regulated) were found in HLHS vs normal iEECs (Figure S5A). Of those, 16 DEGs were shared by both ULV endocardial cells and HLHS iEECs (Figure 5A), including genes related to valve development (FN1, PXDN, SPTBN1, TGM2, LDB2, and MAP4K4) and endocardial markers (HAPLN1 and PLVAP) (Figure S5B). Fibronectin 1 (FN1) was one of the DEGs significantly repressed in both groups (Figure 5A). The suppression of FN1 from scRNA-seq was confirmed by qPCR in 3 HLHS iEEC lines compared with 4 control lines (Figure 5B). In normal human fetal heart, FN1 was universally expressed but enriched in coronary ECs (Figure S5C, left panel), ventricular endocardium (Figure S5C, middle panel), and atrial endocardium (Figure S5C, right panel). Consistent with the reduction at transcript level, FN1 protein expression was significantly reduced in the endocardial layer of HLHS patient hearts compared with control hearts (Figure 5C). FN1 promotes cardiomyocyte proliferation (Hornberger et al., 2000; Ieda et al., 2009), and is required for normal heart development (Mittal et al., 2013), as well as for the left-right asymmetry in vertebrates (Pulina et al., 2011). Therefore, we speculated that FN1 suppression may partially contribute to the ventricular underdevelopment in at least a subset of HLHS cases through the modulation of endocardial cell functions.
Figure 5. Endocardial homeostatic functions and endocardium-cardiomyocyte crosstalk is dependent on FN1, which is absent in HLHS.

(A) Illustration: Overlap of the DEGs from human fetal heart tissue and iPSC-ECs comparing control vs. patient. DEGs from human fetal heart tissue were determined based on the transcriptomic comparison of the endocardial subpopulation from healthy control vs. fetal heart with underdeveloped left ventricle using scRNA-seq analysis. Monte-Carlo simulation indicated the overlapping significance. (B) FN1 gene expression in iEECs from control (n=4) and HLHS patients (n=3). (C) Left panel: Immunostaining of FN1 proteins at endocardial layers in human hearts from control and HLHS patients. Yellow arrowheads indicated positive FN1 staining. V: ventricular chamber. Right panel: Quantification of FN1 positive endocardial cells in control (n=6) and HLHS (n=12) patients. Expression of genes related to the endocardium (D) and EndoMT pathways (E) in left heart endocardial cells with FN1 knock-down. (F) The interactions strength of each endocardium ligand and its cardiomyocyte receptor are displayed in the chord diagram based on day 83 normal human fetal heart scRNA-seq. A ligand-receptor relationship is marked by the color of the ligand (FN1 in red), and interaction strength is shown by the width of chords. Left half circle: Ligands expressed in endocardium; right half circle: receptors expressed in myocardium. (G) Immunofluorescence staining of Ki67 protein in normal iPSC-CMs after co-cultured with primary endocardium with or without FN1 knock-down. DAPI: nucleus; Green: Ki67 positive nucleus; Red: TNNT2 positive cardiomyocytes. Gene expression related to proliferation (H) and cardiac maturation (I) and ion channels (J) in iPSC-CMs from (G). (K) Illustration of iPSC-CM and iEEC co-culture experiments with supplementation of fibronectin (5 μg/ml, 48 hr) to the iPSC-CMs. Contraction velocity measurement (L) and sarcomere organization (M) on D30 iPSC-CMs from (K). (O) Ki67 positive iPSC-CMs on D15 iPSC-CMs from (K). DAPI: nucleus; Green: Ki67 positive nucleus; Red: TNNT2 positive cardiomyocytes. qPCR quantification of gene expressions related to myocardial maturation (N) and proliferation (P). (D-F), n=3. Data shown as the mean ± SEM. *p<0.05, **p<0.01, control vs HLHS, scramble vs siFN1, or HLHS vs HLHS + fibronectin. See also Figures S4, S5 and Table S3.
As iEECs may not fully recapitulate the primary endocardial features, we established a protocol to isolate endocardial cells directly from human fetal heart to test the endocardial-specific effect of FN1 (Figure S5D). Gene expression assessment revealed enriched EC populations as compared to fibroblasts and iPSC-CMs (Figure S5E). Moreover, endocardial and coronary endothelial populations could be separated using our isolation and purification protocol: endocardial markers, NPR3 and HAPLN1 were selectively expressed in human heart endocardial cells compared to coronary ECs or aortic ECs; whereas coronary markers, MGLL and APLN showed higher expression in the isolated coronary ECs (Figure S5F).
Next, we reduced FN1 expression using siRNA in the native endocardial cells isolated from fetal hearts and found significantly decreased endocardial (Figure 5D) and EndoMT genes (Figure 5E), similar to those observed in HLHS iEECs. scRNA-seq from normal fetal heart predicted FN1-interacting partners in the CM population, ITGA5 and ITGB1 (Figure 5F). Suppression of FN1 in the endocardium not only impaired its intrinsic functions, but also significantly impeded iPSC-CM proliferation judged by reduced Ki67+ cells (Figure 5G) and reduced expression of genes related to proliferation (Figure 5H). Additionally, the iPSC-CMs co-cultured with endocardial cells with reduced FN1 had decreased expression of genes related to maturation and ion channels (Figure 5I & J). Intriguingly, addition of fibronectin to the control iPSC-CMs co-cultured with HLHS iEECs significantly improved CMs functions in a dose-dependent manner (Figure 5K-P, Figure S5I-K), including enhanced myocardial contraction velocity (Figure 5L and Figure S5I), sarcomere organization (Figure 5M), Ki67 positive CMs (Figure 5O), and related gene expression augmentation (Figure 5N and P, Figure S5J and K). These data suggest that reduced FN1 in HLHS endocardial cells may underlie the impaired myocardial development in HLHS, which is consistent with the phenotype in iPSC-CMs co-cultured with HLHS iEECs. In addition to endocardium, the expression of FN1 was also found in the coronary endothelium (Figure S5C). Although FN1 expression levels were lower in the HLHS coronary ECs (CECs) from both iPSC-ECs and fetal heart scRNA-seq results (Figure S5G), suppression of FN1 in CECs did not alter CM proliferation in the co-culture system (Figure S5H). This indicates the importance of the endocardium in orchestrating normal myocardial growth during heart development.
HLHS de novo mutations altered endocardial and myocardial functions through transcriptional regulation of endocardial genes
As revealed by the fetal heart scRNA-seq, most of the genes that have been reported to harbor DNMs in HLHS patients were highly expressed in the endocardium/endothelium (Figure 1D). The expression levels of several of these genes in HLHS iEECs were consistently down-regulated compared with controls, such as ETS1, CHD7, KMT2D, and ARID1B, and these are primarily chromatin remodelers and transcription factors ((Figure 6A, Figure S6A). Similarly, it is reported that damaging DNMs accounting for ~10% of the sporadic CHD cases were highly enriched in genes encoding chromatin remodelers (Jin et al., 2017). Reducing levels of ETS1, CHD7, and KMT2D, but not ARID1B (Figure S6B-C) in human primary endocardial cells led to the suppression of endocardial-related genes (Figure 6B) and FN1 expression (Figure 6C). To further understand the role of these mutations in regulating the endocardial gene network, we first focused on one transcription factor ETS1 and one chromatin remodeler CHD7. Chromatin immunoprecipitation assays (ChIP) on iEECs revealed reduced ETS1 and CHD7 binding to the NPR3 promoter region in HLHS iEECs compared to the control cells (Figure 6D). We also applied human umbilical vein endothelial cell (HUVEC) tracks from ENCODE to detect a region in the FN promoter (‘FN1-’, in Figure 6E) and enhancer (‘FN1-b’ in Figure 6E) that we used for the ChIP-qPCR. Both ETS1 and CHD7 showed reduced binding to FN1 promoter and enhancer in HLHS vs control iEECs (Figure 6E). Similarly, their binding capacities to the enhancer regions (i.e., H3K27ac and H3K4me1 high, while H3K4me3 low) of another pivotal endocardial gene, CDH11 in HLHS iPSC-ECs, were consistently reduced (Figure S6D).
Figure 6. HLHS DNMs altered endocardial and myocardial functions through transcriptional regulation of endocardial genes and FN1.

