

Abstract
The focus of this review is on Duchenne muscular dystrophy (DMD), caused by the absence of the protein dystrophin, is characterized as a neuromuscular disease in which muscle weakness, increased susceptibility to muscle injury, and inadequate repair appear to underlie the pathology. Considerable attention has been dedicated to studying muscle fiber damage, but data show that both human patients and animal models for DMD present with fragmented neuromuscular junction (NMJ) morphology. In addition to pre- and post-synaptic abnormalities, studies indicate increased susceptibility of the NMJ to contraction-induced injury, with corresponding functional changes in neuromuscular transmission and nerve-evoked electromyographic activity. Such findings suggest that alterations in the NMJ of dystrophic muscle may play a role in muscle weakness via impairment of neuromuscular transmission. Further work is needed to fully understand the role of the NMJ in the weakness, susceptibility to injury, and progressive wasting associated with DMD.
Keywords: mdx, NMJ, Duchenne muscular dystrophy, eccentric injury
Graphical abstract
1. Introduction
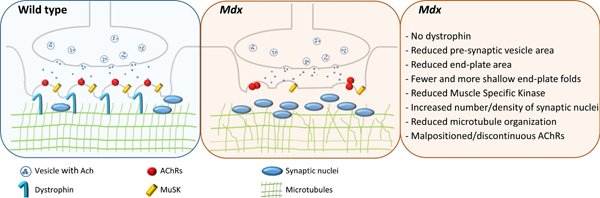
The area of synaptic contact between motor neurons and their target muscle fibers is the neuromuscular junction (NMJ). This synapse occurs at a specialized area of the sarcolemma called the end-plate. The “pretzel-shape” of a typical, healthy end-plate in mammalian muscle (Fig. 1) results from the several twisting branches of the motor neuron. The distal aspect of each branch is enlarged and these expansions form the terminal synaptic boutons, which contain synaptic vesicles filled with the neurotransmitter, acetylcholine (ACh). Boutons are located over invaginations of the sarcolemma called junctional folds [89], at the top of which high-density clusters of acetylcholine receptors (AChRs) reside. When released into the synaptic cleft, ACh binds to its post-synaptic receptors, causing an end-plate potential (EPP), a local depolarization spreading along the muscle fiber as a conducted action potential.
Fig. 1.

Top panels show representative images of neuromuscular junctions (NMJ) stained with α-bungarotoxin conjugated to Alexa-594 in muscles from WT (wild type), mdx (lacking dystrophin), dko (double knockout, lacking both dystrophin and utrophin), and Fiona (lacking dystrophin but upregulation of utrophin) mice. Bottom panels show representative skeletonized images of the corresponding NMJ. Total stained area, total stained perimeter, total area, and total perimeter were calculated using binarized images using Image J. Dispersion index was calculated by total stained area/total area, and is a measure of density of acetylcholine receptors. Discontinuity and number of branching were calculated using the skeletonized images. Number of clusters was calculated by counting the number of separate acetylcholine receptor aggregates. * indicates significant difference from WT, # indicated significant difference from Fiona, p< 0.05; one-Way ANOVA, approximately 10 NMJs in each genotype, with 5 muscles analyzed).
Accumulating evidence has made it clear that the NMJ in mature skeletal muscle is not a fixed permanent structure [27, 28], but instead is continually remodeling, thereby possessing a large degree of functional plasticity [30]. The morphology and physiology of the NMJ can display alterations in synaptic organization due to exercise [26, 89], inactivity [66, 67, 83], denervation [49, 92], aging [24, 41, 76], crushing of the nerve/muscle [44, 80], or the absence of associated proteins [1, 3, 16, 45, 48]. Ultrastructural changes in the NMJ have been documented following muscle disuse and nerve injury or denervation [9, 49].
The motor end-plate (the post-synaptic membrane of NMJ) is a specialized area of the sarcolemma that rapidly and consistently responds to release of a neurotransmitter from the overlying nerve terminal. Neuromuscular transmission is normally highly reliable, as each nerve impulse results in the release of more neurotransmitter (acetylcholine) than is required for evoking an action potential in the muscle fiber. This release of surplus transmitter and consequent excess depolarization of the postsynaptic membrane via AChRs is often referred as the ‘safety factor’ [91], which ensures that a post-synaptic action potential will occur in response to each nerve impulse, at least in healthy tissue. Proper development and organization at the NMJ is necessary for effective neuromuscular transmission [79, 90], but a number of pathological conditions affecting the distribution of AChRs can lead to a reduction in the safety factor and impairment of neuromuscular transmission [63, 91]. Although there are a group of diseases caused by defective synaptic transmission at the NMJ (congential myasthenic syndromes)[61] , this review focuses on muscular dystrophies, specifically Duchenne muscular dystrophy.
2. NMJ morphology is altered in muscular dystrophy
Duchenne muscular dystrophy (DMD), the most common and severe form of muscular dystrophy, is caused by the absence of dystrophin, a structural protein found on the cytoplasmic surface of the sarcolemma. Dystrophin is the central component of a molecular link that connects the contractile apparatus inside the muscle fiber to the extracellular matrix outside the muscle fiber, and binds directly or indirectly to a group of proteins at the sarcolemma collectively known as the dystrophin-associated protein complex (DAPC or DPC) or the dystrophin-glycoprotein complex (DGC).
