

Abstract
Clinical studies implicated an increased risk of intestinal fibrosis in patients with nonalcoholic fatty liver disease (NAFLD). Our previous studies have shown that microcystin-LR (MC-LR) exposure led to altered gut microbiome and increased abundance of lactate producing bacteria and intestinal inflammation in underlying NAFLD. This led us to further investigate the effects of the MC-LR, a PP2A inhibitor in activating the TGF-β fibrotic pathway in the intestines that might be mediated by increased lactate induced redox enzyme NOX2. Exposure to microcystin led to higher lactate levels in circulation and in the intestinal content. The higher lactate levels were associated with NOX2 activation in vivo that led to increased Smad2/3-Smad4 co-localization and high alpha-smooth muscle actin (α-SMA) immunoreactivity in the intestines. Mechanistically, primary mouse intestinal epithelial cells treated with lactate and MC-LR separately led to higher NOX2 activation, phosphorylation of TGFβR1 receptor and subsequent Smad 2/3-Smad4 co-localization inhibitable by apocynin (NOX2 inhibitor), FBA (a peroxynitrite scavenger) and DMPO (a nitrone spin trap), catalase and superoxide dismutase. Inhibition of NOX2-induced redox signaling also showed a significant decrease in collagen protein thus suggesting a strong redox signaling induced activation of an ectopic fibrotic manifestation in the intestines. In conclusion, the present study provides mechanistic insight into the role of microcystin in dysbiosis-linked lactate production and subsequently advances our knowledge in lactate-induced NOX2 exacerbation of the cell differentiation and fibrosis in the NAFLD intestines.
Keywords: MC-LR, PP2A inhibitor, lactate, dysbiosis, peroxynitrite, NOX2, fibrosis, NAFLD
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a condition of excessive accumulation of fat (steatosis) in the liver cells. It is considered a global pandemic that affects 70% of the obese population of the United States, according to the Centre for Disease Control and Prevention. We have shown that microcystin co-exposure in mice advances fatty liver disease. Previous studies have described NAFLD contributes to several ectopic disorders including intestinal inflammation. Emerging data have highlighted the co-existence of NAFLD with inflammatory bowel disease (IBD) (Chao et al., 2016). Previously, we also showed a strong association of NAFLD with colonic inflammation and IBD (Chandrashekaran et al., 2017). Environmental and genetic factors have been shown to be associated with progression of nonalcoholic fatty liver disease (Pappachan et al., 2017).We reported previously that several factors including environmental toxins induce pathology of NAFLD and intestinal complications in the murine model and most importantly found that redox stress as one of the possible mechanisms of intestinal and renal co-morbidities in NAFLD (Alhasson et al., 2015; Chandrashekaran et al., 2017). Importantly, there are several reports of harmful algal blooms present in the fresh and brackish waters, which can release a variety of cyanotoxins that possess a serious threat to humans, as well as other organisms (Funari & Testai, 2008). Microcystin-LR (MC-LR) is a toxin produced mainly by the cyanobacterium Microcystis aeruginosa and some other freshwater cyanobacteria. MC-LR is highly toxic with relatively low doses required for lethal effects (Rao et al., 2002). Being a potential hepatotoxin, MC-LR not only affects the liver but it is also known to affect various other mammalian organs like heart, kidney, and intestines (McLellan & Manderville, 2017). Several studies have reported that MC-LR alters the intestinal gut microbiome, which plays an important role in gut homeostasis (Adamovsky et al., 2018; Chen et al., 2015). Exposure of MC-LR to mice results in accumulation of the cyanotoxin in the small intestinal villi regions and is correlated with increased apoptosis, loss of epithelial cells and decreased intraepithelial lymphocyte count (Botha et al., 2004; Ito et al., 2000; Sedan et al., 2015). In a recently published work, we showed that MC-LR exposure caused an alteration of the gut microbiome, with an abundance of phylum Firmicutes, Proteobacteria and subsequently a decrease in the abundance of phylum Bacteroidetes, compared to the control groups. Similar trends were observed in different microbes in the order, class, family and genus levels. Most importantly, MC-LR exposure in murine NAFLD causes an abundance of lactate producing bacteria like Enterococcus and Lactobacillus which led us to believe in the possible cause of gastrointestinal disturbances in NAFLD. Upon exposure, MC-LR is taken up by the intestinal epithelia and it disrupts the cellular functions by deactivating and inhibiting different enzymes. Studies have reported that MC-LR specifically inhibited protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) that lead to tumorigenic phenotype (Campos & Vasconcelos, 2010; Honkanen et al., 1990; Yoshizawa et al., 1990). MC-LR interacts with the catalytic subunit of the PP2A enzyme and leads to the uncontrolled inhibition of the protein phosphatases (Maynes et al., 2006). The interaction of MC-LR and PP2A follows a two-step mechanism, in which the catalytic subunit of the enzyme PP2A is first inactivated and then it forms covalent bonds between its side chain and amino acid residues (Xing et al., 2006). Interaction of MC-LR and PP2A prevents the enzymatic activity of PP2A, which acts as a potent tumor suppressor (Xing et al., 2006). The PP2A enzyme plays an important role in regulating the transforming growth factor β (TGF-β) fibrotic pathway (Jiang et al., 2014). Being a phosphatase, it dephosphorylates and thereby deactivates the activin like kinase-5 (ALK5/TGF-β receptor 1) to inhibit the downstream signaling pathway (T. Liu & Feng, 2010). In this scenario, MC-LR covalently interacts with protein phosphatase and allowing the kinases to phosphorylate and activate TGF-βR1 for the onset of the TGF-β signaling pathway. The following effects of MC-LR as a PP2A inhibitor are exacerbated in an underlying chronic inflammatory condition like NAFLD. In this context, it becomes increasingly important to study the role of the PP2A inhibitor, (MC-LR) in inducing pathogenesis in the gut in the underlying conditions of NAFLD.
The current study investigates the role of the microcystin co-exposure, a PP2A inhibitor (PP2Ainh) in the onset of fibrotic disease in the murine intestine under an inflammatory phenotype. We hypothesized that MC-LR exposure led to an increase in the abundance of several pathogens and lactate-producing bacteria, which in turn increased the serum lactate levels in the treated mice under the conditions of NAFLD as compared to the control mice. Over-production of lactate was closely associated with the activation of NADPH oxidase 2 (NOX 2) and the release of reactive oxygen species (peroxynitrite) which activated the TGF-β signaling pathway. The enzyme PP2A is known to inhibit the progression of the TGF-β-Smad2/3-Smad4 fibrotic pathway. MC-LR, a PP2A inhibitor, led to the receptor activation of TGF-βR1 by its phosphorylation, which activated the downstream Smad2/3- Smad4 pathway, leading to a fibrotic pathology in the intestine. The current study used a murine model of NAFLD and in vitro studies with mouse primary intestinal epithelial cells to elucidate the mechanisms.
MATERIALS AND METHODS
Materials
Specific PP2A inhibitor (Microcystin, MC-LR) was purchased from Cayman Chemical Company (Ann Arbor, Michigan) and leptin was purchased from BioVision (Milpitas, CA). Anti-TGF-β, anti-collagen I, anti-alpha smooth muscle actin (α-SMA), anti-fibronectin, anti-3 nitrotyrosine primary antibodies were purchased from Abcam (Cambridge, MA). Anti-Smad2/3, anti-Smad 4, anti-vimentin, anti-gp91phox, and anti-p47phox primary antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-TGF-βR1, anti-phospho TGF-βR1 primary antibodies were purchased from Thermofisher Scientific (Waltham, MA). Apocynin, phenylboronic acid, 5,5-Dimethyl-1-pyrroline N-oxide (DMPO), catalase and superoxide dismutase were purchased from Sigma Aldrich. Wild-type and gene-specific knockout (KO) mice including p47phox knock out mice were purchased from The Jackson Laboratories (Bar Harbor, ME). Animal diets were purchased from Research Diets (New Brunswick, NJ). Species-specific biotinylated conjugated secondary antibodies and streptavidin-horse radish peroxidase (Vectastain Elite ABC kit) were purchased from Vector Laboratories (Burlingame, CA). Fluorescence-conjugated (Alexa Fluor) secondary antibodies, ProLong Gold antifade mounting media with DAPI were purchased from Thermofisher Scientific (Grand Island, NY). All other chemicals used in this project were of analytical grade and purchased from Sigma only if otherwise specified. Paraffin-embedding of tissue sections on slides were done by AML laboratories (Baltimore, MD).