(A) qPCR detection of HLHS DNM genes in iEECs from control (n=3) and HLHS patients (n=3). Red denotes high expression, blue minimal expression. Genes not shown mean non-detectable in iEECs. Genes highlighted by red are transcription factors or chromatin modifiers. qPCR validation of the endocardial gene expression (B) and FN1 (C) in primary human fetal endocardial cells after ETS1 or CHD7 knock-down, respectively. (D) Left: RNAPII and histone marker ChIP-seq tracks of the NPR3 gene in HUVECs from the ENCODE. The black box and red shade indicate the designated location of primers for ChIP-qPCR. The red arrow indicates the transcriptional start site (TSS) and direction of transcription. Right: ChIP-qPCR of ETS1 or CHD7 binding to the NPR3 promoter region in control and HLHS iEECs. (E) Left: Histone ChIP-seq tracks of the FN1 gene in HUVECs from the ENCODE. Right: ChIP-qPCR for ETS1 or CHD7 to the FN1 promoter (a) and enhancer (b) regions. *p<0.05, **p<0.01, control (n=4) vs HLHS (n=3). Data are represented as mean ± SEM. (F) EndoMT-related gene expression in endocardial cells after ETS1 knock-down. (G) iPSC-CMs were co-cultured with isolated fetal left heart endocardial cells with or without ETS1 knock-down. Expression of genes related to cardiomyocyte maturation (G) and ion channel (H) in iPSC-CMs were examined by qPCR. (I) Immunostaining of Ki67 (green) and TNNT2 (red) in iPSC-CMs from (G). DAPI: nucleus. (J) qPCR for proliferative genes in iPSC-CMs from (G). (K) Illustration of control iPSC-CM and iEEC co-culture experiments with ETS1 overexpression (5 multiplicity of infection-MOI, 48 hr) in HLHS iEECs (HLHS+ETS1). Contraction velocity measurement (L) and sarcomere organization (M) on day 30 iPSC-CMs from (K). (O) Ki67 positive iPSC-CMs on day 15 iPSC-CMs from (K). DAPI: nucleus; Green: Ki67 positive nucleus; Red: TNNT2 positive cardiomyocytes. qPCR quantification of gene expressions related to myocardial maturation (N) and proliferation (P). n=3 patients in each treatment group. Data shown as the mean ± SEM. *p<0.05, **p<0.01, scramble vs. siRNA knockdown or HLHS vs. HLHS+ETS1. See also Figure S6.
To further understand how the genes associated with HLHS regulate downstream endocardial and myocardial dysfunction, we first focused on ETS1, which was significantly reduced in HLHS iPSC-ECs based on scRNA-seq analysis (Figure S6E). Reduction of ETS1 expression using siRNA in primary human endocardial cells significantly suppressed genes related to the EndoMT pathway (Figure 6F). Mechanistically, knockdown of ETS1 in human endocardial cells diminished RNAPII recruitment to the promoter regions of endocardial genes, such as NPR3, PLVAP, and CDH1,1 but not FN1 (Figure S6F). We speculated that other chromatin modifiers, such as mediator complex or the dynamic RNAPII regulatory steps during transcription initiation might be involved in ETS1-mediated FN1 transcription (Chen et al., 2015b; Jeronimo and Robert, 2017; Steurer et al., 2018). Correspondingly, we analyzed a previously published dataset of ETS1 over-expression (Mod_ETS1 vs Mod_GFP) in HUVECs (Chen et al., 2017). RNAPII binding capacity to endocardial gene (NPR3 and PLVAP) promoter regions were significantly increased with ETS1 over-expression (Figure S6G), which was in line with our ETS1 knock-down panel. Additionally, similar to the HLHS iEECs and the FN1-deficit endocardium, endocardial cells lacking ETS1 impeded CM maturation (Figure 6G & H) and proliferation, evidenced by reduced Ki67+ in CMs (Figure 6I) and lower expression of proliferative genes (Figure 6J). Intriguingly, HLHS iEECs overexpressing ETS1 rescued the defective proliferation and maturation in normal iPSC-CMs co-cultured with HLHS iEECs (Figure 6K-P) in a dose-dependent manner (Figure S6H-J). Similar to FN1 rescue, contraction velocity (Figure 6L), sarcomere organization (Figure 6M), Ki67 positive CMs (Figure 6O), and related gene expressions (Figure 6N and P) were improved with ETS1 overexpression. In conclusion, these DNMs provide a genetic clue to explain the mechanism involved in the endocardial defects and reduced FN1 in HLHS.
Dysfunctional HLHS iPSC-CMs were rescued by FN1 supplementation or ETS1 over-expression
It has been reported that both endocardial and myocardial dysfunction contribute to the pathogenesis of HLHS (Hrstka et al., 2017; Liu et al., 2017; Yang et al., 2017). Thus, we applied the co-culture system to investigate the potential beneficial effect of FN1 and/or ETS1 on HLHS patient iPSC-CMs. Each patient’s iPSC-CMs were tested with their corresponding iEECs with or without FN1/ETS1. Contraction velocity, sarcomere organization, Ki67 positive iPSC-CMs and associated gene expression were measured (Figure S7). We found that both FN1 and ETS1 could improve each patient’s myocardial proliferation and maturation with one exception: Patient 2 ETS1 over-expression failed to enhance CM proliferation (Figure S7B.d and B.e), indicating a patient specific response to various rescues.
Suppression of Ets1 in Xenopus caused reduced endocardial FN1 and impaired heart development
Previously it has been demonstrated that loss of Ets1 in the cardiac mesoderm of Xenopus embryos leads to heart malformations exhibited by reduced ventricular chamber size, similar to that of a HLHS heart (Nie and Bronner, 2015). To determine whether the loss of Ets1 results in dysregulated FN1 expression in Xenopus heart, we examined the expression of FN1 by immunostaining. Ets1 was knocked-down in Xenopus hearts by injecting morpholinos (Ets1-MO) into cardiogenic mesoderm during cleavage stages. The suppression of Ets1 resulted in significantly reduced ventricular chamber size (outlined with a white dashed box) when compared to the controls (Figure 7A & B). In the control hearts, FN was detected in the inner lining of the endocardial tissue (n=4/4). In contrast, FN staining was lost in the endocardium of Ets1 morphant hearts (n=6/6) (Figure 7A & C). This in vivo evidence suggests the existence of an ETS1-FN1 regulatory axis that is sufficient to cause heart ventricular underdevelopment, similar to what is observed in HLHS.
Figure 7. Knock-down of ETS1 in vivo caused FN1 reduction in the endocardium and led to abnormal heart development.

(A) Staining of FN1 in control and Ets1 morphant Xenopus hearts. White dashed lines outline the chamber of the Xenopus ventricle. Yellow dashed box area was further zoomed in to visualize endocardial FN1 expression. V: ventricle. (B) Quantification of the absolute sizes of the heart chamber, ventricle, and the relative chamber size. The ventricle size measured the entire area in the cross-section of heart ventricle, and the chamber size measured the area of the inner cavity inside the ventricle. (C) FN1 fluorescence signal intensity within the endocardium. control=4; Ets1-MO=6. Data shown as the mean ± SEM. *p<0.05, **p<0.01, control vs Ets1-MO. (D) Schematic illustration of the endocardial and myocardial defects in HLHS. In HLHS endocardial cells, ETS1 and CHD7 expression levels were significantly reduced compared to healthy controls. This directly led to the decreased expression and secretion of fibronectin, which further disrupted the fibronectin-integrin interaction with cardiomyocytes. Impaired integrin activation in cardiomyocytes resulted in decreased cell maturation and proliferation, which may contribute to the underdevelopment of the left ventricle. Additionally, decreased ETS1 and CHD7 also led to the suppression of downstream endocardial gene expression such as NPR3 and CDH11. The impaired endocardium showed aberrant endothelial to mesenchymal transition as well as NOTCH signaling pathway, which are critical to cardiac valve development.
Discussion
During heart development, the endocardium serves as a key component in facilitating valve, septum, and coronary artery formation and maturation. Growing evidence suggests that underdevelopment of the left ventricle could also be due to abnormal blood flow, which is secondary to hypoplastic and/or atretic valve structures among all HLHS subtypes (Grossfeld et al., 2019). However, the potential mechanistic role of an endocardial defect in causing ventricular underdevelopment in HLHS remained unknown. It had been reported that genetic ablation of the endocardial cell population during murine cardiac development arrested the growth of the left ventricle, resulting in embryonic lethality by E11 (Snider et al., 2014). Clinically, a subset of HLHS cases is associated with a thickened layer of fibroelastosis lining the cavity of the ventricle, indicative of endocardial injury (Grossfeld et al., 2019). Herein, using patient-specific iPSCs, fetal heart tissue, and single cell transcriptomic analysis, we have provided direct evidence that severely impaired endocardial functions associated with abnormal valve formation and myocardial growth were present in HLHS patient hearts.
Although our current study focused on endocardial defect, we believe that both intrinsic endocardial and myocardial deficiencies are pivotal in the etiology of HLHS. Several studies have demonstrated impaired cardiogenesis, augmented cardiac apoptosis, decreased proliferation, and altered heart development signaling at both transcriptomic and epigenomic levels in HLHS iPSC-CMs (Gaber et al., 2013; Hrstka et al., 2017; Kobayashi et al., 2014; Yang et al., 2017). These cellular observations could be mainly associated with the “slit-like” LV in patients with mitral and aortic atresia. However, if CM hypertrophy could contribute to the thickened LV wall in patients with aortic atresia and mitral stenosis remains unclear. The crosstalk between endocardium and myocardium during the development of HLHS is worth further investigation.