The DGC of the sarcolemma accumulates at the post-synaptic membrane (motor end-plate) of the NMJ, and the absence of associated proteins can cause changes in NMJ structure and function [6]. The NMJ is noticeably disrupted in muscles from mdx and other DMD mouse models [3, 43, 55, 82]. Dystrophin appears to be involved in synaptic homeostasis [64]; it is not required for NMJ formation, but is required for endplate maintenance [45] and likely for endplate remodeling in regenerating fibers. Mdx mice show NMJ fragmentation in adult muscle fibers and excessive nerve sprouting compared to wild-type mice [56], and ultrastructural studies using electron microscopy indicate a loss in the number and depth of synaptic folds of the motor end-plate in mdx muscles [3, 55].
Patients with DMD and mdx mice have muscles with weakness and increased susceptibility to injury compared to their non-dystrophic counterparts. Over time, this damage/degeneration exceeds the ability to repair/regenerate muscle, leading to irreversible muscle wasting throughout life. The increased force loss after contraction-induced injury is typically attributed to structural weakness of the muscle fiber cytoskeleton and changes in signaling within muscle fibers secondary to the loss of dystrophin [53]. However, studies have reported that loss in whole muscle function after injury is also associated with alterations in NMJ end-plate morphology in mdx mice [68–70]. Some studies report strong evidence showing that disrupted NMJ morphology is the consequence of muscle fiber degeneration and regeneration [37, 50, 51, 59], but others have suggested that NMJ fragmentation in mdx muscle is independent of degeneration and regeneration [3, 46]. In support of the latter, NMJ post-synaptic morphology, including AChR area, is normal in fibers of muscle subjected to toxin-induced degeneration [75, 88]. It is thought that the motor neuron in DMD and mdx mice is unaffected, except for axonal sprouting near the NMJ and changes in the terminal bouton. However, recent data suggest that the persistent, chronic NMJ remodeling that occurs over the lifespan of the mdx mouse can adversely affect the motor neuron [47].
Utrophin, short for “ubiquitous dystrophin,” is a protein highly homologous to dystrophin [52]. During fetal muscle development, utrophin is found along the entire muscle sarcolemma. Once dystrophin is expressed, however, utrophin disappears from most of the sarcolemmal membrane so that, in normal adult muscle, it is located only at the neuromuscular and myotendinous junctions [85]. One reason mdx mice do not display pathology equivalent to that seen in DMD may be that utrophin is up-regulated to levels sufficient to compensate, in part, for the lack of dystrophin. Although studies have suggested that NMJ disruption is not dependent on utrophin [88], NMJ morphology appears more disrupted when dystrophin and utrophin are absent [74], and the upregulation of utrophin (through artificial transcription factors) appears to rescue NMJ morphology [65]. In the Fiona mouse, a transgenic mdx mouse in which utrophin is upregulated in skeletal muscles, using established methods [68–71] (detailed methods in supplemental material) we have found a partial rescue of NMJ morphology (Fig. 1).
Muscles from mice that lack genes for other components of the DGC, such as α-dystrobrevin, dystroglycan, or syntrophin, have altered NMJ morphology [1, 6, 33, 35, 39], even though absence of other DGC components, such as α- or ϒ-sarcoglycan, does not appear to affect NMJ morphology [23, 36]. Aberrations in NMJ morphology have also been observed in laminopathies, which are typically caused by mutations in the LMNA gene, resulting in cardiac and skeletal disorders. Patients often have abnormal EMG, indicating myopathy [8, 58]. In mouse models with LMNA mutations, NMJ morphology is disorganized and fragmented, with mislocalized synaptic nuclei [58]. Furthermore, the NMJ disorganization was observed before myopathic phenotype, furthering the notion of NMJ aberrations as a driver of disease phenotype.
3. Disrupted NMJ morphology in dystrophic muscle contributes to impairment of nerve-evoked muscle contraction
Structure is clearly a major determinant of function: as the development of force relies on the controlled overlap of actin and myosin, so does effective neuromuscular transmission on the apposition of the nerve terminal and the underlying motor end-plate[79, 90]. Moreover, maintenance of the neuromuscular apparatus relies on bidirectional communication between muscle and nerve [34]. However, several pathological conditions affecting the distribution of AChRs can lead to a reduction in the safety factor and impairment of neuromuscular transmission[63].