Mouse model
Experimental Models Used
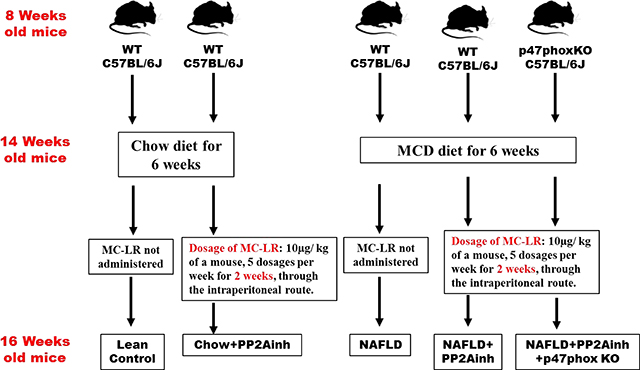
Pathogen-free, adult (8 weeks old), male C57BL/6J wild-type (WT) mice and similar background, p47phox gene deleted (p47phox KO- B6 (Cg)-Ncflm1J/J) mice (Jackson Laboratories, Ban Harbor, ME) were used in the study. The groups used for the experiment were the wild type mice fed with chow diet only (Lean Control), wild type mice fed with methionine choline-deficient and high-fat diet (MCD-HFD) only (NAFLD), wild type mice fed with chow diet and then exposed to the PP2A inhibitor (Chow+ PP2Ainh.), wild type mice fed with methionine choline-deficient diet and then exposed to PP2A inhibitor (NAFLD+ PP2Ainh.), and another group of p47phox KO mice fed with methionine choline-deficient diet and then exposed to PP2A inhibitor (NAFLD+PP2Ainh+p47phox KO). The total number of animals in each group were assessed based on the calculations that ensured enough statistical power of 0.5. There were 6 mice per group that were allocated to their respective cages following the procedure of randomization. A total of 30 mice were used in the experiments for the paper. All mice experiments were carried out based on the approval of the institutional animal ethics committee (IACUC) at the University of South Carolina.
a. Diet-induced NAFLD model
Pathogen-free, male, C57BL/6J (WT) and the p47phox gene deleted mice (p47phoxKO) were fed with MCD-HFD diet (Research diets, New Brunswick, NJ) from 8 weeks to 14weeks for diet-induced NAFLD model until the experimental dosing was over and were euthanized soon after. The mice weighed about 25 grams during the time of euthanizing. All mice had ad libitum access to food and water and were housed in a temperature-controlled room at 23°C −24°C with a 12-hour light/dark cycle. All animals were treated in strict accordance with the NIH Guide for the Humane Care and Use of Laboratory Animals and local IACUC standards.
b. Exposure of NAFLD mice to environmental toxin Microcystin
WT control and gene-specific knockout mice (p47phox KO) were fed with the MCD-HFD diet for 6 weeks and then administered with MC-LR, a PP2A inhibitor (10μg per kg of a mouse, 5 dosages per week), through the intraperitoneal route for two weeks. The dosage was given each afternoon at the same time to eliminate any bias in the study. Since this is a sub-chronic study, the cumulative effect of the PP2A inhibitor on inflammatory phenotypes was considered. The mice were euthanized after the completion of the dosage. Blood serum and small intestine tissue were collected for further processing. The dosage of MC-LR was optimized to 10μg/ kg of the bodyweight of a mouse for 14 days. The time, dosage and route of MC-LR administration were decided according to previous studies and background literature. Heinze et al. demonstrated that the lowest observable effect (LOAEL) in terms of the limitation of the study to be 50 μg/kg/day for MC-LR toxicity. Also, Li et al. demonstrated that 5, 10 and 15μg dosage concentration of MC-LR led to toxicity in the reproductive system of the male rats (Heinze, 1999). Hence the concentration of 10μg/ kg of the body weight/day was selected to be the most favorable dose for MC-LR toxicity in our mice model. A schematic representation of the in vivo experiments is given below:
Intestinal epithelial cell culture and treatments
Primary mouse intestinal epithelial cell line was grown and maintained in complete Dulbecco’s Modified Eagle Medium (DMEM) media containing high glucose, Insulin-Transferrin-Selenium (ITS), epidermal growth factor (EGF) and 2% fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 5% CO2. After overnight serum starvation (0.2% FBS) the cells were then treated with vehicle (Control), mouse leptin (100 ng/mL), lactate (15 ng/μl), apocynin (100 μM), DMPO (100 μM) and phenylboronic acid (100 μM), catalase (50 μg/ml) and superoxide dismutase (100 μg/ml) separately or in combinations for 24 hours. The lactate concentration was determined from the serum samples of mice. Upon completion of treatment, cells and supernatant were processed for western blot and immunofluorescence imaging. All the in vitro experiments were performed three times. The various lactate-treated groups are mentioned in the table below:
| Primary mouse intestinal epithelial cells | Treated with | Group name |
|---|---|---|
| Cells | Vehicle | Control |
| Cells | Mouse leptin (100 ng/mL) | LEP |
| Cells | Lactate (15 ng/μl) | LAC |
| Cells | Mouse leptin+ Lactate | LEP+LAC |
| Cells | Mouse leptin+ lactate+ apocynin (100 μM) | LEP+LAC+APO |
| Cells | Mouse leptin+ Lactate+ DMPO (100 μM) | LEP+LAC+DMPO |
| Cells | Mouse leptin+ Lactate+ phenylboronic acid (100 μM) | LEP+LAC+FBA |
| Cells | Mouse leptin+ Lactate+ catalase (50 μg/ml) | LEP+LAC+CATALASE |
| Cells | Mouse leptin+ Lactate+ superoxide dismutase (100 μg/ml) | LEP+LAC+SOD |
Another group of cells was treated with the MC-LR to determine its effects on the downstream pathway. The treatment was done for 24 hours and after completion of treatment, cells and supernatant were processed for western blot and immunofluorescence imaging. The PP2A inhibitor-treated groups are listed below in the table.
| Primary mouse intestinal epithelial cells | Treated with | Group name |
|---|---|---|
| Cells | Vehicle | Control |
| Cells | Mouse leptin (100 ng/mL) | LEP |
| Cells | PP2A inhibitor (20μM) | PP2Ainh |
| Cells | Mouse leptin+ PP2Ainh | LEP+ PP2Ainh |
| Cells | Mouse leptin+ lactate+ apocynin (100 μM) | LEP+ PP2Ainh +APO |
| Cells | Mouse leptin+ PP2Ainh + DMPO (100 μM) | LEP+ PP2Ainh +DMPO |
| Cells | Mouse leptin+ PP2Ainh + phenylboronic acid (100 μM) | LEP+ PP2Ainh +FBA |
| Cells | Mouse leptin+ PP2Ainh + catalase (50 μg/ml) | LEP+ PP2Ainh +CATALASE |
| Cells | Mouse leptin+ PP2Ainh + superoxide dismutase (100 μg/ml) | LEP+ PP2Ainh +SOD |
Laboratory analysis Picrosirius Red staining
Sections of small intestine tissue from each mouse were collected and fixed in 10% neutral buffered formalin. These formalin-fixed, paraffin-embedded tissues were cut in 5 μm thick sections. Picrosirius red staining of formalin-fixed, paraffin-embedded small intestine tissue was done using NovaUltra™ Picro-Sirius Red Stain Kit following manufacturer’s instructions (IHC-World). Liver sections were observed under a 10X objective of a light microscope. Morphometric analysis of the stained regions of the tissue sections was performed using cellSens software (Olympus). The small intestine fibrosis was defined, for a small intestine section, as the ratio of the area of stained collagen fibers versus the total area of the small intestine section, expressed in percent.
Immunohistochemistry
Formalin-fixed, paraffin-embedded small intestine tissue sections were deparaffinized using standard laboratory protocol. Epitope retrieval solution and steamer (IHC-World, Woodstock, MD, USA) were used for antigen epitope retrieval of the tissue sections. 3% H2O2 solution was used to block the endogenous peroxidase activity for 5 minutes, followed by serum blocking (5% goat serum, 1hr). The primary antibody for α-SMA (Abcam, Cambridge, MA), TGF-β, vimentin (Santa Cruz Biotechnology, Dallas, TX) were used overnight in recommended dilutions (1:300). Species-specific biotinylated conjugated secondary antibody and streptavidin-conjugated with HRP were used to perform antigen-specific immunohistochemistry according to the manufacturer’s standard protocols. 3, 3’ Diaminobenzidine (DAB) (Sigma-Aldrich, St. Louis, and MA) was used as a chromogenic substrate. Tissue sections were counter-stained with Mayer’s hematoxylin (Sigma- Aldrich). Tissue sections were washed with 1X PBS-T (Phosphate buffered saline+ 0.05% Tween 20) between the steps. Sections were finally mounted in Aqua mount (Fisher Scientific) and observed under a 20X objective using an Olympus BX43 microscope (Olympus, America). Morphometric analysis was done using CellSens Software from Olympus America (Center Valley, PA).
Immunofluorescence staining and microscopy
In vivo, formalin-fixed, paraffin-embedded small intestine tissue sections were subjected to deparaffinization according to standard instructions. Epitope retrieval of the deparaffinized sections was done with an epitope retrieval solution and steamer (IHC World) according to the manufacturer’s protocol. The primary antibodies anti-gp91phox, anti-p47phox, anti-smad2/3, anti-smad4, anti-3-nitrotyrosine, anti-collagen I, and anti-fibronectin were used at recommended dilutions (1:300). Species-specific anti-IgG secondary antibodies conjugated with Alexa Fluor 488 or 633 (Invitrogen) were used. The sections were mounted in a ProLong Gold antifade reagent with DAPI (Life Technologies, Carlsbad, CA). Images were taken under 20X, 40X and 60X magnification with Olympus BX43 and BX63 microscope. In vitro, after completion of the treatments under serum-starved conditions as in cell culture, cells attached to coverslips were fixed with 10% neutral buffered saline. After the cells were washed with PBS containing 0.1% Triton X (Sigma), they were blocked with 3% BSA, 0.2% Tween (Fisher), 10% FBS in PBS. Cells were incubated with primary antibodies anti-gp-91-phox, anti-p47phox, anti-Smad2/3, anti-Smad4, anti-3-nitrotyrosine followed by species-specific Alexa Fluor 633 or 488. The stained cells attached to the coverslips were mounted on slides with ProLong Gold antifade reagent with DAPI (Life Technologies) and viewed under 40X and 60X magnifications with Olympus BX43 and BX63 microscope.