The genetics underlying HLHS are complex. Pediatric Cardiac Genomics Consortium (PCGC) carried out whole exome sequencing (WES) in over 10,000 CHD probands, and revealed 27 DNMs that were associated with HLHS (Homsy et al., 2015; Zaidi et al., 2013). Interestingly, one HLHS patient carried only one of these DNMs. We hypothesized that there may be a common gene or pathway downstream of the genes harboring these DNMs that leads to the development of HLHS. Our study first focused on two mutations: a transcription factor (ETS1) and a chromatin remodeler (CHD7). A loss-of-function mutation in ETS1 has been identified in patients with hypoplastic left ventricle and other features that were seen in Jacobsen syndrome (Glessner et al., 2014; Tootleman et al., 2019; Ye et al., 2010). Genetic ablation of Ets1 specifically in the cardiac mesoderm in Xenopus produced a HLHS like phenotype associated with loss of endocardial cells (Nie and Bronner, 2015). Likewise, systemic deletion of Ets1 in mice caused a non-apex forming left ventricle phenotype (Ye et al., 2010), one of the hallmarks of HLHS. However, the penetrance of the ETS1 mutation is low, indicating that there might be several other rare or even common variants that are responsible for the HLHS phenotypes. A loss-of-function mutation in CHD7 was related to severe heart defects in CHARGE syndrome, with overrepresentation of atrioventricular septal defects and outflow tract defects (Corsten-Janssen and Scambler, 2017). Because there is a large gene network downstream of ETS1 and CHD7, we first narrowed down the pool of candidate genes by focusing on the ones that were differentially expressed between control vs. HLHS endocardial cells. FN1 was the most strikingly down-regulated gene in HLHS endocardium. Through ChIP-qPCR analysis, we found that the binding of both ETS1 and CHD7 to the promoter and enhancer regions of FN1 was significantly decreased in HLHS iEECs compared with controls. It is encouraging that in the Xenopus model, we confirmed that Ets1-knockdown could reduce Fn1 expression in the endocardial layer. So far, there is no in vivo study showing how Ets1 controls endocardial Fn1 expression in the mouse model. Therefore, further studies in an Ets1-targeted mouse model perhaps in different genetic backgrounds, merit consideration.
FN1 is one of the major extracellular matrix (ECM) components that plays an active role in cardiac development. It was reported that proliferating human fetal CMs synthesize fibronectin, while inhibition of its production arrests CM growth (Hornberger et al., 2000). Furthermore, coating a culture dish with fibronectin augments embryonic CM proliferation (Ieda et al., 2009). Therefore, FN1 may act as an extracellular factor to trigger a cascade of signaling pathways that modulate CM growth. There is accumulating evidence that supports integrin α5β1 as the key receptor involved in FN1 signaling in CMs (Ieda et al., 2009; Mittal et al., 2013). We further confirmed this in our ligand-receptor interaction analysis using our human fetal heart scRNA-seq data. We showed that the endocardium is an important source of FN1 to facilitate normal heart development; when FN1 is lacking, impaired iPSC-CM growth can be the consequence of disrupting the FN1-integrin α5β1 interaction. When FN1 is supplemented to the HLHS iEECs and HLHS iPSC-CMs in co-culture, the decreased proliferation and maturation of the CMs was rescued.
Although several studies have shown that global or cell type-specific (Isl1-Cre or Tfap2a-Cre) knockout of Fn1 in mice significantly impaired ventricle development (Mittal et al., 2013), cardiac outflow tract growth, incidence of ventricular septum defects (Chen et al., 2015a), and left-right embryonic body plan (Pulina et al., 2011), there is no report that of FN1 directly relating to HLHS phenotypes. Based on our current study, it would be interesting to further investigate the cardiac phenotype in a mouse model with an endocardium-specific deletion of Fn1. This will allow us to better comprehend the therapeutic potential of fibronectin in HLHS.
In most of the HLHS patients, they complete the major cardiovascular development with the right ventricle, tricuspid valve, and pulmonic valve, so the endocardial defect appears to affect only development of the left heart. Surprisingly, we were able to reproduce a striking endocardial impairment mimicking the left heart event with purified HLHS iEECs. Similarly, previous HLHS iPSC works confirmed the intrinsic myocardial dysfunctions even in patients with normal RV development at birth (Gaber et al., 2013; Hrstka et al., 2017; Yang et al., 2017). It seems that the in vitro system favors the left-side heart defect, nevertheless the underlying mechanisms are unknown. It is possible that there is some compensatory mechanism occurring only in vivo that allows the right side of the heart to development normally. This requires further investigation using mouse model with heart-field specific endocardial-manipulation.
Previously published scRNA-seq work delineating human heart development identified four different cardiac EC populations, including free wall endocardium, coronary vascular ECs, vascular ECs, and valvar ECs (Cui et al., 2019). However, due to the low cell numbers captured in their EC population, it would be difficult to perform in-depth EC sub-cluster analysis. In our current study, we specifically enriched cardiac EC populations and established a high resolution human cardiac EC atlas including coronary artery ECs, vein ECs, capillary ECs, endocardial ECs, valvular ECs, and lymphatic ECs. Our study thus provides a database to study the differences between EC subtypes in congenital heart disease.
Collectively, our studies based on scRNA-seq analysis using patient-specific iPSC-ECs and human fetal heart tissue reveal robust endocardial functional defects and aberrant endocardium-myocardium crosstalk in HLHS. By modulating HLHS related gene variants and ChIP-qPCR analyses, we further elucidate molecular mechanisms underlying the dysfunctional endocardium (Figure 7D). Our study provides a comprehensive platform to improve our understanding of HLHS etiology in a patient-specific manner, and paves the way to develop new therapies for this condition in the future.
Limitations of the Study
In the single cell study, we have analyzed 1482 cells from one control (control 1) and 901 cells from one patient (patient 3) to provide some indications for further biological validation. We acknowledged that one sample may not be representative of a whole cohort. Thus, we further verified our findings in four control and three HLHS iEEC lines, fetal heart tissue sections from six controls and twelve HLHS patients, fetal heart tissue with an underdeveloped left ventricle, as well as an Ets1 knock-down Xenopus model. The variation amongst different samples were minimal, and did not affect statistical analysis.
Our current differentiation protocol generates heterogeneous iPSC-ECs with high efficiency and low variation (Gu, 2018; Sa et al., 2017). However, deep understanding of the endocardial defect in HLHS patients requires an effective protocol to differentiate patient-specific iPSCs into endocardial cells. Several groups have aimed to generate endocardial-like ECs from iPSCs through genetic-manipulation or cell sorting (Bao et al., 2017; Neri et al., 2019; Vander Roest and Merryman, 2017). However, to date, there is no published protocol that could very-well recapitulate the in vivo endocardial features. Despite this limitation, we were able to utilize scRNA-seq to assess the endocardial specific gene expression abnormalities in HLHS by separating different EC subpopulation, and further confirmed our findings in FACS-sorted, NPR3+ iEECs. To dissect the endocardial dysfunction independent of the endocardial abundance, we utilized an equal number of NPR3+ iEECs from control and HLHS groups for gene expression and functional analyses.
STAR Methods:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Mingxia Gu, MD, PhD. (Mingxia.Gu@cchmc.org).
Material Availability
This study did not generate new unique plasmids/mouse lines/iPSC lines/reagents so there is no restriction for use of any material in this study.
Data and Code Availability
All sequencing data have been deposited into GEO database under GEO accession number GSE138979 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138979).
Data visualization could be accessed through the Broad Institute Single Cell Portal: Heart tissue: https://singlecell.broadinstitute.org/single_cell/study/SCP1020 iPSC-EC: https://singlecell.broadinstitute.org/single_cell/study/SCP1021 All R codes have been uploaded to GitHub (https://github.com/Lei-Tian/HLHS).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human cell lines
Clinicopathological baseline characteristics of patients and donors are given in Table S1. De-identified HLHS patients’ peripheral blood mononuclear cells (PBMCs) were collected at Mayo Clinic through the Todd and Karen Wanek Family Program for Hypoplastic Left Heart Syndrome. HLHS iPSC lines 1, 2, and 3 were reprogrammed by ReGen Theranostics, Inc Rochester, MN. Skin biopsies from healthy control individuals (control cell line 1, 3, and 4) were obtained from Stanford Transplant Center, and skin fibroblasts were further reprogrammed at Stanford Cardiovascular Institute (SCVI) Biobank. Control cell line 2 was purchased from CIRM iPSC Repository (Cat# CW60359EE1). The derivation protocol was modified based on previous publications (Burridge et al., 2014; Gu et al., 2017). All the iPSC cell lines have been cultured in Essential 8 or mTeSR medium. Consent was obtained from both control and patients under approved IRBs: Mayo Clinic: 10-006845; Stanford: IRB 5443.
Human fetal heart tissue
De-identified human fetal heart tissues (Normal heart, day 83, sex unknow; Underdeveloped left heart, day 84, sex male) were obtained in collaboration with the University of Washington Birth Defects Research Lab (BDRL, IRB STUDY00000380). All samples were collected from patients who provided signed informed consent according to IRB guidelines in accordance with all university, state, and federal regulations. Fetal tissue was transferred from BDRL to Stanford under Material Transfer Agreement #44556A.
Human heart tissue sections
Samples from the heart free wall were acquired from the pathology department at the time of fetal autopsies performed as soon as possible after pregnancy termination or fetal demise. All investigations were conducted according to the Declaration of Helsinki principles. Studies were approved by the Hospital for Sick Children and the Mount Sinai Hospital institutional review boards, and written informed consent was obtained from study participants (pregnant mothers) before inclusion in the study. Myocardial tissues were fixed and sectioned (5 μm) at University of Toronto and sent to Stanford University for staining. Clinicopathological baseline characteristics of patients were shown in Table S4.