The increased muscle weakness and susceptibility to injury (exaggerated loss in force to a mechanical insult) in patients with DMD and in the mdx murine model, is hypothesized to be due to structural weakness of the cytoskeleton or changes in signaling secondary to the loss of dystrophin [53], but additional mechanisms are still being clarified. A mechanical model [4, 7] suggests that the absence of dystrophin results in structural fragility of the cytoskeleton [53]. In an attempt to understand mechanisms underlying force loss, much of the focus has been on structural damage within the myofiber. The NMJ is clearly disrupted in muscles from DMD mouse models and associated deficits in neuromuscular function have now also been identified [3, 14]. We have reported that loss in whole muscle contractile force after muscle injury in mdx mice is also associated with alterations in NMJ morphology, such as increased discontinuity and branching, and corresponding aberrant synaptic transmission, such as decreased EMG amplitudes and increased neuromuscular transmission failure [68–70]. Other groups have also shown disruption at the NMJ in dystrophic mouse muscles with corresponding changes in function, such as reduced amplitude of miniature endplate potentials, reduction of postsynaptic sensitivity for the neurotransmitter acetylcholine and exhaustion of presynaptic acetylcholine release during intense synaptic activity [88]. After contraction-induced injury, there is a loss of force in both the WT and mdx muscles, but only mdx mice show significant additional changes in NMJ morphology, neuromuscular transmission, and EMG activity [68, 69]. Although other factors, such as sarcolemma damage could be a factor, these findings suggest that NMJ structural and functional changes may contribute to the greater force loss seen after injury in dystrophic muscle. While some studies attribute changes in myofiber action potential conductivity as the primary reason for reduced muscle excitability in mdx muscles after injury, these studies typically utilize a low-strain, high-repetition injury protocol in distal muscles [5, 11, 77]; other studies utilizing a high-strain, lower repetition injury protocol in proximal muscles have attributed reduced muscle excitability to changes in the NMJ [68, 69].
Synaptic transmission becomes more variable with age in the mdx mouse model of DMD [12, 44], which could provide one explanation why, despite the consistent lack of dystrophin, mdx skeletal muscle generates less specific force and becomes more susceptible to damage with age [15]. EMG changes are measurable in patients with various muscular dystrophies, including DMD [18, 31, 72]. Some animal studies report changes in EMG activity between WT and mdx mice [38, 68], while others do not [13]. The study of different muscles might be one reason for such conflicting data; for example, proximal muscles are affected earlier and to a greater extent in DMD [17, 57] and a similar pattern of increased damage in more proximal muscles has been documented in young mdx mice [60].
Patients with DMD have similar intramuscular fatigability, neuromuscular transmission, and central activation as controls [81]. Such observations might argue against a significant role for NMJ dysfunction in DMD in the absence of injury, however increased sensitivity to neuromuscular blocking drugs (and a slower recovery from them) indicates NMJ is vulnerable in DMD, with more obvious findings in older-aged and ambulatory patients with DMD [88]. Furthermore, although the reduced safety factor and miniature end-plate potentials might not be sufficient to result in overt changes to the compound muscle action potentials (CMAPs), they could render the NMJ dysfunctional under certain intense activity [88].
4. Therapeutic approaches to affect NMJ form and function
Several approaches of gene therapy are being examined to ameliorate the pathophysiology that results from the absence of dystrophin. Although transgenic expression of a short isoform of dystrophin in mdx muscle can prevent muscle degeneration, NMJ morphology remains fragmented [3]. Indeed, the threshold of dystrophin lies between 19% and 50% for correction of NMJ morphology in muscles of mdx mice [87].
Muscle-specific Kinase (MuSK) is a transmembrane tyrosine kinase crucial for forming and maintaining the neuromuscular junction, and activation of the MuSK complex that drives AChR clustering [10, 32]. MuSK inactivation at the NMJ of adult muscle is known to cause a reduction in AChR density and a change in the gross synaptic arborization of the endplate, which can lead to the complete loss of AChRs and disappearance of the synaptic structure [40]. MuSK levels differ between various adult skeletal muscles, which may correlate with muscle specific differences in the response to agrin [25, 73]. In our previous work, we assessed transcripts for the multi-protein MuSK signaling complex responsible for AChR clustering in WT and mdx mice [69]; interestingly, the only significant difference between WT and mdx muscles was a decrease in MuSK in the latter. Since MuSK plays a critical role in the aggregation, or clustering, of NMJs, it is conceivable that this reduction directly contributes to the altered morphology of mdx NMJs and is therefore a potential therapeutic target. Indeed, increasing expression of MuSK or rapsyn (a cytoplasmic MuSK effector protein) with adeno-associated viral vectors protected mdx muscles from contraction-induced injury [86].
Utrophin upregulation results in a positive effect on NMJ post-synaptic morphology, including an increase in the number of AChRs and improved NMJ morphology [65]. This is accompanied by an improvement in muscle contractility, but it is difficult to tease apart the contribution of the NMJ versus other changes in the cell (e.g., improvement in sarcolemma stability, mechanotransduction of force, etc.). Interestingly, drugs commonly used for myasthenia gravis and myasthenic syndromes (autoimmune disease and inherited conditions, respectively, affecting the NMJ) do not seem to have a significant benefit for muscular dystrophies.
Exercise can exert beneficial effects not only on muscle, but also to the NMJ morphology and function [22, 78]. Endurance training affects the morphology of NMJs in young adults, and has been studied as a measure to counter changes in the NMJ with aging [2]. Specific adaptations to exercise training include increases in the length and number of nerve terminal branches, a higher number of pre-synaptic vesicles, and increase number and distribution of AChRs [20, 22, 78, 89]. Thus, the benefits attained from endurance training are likely beyond just muscle fiber remodeling and extend to the NMJ. Exercise can induce activation of neurotrophic factors and other molecules, which have a positive impact on NMJ morphology [29, 62]. Furthermore, alterations of structure induced by endurance training are associated with significant NMJ functional changes, such as synaptic transmission. Resistance exercise appears to yield similar benefits for the NMJ, but to a lesser degree [19, 21].