Morphometric analysis
The morphometric analysis was done in five different microscopic fields by using the Cell Sens Software and the dataset was generated by the software. The co-localization events were calculated by the relative yellow area of immunofluorescence in the respective fields. Morphometry for the separate red and green channels was carried out by using the above-mentioned CellSens software which generated an unbiased dataset based on the relative area of immunofluorescence.
Western blot analysis
Protein lysates from respective small intestine tissues and cells were extracted using RIPA lysis buffer supplemented with protease and phosphatase inhibitor cocktail and subsequently quantified by the BCA assay kit (Thermo Fisher Scientific, Rockford, IL). Approximately, 30 μg of denatured protein was loaded on per well of Novex 4%–12% bis-tris gradient gel and subjected to standard SDS-PAGE. Resolved protein bands were then transferred to a nitrocellulose membrane using the Trans-Blot Turbo transfer system (Bio-rad, Hercules, CA). After Ponceau S staining, the membrane was blocked with 5% bovine serum albumin (BSA) for 1 hour, followed by incubation with primary antibody overnight at 4°C. Primary antibodies of anti-TGF-βR1, anti-phospho-TGF-βR1 (ThermoFisher Scientific), anti-α-SMA (Abcam), anti-β-actin (Santa Cruz Biotechnology) were used at recommended dilutions, and compatible horseradish peroxidase-conjugated secondary antibodies were used. Pierce ECL Western Blotting substrate (Thermo Fisher Scientific, Rockford, IL) was used for detection. The blot was imaged using G: BoxChemi XX6 (Syngene imaging systems) and subjected to densitometry analysis using Image J. software.
Lactate assay
Serum lactate was measured in the blood serum of the murine models using the Lactate Assay kit from Sigma Aldrich using the manufacturer’s protocol. Serum was collected from the samples of lean mouse control (Lean Control), chow diet-fed mice treated with MC-LR referred in the figures as PP2Ainh (Chow+ PP2Ainh), the high-fat diet fed mice (NAFLD), and high-fat diet fed mice treated with PP2A inhibitor (NAFLD+ PP2Ainh). The experiment was repeated thrice with triplicates of each sample. Lactate concentration was also measured from fecal pellets from the samples Lean Control, (Chow+ PP2Ainh), NAFLD and (NAFLD+ PP2Ainh) groups, by homogenizing the samples with sterile PBS. The pellet samples were spun down at 4500 rpm for 15 minutes at 4°C, the supernatant was collected and filtered through a 0.45μm syringe filter. The purified supernatant was then used for the lactate assay. The experiment was repeated thrice with triplicates of each sample.
Proximity ligation assay
The small intestine sections were deparaffinized using a standard protocol used in immunofluorescence and immunohistochemistry procedures. Epitope retrieval of the deparaffinized sections was done with an epitope retrieval solution and steamer (IHC World) according to the manufacturer’s protocol. The tissues were then blocked for 60 minutes at 37°C by DuoLink blocking solution (Sigma Aldrich). The primary antibodies were then diluted (1:250) using the provided DuoLink antibody diluent (Sigma Aldrich) and incubated for overnight at 4°C. The tissues were then washed using Wash Buffer A (Sigma Aldrich) as recommended and were incubated for 60 minutes at 37°C with the PLA PLUS and MINUS probes (1:5 dilution) (Sigma Aldrich), according to the manufacturer’s protocol. After subsequent washes, the Ligase was diluted (1:40) in DuoLink Ligation Buffer (Sigma Aldrich) and incubated for 30 minutes at 37°C. The polymerase diluted (1:80) in the DuoLink Amplification Buffer (Sigma Aldrich) was added to the tissues and incubated for 100 minutes at 37°C. The tissues were then washed with Wash Buffer B according to the manufacturer’s protocol and mounted with ProLong Gold antifade reagent DAPI (Life Technologies, Carlsbad, CA). The tissues were viewed under 40X magnification with an Olympus BX43 microscope.
Statistical analyses
All in vivo experiments were repeated three times with at least six mice per group (n=6) data from each group of six mice were pooled). The statistical analysis was carried out by unpaired t-test and analysis of variance (ANOVA) for assessing the difference between multiple groups. For all analyses p < 0.05 was considered statistically significant. For experiments involving 2 groups where the distribution of data was not clearly parametric, Mann-Whitney U tests were performed with GraphPad Prism Software Inc, CA, and Version 5.03. For experiments involving 3 or more groups, data were evaluated using one-way ANOVA with multiple comparison Bonferroni Dunn post hoc analysis. Data are expressed as mean ± SEM, or as an absolute number or percentage for categorical variables. The significance level was set at α = 5% for all comparisons. Correlation analysis of serum and fecal lactate levels with NOX2 activation was carried out by estimating the R-value following individual data points from 6 samples.
RESULTS
Pathology of the murine small intestine following exposure to microcystin (referred to hereafter as PP2A inhibitor) under the conditions of NAFLD
In our study, we observed that specific inhibition by MC-LR (PP2A inhibitor, PP2Ainh) led to a fibrotic pathology in the murine small intestine. Small intestine tissues were analyzed for fibrosis by Picro Sirius red staining. Increased collagen fiber deposition in the (NAFLD+PP2Ainh) group of mice was observed compared to the NAFLD mice (*p <0.05) (Fig. 1A (i–iv), C). A slight increase in the collagen fiber deposition in the (Chow+PP2Ainh) group compared to the Lean Control was also observed (Fig. 1A (i–iv), C). Several studies have revealed that the expression of vimentin is required for epithelial to mesenchymal transition (Wang et al., 2018). It is also known to play an important role in the development of fibrosis by stabilizing the collagen mRNA (Challa & Stefanovic, 2011). Our results showed a significant increase in the vimentin expression in the (NAFLD+PP2Ainh) group compared to the NAFLD mice control (*p <0.05) (Fig. 1B (i–iv), D). A similar marked increase of vimentin expression was observed in the (Chow+PP2Ainh) group compared to the Lean Control group (Fig. 1B (i–iv), D), indicating a condition for the development of fibrosis in the small intestine. Our results showed a significant increase in the serum lactate levels in both Chow and NAFLD mice exposed to the PP2A inhibitor as compared to the Lean Control and NAFLD groups respectively (0.05<**p < 0.01) (Fig.1E). Also, a significant increase of the lactate concentration determined from the fecal pellets in (Chow+PP2Ainh) and (NAFLD+ PP2Ainh) mice groups were observed as compared to the Lean Control and the NAFLD only group of mice (#p< 0.01) (Fig. 1F). The results of a significant increase in lactate levels in both serum and fecal pellet were consistent with our recently published study that showed a higher abundance of lactate producing bacteria following PP2A inhibition by MC-LR (Sarkar et al., 2019).
Fig. 1.

Formalin-fixed, paraffin-embedded 5 μm intestinal slices from Lean control, wild type mouse control fed with MCD-HFD (NAFLD), wild type mouse group fed with chow diet exposed to PP2A inhibitor (Chow+PP2Ainh) and another NAFLD group of mice exposed with PP2A inhibitor (NAFLD+ PP2Ainh) were used for Picrosirus Red imaging. (A)(i-iv) Representative Picrosirius Red stained (PSR) images of intestinal sections showed collagen fiber deposition in the small intestine. Images were taken at 20X magnification. (B)(i-iv) Representative Immunohistochemistry images depicting Vimentin immunoreactivity in the small intestine of the mice groups. Images were taken at 20X magnification. (C) Automatic digital imaging quantification of % fibrosis area (*p < 0.05). (D) Morphometric analysis of Vimentin immunoreactivity (*p < 0.05). (E) Measurement of serum lactate concentration (ng/μL) in the Lean Control, (Chow+PP2Ainh), (NAFLD) and (NAFLD+ PP2Ainh) groups. (F) Measurement of lactate concentration (ng/μL) from the fecal pellets in the Lean Control, (Chow+PP2Ainh), (NAFLD) and (NAFLD+ PP2Ainh) groups. Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
Exposure to MC-LR alters the redox status of the murine intestine
Previously, we reported that alteration of the gut microbiome induced activation of the NADPH oxidase 2 (NOX2) which led to increased oxidative stress in the intestine (Sarkar et al., 2019). NOX2 is activated when the cytosolic p47phox subunit associates with its membrane-bound counterpart gp91phox subunit. We used immunofluorescence staining to study the membrane association of the two subunits in mice small intestine. The results were analyzed based on the number of co-localization events (yellow) (Fig 2A (i–iv), C). We observed that the (NAFLD+PP2Ainh) and (Chow+PP2Ainh) groups had a significant increase in the co-localization events when compared to the NAFLD only group and the Lean Control group respectively (**0.05< p< 0.01) (Fig 2A (i–iv), C). A proximity ligation assay was performed to further confirm the activation of NOX2 followed by the inhibition of PP2A. The proximity ligation assay is a method that extends the efficiency of traditional immunoassays in the detection of protein interactions, modifications, and specificity (Bagchi et al., 2015). Interactions between the gp91phox and p47phox proteins due to close proximity were shown by red spots that were detected by immunofluorescence microscopy. Results showed a significant increase in the red spots in (NAFLD+PP2Ainh) group when compared to the NAFLD only group (#p <0.01) (Fig 2B (i–iv), D), thereby confirming the activation of NOX2 on exposure to the PP2A inhibitor. Fecal lactate levels derived from intestinal content and serum lactate level were correlated with co-localization events (gp91phox and p47phox) with signifying NOX2 activation. Results showed that both fecal and serum lactate levels strongly correlated with increased NOX2 activation in vivo (Fig. 2E and 2F).