Frog embryo
The Xenopus laevis embryos were obtained by in vitro fertilization from wild type adult frogs (Nasco). The sex of embryos was not determined. The developmental stages of the embryos were specified in the METHOD DETAILS. Embryos of the same age were randomly assigned to experimental groups. All experimental procedures involving frog embryos were performed according to USDA Animal Welfare Act Regulations and have been approved by Institutional Animal Care and Use Committee, in compliance of Public Health Service Policy.
METHOD DETAILS
Differentiation of cardiomyocyte and ECs from iPSCs
For differentiation of endocardial/endothelial cells from iPSCs, we used our previously published protocol (Gu, 2018; Gu et al., 2017). iPSCs at 80% confluence were placed in differentiation medium (RPMI and B27 supplement minus insulin, Gibco) with 6 μM CHIR-99021 (Selleck Chemicals) for two days, followed by 3μM CHIR-99021 for another two days. Then medium was switched to EGM-2 EC differentiation medium (Lonza) with 50 ng/mL vascular endothelial growth factor (VEGF, Gemini) and 10 ng/mL basic fibroblast growth factor (bFGF, Gemini) for 5 days. On the last day of differentiation, cells were dissociated and sorted for CD144+ population using a MACS sorter (Miltenyi Biotec). After sorting, the cells were plated on 0.2% gelatin (Sigma)-coated plate with EGM-2 EC culture medium until 80%-90% confluence. NPR3 positive iEECs were further sorted by FACS and MACS from iPSC-ECs. Specifically, 100 ul freshly prepared cells (5X106) were incubated with anti-mouse NPR3 antibody (OriGene) to a final concentration of 5 ug/ml for 1 hr on ice. Then secondary antibody detecting mouse IgG (FACS: anti-mouse secondary antibody, Alexa 594, 1:100 dilution; MACS: anti-mouse IgG microbeads, 1:5 dilution) was conjugated for downstream cell sorting. iPSC-ECs and iEECs used in the current study were between passage 2-4.
For differentiation of cardiomyocytes, we used previous published protocol (Burridge et al., 2014). iPSCs were maintained in E8 media (Gibco). At a confluency of 70-75%, iPSCs were passaged at a 1:12 ratio with Accutase (Gibco) or 0.5mM EDTA (Gibco) and evenly plated on 6-well plates. At a confluency of 95-98% on Day 0, E8 media was changed to RPMI 1640 (Gibco) containing 6 μM CHIR-99021 (Selleck Chemicals) and B27 Minus Insulin (Gibco). On Day 2, media was changed to RPMI 1640 supplemented with B27 Minus Insulin. On Day 3, media was changed to RPMI containing 2 μM C59 (Selleck Chemicals) and B27 Minus Insulin. On day 5, cells were rinsed with 1x PBS, and fed with RPMI 1640 supplemented with B27 Minus Insulin. On day 7, media was changed to RPMI 1640 supplemented with B27 Plus Insulin (Gibco). By day 9, beating cells could be observed and cells were glucose starved to enrich for cardiomyocytes by two-day treatment with RPMI 1640 Minus Glucose supplemented with B27 Plus Insulin. On day 12-13, cardiomyocytes were replated using 10X TrypLE (Gibco) Select for 10 min. Cells were detached from wells by gentle pipetting and neutralization of TrypLE Select by cardiomyocyte replating media consisting of 80% RPMI 1640, 20% Knockout Serum (Gibco), B27 Plus Insulin, and 10 μM Y-27632 (Tocris). The day after replating, cardiomyocytes were subsequently maintained in RPMI 1640 with B27 Plus Insulin by media changes every two days.
Frog embryo manipulations
Xenopus laevis embryos were obtained and microinjected with Ets1-MO as previously described (Nie and Bronner, 2015). Ets1-MO (5’-TAAGGTCTAGTGCAGCTTTCATGGC-3’) was injected into both dorsal vegetal blastomeres at 8-cell stage to target cardiac mesodermal tissue.
All experimental procedures were performed according to USDA Animal Welfare Act Regulations and have been approved by Institutional Animal Care and Use Committee, in compliance of Public Health Service Policy.
Isolation of human fetal heart ECs
This protocol was modified based on endocardium isolation protocol from rat (Klein et al., 2020). Human fetal heart was obtained from aborted fetuses with parents’ consent at the University of Washington Birth Defects Research Laboratory and preserved in Belzer UW® Cold Storage Solution (Thermo Fisher) for overnight shipping. The heart was kept on ice throughout processing. 1X HBSS (Gibco) was used to remove excess blood cells, the heart was then micro-dissected into aorta, pulmonary artery, left ventricle free wall, right ventricle free wall, and septum. Digestion buffer was prepared in DMEM medium with each component as the following final concentration: Liberase TM, 0.5 mg/ml; DNase I, 20 μg/ml; HEPEs, 10mM.
For endocardial cell isolation, the inner side of the ventricular free wall was placed face down in the culture dish with approximately 500 μl of digestion buffer to ensure that only the endocardial layer was immersed in the buffer. After digesting for 5 min at 37°C, the endocardium was washed with EGM-2 medium prior to centrifugation to pellet the cells. For coronary EC isolation, the outer layer of the ventricular free wall was carefully cut out and collected to avoid contamination with endocardial cells. Tissues were minced into 1 mm3 pieces and digested in 1.5 ml digestion buffer for 10-15 mins at 37°C. The reaction was quenched with EGM-2 medium and centrifuged to pellet cells. For aortic ECs, the connective tissue from the aortic ring was removed. The aorta was immediately digested at 37°C for 10 min, then EGM-2 medium was used to flush the inner luminal layer of the vessel prior to centrifugation to pellet the cells. All of the above cell pellets were resuspended with EGM-2 containing an additional 50 ng/ml VEGF and 10 ng/ml bFGF, cell clumps were separated through a 40 μm cell strainer, and seeded on a collagen I (Sigma)-coated plate. After reaching 90% confluence, cells were sorted using a MACS sorter with a CD144 antibody (Miltenyi Biotec). Endocardial and endothelial cells populations were tested for CD144+ by flow cytometry and purity by quantitative PCR.
Chromatin immunoprecipitation
ChIP assays were performed as previously described (Miao et al., 2018). Briefly, 1×107 human fetal endocardial cells or iPSC-ECs/iEECs were treated with 0.75% formaldehyde for 20 min at room temperature. Afterwards, fixation was stopped by adding 125 mM glycine and cells were collected. The pelleted cells were lysed and sonicated using default parameter by Bioruptor® Pico (Diagenode Inc.) for 10 times using ‘30 sec ON’ 30 sec OFF, program at 4°C. The sonicated samples were then centrifuged and 1% of supernatant was taken as input. After sonication, the chromatin was immunoprecipitated by various antibodies (ETS1, #39580, Active Motif-4μg per ChIP; RNA Pol II, #39097, Active Motif-4μg per ChIP; CHD7, #6505S, Cell Signaling Technology-4μg per ChIP) conjugated to pre-washed Protein A or Protein G Dynabeads. Protein and RNA were digested by proteinase K and RNase A, respectively. The purified chromatin DNA was then used as the template for a quantitative polymerase chain reaction. As an isotype control, non-specific IgG derived from the same species as specific antibodies were used in ChIP.
Quantitative reverse-transcription PCR
Our detailed protocol was previously published (Gu et al., 2017). Briefly, total RNA was extracted, purified, and quantified for reverse transcription using High Capacity RNA to cDNA Kit (Applied Biosystems) according to the manufacturer’s instructions. qPCR was carried out using 5 ng cDNA and 6 μl SYBR green master mix (Applied Biosystems). Primers were listed in Table S5. Each measurement was performed in triplicate.
Immunofluorescence and immunohistochemistry
iPSC-CMs or iEECs were fixed in 4% paraformaldehyde for 5 min and blocked with 2% BSA for 1 hr. Cells were stained with Ki67 (1:5000, Abcam, ab11580) and TNNT2 (1:200, ThermoFisher, MS-295-P) or TNNT2 (1:400, Abcam, ab45932) and α-actinin (1:400, Sigma-Aldrich, A7811) or α-SMA (1:200, Abcam, ab5694) at 4°C overnight. AlexaFluor-conjugated secondary antibody (Life Technologies) was then used and co-stained with DAPI (Vector Laboratories). Imaging was captured with a confocal microscope (Leica) and analyzed with ImageJ software.
The human control and HLHS heart paraffin sections were dewaxed, rehydrated and antigens were retrieved using Tris-based buffer (Vector Laboratories) for 20 min in the microwave. The samples were pre-treated with 0.5% H2O2 (Fisher Scientific) for 30 min before blocked with 3% BSA (Sigma) for 1 hr. CDH11 (1:50, Invitrogen, 32-1700), CD31 (1:50, Abcam, ab9498), and FN1 (1:50, Abcam, ab6328) were used for overnight staining, followed by HRP-ABC development (Vector Laboratories) and hematoxylin (Vector Laboratories) nuclear staining. Imaging was captured using a bright field microscope (Leica) and analyzed using ImageJ software. All the tissue section information for each patient was included in Table S4.
Frog embryos were fixed at stage 46 in the fixative MEMFA and dehydrated in ethanol and then in methanol overnight. The embryos were then rehydrated gradually into PBS and equilibrated in 20% sucrose solution. The embryos were next changed into OCT through graded washes, embedded in OCT, and sectioned at 12 microns. Sections through the heart was stained with anti-FN antibody (1:50, Developmental studies hybridoma bank-DSHB, 4H2) as previously described (Garmon et al., 2018). For FN signal intensity, a line was drawn across the ventricles and pixel intensities at the peaks of the plot were considered as FN signal at the inner lining of the ventricular chamber. The ventricle size measures the entire area in the cross-section of heart ventricle, while the chamber size measures the area of the inner cavity inside the ventricle.
