

Abstract
Advanced ovarian cancers are a leading cause of cancer-related death in women and are currently treated with surgery and chemotherapy. This standard of care is often temporarily successful but exhibits a high rate of relapse after which treatment options are few. Here we investigate whether biomarker-guided use of multiple targeted therapies, including small molecules and antibody-drug conjugates, is a viable alternative. A panel of patient-derived ovarian cancer xenografts (PDX), similar in genetics and chemotherapy responsiveness to human tumors, was exposed to 21 mono and combination therapies. Three monotherapies and one combination were found to be active in different subsets of PDX. Analysis of gene expression data identified biomarkers associated with responsiveness to each of the three targeted therapies, none of which directly inhibits an oncogenic driver. While no single treatment had as high a response rate as chemotherapy, nearly 90% of PDX were eligible for and responded to at least one biomarker-guided treatment, including tumors resistant to standard chemotherapy. The distribution of biomarker positivity in TCGA data suggests the potential for a similar precision approach in human patients.
Keywords: Ovarian cancer, PDX, precision medicine, PI3K inhibitor, MEK inhibitor
INTRODUCTION
Epithelial ovarian cancer is among the leading causes of cancer-related death in women. The high mortality of this cancer reflects the fact that it is often at an advanced stage when diagnosed and that few effective therapies exist for recurrent disease (1). The most common treatment for advanced-stage epithelial ovarian cancer is surgical resection followed by taxane/platinum-based chemotherapy; surgery is not always possible and some patients proceed directly to chemotherapy. A high percentage of patients respond initially but the disease usually relapses, resulting in 5-year survival of around 30% (2,3). Angiogenesis and PARP inhibitors have improved overall survival in ovarian cancer (4,5), and when used in combination, the drugs have recently been shown to significantly increase the duration of tumor control (6) although rates of long term remission are not yet known. The FDA recently approved Olaparib plus Bevacizumab as first-line maintenance treatment for homologous recombination deficient ovarian cancers (7), making the combination one of the first demonstrated successes of targeted therapy in a first-line setting for ovarian cancer. Thus, after decades that have witnessed relatively few new therapeutic strategies, there is increasing promise in the use of targeted therapies as first-line treatment.
The difficulty in developing targeted therapies for epithelial ovarian cancers arises in part because the mutational spectrum of the disease does not exhibit many recurrent mutations in genes that might make attractive therapeutic targets; this is in contrast to other types of cancer for which targeted therapies have been developed for specific, biomarker-defined subtypes (e.g. lung adenocarcinoma, chronic and acute myeloid leukemia, melanoma, HER2+ breast cancer). In ovarian cancers, mutations in the P53 and homologous recombination repair pathways are common (96% and 22% of tumors respectively), but few patients have additional mutations that can be used to guide therapy (reviewed in Coward et al, 2015 (4)). Ovarian cancers nonetheless display substantial inter-patient heterogeneity in therapeutic response, suggesting the existence of as-yet unrecognized determinants of drug sensitivity and resistance and the potential for personalized treatment regimens. Preclinical models that reflect the heterogeneous biology of this cancer have the potential to improve treatment by providing data on therapeutic vulnerabilities that can be associated with specific biomarkers.
In contrast to cell lines and conventional xenografts, patient-derived xenografts (PDXs) often preserve histopathologic and genetic features of the original tumors at the time of their resection (8–14). PDX models are not an ideal mimic of human disease, in part because xenografting human tumors requires the use of mice that lack functional adaptive immune systems, and partly due to genomic aberrations such as copy number alternations acquired during PDX passaging (15). Nonetheless, PDX models remain one of the best experimental systems in which changes in tumor volume and duration of progression-free survival (PFS) can be assessed across a panel of genetically diverse tumors. Conventional xenografts and genetically engineered mouse models (GEMMs) lack this genetic diversity and studies with cell lines do not provide data on tumor progression. Informative PDX-based preclinical studies of patient to-patient variability in drug response have been reported for lung, breast, colon, melanoma and pancreatic cancers, including a previously described encyclopedia of >1000 PDXs (10,16,17). Moreover, the data in the current study demonstrate that ovarian tumor PDXs can recapitulate the clinically known association between BRCA loss-of-function mutations and sensitivity to the PARP inhibitor Olaparib, an association that is not reproduced in panels of ovarian cancer cell lines (Supplementary Fig. S1 (18–20)).