It is established that heavy resistance training has deleterious effects on dystrophic skeletal muscle, particularly if it involves eccentric contractions [54]; in addition to the risk of further muscle damage, there is no evidence of beneficial adaptation to heavy resistance training in dystrophic animals, or in humans with muscular dystrophy. Although historically exercise has been used sparingly in the treatment of muscular dystrophies, evidence suggests beneficial impact from moderate exercise [42]. Several well-controlled studies indicate that light to moderate exercise can have beneficial effects in patients with muscular dystrophies, such as increased strength. Unfortunately, there are relatively few controlled studies available that are easily translated to the human population. There is therefore a great need for careful studies to determine the forms of exercise that are most beneficial to patients with different types of muscle diseases, and the effects of exercise on the NMJ in dystrophic muscle.
5. Conclusions
Knowledge of NMJ dysfunction in DMD animal models is incomplete, and sometimes conflicting, but findings suggest that, in addition to mechanical damage to the myofiber, structural and functional changes at the NMJ may be another contributor to the greater force loss seen after injury in dystrophic muscle. In terms of functional impact, the relative roles of muscle fiber injury, degeneration, and denervation that contribute to the changes seen in NMJ morphology of dystrophic muscles are not yet known, but will be important to determine, as NMJ dysfunction is fundamental to understanding impairment of muscle. The notion that structure determines function is a key tenet in biology, yet it is possible that disruption of NMJ morphology in dystrophic muscle represents an effective repair process that maintains efficacy; there is still no clear evidence showing correlation between the degree of NMJ fragmentation and the efficacy of transmission [84]. Continued experimentation is paramount for elucidating mechanisms underlying the dystrophic progression. The specific role of the NMJ in neuromuscular health and its relationship to dystrophy is still being defined, but might play a role in the exacerbated response to injury.
The electrophysiological NMJ features of one muscle might not necessarily be identical in all skeletal muscles [88], but there is now sound evidence indicating a role for the NMJ in muscle weakness, susceptibility to damage, and loss of functional performance in dystrophic muscle. Future work is needed to systematically study constituents of the MuSK signaling complex, compare various muscles, and to follow the morphology and function of the NMJ at various time points after injury and throughout the lifespan.
Supplementary Material
Highlights.
Duchenne muscular dystrophy (DMD), the most common and severe form of muscular dystrophy, is caused by the lack of dystrophin, the protein encoded by the DMD gene
The neuromuscular junction (NMJ) does not have a fixed, permanent structure, but instead shows plasticity in response to muscle injury, exercise, and aging
NMJs in muscles from DMD patients and mdx mice (a preclinical animal model) show aberrant changes in both pre- and post-synaptic NMJ structure, which could influence neuromuscular transmission and ultimately muscle function
The hypothesis that the NMJ contributes to functional deficits in DMD represents a paradigm shift from more prevalent myo-centric perspectives
Acknowledgments
This work was supported by grants to RML from the National Institutes of Health (R56AR073193), to SRI from the National Institutes of Health (K01AR074048) and the Muscular Dystrophy Association development grant (MDA 577897), and to KD from the Medical Research Council, UK
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, Froehner SC, Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin, J.Cell Biol 150 (2000) 1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Andonian MH, Fahim MA, Effects of endurance exercise on the morphology of mouse neuromuscular junctions during ageing, J Neurocytol 16 (1987) 589–599. [DOI] [PubMed] [Google Scholar]
- [3].Banks GB, Chamberlain JS, Froehner SC, Truncated dystrophins can influence neuromuscular synapse structure 1, Mol.Cell Neurosci 40 (2009) 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Batchelor CL, Winder SJ, Sparks, signals and shock absorbers: how dystrophin loss causes muscular dystrophy, Trends Cell Biol 16 (2006) 198–205. [DOI] [PubMed] [Google Scholar]