Fig. 2.

Formalin-fixed, paraffin-embedded 5 μm intestinal slices from Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh) groups were used for immunofluorescence imaging. (A) (i-iv) Immunofluorescence images depicting gp91phox (red) and p47phox (green) co-localization events in the small intestine, counterstained with DAPI (blue) of Lean Control, (Chow +PP2Ainh), NAFLD and (NAFLD+PP2Ainh) mice groups. Images were taken at 40X magnification, (B)(i-iv) Immunofluorescence images depicting the proximity ligation (red) of gp91phox and p47phox proteins in the small intestine, counterstained with DAPI (blue) of Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh) mice groups. Images were taken at 40X magnification. (C) Morphometric analysis of gp91phox- p47phox co-localization events. Y-axis shows % positive immunoreactive area (% ROI) (n=3, analysis from three separate microscopic fields) (0.05<**p < 0.01). (D) Morphometric analysis of gp91phox and p47phox proximity ligation. Y-axis shows % positive immunoreactive area (%ROI) (n=3, analysis from three separate microscopic fields) (#p < 0.01). (E and F) Correlation analysis of fecal and serum lactate with NOX2 activation. Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
Activation of NADPH Oxidase 2 regulates the TGF-β receptor activation following MC-LR exposure
TGF-β has been shown to play an important role in the activation of the fibrotic pathway by various research groups (Biernacka et al., 2011; Branton & Kopp, 1999; Meng et al., 2016; Nakerakanti, 2012). To determine the role of NOX2 in inducing the TGF-β expression following PP2A inhibitor exposure, p47phox gene deleted (p47phox knockout mice) mice group was used. Immunohistochemistry was performed to study the TGF-β occurrence in the murine intestine induced by the increased oxidative stress. Results showed an increased, although non-significant immunoreactivity of TGF-β in (NAFLD+PP2Ainh) group compared to the NAFLD only group (Fig 3A (i–iv), B). A significant increase in TGF-β immunoreactivity was observed in the (Chow+PP2Ainh) group when compared to the Lean Control group (#p <0.01) (Fig 3A (i–ii), B). Also, a decrease in TGF-β immunoreactivity was observed in the (NAFLD+PP2Ainh+p47phox KO) mice group (#p <0.01) (Fig 3A (v), B) when compared to (NAFLD+PP2Ainh) mice group, suggesting the role of NOX2 in the activating the TGF-β signaling pathway. The TGF-β molecule binds to the TGF-βRII receptor complex and activates the activin receptor-like kinase 5 (ALK5) by phosphorylating the TGF-βR1 receptor (T. Liu & Feng, 2010). We performed western blot to analyze the protein expression of phospho-TGF-βR1 and total-TGF-βR1 in small intestine tissue lysates. Our results showed that there was a significant increase in phospho-TGF-βR1 levels in the (NAFLD+PP2Ainh) group as compared to the NAFLD only group (#p< 0.01) (Fig. 3C, D). Although, we observed an increase in phospho-TGF-βR1 level in the (Chow+PP2Ainh) group when compared to the Lean Control group, it was non-significant. A significant decrease was observed in the (NAFLD+PP2Ainh+p47phox KO) group of mice compared to the (NAFLD+PP2Ainh) group, suggesting a crucial role of NOX2 in activating the downstream TGF-β signaling pathway (#p< 0.01) (Fig. 3C, D). The fold change was observed by quantifying the protein bands of phospho-TGF-βR1 with respect to the total-TGF-βR1 (#p< 0.01) (Fig. 3C, D).
Fig. 3.

Formalin-fixed, paraffin-embedded 5 μm intestinal slices from Lean Control, wild type mouse group fed with chow diet exposed to PP2A inhibitor (Chow+PP2Ainh), wild type mouse control fed with MCD-HFD (NAFLD), a NAFLD group of mice exposed with PP2A inhibitor (NAFLD+PP2Ainh) and another NAFLD+PP2Ainh group with p47phox gene knock out (NAFLD+PP2Ainh+p47phox KO) were used for immunohistochemistry imaging. (A)(i-v) Immunohistochemistry images depicting TGF-β immunoreactivity in the small intestine of Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+ PP2Ainh) and (NAFLD+PP2Ainh+p47phox KO) groups. Images were taken at 20X magnification. (B) Morphometric analysis depicting TGF-β immunoreactivity. Y-axis shows % positive immunoreactive area (% ROI) (n=3, analysis from three separate microscopic fields) (#p< 0.01, ns: non-significant)). (C) Western blot analysis of phospho-TGF-βR1 and total-TGF-βR1 protein levels in the small intestine tissue lysate. Lanes 1–5 represent Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh) and (NAFLD+PP2Ainh+p47phox KO) groups, respectively. (D) Band quantification of phospho-TGF-βR1 immunoblot in Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh) and (NAFLD+PP2Ainh+p47phox KO) groups of mice, data were normalized against total- TGF-βR1 (#p< 0.01, ns: non-significant). Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
NADPH oxidase 2 mediated TGF-βR1 activation regulates the translocation of Smad2/3-Smad4 assembly in the nucleus
Activated TGF-β signaling pathway induces the association of downstream Smad2/3-Smad4 complex and inhibition of Smad7 leading to collagen deposition (Verrecchia & Mauviel, 2007). The activated TGF-βR1 phosphorylates the Smad2/3 (Ray et al., 2010), which then binds with Smad4 and translocate to the nucleus leading to the TGF-β signal transduction (Dattaroy et al., 2015). Mouse small intestine tissues were used to study the immunoreactivity of Smad2/3 and Smad4, and the co-localization events were studied using immunofluorescence microscopy. Our results showed that the nuclear co-localization events were significantly higher in the (NAFLD+PP2Ainh) group compared to the NAFLD only group (*p <0.05) (Fig. 4A(i–iv), C), and also a similar significant increase was observed in the (Chow+PP2Ainh) group compared to the Lean Control group (#p <0.01) (Fig. 4A (i–iv), C). Proximity ligation assay was used to confirm the Smad2/3 and Smad4 protein interactions in the nucleus due to proximity, and it was visible as red spots when detected by immunofluorescence microscopy. Results showed a significant increase in the red spots in the (NAFLD+PP2Ainh) group compared to the NAFLD only group (*p <0.05), and a marked increase in the (Chow+PP2Ainh) group compared to the Lean Control group (Fig. 4B (i–iv), D). A significant decrease in the Smad2/3-Smad4 co-localization (#p <0.01) (Fig 4A(v), C) and a subsequent significant decrease in the red spots (** 0.05< p< 0.01) (Fig 4B(v), D) in the (NAFLD+PP2Ainh+p47phox KO) mice group, compared to the (NAFLD+ PP2Ainh) group was observed.
Fig. 4.

Formalin-fixed, paraffin-embedded 5 μm intestinal slices from Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh), and (NAFLD+PP2Ainh+p47phox KO) groups were used for immunofluorescence imaging. (A)(i-iv) Immunofluorescence images depicting Smad2/3 (red) and Smad4 (green) co-localization events in the small intestine, counterstained with DAPI (blue) of Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh), and (NAFLD+PP2Ainh+p47phox KO) mice groups. Images were taken at 20X magnification, (B)(i-v) Immunofluorescence images depicting the proximity ligation (red) of Smad2/3 and Smad4 proteins in the small intestine, counterstained with DAPI (blue) of Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh), and (NAFLD+PP2Ainh+p47phox KO) groups of mice. Images were taken at 20X magnification. (C) Morphometric analysis of Smad2/3-Smad4 co-localization events. Y-axis shows % positive immunoreactive area (% ROI) (n=3, analysis from three separate microscopic fields) (*p < 0.05, #p< 0.01). (D) Morphometric analysis of Smad2/3 and Smad4 proximity ligation. Y-axis shows % positive immunoreactive area (%ROI) (n=3, analysis from three separate microscopic fields) (*p < 0.05, 0.05<**p < 0.01). Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
TGF-β signal transduction due to nuclear translocation of the Smad2/3-Smad4 assembly cause extracellular matrix protein deposition and fibrosis
Fibronectin is a component of the extracellular matrix present in the cell (X. Y. Liu et al., 2016; Matsui et al., 1997; Xu et al., 1997), which is required for collagen matrix assembly and plays an important role in fibrosis. Expression of fibronectin is often considered as an important marker for extracellular matrix deposition (Krzyzanowska-Go?ab et al., 2007; Roeb & Matern, 1993; Sottile & Hocking, 2002; Velling et al., 2002). The expression of α-smooth muscle actin (α-SMA) also denotes the presence of myofibroblast cell phenotype, since it is considered as a marker for collagen-producing fibroblasts and has been known to play a crucial role in fibrosis (Carpino et al., 2005). Immunohistochemical analysis and immunofluorescence staining were done using mice small intestine tissues to detect α-SMA and fibronectin immunoreactivity, respectively. Our results showed a significant increase in α-SMA immunoreactivity in the (NAFLD+PP2Ainh) group compared to the NAFLD only group (#p < 0.01) (Fig 5A(i–iv), C). Similar trends of a significant increase in fibronectin immunoreactivity in (NAFLD+PP2Ainh) and (Chow+PP2Ainh) groups were observed compared to the NAFLD only and the Lean Control groups respectively (*p < 0.05) (Fig 5B (i–iv), D). A significant decrease in both α-SMA (#p <0.01) (Fig 5A (v), C) and fibronectin (#p <0.01) (Fig 5B (v), D) immunoreactivity was observed in the (NAFLD+PP2Ainh+p47phox KO) mice group compared to the PP2A inhibitor exposed group.