Angiogenesis assay
Cells were starved in medium containing 0.2% FBS overnight and seeded at the density of 20,000 cells per 24-well plate in growth factor reduced Matrigel (Corning). The tube numbers were counted at 6 hr and 12 hr afterwards in three random fields. We defined a tube and quantified the number of tubes as described (Nickel et al., 2015).
Cell adhesion assay
Cell adhesion was carried out as previously described (de Jesus Perez et al., 2012). iEECs (10,000/well) were seeded on to a 24-well plate either uncoated (plastic) or coated with collagen IV (BD BioCoat™), gelatin (0.2%, Sigma), laminin (BD BioCoat™) or fibronectin (0.1 mg/ml, Sigma) and allowed to adhere for 1 hr. Non-adhesive cells were then washed away using PBS. The remaining adherent cells were stained with DAPI and imaged. The average number of cells was calculated by counting the total number of cells in six random views per well (10X magnification). All assays were repeated in triplicate.
Co-culture of iPSC-CMs and ECs
Control iPSC-CMs at Day 15-30 of differentiation were used for the co-culture assays. iPSC-CMs were seeded on the bottom well that was pre-coated with Matrigel overnight. Native endocardial cells or iPSC-ECs/iEECs were seeded onto the 0.4-μm pore cell culture insert (Corning) at 90% confluence. After co-cultured for 48 hr, iPSC-CMs were either fixed for immunostaining, harvested for RNA extraction (Zymo Research), or rescue experiments. Fibronectin (Sigma-Aldrich) was mixed with B27 medium (final concentration: 5 μg/ml) and added to the iPSC-CMs co-cultured with HLHS iEECs for 48 hr. HLHS iEECs were infected with either GFP or ETS1 overexpression lentivirus (kindly provided by Dr. William T. Pu) for 48 hr and reseeded into the transwell to co-culture with the iPSC-CMs for 48 hr.
Contractility assay
Contractility was measured using the Sony SI8000 cell motion imaging system, in the presence of 5% CO2 and at a temperature of 37°C. Prior to initiation of recording, the cells were given time to equilibrate for 15 min at the given setting. Video imaging of the beating iPSC-CMs monolayers were recorded from 2-3 separate locations in each well (center and lateral edge), spatially adjusted in an automated recognized fashion, for 10 sec, at a frame rate of 75 fps, a resolution of 1024 × 1024 pixels, and a depth of 8 bits using a 10×objective.
Quantification of sarcomere organization
A prominent feature of mature cardiomyocyte is the regular lateral alignment of adjacent myofibrils that are stabilized by Z-disc proteins including α-actinin. To determine the extent of sarcomere organization, the orientation angle of Z-disc with respect to myofibrils was calculated. First, iPSC-CMs were immunostained with rabbit cardiac troponin T antibody (Abcam, Cat. ab45932) and mouse α-actinin antibody (Sigma-Aldrich, Cat. A7811), followed by Alexa-488 anti mouse antibody and Alexa-594 anti rabbit antibody. Cardiac troponin T channel of immunofluorescent images was used to define the region of interest (ROI) that contains myofibrils parallel to the y-axis. Next, immunofluorescent sarcomeric α-actinin of the ROIs was segmented into individual Z-disc particles by morphological reconstruction method. Since the segmented Z-disc particle is elliptical, the angle of the long axis of the particle with respect to x-axis was calculated. When y-axis is the orientation of a myofibril, sarcomeric α-actinin parallel to x-axis is considered as aligned sarcomere, whose percentage is eventually plotted for comparison. In this study, the segmented Z-disc particles with the angle within ±30° was considered aligned. Quantification was performed as double blinded as one person generates ROIs and another person performs the quantification without knowing the sample groups.
siRNA transfection
siRNA transfection was carried out according to manufacturer’s instruction. ON-TARGET plus scramble, ETS1, FN1, CHD7, KMT2D, FOXM1, and ARID1B siRNA (GE Dharmacon) were transfected at 25 nM into subconfluent early passage native endocardium using Lipofectamine RNAiMax (Invitrogen). 48 hr after transfection, cells were either fixed for staining or subjected to RNA extraction.
Bulk RNA-seq data analysis
The raw 150 bp paired-end reads were first trimmed for quality control by TrimGalore v0.4.4 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to exclude adapter sequences and bases with Phred scoring less than 20, reflecting a low probability of base-calling errors (p<1%), equivalent to >99% accuracy. Then, the trimmed pair-end reads were mapped to hg38 using STAR software (2.5.4 a) (https://github.com/alexdobin/STAR) (Dobin et al., 2013) with ENCODE options. FeatureCounts (Liao et al., 2014) v1.5.1 (http://subread.sourceforge.net/) was used to count the number of reads in each gene annotated by GENCODE (Harrow et al., 2012) v25. DESeq2 (Love et al., 2014) R package was used to detect DEGs between iPSC-CMs co-cultured with control or HLHS iPSC-ECs using the likelihood ratio test. Full model was set as full=~treatment+1, and the reduced model was reduce =~1. Genes with p-value<5% and fold-change ≥1.2 were define as DEGs. We used DESeq2 R package to normalize the raw counts from bulk RNA-seq data. The expression values of each candidate gene were extracted and followed by z-score normalization across all samples using “heatmap.2” function in R (https://www.rdocumentation.org/packages/gplots/versions/3.0.1.1/topics/heatmap.2). The heatmap shows the z-score values of each gene in each sample. Gene Ontology (Ashburner et al., 2000) (GO) term enrichment analysis of differentially expressed genes was performed using the R package “GeneAnswers” (Feng, 2018). GO terms with a false discovery rate (FDR)<5% were considered as significantly enriched. P-value before the FDR correction for each functional annotated term was calculated from the hypergeometric test as follows:
Where N is the total number of genes in any GO term; n is the number of differentially expressed genes in any GO term; M is the number of genes annotated for a particular GO term; m is the number of differentially expressed genes annotated for a particular GO term. Z score of a GO term was calculated as follows:
Where count is the total regulated gene number, up and down are the number of up-regulated or down-regulated differentially expressed genes, respectively. A custom R script was used for the data visualization.
Single-cell RNA-seq on iPSC-EC and human fetal heart
iPSC-ECs from healthy control (control 1) and HLHS patient (HLHS 3) were dissociated using Accutase (Gibco) and resuspend in EGM-2 medium to maintain cell viability >90%. Human fetal heart was micro-dissected into left and right ventricular free wall, minced, and digested using Liberase (Sigma). Following digestion for 10 min, EGM-2 medium was added to stop the reaction and cells were transferred through a 100 μm cell strainer and further purified to CD144+ and CD144− populations using the MACS sorter (Miltenyi Biotec). Cells with viability>90% were sent to Stanford Functional Genomics Facility (SFGF) for 10X chromium single-cell RNA-seq paired end library preparation using V2 version (10X Genomics). All samples were uniquely indexed, mixed, and evenly distributed into the Illumina HiSeq 4000 for sequencing.
Single-cell RNA-seq data analysis
Cell ranger for data pre-processing
Chromium Single Cell Software Suite v2.1.1 (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/2.1/what-is-cell-ranger) was used for pre-processing the single cell RNA-seq data produced in the 10x Chromium Platform, which includes sample demultiplexing, read alignment, barcode processing, and UMI counting. “Cellranger mkfastq” was used to generate FASTQ files from BCL files. Specifically, the Illumina BCL output folder was used for sample demultiplexing, which is based on the 8 bp sample index read, and generating FASTQ files for the paired-end reads as well as the sample index by Illumina bcl2fastq. Only the first indexes were analyzed on the dual-indexed flowcell. Next, “cellranger count” was used to generate single-cell gene counts for a single library. Reads in the FASTQ files were mapped to the human reference genome (NCBI build38/UCSC hg38) with STAR software. Of note, for reads that aligned to a single exonic locus but also aligned to one or more non-exonic loci, only the exonic locus was prioritized. Read were considered to be confidently mapped to the exonic loci with MAPQ 255. Reads that mapped to the genome and transcriptome were delivered to a BAM file. Chromium cellular barcodes were used to generate gene-barcode matrices. Only reads that were confidently (uniquely) mapped to the transcriptome were used for the UMI count. Filtered gene-barcode matrices containing only cellular barcodes in the MEX format were used for downstream analysis. Retrieved cell numbers for each sample were listed in Table S2 and S3.