- [5].Baumann CW, Warren GL, Lowe DA, Plasmalemma Function Is Rapidly Restored in Mdx Muscle after Eccentric Contractions, Med Sci Sports Exerc 52 (2020) 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Belhasan DC, Akaaboune M, The role of the dystrophin glycoprotein complex on the neuromuscular system, Neurosci Lett 722 (2020) 134833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bloch RJ, Gonzalez-Serratos H, Lateral force transmission across costameres in skeletal muscle, Exerc.Sport Sci.Rev 31 (2003) 73–78. [DOI] [PubMed] [Google Scholar]
- [8].Bonne G, Quijano-Roy S, Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies, Handb Clin Neurol 113 (2013) 1367–1376. [DOI] [PubMed] [Google Scholar]
- [9].Brown MC, Hopkins WG, Keynes RJ, Importance of pathway formation for nodal sprout production in partly denervated muscles 15, Brain Res 243 (1982) 345–349. [DOI] [PubMed] [Google Scholar]
- [10].Burden SJ, SnapShot: Neuromuscular Junction 7, Cell 144 (2011) 826–826. [DOI] [PubMed] [Google Scholar]
- [11].Call JA, Warren GL, Verma M, Lowe DA, Acute failure of action potential conduction in mdx muscle reveals new mechanism of contraction-induced force loss 1, J.Physiol 591 (2013) 3765–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Carlson CG, Roshek DM, Adult dystrophic (mdx) endplates exhibit reduced quantal size and enhanced quantal variation 2, Pflugers Arch 442 (2001) 369–375. [DOI] [PubMed] [Google Scholar]
- [13].Carter GT, Longley KJ, Entrikin RK, Electromyographic and nerve conduction studies in the mdx mouse 2, Am.J.Phys.Med.Rehabil 71 (1992) 2–5. [DOI] [PubMed] [Google Scholar]
- [14].Chamberlain JS, Metzger J, Reyes M, Townsend D, Faulkner JA, Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma 3, FASEB J 21 (2007) 2195–2204. [DOI] [PubMed] [Google Scholar]
- [15].Chan S, Head SI, Morley JW, Branched fibers in dystrophic mdx muscle are associated with a loss of force following lengthening contractions, Am.J.Physiol Cell Physiol 293 (2007) C985–C992. [DOI] [PubMed] [Google Scholar]
- [16].Chipman PH, Franz CK, Nelson A, Schachner M, Rafuse VF, Neural cell adhesion molecule is required for stability of reinnervated neuromuscular junctions 1, Eur.J.Neurosci 31 (2010) 238–249. [DOI] [PubMed] [Google Scholar]
- [17].Cros D, Harnden P, Pellissier JF, Serratrice G, Muscle hypertrophy in Duchenne muscular dystrophy. A pathological and morphometric study 1, J.Neurol 236 (1989) 43–47. [DOI] [PubMed] [Google Scholar]
- [18].Derry KL, Venance SL, Doherty TJ, Decomposition-based quantitative electromyography in the evaluation of muscular dystrophy severity 1, Muscle Nerve 45 (2012) 507–513. [DOI] [PubMed] [Google Scholar]
- [19].Deschenes MR, Judelson DA, Kraemer WJ, Meskaitis VJ, Volek JS, Nindl BC, Harman FS, Deaver DR, Effects of resistance training on neuromuscular junction morphology, Muscle Nerve 23 (2000) 1576–1581. [DOI] [PubMed] [Google Scholar]
- [20].Deschenes MR, Maresh CM, Armstrong LE, Covault J, Kraemer WJ, Crivello JF, Endurance and resistance exercise induce muscle fiber type specific responses in androgen binding capacity, J.Steroid Biochem.Mol.Biol 50 (1994) 175–179. [DOI] [PubMed] [Google Scholar]
- [21].Deschenes MR, Maresh CM, Crivello JF, Armstrong LE, Kraemer WJ, Covault J, The effects of exercise training of different intensities on neuromuscular junction morphology, J Neurocytol 22 (1993) 603–615. [DOI] [PubMed] [Google Scholar]
- [22].Deschenes MR, Roby MA, Glass EK, Aging influences adaptations of the neuromuscular junction to endurance training, Neuroscience 190 (2011) 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der MJ, Holt KH, Lim LE, Sanes JR, Davidson BL, Faulkner JA, Williamson R, Campbell KP, Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice, Journal of Cell Biology 142 (1998) 1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Elkerdany MK, Fahim MA, Age changes in neuromuscular junctions of masseter muscle 9, Anat.Rec 237 (1993) 291–295. [DOI] [PubMed] [Google Scholar]
- [25].Eusebio A, Oliveri F, Barzaghi P, Ruegg MA, Expression of mouse agrin in normal, denervated and dystrophic muscle 1, Neuromuscul.Disord 13 (2003) 408–415. [DOI] [PubMed] [Google Scholar]
- [26].Fahim MA, Endurance exercise modulates neuromuscular junction of C57BL/6NNia aging mice 1, J.Appl.Physiol 83 (1997) 59–66. [DOI] [PubMed] [Google Scholar]
- [27].Fambrough DM, Control of acetylcholine receptors in skeletal muscle 7, Physiol Rev 59 (1979) 165–227. [DOI] [PubMed] [Google Scholar]
- [28].Fambrough DM, Devreotes PN, Gardner JM, Card DJ, The life history of acetylcholine receptors 6, Prog.Brain Res 49 (1979) 325–334. [DOI] [PubMed] [Google Scholar]