Fig. 5.

Formalin-fixed, paraffin-embedded 5 μm intestinal slices from Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh), and (NAFLD+PP2Ainh+p47phox KO) groups were used for immunohistochemistry and immunofluorescence imaging. (A)(i-v) mmunohistochemistry images depicting α-SMA immunoreactivity in the small intestine of Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh), and (NAFLD+PP2Ainh+p47phox KO) mice groups. Images were taken at 20X magnification. (B)(i-v) Immunofluorescence images depicting fibronectin (green) immunoreactivity, counterstained with DAPI (blue) in the small intestine of Lean Control, (Chow+PP2Ainh), NAFLD, (NAFLD+PP2Ainh), and (NAFLD+PP2Ainh+p47phox KO) mice groups. Images were taken at 20X magnification. (C) Morphometric analysis of α-SMA immunoreactivity. Y-axis shows % positive immunoreactive area (% ROI) (n=3, analysis from three separate microscopic fields) (#p< 0.01). (D) Morphometric analysis of fibronectin immunoreactivity. Y-axis shows % positive immunoreactive area (% ROI) (n=3, analysis from three separate microscopic fields) (*p< 0.05, #p< 0.01). Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
Lactate causes the formation of peroxynitrite and increased oxidative stress due to NADPH oxidase 2 activation in mouse primary intestinal epithelial cells
Primary mouse intestinal epithelial cells were exposed to leptin to create the underlying conditions of NAFLD, followed by stimulation with lactate, which is believed to be present in the system due to alteration of the gut microbiome. Results showed that cells primed with leptin and then stimulated with lactate (LEP+LAC) showed significantly increased NOX2 activation when compared to the cells only, leptin only and lactate only treated cells (#p <0.01) (Fig 6A(i–iv), C). Activation of NOX2 leads to increased oxidative stress through the formation of peroxynitrite. To estimate peroxynitrite formation in the intestinal epithelial cells, they were stained for 3-nitrotyrosine (3-NT) immunoreactivity. Results showed a significantly increased reactivity in the (LEP+LAC) group although having lesser cell density compared to the cells only, leptin only, and lactate only groups (#p <0.01) (Fig 6B (i–iv), D). Significantly increased 3-NT production was also observed in the lactate only group which denoted that lactate released due to altered gut microbiome itself can generate oxidative stress in the murine intestine. Incubation with the peroxynitrite scavenger phenylboronic acid (FBA), NOX2 inhibitor apocynin, nitrone spin trap DMPO, hydrogen peroxide inhibitor catalase, and the superoxide scavenger superoxide dismutase significantly decreased the 3-NT formation (#p <0.01) (Fig 6B(v–ix), D) compared to the (LEP+LAC) group.
Fig. 6.

Mouse primary intestinal epithelial cell line was used as control (Control), NAFLD was induced in one group of cells with leptin (LEP), a group of cells was treated with lactate (LAC), and another group of cells was treated with both leptin and lactate (LEP+LAC). The remaining groups of mouse primary intestinal epithelial cell cells were blocked by Apocynin (LEP+LAC+APO), FBA (LEP+LAC+FBA), DMPO (LEP+LAC+DMPO), catalase (LEP+LAC+CATALASE), superoxide dismutase (LEP+LAC+SOD) and then treated with leptin and lactate. The cell groups were used for immunofluorescence imaging and western blot. (A)(i-iv) Immunofluorescence images depicting gp91phox (red) and p47phox (green) co-localization (in yellow, marked by white circles), counterstained with DAPI (blue) in Control, LEP, LAC, (LEP+LAC) groups of cells. Images were taken at 60X magnification. (B)(i-ix) Immunofluorescence images depicting 3-Nitrotyrosine immunoreactivity (in green, marked by white arrows), counterstained with DAPI (blue) in Control, LEP, LAC, (LEP+LAC), (LEP+LAC+APO), (LEP+LAC+FBA), (LEP+LAC+DMPO), (LEP+LAC+CATALASE), and (LEP+LAC+SOD) groups of cells. Images were taken at 60X magnification. (C) Morphometric analysis of gp91phox-p47phox co-localized immunoreactivity. Y-axis shows % positive immunoreactive area (% ROI) (n=5, analysis from five separate microscopic fields) (#p< 0.01). (D) Morphometric analysis of 3-Nitrotyrosine immunoreactivity. Y-axis shows % positive immunoreactive area (% ROI) (n=5, analysis from five separate microscopic fields) (#p< 0.01). Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
Increased oxidative stress up-regulates the TGF-β levels leading to nuclear translocation of R-SMAD assembly
Activation of NOX2 and increased oxidative stress leads to the TGF-β signaling pathway which subsequently induces the formation of the Smad2/3-Smad4 complex. Western blot was performed to detect the expression of the phosphorylated TGF-βR1 with respect to total TGF-βR1 by using the intestinal epithelial cell lysates. We observed that there was a significant increase of relative phosphorylated TGF-βR1 expression in the (LEP+LAC) group when compared to the cells only, leptin only or lactate only groups (#p< 0.01, **0.05< p< 0.01) (Fig7A, C). Interestingly, phosphorylated TGF-βR1 expression was significantly decreased in the following groups in the presence of inhibitors like apocynin, FBA, DMPO, catalase, and SOD. The fold change was observed by quantifying the protein bands of phospho-TGF-βR1 with respect to the total-TGF-βR1(#p< 0.01, **0.05< p< 0.01) (Fig 7C). Following the activation of the TGF-βR1, a significant increase in the Smad2/3-Smad4 co-localization (represented by yellow dots in the cells) was observed in the groups treated with (LEP+LAC) compared to the cells only and lactate only groups (#p< 0.01) (Fig 8A (i–iv), C). The co-localization events were significantly decreased in the groups that were incubated with the inhibitors apocynin, FBA, DMPO, Catalase and SOD (#p< 0.01) (Fig 8A (v–ix), C), suggesting the role of the reactive oxygen species in activating the TGF-β downstream signaling pathway.
Fig. 7.

(A) Western blot analysis of α-SMA, phospho-TGF-βR1 and total TGF-βR1 protein levels in the mouse primary intestinal epithelial cell lysates. Lanes 1–9 represent Control, LEP, LAC, (LEP+LAC), (LEP+LAC+APO), (LEP+LAC+FBA), (LEP+LAC+DMPO), (LEP+LAC+CATALASE), and (LEP+LAC+SOD) groups of cells respectively. Band quantification of α-SMA immunoblot (B) normalized against β-actin (#p< 0.01), phospho-TGF-βR1 immunoblot (C) normalized against total TGF-βR1 (0.05<**p < 0.01, #p< 0.01) in the above-mentioned groups of mouse primary intestinal epithelial cells. Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
Fig. 8.

(A)(i-ix) Immunofluorescence images depicting Smad2/3 (red) and Smad4 (green) co-localization (in yellow) events, counterstained with DAPI (blue) in Control, LEP, LAC, (LEP+LAC), (LEP+LAC+APO), (LEP+LAC+FBA), (LEP+LAC+DMPO), (LEP+LAC+Catalase), and (LEP+LAC+SOD) groups of cells. Images were taken at 60X magnification. (B) (i-ix) Immunofluorescence images depicting collagen I immunoreactivity (in red), counterstained with DAPI (blue) in Control, LEP, LAC, (LEP+LAC), (LEP+LAC+APO), (LEP+LAC+FBA), (LEP+LAC+DMPO), (LEP+LAC+CATALASE), and (LEP+LAC+SOD) groups of cells. Images were taken in 40X magnification. (C) Morphometric analysis of Smad2/3-Smad4 co-localization events. Y-axis shows % positive immunoreactive area (% ROI) (n=5, analysis from five separate microscopic fields) (#p< 0.01, ns: non-significant). (D) Morphometric analysis of collagen I immunoreactivity, Y-axis shows % positive immunoreactive area (% ROI) (n=5, analysis from five separate microscopic fields) (0.05<**p < 0.01, #p< 0.01). Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
Activation of the TGF-β signaling pathway causes collagen deposition and a change of the epithelial to the myofibroblastic phenotype of the cells
Following our observation of TGF-β signaling cascade activation by exogenous PP2A inhibitor we studied the expression of α-SMA by western blot to detect myofibroblast cells. Results showed that there was a significant increase in α-SMA expression in the (LEP+LAC) group compared to the cells only, leptin only, and lactate only groups (#p <0.01) (Fig 7A, B). Collagen 1 immunoreactivity was detected through immunofluorescence staining to analyze the expression of collagen (represented by red in the cells) which indicated the formation of the fibrotic phenotype. Results suggested a significant increase in the collagen levels in the (LEP+LAC) group, compared to the cells only (#p <0.01) (Fig 8B (i–iv), D). There was a significant decrease in the α-SMA expression (#p <0.01) (Fig 7A,B) and collagen I immunoreactivity (Fig 8B (v–ix), D) when the cells were incubated with the inhibitors apocynin (**0.05< p< 0.01), FBA (**0.05< p< 0.01), DMPO (#p< 0.01), catalase (#p <0.01) and SOD (#p <0.01).