Cellular clustering
The filtered gene-barcode matrix from the cell ranger pipeline for each sample was fed into the Seurat(Butler et al., 2018) R package for downstream analyses. We set the cluster resolution as 0.8 for our 2383 iPSC-ECs according to Seurat online tutorial that “resolution parameter between 0.4-1.2 typically returns good results for single-cell datasets of around 3K cells. Increasing the cluster resolution from 0.8 to 1.8 in “FindClusters” function in the Seurat R package for cell clustering did not change endocardial cell clustering. Genes expressed in < 3 cells and cells with <500 detected genes or the percentage of mitochondrial genes >20% were removed. We used “MergeSeurat” command to combine samples. The raw read counts generated by cellranger were further normalized by a global-scaling normalization method, “LogNormalize”, that normalizes the gene expression measurements for each cell by the total read counts. The log-transformed normalized single cell expression values were used for visualizations (violin and feature plots) and differential expression tests. Variable genes with average expression in the interval [0.0125, 3], and dispersion≥0.5 were selected for Principal Component Analysis. Technical noise, batch effects and biological sources of variation such as number of UMI and the percentage of mitochondrial genes, were regressed to improve downstream dimensionality reduction and clustering. The scaled z-scored residuals were used for principal component analysis (PCA). The top 20 principal components were used for Uniform Manifold Approximation and Projection (UMAP). DEGs among clusters were detected by comparing cells in each cluster against all other cells using the Wilcoxon Rank Sum test. A gene is defined as a DEG in cluster X if it can be detected in ≥25% cells in cluster X, with adjusted p-value <5% and fold-change ≥1.5 between cell in cluster X and all other cells. Methods for pathway enrichment analysis was the same as described in the bulk RNA-Seq data analysis. In the iPSC-EC pathway enrichment, only up-regulated genes in each cell cluster were selected, therefore no z score was expected. We followed the steps described in Paik et al. (Paik et al., 2018) for cell-cell communication analysis. For each ligand-receptor pair, the mean expression of the ligand gene was calculated for all cells in the clusters classified as endocardium, and the mean expression of the receptor gene was calculated for all cells in the clusters classified as cardiomyocyte. The interaction value was estimated as the product of the two mean values: (mean expression of ligand gene in endocardial population) X (mean expression of receptor gene in myocardial population) and reflected as width of chords on circular plot using customized R package.
To generate the DNMs gene expression heatmap, we calculated their average expression values within one cluster, which were further normalized to z-scores across clusters using “heatmap.2” function in R. The heatmap shows the z-score values of each gene in each cluster.
ChIP-seq peak annotation and pathway analysis
ChIP-seq peaks were downloaded from GSE93030, ETS1 over expression induced ETS1/RNAPII binding peaks were generated by subtracting mod_GFP from mod_ETS1 in ETS1/RNAPII ChIP-Seq peaks. Bedtools (https://bedtools.readthedocs.io/en/latest/) with -A parameter (i.e. remove entire peak if any overlap) was used for peak subtraction. Peaks were annotated using homer (Heinz et al., 2010) annotatePeaks.pl script set to the default parameters.
QUANTIFICATION AND STATISTICAL ANALYSIS
First, normal distribution from each group was confirmed using χ2 test before any comparison between groups. Statistical analysis was then performed using Student’s t-test (two-sided) between two groups or ANOVA followed by Bonferroni post-test for multiple groups comparisons. If variances between two groups were significantly different (F-test), nonparametric Mann-Whitney test was applied. P<0.05 was considered as statistically significant. Statistical details can be found in the figure legends. As for all the experiments, at least three independent experiments were performed to reach the minimal requirement for statistical significance, unless otherwise specified. Blinding or randomization was not performed unless otherwise specified. Exclusion is not applied in this manuscript. Analyses were carried out using GraphPad Prism 8.0 (GraphPad Software Inc., La Jolla, CA).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD144 (VE-Cadherin) microBeads | Miltenyi Biotec | Cat#130-097-857 |
| Anti-mouse IgG microbeads | Miltenyi Biotec | Cat# 130-048-401 |
| Rabbit polyclonal anti-ETS1 | Active Motif | Cat#39580; RRID: AB_2793266 |
| Mouse monoclonal anti-RNA pol II (Clone: 4H8) | Active Motif | Cat#39097; RRID: AB_2732926; |
| Rabbit monoclonal anti-CHD7 (Clone: D3F5) | Cell Signaling Technology | Cat#6505; RRID: AB_11220431 |
| Mouse monoclonal anti-NPR3 (Clone: OTI11B5) | OriGene | Cat# TA501079; RRID: AB_11127026 |
| Rabbit polyclonal anti-α-SMA | Abcam | Cat#ab5694; RRID: AB_2223021 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Mouse monoclonal anti-TNNT2 (Clone: 13- 11) | Thermo Scientific | Cat#MS-295-P; RRID: AB_61806; |
| Rabbit polyclonal anti-TNNT2 | Abcam | Cat#ab45932; RRID: AB_956386 |
| Mouse monoclonal anti-α-actinin (Clone: EA- 53) | Sigma-Aldrich | Cat#A7811; RRID: AB_476766 |
| Mouse monoclonal anti-CDH11 (Clone: 5B2H5) | Invitrogen | Cat#321700; RRID: AB_2533068 |
| Mouse monoclonal anti-CD31 (Clone: JC/70A) | Abcam | Cat#ab9498; RRID: AB_307284 |
| Mouse monoclonal anti-FN1 (Clone: IST-9) | Abcam | Cat#ab6328; RRID: AB_305428 |
| Mouse monoclonal anti-FN1- Xenopus laevis | Developmental studies hybridoma bank | Cat#4H2S; RRID: AB_2721949; |
| Donkey anti-Mouse secondary antibody, Alexa 488 | Invitrogen | Cat#A21202; RRID: AB_141607 |
| Donkey anti-Rabbit secondary antibody, Alexa 488 | Invitrogen | Cat#A21206; RRID: AB_2535792 |
| Donkey anti-Mouse secondary antibody, Alexa 594 | Invitrogen | Cat#A21203; RRID: AB_2535789 |
| Donkey anti-Rabbit secondary antibody, Alexa 594 | Invitrogen | Cat#A21207; RRID: AB_141637 |
| Bacterial and Virus Strains | ||
| GFP lentivirus | (Chen et al., 2017) | NA |
| ETS1 lentivirus | (Chen et al., 2017) | NA |
| Biological Samples | ||
| Human fetal heart samples | University of Washington Birth Defects Research Lab, USA. | NA |
| Human heart tissue sections | Sick Children and the Mount Sinai Hospital, University of Toronto, Canada. | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EGM-2 MV BulletKit (CC-3156 and CC-4147) | Lonza | Cat# CC-3162 |
| Essential 8 Medium | Gibco | Cat# A1517001 |
| mTeSR Medium | STEMCELL | Cat# 85850 |
| B-27 Supplement, minus insulin | Gibco | Cat# A1895601 |
| B-27 Supplement | Gibco | Cat# 17504044 |
| RPMI 1640 Medium | Gibco | Cat# 11875093 |
| RPMI 1640 Medium, minus glucose | Gibco | Cat# 11879020 |
| StemPro Accutase Cell Dissociation Reagent | Gibco | Cat# A11105-01 |
| DMEM, high glucose | Gibco | Cat# 11965118 |
| HBSS, calcium, magnesium, no phenol red | Gibco | Cat# 14025092 |
| Knockout™ DMEM | Gibco | Cat# 10829-018 |
| HEPES (1M) | Gibco | Cat# 15630080 |
| TrypLE™ Select Enzyme (10X) | Gibco | Cat# A1217703 |
| KnockOut™ Serum Replacement | Gibco | Cat# 10828028 |
| UltraPure 0.5M EDTA, pH 8.0 | Invitrogen | Cat# 15575-020 |
| 1X RBC lysis buffer | Invitrogen | Cat# 433357 |
| FcR Blocking Reagent, human | Miltenyi Biotec | Cat# 130-059-901 |
| Human VEGF-165 | Gemini | Cat#300196P100UG |
| Human FGF-basic 154 | Gemini | Cat#300113P050UG |
| CHIR-99021 (CT99021) HCl | Selleck Chemicals | Cat# S2924 |
| C59 | Selleck Chemicals | Cat# S7037 |
| Matrigel™ hESC-qualified Matrix, *LDEV- Free, 5mL | Corning | Cat# 354277 |
| Matrigel™ Membrane Matrix, Growth Factor Reduced | Corning | Cat# 354230 |
| Corning® BioCoat™ Laminin Plates | Corning | Cat# 354412 |
| Gelatin solution, Type B | Sigma-Aldrich | Cat# G1393-100mL |
| Fibronectin human plasma | Sigma-Aldrich | Cat# F2006 |
| Liberase TM | Sigma-Aldrich | Cat# 5401127001 |
| Y-27632 dihydrochloride | Tocris | Cat# 1254 |
| Recombinant Human TGF-β2 | PeproTech | Cat# 100-35B |
| Antigen Unmasking Solution, Tris-Based | Vector Lab | Cat# H-3301 |
| DNase I | Worthington | Cat# LK003172 |
| Belzer UW® Cold Storage Solution | Thermo Fisher | Cat# NC0952695 |
| Corning™ BioCoat™ Collagen IV Multiwell Plates | Thermo Fisher | Cat# 08-774-29 |
| Critical Commercial Assays | ||
| Corning® Transwell® polyester membrane cell culture inserts | Sigma-Aldrich | Cat# CLS3450-24EA |
| RNeasy Plus Micro Kit | Qiagen | Cat# 74034 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | Cat# A25742 |
| High-Capacity RNA-to-cDNA™ Kit | Applied Biosystems | Cat# 4387406 |
| Deposited Data | ||
| Raw data files for scRNA-seq and bulk RNA- seq | NCBI GEO | GEO: GSE138979 |
| Experimental Models: Cell Lines | ||
| Control iPSC line 2 | CIRM Human Induced Pluripotent Stem Cell Repository | Cat# CW60359E |
| Control iPSC lines 1, 3, and 4 | Stanford University | NA |
| HLHS iPSC lines 1-3 | Mayo Clinic | NA |
| Experimental Models: Organisms/Strains | ||
| Xenopus laevis embryos | Nasco | Cat# LM00490 |
| Oligonucleotides | ||
| ON-TARGETplus Human ETS1 (2113) siRNA -SMARTpool | Dharmacon | Cat# L-003887-00-0005 |
| ON-TARGETplus Human CHD7 (55636) siRNA - SMARTpool | Dharmacon | Cat# L-025947-01-0005 |
| ON-TARGETplus Human FN1 (2335) siRNA - SMARTpool | Dharmacon | Cat# L-009853-00-0005 |
| ON-TARGETplus Human ARID1B (57492) siRNA - SMARTpool | Dharmacon | Cat# L-013970-01-0005 |
| ON-TARGETplus Human KMT2D (8085) siRNA - SMARTpool | Dharmacon | Cat# L-004828-00-0005 |
| Morpholino: MO-Ets1 5’-TAAGGTCTAGTGCAGCTTTCATGG C-3’ | This paper | NA |
| SYBR Green primers | See Table S5 | NA |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/indeex.html |
| GraphPad Prism | GraphPad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| TrimGalore v0.4.4 | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ | |
| STAR (2.5.4 a) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| FeatureCounts | (Liao et al., 2014) | http://subread.sourceforge.net/ |
| Bioconductor package DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Heatmap.2 in R package | https://www.rdocumentation.org/packages/gplots/versions/3.0.1.1/topics/heatmap.2 | |
| 10X Chromium Single Cell Software Suite v2.1.1 | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/2.1/what-is-cell-ranger | |
| scRNA-seq dimensional reduction, clustering, visualization, differential expression detection- Seurat R packages | (Butler et al., 2018) | https://satijalab.org/seurat |
| Bioconductor package GeneAnswers | (Feng, 2018) | https://www.bioconductor.org/packages/release/bioc/html/GeneAnswers.html |
| Bedtools | https://bedtools.readthedocs.io/en/latest | |
| Homer annotatePeaks.pl script | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ngs/annotation.html |
| Customized R packages | This paper | https://github.com/Lei-Tian/HLHS |
| Other | ||
| QuadroMACS™ Separator and Starting Kits (LS) | Miltenyi Biotec | Cat# 130-091-051 |
| Bioruptor® Pico sonication device | Diagenode | Cat# B01060010 |
Highlights.