- [29].Ferraiuolo L, De Bono JP, Heath PR, Holden H, Kasher P, Channon KM, Kirby J, Shaw PJ, Transcriptional response of the neuromuscular system to exercise training and potential implications for ALS, J Neurochem 109 (2009) 1714–1724. [DOI] [PubMed] [Google Scholar]
- [30].Ferre J, Mayayo E, Brunet R, Morphometric study of the neuromuscular synapses in the adult rat with special reference to the remodelling concept 1, Biol.Cell 60 (1987) 133–144. [DOI] [PubMed] [Google Scholar]
- [31].Frascarelli M, Rocchi L, Feola I, EMG computerized analysis of localized fatigue in Duchenne muscular dystrophy 3, Muscle Nerve 11 (1988) 757–761. [DOI] [PubMed] [Google Scholar]
- [32].Ghazanfari N, Fernandez KJ, Murata Y, Morsch M, Ngo ST, Reddel SW, Noakes PG, Phillips WD, Muscle specific kinase: organiser of synaptic membrane domains 1, Int.J.Biochem.Cell Biol 43 (2011) 295–298. [DOI] [PubMed] [Google Scholar]
- [33].Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR, Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin--glycoprotein complex, Neuron 25 (2000) 279–293. [DOI] [PubMed] [Google Scholar]
- [34].Grinnell AD, Dynamics of nerve-muscle interaction in developing and mature neuromuscular junctions 1, Physiol Rev 75 (1995) 789–834. [DOI] [PubMed] [Google Scholar]
- [35].Gumerson JD, Davis CS, Kabaeva ZT, Hayes JM, Brooks SV, Michele DE, Muscle-specific expression of LARGE restores neuromuscular transmission deficits in dystrophic LARGE(myd) mice, Hum Mol Genet 22 (2013) 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hack AA, Ly CT, Jiang F, Clendenin CJ, Sigrist KS, Wollmann RL, McNally EM, Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin, J.Cell Biol 142 (1998) 1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Haddix SG, Lee YI, Kornegay JN, Thompson WJ, Cycles of myofiber degeneration and regeneration lead to remodeling of the neuromuscular junction in two mammalian models of Duchenne muscular dystrophy, PLoS One 13 (2018) e0205926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Han JJ, Carter GT, Ra JJ, Abresch RT, Chamberlain JS, Robinson LR, Electromyographic studies in mdx and wild-type C57 mice 3, Muscle Nerve 33 (2006) 208–214. [DOI] [PubMed] [Google Scholar]
- [39].Herbst R, Iskratsch T, Unger E, Bittner RE, Aberrant development of neuromuscular junctions in glycosylation-defective Large(myd) mice, Neuromuscul Disord 19 (2009) 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hesser BA, Henschel O, Witzemann V, Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK 1, Mol.Cell Neurosci 31 (2006) 470–480. [DOI] [PubMed] [Google Scholar]
- [41].Jang YC, Van Remmen H, Age-associated alterations of the neuromuscular junction 2, Exp.Gerontol 46 (2011) 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jansen M, van Alfen N, Geurts AC, de Groot IJ, Assisted bicycle training delays functional deterioration in boys with Duchenne muscular dystrophy: the randomized controlled trial “no use is disuse”, Neurorehabil Neural Repair 27 (2013) 816–827. [DOI] [PubMed] [Google Scholar]
- [43].Jerusalem F, Engel AG, Gomez MR, Duchenne dystrophy. II. Morphometric study of motor end-plate fine structure, Brain 97 (1974) 123–130. [DOI] [PubMed] [Google Scholar]
- [44].Kawabuchi M, Tan H, Wang S, Age affects reciprocal cellular interactions in neuromuscular synapses following peripheral nerve injury 1, Ageing Res.Rev 10 (2011) 43–53. [DOI] [PubMed] [Google Scholar]
- [45].Kong J, Anderson JE, Dystrophin is required for organizing large acetylcholine receptor aggregates 1, Brain Res 839 (1999) 298–304. [DOI] [PubMed] [Google Scholar]
- [46].Kong J, Yang L, Li Q, Cao J, Yang J, Chen F, Wang Y, Zhang C, The absence of dystrophin rather than muscle degeneration causes acetylcholine receptor cluster defects in dystrophic muscle, Neuroreport 23 (2012) 82–87. [DOI] [PubMed] [Google Scholar]
- [47].Krishnan VS, Aartsma-Rus A, Overzier M, Lutz C, Bogdanik L, Grounds MD, Implications of increased S100beta and Tau5 proteins in dystrophic nerves of two mdx mouse models for Duchenne muscular dystrophy, Mol Cell Neurosci (2020) 103484. [DOI] [PubMed]
- [48].Kulakowski SA, Parker SD, Personius KE, Reduced TrkB expression results in precocious age-like changes in neuromuscular structure, neurotransmission, and muscle function 1, J.Appl.Physiol 111 (2011) 844–852. [DOI] [PubMed] [Google Scholar]
- [49].Labovitz SS, Robbins N, Fahim MA, Endplate topography of denervated and disused rat neuromuscular junctions: comparison by scanning and light microscopy 1, Neuroscience 11 (1984) 963–971. [DOI] [PubMed] [Google Scholar]