Exposure of MC-LR to intestinal epithelial cells led to the fibrotic pathology in the murine intestine
Treatment of the mouse primary intestinal epithelial cells with MC-LR, a PP2Ainh led to the activation of NOX2. Subsequently, NOX2 activation by PP2Ainh triggered the release of reactive oxygen species in the cellular system, which was then detected by the immunoreactivity of 3-NT. Results showed that there was a significant increase in the NOX2 activation in the (LEP+PP2Ainh) group (#p <0.01), compared to the control, leptin only, and the PP2Ainh only groups (Fig 9A (i–iv), C). The immunoreactivity was observed by detecting the co-localization events of membrane-bound gp91phox and cytosolic p47phox subunits of NOX2. The results also depicted a significant increase in the 3-NT immunoreactivity in the (LEP+PP2Ainh) group compared to the control, leptin only, and the PP2Ainh only group (#p< 0.01) (Fig 9B (i–iv) and D). We observed that the effect of the PP2A inhibitor alone on the primary intestinal epithelial cells was not as pronounced as the leptin-primed PP2A inhibitor-treated cells, suggesting that there was a higher possibility of intestinal fibrotic pathology under inflammatory conditions of NAFLD. The 3-NT formation was significantly decreased when the cells were incubated with the inhibitors FBA, apocynin, DMPO, catalase and superoxide dismutase (#p< 0.01) (Fig 9B (v– ix), D). Immunoblot results implicated that exposure of the cells to PP2Ainh showed a significant increase of relative phosphorylated TGF-βR1 expression in the (LEP+PP2Ainh) group compared to the control and leptin only, and PP2A inhibitor only groups (#p< 0.01) (Fig 10A, B). Also, relative phosphorylated TGF-βR1 expression was observed to significantly decreased in the groups incubated with apocynin, FBA, DMPO, catalase and SOD (#p <0.01) (Fig 10A, B). Immunofluorescence results also showed a significant increase in the Smad2/3-Smad4 co-localization (represented by yellow dots in the cell) in the group treated with (LEP+PP2Ainh) compared to the control, leptin only, and PP2Ainh only groups (#p <0.01) (Fig 11A (i–iv), C). The co-localization events were significantly decreased in the groups incubated with the inhibitors apocynin, FBA, DMPO, catalase and SOD (#p< 0.01) (Fig 11A(v–ix), C), suggesting that inhibiting the reactive oxygen species might help in attenuating the onset of the TGF-β fibrotic pathway. Similar results of significant increase were observed for the collagen I immunoreactivity (represented by red dots in the cell) in the (LEP+PP2Ainh) group when compared to the control and PP2Ainh only group (*p <0.05) (Fig 11B (i–iv), D). The immunoreactivity for collagen I was significantly decreased when the cells were incubated with the inhibitors apocynin, FBA, DMPO, catalase and SOD (*p <0.05) (Fig 11B (v–ix), D).
Fig. 9.

Mouse primary intestinal epithelial cell line was used as control (Control), NAFLD was induced in one group of cells with leptin (LEP), a group of cells was treated with MC-LR, the PP2A inhibitor (PP2Ainh), and another group of cells was treated with both leptin and PP2Ainh (LEP+PP2Ainh). The remaining groups of mouse primary intestinal epithelial cell cells were blocked by Apocynin (LEP+PP2Ainh+APO), FBA (LEP+PP2Ainh+FBA), DMPO (LEP+PP2Ainh+DMPO), catalase (LEP+PP2Ainh+CATALASE), superoxide dismutase (LEP +PP2Ainh+SOD) and then treated with leptin and PP2Ainh. The cell groups were used for immunofluorescence imaging and western blot. (A)(i-iv) Immunofluorescence images depicting gp91phox (red) and p47phox (green) co-localization (in yellow, marked by white arrows), counterstained with DAPI (blue) in Control, LEP, PP2Ainh, (LEP+PP2Ainh) groups of cells. Images were taken at 60X magnification. (B)(i-ix) Immunofluorescence images depicting 3-Nitrotyrosine immunoreactivity (in green, marked by white arrows), counterstained with DAPI (blue) in Control, LEP, PP2Ainh, (LEP+PP2Ainh), (LEP+PP2Ainh+APO), (LEP+PP2Ainh+FBA), (LEP+PP2Ainh+DMPO), (LEP+PP2Ainh+CATALASE), and (LEP+ PP2Ainh+SOD) groups of cells. Images were taken at 60X magnification. (C) Morphometric analysis of gp91phox-p47phox co-localized immunoreactivity. Y-axis shows % positive immunoreactive area (% ROI) (n=5, analysis from five separate microscopic fields) (#p< 0.01). (D) Morphometric analysis of 3-Nitrotyrosine immunoreactivity. Y-axis shows % positive immunoreactive area (% ROI) (n=5, analysis from five separate microscopic fields) (#p< 0.01). Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
Fig. 10.

(A) Western blot analysis of phospho-TGF-βR1 and total TGF-βR1 protein levels in the mouse primary intestinal epithelial cell lysates. Lanes 1–9 represent Control, LEP, PP2Ainh, (LEP+PP2Ainh), (LEP+PP2Ainh+APO), (LEP+PP2Ainh+FBA), (LEP+PP2Ainh+DMPO), (LEP+PP2Ainh+CATALASE), and (LEP+PP2Ainh+SOD) groups of cells respectively. (B) Band quantification of phospho-TGF-βR1 immunoblot normalized against total TGF-βR1 in the above-mentioned groups of mouse primary intestinal epithelial cells (#p< 0.01). Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
Fig. 11.

(A)(i-ix) Immunofluorescence images depicting Smad2/3 (red) and Smad4 (green) co-localization events (in yellow), counterstained with DAPI (blue) in Control, LEP, PP2Ainh, (LEP+PP2Ainh), (LEP+PP2AInh+APO), (LEP+PP2AInh+FBA), (LEP+PP2AInh+DMPO), (LEP+PP2AInh+CATALASE), and (LEP+PP2AInh+SOD) groups of cells. Images were taken at 60X magnification. (B) (i-ix) Immunofluorescence images depicting collagen I immunoreactivity (in red), counterstained with DAPI (blue) in in Control, LEP, PP2Ainh, (LEP+PP2AInh), (LEP+PP2AInh+APO), (LEP+PP2AInh+FBA), (LEP+PP2AInh+DMPO), (LEP+PP2AInh+CATALASE), and (LEP+PP2AInh+SOD) groups of cells. Images were taken in 40X magnification, (C) Morphometric analysis of Smad2/3-Smad4 co-localization events. Y-axis shows % positive immunoreactive area (% ROI) (n=5, analysis from five separate microscopic fields) (#p< 0.01). (D) Morphometric analysis of collagen I immunoreactivity, Y-axis shows % positive immunoreactive area (% ROI) (n=5, analysis from five separate microscopic fields) (*p < 0.05). Significance was tested by performing unpaired t-test between the groups (*p< 0.05, 0.05<**p < 0.01, #p< 0.01), followed by Bonferroni Dunn Post hoc corrections. Results were expressed as mean ± SEM.
DISCUSSION
The present study reports a novel effect of the MC-LR exposure in an underlying condition of NAFLD. We show that increased circulatory lactate mediated NOX2 activation led to the fibrotic phenotype in the murine small intestine, a frequent co-morbidity in the diagnostic pathology of the human NAFLD. The higher lactate in the serum was presumably due to the result of dysbiosis observed in our previously reported study that caused an increased abundance of lactate producing bacteria following MC-LR exposure. This assumption is based on two main findings. The higher fecal lactate, an evidence of lactate in the intestinal lumen and serum lactate that represents a likely higher lactate sequestration in the circulatory system though the portal vein from the lumen. The results that there is no stoichiometric assessment of the concentration of lactate that actually is produced by the gut bacteria limits the ability of this study to conclude clearly that an overall increase in fecal lactate or serum lactate is a direct consequence of gut dysbiosis. It can be speculated that increased sophistication of techniques primarily via metabolomic analysis and tracing techniques might help clearly define the cause and effect relationship of dysbiosis associated lactate sequestration following MC-LR exposure. Our previous studies suggested that the exposure of MC-LR to mice led to the activation of an inflammatory pathway and disruption of the intestinal barrier thereby forming a leaky gut (Sarkar et al., 2019). MC-LR also led to a significant increase in the expression of lactate producing bacteria Lactobacillus and Enterococcus (Sarkar et al., 2019). Our previous results of increased lactate production in the same model correlated well to the increase of the serum lactate levels in the mice which were exposed to MC-LR (Sarkar et al., 2019). Latsenko et al. reported that the intestinal NOX2 enzyme was activated by lactate which was produced in drosophila (Iatsenko et al., 2018). This led us to study the potential role of lactate in activating NOX2 in the diseased model of NAFLD following MC-LR exposure. The lactate thus released is believed to cause the activation of the NOX2 enzyme that leads to increased oxidative stress in the murine intestine as indicated by our data that showed an R-value of >0.7 in the correlation analysis (Fig. 2E and 2F). NOX2 knockout mice with p47phox gene deleted were used to prove the role of NOX2 in activating the downstream signaling pathway leading to the fibrotic phenotype. In our previous studies, we showed that NOX2 led to the generation of peroxynitrite that caused increased oxidative stress (Das et al., 2015). In our present study, serum lactate activated NOX2following MC-LR exposure led to the formation of the peroxynitrite species. There was no peroxynitrite generation observed in the p47phox knockout mice thereby confirming that NOX 2 had a significant role to play in activating the important proinflammatory reactive nitrogen species. The use of specific peroxynitrite scavenger phenylboronic acid (FBA) (Zielonka et al., 2010) also showed a significant decrease in the peroxynitrite generation, thereby confirming its formation.