Single cell RNA-seq identifies an abnormal endocardial population in HLHS.
HLHS endocardial cells show impaired endothelial to mesenchymal transition.
Endocardial defects in HLHS contribute to suppressed myocardial growth and maturation.
Reduction of FN1 underlies endocardial and myocardial dysfunctions in HLHS.
Acknowledgments
We thank Drs. Jingjing Li, Qing Liu, Michael P. Snyder and Mr. Eric Wei for DNM analysis and scRNA-seq sample preparation. We thank Ms. Julia Ryan for technical assistance with generation of iPSC-CMs. We greatly appreciate the administrative help of Ms. Michelle Fox. We also thank Drs. Mark Mercola, Kristy Red-Horse, Kyle Loh, Virginia D. Winn, and James Wells for providing intellectual consultant to the project. We also thank ReGen Theranostics, Inc Rochester, MN as the manufacturer for the iPSC lines and Dr. William Pu from Boston Children’s Hospital, Harvard Medical School who provided GFP and ETS1 overexpression lentivirus. This work was supported by the Hoffman/Schroepfer single ventricle gift fund from Stanford University, Todd and Karen Wanek Family Program for Hypoplastic Left Heart Syndrome, NIH 2R24HD000836-52 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (The University of Washington Birth Defects Research Laboratory), and AHA 20POST35210924 (LT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
References:
- Ahmed RE, Anzai T, Chanthra N, and Uosaki H (2020). A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front Cell Dev Biol 8, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Bhute VJ, Han T, Qian T, Lian X, and Palecek SP (2017). Human pluripotent stem cell-derived epicardial progenitors can differentiate to endocardial-like endothelial cells. Bioeng Transl Med 2, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boselli F, Freund JB, and Vermot J (2015). Blood flow mechanics in cardiovascular development. Cell Mol Life Sci 72, 2545–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto LD, Correa A, and Erickson JD (2001). Racial and temporal variations in the prevalence of heart defects. Pediatrics 107, E32. [DOI] [PubMed] [Google Scholar]
- Bressan M, Yang PB, Louie JD, Navetta AM, Garriock RJ, and Mikawa T (2014). Reciprocal myocardial-endocardial interactions pattern the delay in atrioventricular junction conduction. Development 141, 4149–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner ME, Hillis LD, and Lange RA (2000). Congenital heart disease in adults. First of two parts. N Engl J Med 342, 256–263. [DOI] [PubMed] [Google Scholar]
- Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, et al. (2014). Chemically defined generation of human cardiomyocytes. Nat Methods 11, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey DJ (1991). Control of growth and differentiation of vascular cells by extracellular matrix proteins. Annu Rev Physiol 53, 161–177. [DOI] [PubMed] [Google Scholar]
- Chen D, Wang X, Liang D, Gordon J, Mittal A, Manley N, Degenhardt K, and Astrof S (2015a). Fibronectin signals through integrin alpha5beta1 to regulate cardiovascular development in a cell type-specific manner. Dev Biol 407, 195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Gao X, and Shilatifard A (2015b). Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev 29, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Fu Y, Day DS, Sun Y, Wang S, Liang X, Gu F, Zhang F, Stevens SM, Zhou P, et al. (2017). VEGF amplifies transcription through ETS1 acetylation to enable angiogenesis. Nat Commun 8, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs MD, and Yutzey KE (2009). VEGF and RANKL regulation of NFATc1 in heart valve development. Circ Res 105, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsten-Janssen N, and Scambler PJ (2017). Clinical and molecular effects of CHD7 in the heart. Am J Med Genet C Semin Med Genet 175, 487–495. [DOI] [PubMed] [Google Scholar]
- Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, Hu Y, Dong J, Li Q, Wu X, et al. (2019). Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep 26, 1934–1950 e1935. [DOI] [PubMed] [Google Scholar]
- D'Amato G, Luxan G, and de la Pompa JL (2016). Notch signalling in ventricular chamber development and cardiomyopathy. FEBS J 283, 4223–4237. [DOI] [PubMed] [Google Scholar]
- de Jesus Perez VA, Yuan K, Orcholski ME, Sawada H, Zhao M, Li CG, Tojais NF, Nickel N, Rajagopalan V, Spiekerkoetter E, et al. (2012). Loss of adenomatous poliposis coli-alpha3 integrin interaction promotes endothelial apoptosis in mice and humans. Circ Res 111, 1551–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Pompa JL, and Epstein JA (2012). Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell 22, 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg LM, and Markwald RR (1995). Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res 77, 1–6. [DOI] [PubMed] [Google Scholar]
- Feng GD,P; Kibbe AW; Lin S (2018). GeneAnswers, Integrated Interpretation of Genes. Bioconductor. [Google Scholar]
- Gaber N, Gagliardi M, Patel P, Kinnear C, Zhang C, Chitayat D, Shannon P, Jaeggi E, Tabori U, Keller G, et al. (2013). Fetal reprogramming and senescence in hypoplastic left heart syndrome and in human pluripotent stem cells during cardiac differentiation. Am J Pathol 183, 720–734. [DOI] [PubMed] [Google Scholar]
- Garmon T, Wittling M, and Nie S (2018). MMP14 Regulates Cranial Neural Crest Epithelial-to-Mesenchymal Transition and Migration. Dev Dyn 247, 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Bick AG, Ito K, Homsy J, Rodriguez-Murillo L, Fromer M, Mazaika E, Vardarajan B, Italia M, Leipzig J, et al. (2014). Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ Res 115, 884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossfeld P, Nie S, Lin L, Wang L, and Anderson RH (2019). Hypoplastic Left Heart Syndrome: A New Paradigm for an Old Disease? J Cardiovasc Dev Dis 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M (2018). Efficient Differentiation of Human Pluripotent Stem Cells to Endothelial Cells. Curr Protoc Hum Genet, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Shao NY, Sa S, Li D, Termglinchan V, Ameen M, Karakikes I, Sosa G, Grubert F, Lee J, et al. (2017). Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 20, 490–504 e495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris IS, and Black BL (2010). Development of the endocardium. Pediatr Cardiol 31, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JI, and Kaplan S (2002). The incidence of congenital heart disease. J Am Coll Cardiol 39, 1890–1900. [DOI] [PubMed] [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. (2015). De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberger LK, Singhroy S, Cavalle-Garrido T, Tsang W, Keeley F, and Rabinovitch M (2000). Synthesis of extracellular matrix and adhesion through beta(1) integrins are critical for fetal ventricular myocyte proliferation. Circ Res 87, 508–515. [DOI] [PubMed] [Google Scholar]
- Hrstka SC, Li X, Nelson TJ, and Wanek Program Genetics Pipeline, G. (2017). NOTCH1-Dependent Nitric Oxide Signaling Deficiency in Hypoplastic Left Heart Syndrome Revealed Through Patient-Specific Phenotypes Detected in Bioengineered Cardiogenesis. Stem Cells 35, 1106–1119. [DOI] [PubMed] [Google Scholar]
- Hulin A, Hortells L, Gomez-Stallons MV, O'Donnell A, Chetal K, Adam M, Lancellotti P, Oury C, Potter SS, Salomonis N, et al. (2019). Maturation of heart valve cell populations during postnatal remodeling. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, and Srivastava D (2009). Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell 16, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, and Robert F (2017). The Mediator Complex: At the Nexus of RNA Polymerase II Transcription. Trends Cell Biol 27, 765–783. [DOI] [PubMed] [Google Scholar]
- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 49, 1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A, Bayrau B, Miao Y, and Gu M (2020). Isolation of Endocardial and Coronary Endothelial Cells from the Ventricular Free Wall of the Rat Heart. J Vis Exp. [DOI] [PubMed] [Google Scholar]