- [50].Li Y, Lee Y, Thompson WJ, Changes in aging mouse neuromuscular junctions are explained by degeneration and regeneration of muscle fiber segments at the synapse 1, J.Neurosci 31 (2011) 14910–14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Li Y, Thompson WJ, Nerve terminal growth remodels neuromuscular synapses in mice following regeneration of the postsynaptic muscle fiber 1, J.Neurosci 31 (2011) 13191–13203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Love DR, Hill DF, Dickson G, Spurr NK, Byth BC, Marsden RF, Walsh FS, Edwards YH, Davies KE, An autosomal transcript in skeletal muscle with homology to dystrophin, Nature 339 (1989) 55–58. [DOI] [PubMed] [Google Scholar]
- [53].Lovering RM, Michaelson L, Ward CW, Malformed mdx myofibers have normal cytoskeletal architecture yet altered EC coupling and stress-induced Ca2+ signaling, Am.J.Physiol Cell Physiol 297 (2009) C571–C580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lovering RM, Porter NC, Bloch RJ, The muscular dystrophies: from genes to therapies, Phys.Ther 85 (2005) 1372–1388. [PMC free article] [PubMed] [Google Scholar]
- [55].Lyons PR, Slater CR, Structure and function of the neuromuscular junction in young adult mdx mice 1, J.Neurocytol 20 (1991) 969–981. [DOI] [PubMed] [Google Scholar]
- [56].Marques MJ, Taniguti AP, Minatel E, Neto HS, Nerve terminal contributes to acetylcholine receptor organization at the dystrophic neuromuscular junction of mdx mice 2, Anat.Rec.(Hoboken.) 290 (2007) 181–187. [DOI] [PubMed] [Google Scholar]
- [57].Mathur S, Lott DJ, Senesac C, Germain SA, Vohra RS, Sweeney HL, Walter GA, Vandenborne K, Age-related differences in lower-limb muscle cross-sectional area and torque production in boys with Duchenne muscular dystrophy 2, Arch.Phys.Med.Rehabil 91 (2010) 1051–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mejat A, Decostre V, Li J, Renou L, Kesari A, Hantai D, Stewart CL, Xiao X, Hoffman E, Bonne G, Misteli T, Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy, J Cell Biol 184 (2009) 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Minatel E, Santo NH, Marques MJ, Acetylcholine receptors and neuronal nitric oxide synthase distribution at the neuromuscular junction of regenerated muscle fibers 1, Muscle Nerve 24 (2001) 410–416. [DOI] [PubMed] [Google Scholar]
- [60].Muntoni F, Mateddu A, Marchei F, Clerk A, Serra G, Muscular weakness in the mdx mouse 2, J.Neurol.Sci 120 (1993) 71–77. [DOI] [PubMed] [Google Scholar]
- [61].Nicole S, Azuma Y, Bauche S, Eymard B, Lochmuller H, Slater C, Congenital Myasthenic Syndromes or Inherited Disorders of Neuromuscular Transmission: Recent Discoveries and Open Questions, J Neuromuscul Dis 4 (2017) 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nishimune H, Stanford JA, Mori Y, Role of exercise in maintaining the integrity of the neuromuscular junction, Muscle Nerve 49 (2014) 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Personius KE, Sawyer RP, Variability and failure of neurotransmission in the diaphragm of mdx mice, Neuromuscul Disord 16 (2006) 168–177. [DOI] [PubMed] [Google Scholar]
- [64].Pilgram GS, Potikanond S, Baines RA, Fradkin LG, Noordermeer JN, The roles of the dystrophin-associated glycoprotein complex at the synapse, Mol Neurobiol 41 (2010) 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Pisani C, Strimpakos G, Gabanella F, Di Certo MG, Onori A, Severini C, Luvisetto S, Farioli-Vecchioli S, Carrozzo I, Esposito A, Canu T, Mattei E, Corbi N, Passananti C, Utrophin up-regulation by artificial transcription factors induces muscle rescue and impacts the neuromuscular junction in mdx mice, Biochim Biophys Acta Mol Basis Dis 1864 (2018) 1172–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Prakash YS, Miyata H, Zhan WZ, Sieck GC, Inactivity-induced remodeling of neuromuscular junctions in rat diaphragmatic muscle 1, Muscle Nerve 22 (1999) 307–319. [DOI] [PubMed] [Google Scholar]
- [67].Prakash YS, Zhan WZ, Miyata H, Sieck GC, Adaptations of diaphragm neuromuscular junction following inactivity 4, Acta Anat.(Basel) 154 (1995) 147–161. [DOI] [PubMed] [Google Scholar]
- [68].Pratt SJ, Shah SB, Ward CW, Inacio MP, Stains JP, Lovering RM, Effects of in vivo injury on the neuromuscular junction in healthy and dystrophic muscles 1, J.Physiol 591 (2013) 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Pratt SJ, Shah SB, Ward CW, Kerr JP, Stains JP, Lovering RM, Recovery of altered neuromuscular junction morphology and muscle function in mdx mice after injury, Cell Mol.Life Sci (2014). [DOI] [PMC free article] [PubMed]
- [70].Pratt SJ, Valencia AP, Le GK, Shah SB, Lovering RM, Pre- and postsynaptic changes in the neuromuscular junction in dystrophic mice, Front Physiol 6 (2015) 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Pratt SJP, Iyer SR, Shah SB, Lovering RM, Imaging Analysis of the Neuromuscular Junction in Dystrophic Muscle, Methods Mol.Biol 1687 (2018) 57–72. [DOI] [PubMed] [Google Scholar]