The present study is also of significance because it establishes the effects of the MC-LR in a downstream TGF-β signaling pathway in the intestinal epithelia with lactate induced NOX2 being the primary mediator. The activated NOX2 enzyme then leads to the formation of reactive oxygen species that transforms the latent form of TGF-β molecule to the active form as described before (Barcellos-Hoff & Dix, 1996). The activated TGF-beta then binds to the TGF-β receptor leading to the likely activation of the fibrotic pathway. Our studies showed an increased TGF-β immunoreactivity in the small intestine of mice group fed with MCD-HFD and exposed to MC-LR, while it was significantly decreased in the p47phox knockout mice group, suggesting a role of NOX2 derived oxidative stress leading to TGF-β production. Since PP2A inhibitors act as major serine-threonine phosphatases, it is likely that MC-LR being a PP2A inhibitor covalently binds with the protein phosphatase 2A and inhibits it from dephosphorylating the TGF-βR1. The TGF-βR1, therefore, remains activated and leads to the phosphorylation of Smad2/3 and its subsequent association with Smad4. The associated moiety then translocates to the nucleus for its effect on triggering the profibrotic mechanisms. In our results, we observed that there was a very significant colocalization of both Smad2/3 and Smad4 proteins in the mice treated with the MC-LR, while the colocalization significantly decreased in the p47phox knockout group suggesting the prime role of NOX2 in triggering the profibrotic pathway following MC-LR exposure. Finally, we also observed increased levels of α-SMA, collagen I, vimentin, and fibronectin, denoting the formation of myofibroblast cell phenotypes and also extracellular matrix protein deposition in the MC-LR-treated group compared to the control group. These levels were significantly decreased in the mice with the p47phox gene deleted mice. The fibrotic phenotype in the murine intestine was also observed in the treated groups via Picrosirius Red staining, where increased collagen fiber deposition was observed, compared to the control group. The mechanisms were also confirmed in the in vitro experiments where we mimicked the profibrotic microenvironment by exposing intestinal epithelial cells to leptin and MC-LR. The fact that the use of Apocynin, DMPO and FBA significantly decreased SMAD2–3/4 colocalizations is further proof of a likely NOX-2 mediated redox mechanism for fibrosis induced my MC-LR in an ectopic site.
In conclusion, in this study, we show that exposure to MC-LR under chronic inflammatory conditions of NAFLD can lead to a fibrotic phenotype in the murine intestine, through the activation of the NADPH oxidase 2 enzyme. NOX2, in turn, caused the phosphorylation of TGF-βR1 and downstream fibrotic pathology presumably by generating redox species dominated by peroxynitrite through independent species such as superoxide radicals or hydrogen peroxide cannot be ruled out. Both in vivo and in vitro evidence point to the fact that PP2A inhibition in NAFLD is mediating the fibrotic pathology via likely higher lactate from the gut and subsequent NOX2 activation. More direct studies should be conducted to find a concrete role of environmental toxins-induced microbiome alterations and its effect on higher lactate production, and subsequent NOX2 activation via metabolomic analysis and tracing techniques to understand disease pathology. By far our present study is first of its kind which tries to establish a strong connection between our previously reported microcystin-induced dysbiosis, and higher lactate-associated redox pathology as increased emphasis is placed on the role of environmental toxins and microbiome alterations in fatty liver disease. Furthermore, our results also show the importance of redox signaling inhibitors and broadly, antioxidants as potential candidates for therapeutic applications in chronic inflammatory and fibrotic pathology that establish the role of lactate induced NOX2 in such conditions. However, one limitation of the present study is the non-inclusion of an IBD model to study whether a similar dysbiosis associated and lactate-linked redox activation of NOX2 exists to cause ectopic inflammatory manifestations in the liver. We understand that it is very crucial to study the above pathway to help create an effective strategy of cure NAFLD complications in IBD patients.
Highlights.
Co-exposure to microcystin led to higher lactate levels in circulation and intestine.
The higher lactate levels were associated with NOX2 activation in vivo
Lactate increased Smad2/3-Smad4 co-localization and early fibrosis.
Study provides insight into the role of microcystin in dysbiosis-linked lactate production in intestinal fibrosis
Advances our knowledge in lactate-induced NOX2 exacerbation of intestinal fibrosis
Acknowledgments
The authors gratefully acknowledge the technical services of Benny Davidson at the IRF, University of South Carolina School of Medicine and AML Labs (Baltimore MD). We also thank the Instrumentation resource facility (IRF) at the University of South Carolina for equipment usage and consulting services. We also thank Second Genome and School of Medicine, the University of South Carolina for carrying out the Microbiome analysis.
Funding: This work has been supported by NIH Awards 2P20GM103641-06,1P01ES028942-01 and P01AT003961 to Saurabh Chatterjee, 1P01ES028942-01 to Dwayne Porter and Geoff I Scott, P01AT003961, P20GM103641, R01AT006888, R01ES019313, R01MH094755 to Mitzi Nagarkatti and Prakash S. Nagarkatti.
Footnotes
Competing Interests: The author(s) declare no competing interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability: All data generated or analyzed during this study are included in this published article. Any additional datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamovsky O, Buerger AN, Wormington AM, Ector N, Griffitt RJ, Bisesi JH, & Martyniuk CJ (2018). The gut microbiome and aquatic toxicology: An emerging concept for environmental health. Environmental Toxicology and Chemistry, 37(11), 2758–2775. 10.1002/etc.4249 [DOI] [PubMed] [Google Scholar]
- Alhasson F, Dattaroy D, Das S, Chandrashekaran V, Seth RK, Schnellmann RG, & Chatterjee S (2015). NKT cell modulates NAFLD potentiation of metabolic oxidative stress-induced mesangial cell activation and proximal tubular toxicity. American Journal of Physiology - Renal Physiology, 310(1), F85–F101. 10.1152/ajprenal.00243.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi S, Fredriksson R, & Wallén-Mackenzie Å (2015). In Situ Proximity Ligation Assay (PLA) BT - ELISA: Methods and Protocols (Hnasko R (ed.); pp. 149–159). Springer; New York: 10.1007/978-1-4939-2742-5_15 [DOI] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, & Dix TA (1996). Redox-mediated activation of latent transforming growth factor-β1. Molecular Endocrinology, 10(9), 1077–1083. 10.1210/me.10.9.1077 [DOI] [PubMed] [Google Scholar]
- Biernacka A, Dobaczewski M, & Frangogiannis NG (2011). TFG-b signaling in fibrosis. Growth Factors, 29(5), 196–201. 10.3109/08977194.2011.595714.TGF- [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botha N, Van De Venter M, Downing TG, Shephard EG, & Gehringer MM (2004). The effect of intraperitoneally administered microcystin-LR on the gastrointestinal tract of Balb/c mice. Toxicon, 43(3), 251–254. 10.1016/j.toxicon.2003.11.026 [DOI] [PubMed] [Google Scholar]
- Branton MH, & Kopp JB (1999). TGF-β and fibrosis. Microbes and Infection, 1(15), 1349–1365. 10.1016/S1286-4579(99)00250-6 [DOI] [PubMed] [Google Scholar]
- Campos A, & Vasconcelos V (2010). Molecular mechanisms of microcystin toxicity in animal cells. International Journal of Molecular Sciences, 11(1), 268–287. 10.3390/ijms11010268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpino G, Morini S, Ginanni Corradini S, Franchitto A, Merli M, Siciliano M, Gentili F, Onetti Muda A, Berloco P, Rossi M, Attili AF, & Gaudio E (2005). Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Digestive and Liver Disease, 37(5), 349–356. 10.1016/j.dld.2004.11.009 [DOI] [PubMed] [Google Scholar]
- Challa AA, & Stefanovic B (2011). A Novel Role of Vimentin Filaments: Binding and Stabilization of Collagen mRNAs. Molecular and Cellular Biology, 31(18), 3773–3789. 10.1128/mcb.05263-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrashekaran V, Seth RK, Dattaroy D, Alhasson F, Ziolenka J, Carson J, Berger FG, Kalyanaraman B, Diehl AM, & Chatterjee S (2017). HMGB1-RAGE pathway drives peroxynitrite signaling-induced IBD-like inflammation in murine nonalcoholic fatty liver disease. Redox Biology, 13(May), 8–19. 10.1016/j.redox.2017.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CY, Battat R, Al Khoury A, Restellini S, Sebastiani G, & Bessissow T (2016). Co-existence of non-alcoholic fatty liver disease and inflammatory bowel disease: A review article. World Journal of Gastroenterology, 22(34), 7727–7734. 10.3748/wjg.v22.i34.7727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Xie P, Lin J, He J, Zeng C, & Chen J (2015). Effects of microcystin-LR on gut microflora in different gut regions of mice. Journal of Toxicological Sciences, 40(4), 485–494. 10.2131/jts.40.485 [DOI] [PubMed] [Google Scholar]