- Kobayashi J, Yoshida M, Tarui S, Hirata M, Nagai Y, Kasahara S, Naruse K, Ito H, Sano S, and Oh H (2014). Directed differentiation of patient-specific induced pluripotent stem cells identifies the transcriptional repression and epigenetic modification of NKX2-5, HAND1, and NOTCH1 in hypoplastic left heart syndrome. PLoS One 9, e102796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu X, Yagi H, Saeed S, Bais AS, Gabriel GC, Chen Z, Peterson KA, Li Y, Schwartz MC, Reynolds WT, et al. (2017). The complex genetics of hypoplastic left heart syndrome. Nat Genet 49, 1152–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxan G, D'Amato G, MacGrogan D, and de la Pompa JL (2016). Endocardial Notch Signaling in Cardiac Development and Disease. Circ Res 118, e1–e18. [DOI] [PubMed] [Google Scholar]
- MacGrogan D, Nus M, and de la Pompa JL (2010). Notch signaling in cardiac development and disease. Curr Top Dev Biol 92, 333–365. [DOI] [PubMed] [Google Scholar]
- Miao Y, Ajami NE, Huang TS, Lin FM, Lou CH, Wang YT, Li S, Kang J, Munkacsi H, Maurya MR, et al. (2018). Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat Commun 9, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A, Pulina M, Hou SY, and Astrof S (2013). Fibronectin and integrin alpha 5 play requisite roles in cardiac morphogenesis. Dev Biol 381, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri T, Hiriart E, van Vliet PP, Faure E, Norris RA, Farhat B, Jagla B, Lefrancois J, Sugi Y, Moore-Morris T, et al. (2019). Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat Commun 10, 1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel NP, Spiekerkoetter E, Gu M, Li CG, Li H, Kaschwich M, Diebold I, Hennigs JK, Kim KY, Miyagawa K, et al. (2015). Elafin Reverses Pulmonary Hypertension via Caveolin-1-Dependent Bone Morphogenetic Protein Signaling. Am J Respir Crit Care Med 191, 1273–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie S, and Bronner ME (2015). Dual developmental role of transcriptional regulator Ets1 in Xenopus cardiac neural crest vs. heart mesoderm. Cardiovasc Res 106, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik DT, Tian L, Lee J, Sayed N, Chen IY, Rhee S, Rhee JW, Kim Y, Wirka RC, Buikema JW, et al. (2018). Large-Scale Single-Cell RNA-Seq Reveals Molecular Signatures of Heterogeneous Populations of Human Induced Pluripotent Stem Cell-Derived Endothelial Cells. Circ Res 123, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papoutsi T, Luna-Zurita L, Prados B, Zaffran S, and de la Pompa JL (2018). Bmp2 and Notch cooperate to pattern the embryonic endocardium. Development 145. [DOI] [PubMed] [Google Scholar]
- Post A, Wang E, and Cosgriff-Hernandez E (2019). A Review of Integrin-Mediated Endothelial Cell Phenotype in the Design of Cardiovascular Devices. Ann Biomed Eng 47, 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulina MV, Hou SY, Mittal A, Julich D, Whittaker CA, Holley SA, Hynes RO, and Astrof S (2011). Essential roles of fibronectin in the development of the left-right embryonic body plan. Dev Biol 354, 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa S, Gu M, Chappell J, Shao NY, Ameen M, Elliott KA, Li D, Grubert F, Li CG, Taylor S, et al. (2017). Induced Pluripotent Stem Cell Model of Pulmonary Arterial Hypertension Reveals Novel Gene Expression and Patient Specificity. Am J Respir Crit Care Med 195, 930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JA, Jackson LF, Lee DC, and Camenisch TD (2003). Form and function of developing heart valves: coordination by extracellular matrix and growth factor signaling. J Mol Med (Berl) 81, 392–403. [DOI] [PubMed] [Google Scholar]
- Sharma A, Zhang Y, Buikema JW, Serpooshan V, Chirikian O, Kosaric N, Churko JM, Dzilic E, Shieh A, Burridge PW, et al. (2018). Stage-specific Effects of Bioactive Lipids on Human iPSC Cardiac Differentiation and Cardiomyocyte Proliferation. Sci Rep 8, 6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short SM, Talbott GA, and Juliano RL (1998). Integrin-mediated signaling events in human endothelial cells. Mol Biol Cell 9, 1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P, Simmons O, Wang J, Hoang CQ, and Conway SJ (2014). Ectopic Noggin in a Population of Nfatc1 Lineage Endocardial Progenitors Induces Embryonic Lethality. J Cardiovasc Dev Dis 1, 214–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steurer B, Janssens RC, Geverts B, Geijer ME, Wienholz F, Theil AF, Chang J, Dealy S, Pothof J, van Cappellen WA, et al. (2018). Live-cell analysis of endogenous GFP-RPB1 uncovers rapid turnover of initiating and promoter-paused RNA Polymerase II. Proc Natl Acad Sci U S A 115, E4368–E4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T, Stanley G, Sinha R, D'Amato G, Das S, Rhee S, Chang AH, Poduri A, Raftrey B, Dinh TT, et al. (2018). Single-cell analysis of early progenitor cells that build coronary arteries. Nature 559, 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoris CV, Li M, White MP, Liu L, He D, Pollard KS, Bruneau BG, and Srivastava D (2015). Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 160, 1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Pu WT, and Zhou B (2015). Cellular origin and developmental program of coronary angiogenesis. Circ Res 116, 515–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tootleman E, Malamut B, Akshoomoff N, Mattson SN, Hoffman HM, Jones MC, Printz B, Shiryaev SA, and Grossfeld P (2019). Partial Jacobsen syndrome phenotype in a patient with a de novo frameshift mutation in the ETS1 transcription factor. Cold Spring Harb Mol Case Stud 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Roest MJ, and Merryman WD (2017). A developmental approach to induced pluripotent stem cells-based tissue engineered heart valves. Future Cardiol 13, 1–4. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wu B, Chamberlain AA, Lui W, Koirala P, Susztak K, Klein D, Taylor V, and Zhou B (2013). Endocardial to myocardial notch-wnt-bmp axis regulates early heart valve development. PLoS One 8, e60244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Hu H, Isaji T, and Dardik A (2019). Molecular identity of arteries, veins, and lymphatics. J Vasc Surg 69, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Xu Y, Yu M, Lee D, Alharti S, Hellen N, Ahmad Shaik N, Banaganapalli B, Sheikh Ali Mohamoud H, Elango R, et al. (2017). Induced pluripotent stem cell modelling of HLHS underlines the contribution of dysfunctional NOTCH signalling to impaired cardiogenesis. Hum Mol Genet 26, 3031–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Coldren C, Liang X, Mattina T, Goldmuntz E, Benson DW, Ivy D, Perryman MB, Garrett-Sinha LA, and Grossfeld P (2010). Deletion of ETS-1, a gene in the Jacobsen syndrome critical region, causes ventricular septal defects and abnormal ventricular morphology in mice. Hum Mol Genet 19, 648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. (2013). De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Huang X, Liu K, Tang J, He L, Pu W, Liu Q, Li Y, Tian X, Wang Y, et al. (2017). Fibroblasts in an endocardial fibroelastosis disease model mainly originate from mesenchymal derivatives of epicardium. Cell Res 27, 1157–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pu W, Li G, Huang X, He L, Tian X, Liu Q, Zhang L, Wu SM, Sucov HM, et al. (2016). Endocardium Minimally Contributes to Coronary Endothelium in the Embryonic Ventricular Free Walls. Circ Res 118, 1880–1893. [DOI] [PubMed] [Google Scholar]
- Zhao L, Ben-Yair R, Burns CE, and Burns CG (2019). Endocardial Notch Signaling Promotes Cardiomyocyte Proliferation in the Regenerating Zebrafish Heart through Wnt Pathway Antagonism. Cell Rep 26, 546–554 e545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data have been deposited into GEO database under GEO accession number GSE138979 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138979).
Data visualization could be accessed through the Broad Institute Single Cell Portal: Heart tissue: https://singlecell.broadinstitute.org/single_cell/study/SCP1020 iPSC-EC: https://singlecell.broadinstitute.org/single_cell/study/SCP1021 All R codes have been uploaded to GitHub (https://github.com/Lei-Tian/HLHS).