- [72].Priez A, Duchene J, Goubel F, Duchenne muscular dystrophy quantification: a multivariate analysis of surface EMG 1, Med.Biol.Eng Comput 30 (1992) 283–291. [DOI] [PubMed] [Google Scholar]
- [73].Punga AR, Maj M, Lin S, Meinen S, Ruegg MA, MuSK levels differ between adult skeletal muscles and influence postsynaptic plasticity 3, Eur.J.Neurosci 33 (2011) 890–898. [DOI] [PubMed] [Google Scholar]
- [74].Rafael JA, Townsend ER, Squire SE, Potter AC, Chamberlain JS, Davies KE, Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure, Hum Mol Genet 9 (2000) 1357–1367. [DOI] [PubMed] [Google Scholar]
- [75].Rich M, Lichtman JW, Motor nerve terminal loss from degenerating muscle fibers, Neuron 3 (1989) 677–688. [DOI] [PubMed] [Google Scholar]
- [76].Robbins N, Compensatory plasticity of aging at the neuromuscular junction 10, Exp.Gerontol 27 (1992) 75–81. [DOI] [PubMed] [Google Scholar]
- [77].Roy P, Rau F, Ochala J, Messeant J, Fraysse B, Laine J, Agbulut O, Butler-Browne G, Furling D, Ferry A, Dystrophin restoration therapy improves both the reduced excitability and the force drop induced by lengthening contractions in dystrophic mdx skeletal muscle, Skelet Muscle 6 (2016) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rudolf R, Khan MM, Labeit S, Deschenes MR, Degeneration of neuromuscular junction in age and dystrophy, Front Aging Neurosci 6 (2014) 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sanes JR, Lichtman JW, Development of the vertebrate neuromuscular junction, Annu.Rev.Neurosci 22 (1999) 389–442. [DOI] [PubMed] [Google Scholar]
- [80].Santo NH, Martins AJ, Minatel E, Marques MJ, Axonal sprouting in mdx mice and its relevance to cell and gene mediated therapies for Duchenne muscular dystrophy 1, Neurosci.Lett 343 (2003) 67–69. [DOI] [PubMed] [Google Scholar]
- [81].Sharma KR, Mynhier MA, Miller RG, Muscular fatigue in Duchenne muscular dystrophy, Neurology 45 (1995) 306–310. [DOI] [PubMed] [Google Scholar]
- [82].Shiao T, Fond A, Deng B, Wehling-Henricks M, Adams ME, Froehner SC, Tidball JG, Defects in neuromuscular junction structure in dystrophic muscle are corrected by expression of a NOS transgene in dystrophin-deficient muscles, but not in muscles lacking alpha- and beta1-syntrophins 1, Hum.Mol.Genet 13 (2004) 1873–1884. [DOI] [PubMed] [Google Scholar]
- [83].Sieck DC, Zhan WZ, Fang YH, Ermilov LG, Sieck GC, Mantilla CB, Structure-activity relationships in rodent diaphragm muscle fibers vs. neuromuscular junctions 1, Respir.Physiol Neurobiol 180 (2012) 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Slater CR, ‘Fragmentation’ of NMJs: a sign of degeneration or regeneration? A long journey with many junctions, Neuroscience 439 (2020) 28–40. [DOI] [PubMed] [Google Scholar]
- [85].Tinsley J, Deconinck N, Fisher R, Kahn D, Phelps S, Gillis JM, Davies K, Expression of full-length utrophin prevents muscular dystrophy in mdx mice, Nat.Med 4 (1998) 1441–1444. [DOI] [PubMed] [Google Scholar]
- [86].Trajanovska S, Ban J, Huang J, Gregorevic P, Morsch M, Allen DG, Phillips WD, Muscle specific kinase protects dystrophic mdx mouse muscles from eccentric contraction-induced loss of force-producing capacity, J Physiol 597 (2019) 4831–4850. [DOI] [PubMed] [Google Scholar]
- [87].van der Pijl EM, van Putten M, Niks EH, Verschuuren J, Aartsma-Rus A, Plomp JJ, Low dystrophin levels are insufficient to normalize the neuromuscular synaptic abnormalities of mdx mice, Neuromuscul Disord 28 (2018) 427–442. [DOI] [PubMed] [Google Scholar]
- [88].van der Pijl EM, van Putten M, Niks EH, Verschuuren JJ, Aartsma-Rus A, Plomp JJ, Characterization of neuromuscular synapse function abnormalities in multiple Duchenne muscular dystrophy mouse models, Eur J Neurosci 43 (2016) 1623–1635. [DOI] [PubMed] [Google Scholar]
- [89].Wilson MH, Deschenes MR, The neuromuscular junction: anatomical features and adaptations to various forms of increased, or decreased neuromuscular activity 2, Int.J.Neurosci 115 (2005) 803–828. [DOI] [PubMed] [Google Scholar]
- [90].Wood SJ, Slater CR, The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast- and slow-twitch muscles, J.Physiol 500 ( Pt 1) (1997) 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wood SJ, Slater CR, Safety factor at the neuromuscular junction, Prog.Neurobiol 64 (2001) 393–429. [DOI] [PubMed] [Google Scholar]
- [92].Xu R, Salpeter MM, Acetylcholine receptors in innervated muscles of dystrophic mdx mice degrade as after denervation 2, J.Neurosci 17 (1997) 8194–8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.