- Das S, Alhasson F, Dattaroy D, Pourhoseini S, Seth RK, Nagarkatti M, Nagarkatti PS, Michelotti GA, Diehl AM, Kalyanaraman B, & Chatterjee S (2015). NADPH Oxidase-Derived Peroxynitrite Drives Inflammation in Mice and Human Nonalcoholic Steatohepatitis via TLR4-Lipid Raft Recruitment. American Journal of Pathology, 185(7), 1944–1957. 10.1016/j.ajpath.2015.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dattaroy D, Pourhoseini S, Das S, Alhasson F, Seth RK, Nagarkatti M, Michelotti GA, Diehl AM, & Chatterjee S (2015). Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. American Journal of Physiology - Gastrointestinal and Liver Physiology, 308(4), G298–G312. 10.1152/ajpgi.00346.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funari E, & Testai E (2008). Human health risk assessment related to cyanotoxins exposure. Critical Reviews in Toxicology, 38(2), 97–125. 10.1080/10408440701749454 [DOI] [PubMed] [Google Scholar]
- Heinze R (1999). Toxicity of the cyanobacterial toxin microcystin-LR to rats after 28 days intake with the drinking water. Environmental Toxicology, 14(1), 57–60. 10.1002/(SICI)1522-7278(199902)14:1<57::AID-TOX9>3.0.CO;2-J [DOI] [Google Scholar]
- Honkanen RE, Zwiller J, Moore RE, Daily SL, Khatra BS, Dukelow M, & Boynton AL (1990). Characterization of microcystin-LR, a potent inhibitor of type 1 and type 2A protein phosphatases. Journal of Biological Chemistry, 265(32), 19401–19404. [PubMed] [Google Scholar]
- Iatsenko I, Boquete JP, & Lemaitre B (2018). Microbiota-Derived Lactate Activates Production of Reactive Oxygen Species by the Intestinal NADPH Oxidase Nox and Shortens Drosophila Lifespan. Immunity, 9(5), 929–942.e5. 10.1016/j.immuni.2018.09.017 [DOI] [PubMed] [Google Scholar]
- Ito E, Kondo F, & Harada KI (2000). First report on the distribution of orally administered microcystin-LR in mouse tissue using an immunostaining method. Toxicon, 38(1), 37–48. 10.1016/S0041-0101(99)00084-7 [DOI] [PubMed] [Google Scholar]
- Jiang F, Liu GS, Dusting GJ, & Chan EC (2014). NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biology, 2(1), 267–272. 10.1016/j.redox.2014.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzanowska-Go?ab D, Lema?ska-Perek A, & Katnik-Prastowska I (2007). Fibronectin as an active component of the extracellular matrix | Fibronektyna jako aktywny sk?adnik macierzy pozakom?rkowej. Postpy Higieny i Medycyny Do?Wiadczalnej (Online), 61, 655–663. [PubMed] [Google Scholar]
- Liu T, & Feng XH (2010). Regulation of TGF-β signalling by protein phosphatases. Biochemical Journal, 430(2), 191–198. 10.1042/BJ20100427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XY, Liu RX, Hou F, Cui LJ, Li CY, Chi C, Yi E, Wen Y, & Yin CH (2016). Fibronectin expression is critical for liver fibrogenesis in vivo and in vitro. Molecular Medicine Reports, 14(4), 3669–3675. 10.3892/mmr.2016.5673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui S, Takahashi T, Oyanagi Y, Takahashi S, Boku S, Takahashi K, Furukawa K, Arai F, & Asakura H (1997). Expression, localization and alternative splicing pattern of fibronectin messenger RNA in fibrotic human liver and hepatocellular carcinoma. Journal of Hepatology, 27(5), 843–853. 10.1016/S0168-8278(97)80322-4 [DOI] [PubMed] [Google Scholar]
- Maynes JT, Luu HA, Cherney MM, Andersen RJ, Williams D, Holmes CFB, & James MNG (2006). Crystal structures of protein phosphatase-1 bound to motuporin and dihydromicrocystin-LA: Elucidation of the mechanism of enzyme inhibition by cyanobacterial toxins. Journal of Molecular Biology, 356(1), 111–120. 10.1016/j.jmb.2005.11.019 [DOI] [PubMed] [Google Scholar]
- McLellan NL, & Manderville RA (2017). Toxic mechanisms of microcystins in mammals. Toxicology Research, 6(4), 391–405. 10.1039/c7tx00043j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Nikolic-Paterson DJ, & Lan HY (2016). TGF-β: the master regulator of fibrosis. Nature Reviews Nephrology, 12(6), 325–338. 10.1038/nrneph.2016.48 [DOI] [PubMed] [Google Scholar]
- Nakerakanti S (2012). The Role of TGF-β Receptors in Fibrosis. The Open Rheumatology Journal, 6(1), 156–162. 10.2174/1874312901206010156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappachan JM, Babu S, Krishnan B, & Ravindran NC (2017). Non-alcoholic Fatty Liver Disease: A Clinical Update. Journal of Clinical and Translational Hepatology, 5 (2017), pp. 384–393. 10.14218/jcth.2017.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PVL, Gupta N, Bhaskar ASB, & Jayaraj R (2002). Toxins and bioactive compounds from cyanobacteria and their implications on human health. Journal of Environmental Biology, 23(3), 215–224. [PubMed] [Google Scholar]
- Ray BN, Lee NY, How T, & Blobe GC (2010). ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis, 31(3), 435–441. 10.1093/carcin/bgp327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeb E, & Matern S (1993). [Fibronectin--a key substance in pathogenesis of liver cirrhosis?]. Leber, Magen, Darm, 23(6), 239–242. [PubMed] [Google Scholar]
- Sarkar S, Kimono D, Albadrani M, Seth RK, Busbee P, Alghetaa H, Porter DE, Scott GI, Brooks B, Nagarkatti M, Nagarkatti P, & Chatterjee S (2019). Environmental microcystin targets the microbiome and increases the risk of intestinal inflammatory pathology via NOX2 in underlying murine model of Nonalcoholic Fatty Liver Disease. Scientific Reports, 9(1), 1–22. 10.1038/s41598-019-45009-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedan D, Laguens M, Copparoni G, Aranda JO, Giannuzzi L, Marra CA, & Andrinolo D (2015). Hepatic and intestine alterations in mice after prolonged exposure to low oral doses of Microcystin-LR. Toxicon, 104, 26–33. 10.1016/j.toxicon.2015.07.011 [DOI] [PubMed] [Google Scholar]
- Shi Y (2009). Serine/Threonine Phosphatases: Mechanism through Structure. Cell, 139(3), 468–484. 10.1016/j.cell.2009.10.006 [DOI] [PubMed] [Google Scholar]
- Sottile J, & Hocking DC (2002). Fibronectin Polymerization Regulates the Composition and Stability of Extracellular Matrix Fibrils and Cell-Matrix Adhesions. Molecular Biology of the Cell, 13(10), 3546–3559. 10.1091/mbc.e02-01-0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velling T, Risteli J, Wennerberg K, Mosher DF, & Johansson S (2002). Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins α11β1 and α2β1. Journal of Biological Chemistry, 277(40), 37377–37381. 10.1074/jbc.M206286200 [DOI] [PubMed] [Google Scholar]
- Verrecchia F, & Mauviel A (2007). Transforming growth factor-beta and fibrosis. World Journal of Gastroenterology, 13(22), 3056–3062. 10.3748/wjg.v13.i22.3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Divanyan A, Jourd’heuil FL, Goldman RD, Ridge KM, Jourd’heuil D, & Lopez-Soler RI (2018). Vimentin expression is required for the development of EMT-related renal fibrosis following unilateral ureteral obstruction in mice. American Journal of Physiology - Renal Physiology, 315(4), F769–F780. 10.1152/ajprenal.00340.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Xu Y, Chen Y, Jeffrey PD, Chao Y, Lin Z, Li Z, Strack S, Stock JB, & Shi Y (2006). Structure of Protein Phosphatase 2A Core Enzyme Bound to Tumor-Inducing Toxins. Cell, 127(2), 341–353. 10.1016/j.cell.2006.09.025 [DOI] [PubMed] [Google Scholar]
- Xu G, Niki T, Virtanen I, Rogiers V, De Bleser P, & Geerts A (1997). Gene expression and synthesis of fibronectin isoforms in rat hepatic stellate cells. Comparison with liver parenchymal cells and skin fibroblasts. Journal of Pathology, 183(1), 90–98. 10.1002/(SICI)1096-9896(199709)183:1<90::AID-PATH1105>3.0.CO;2-J [DOI] [PubMed] [Google Scholar]
- Yoshizawa S, Matsushima R, Watanabe MF, Harada K, Ichihara A, Carmichael WW, & Fujiki H (1990). Inhibition of protein phosphatases by microcystis and nodularin associated with hepatotoxicity. Journal of Cancer Research and Clinical Oncology, 116(6), 609–614. 10.1007/BF01637082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielonka J, Sikora A, Joseph J, & Kalyanaraman B (2010). Peroxynitrite is the major species formed from different flux ratios of co-generated nitric oxide and superoxide: Direct reaction with boronate-based fluorescent probe. Journal of Biological Chemistry, 285(19), 14210–14216. 10.1074/jbc.M110.110080 [DOI] [PMC free article] [PubMed] [Google Scholar]