Here we focus on the effects of 29 well-characterized ovarian PDX models to 21 different drugs used as single agents and in combinations; to our knowledge, no similar study has been reported for this disease. The tumors derive from an existing PDX encyclopedia generate using anonymously donated specimens and span the range of mutations observed in human cohorts. We associate differences in response with differences in tumor gene expression, making it possible to nominate potential biomarkers for ovarian cancer therapies. We find that a majority of ovarian PDX tumors have transcriptional or genomic biomarkers that can be used to guide the selection one or more targeted treatments and that these treatments elicit tumor regression as great (by volume changes) as those associated with standard of care combination chemotherapy. Moreover, the great majority of ovarian cancers in TCGA are positive for at least one of the biomarkers uncovered in our study. Whether these biomarkers will translate into humans remains unknown, but our work serves as a proof-of-concept for the use of multiple biomarkers to guide treatment of ovarian cancer with a panel of drugs having diverse mechanisms of action. The drug sensitivity biomarkers we identify do not correspond to oncogenic drivers, in contrast to genome-informed stratification of disease such as non-small cell lung cancer (21), and our approach is therefore potentially applicable to other cancers with few ‘druggable’ oncogenes.
METHODS
Generation and profiling of patient-derived tumor xenografts for PDX trials
The following procedures were performed as described previously (10) and for completeness are described in full in Supplementary Methods: 1) Generation of ovarian patient-derived tumor xenografts, 2) Ovarian PDX subtype characteristics and take rate, 3) Histopathological characterization of established ovarian PDXs, 4) Genomic profiling of ovarian PDXs, 5) PDX Clinical Trial (PCT) and drug treatment. This includes definitions of tumor response and progression, and treatment dosages and duration for the therapies studied here (Supplementary Table 1).
Mice were maintained and handled in accordance with the Novartis Institutes for BioMedical Research (NIBR) Animal Care and Use Committee protocols and regulations. Patient tumor specimens were obtained from: the National Disease Research Interchange, Philadelphia, PA, USA; the Cooperative Human Tissue Network funded by National Cancer Institute, Rockville, MD, USA; Maine Medical Center, Portland, ME, USA. All patients provided written informed consent, samples were procured and the study was conducted under the approval of the review boards of each institution. Clinical and pathologic data were entered and maintained in appropriate databases.
From 247 anonymously donated treatment-naïve patient tumors, 49 were successfully engrafted (Supplementary Methods), and 37 underwent extensive genomic sequencing (summarized in Supplementary Figure S1; raw data in Supplementary Table 2). Among these models, which had variable growth characteristics and latency periods, 29 models with robust growth were propagated for therapeutic testing.
Note no cell lines were used to perform experiments study; previously released cell line data was re-analyzed to assist with characterization of the PDX models.
Identifying drug combinations with stronger than independent action
Both PFS and tumor volume measurements were analyzed to determine if any drug combinations achieved a response better than their best constituent single-agent response, which is a standard that few combination therapies surpass in vivo (22,23) and is evidence of additive or synergistic effect. Drug additivity and synergy can be defined rigorously in the setting of dose-response studies in cell lines (24,25), but there is currently no methodology for distinguishing additive from synergistic drug responses using tumor volume or survival data (since dose responses are not measured in this context). Our analysis of combination drug action therefore assesses whether drug activity is consistent with a null hypothesis of independent action, which corresponds to tumors responding to the best single agent. If the null hypothesis can be rejected, then evidence exists that tumors have a stronger response to combination therapy than the best single agent, which can involve either additivity or synergy (22).
This analysis was performed for the nine combinations that also had response data for their constituent monotherapies. For the PFS analysis, a survival distribution was generated for the two PDX monotherapy responses that constituted a combination. To simulate the combination response expected by independent action (22), a value was randomly sampled from each of the monotherapy response distributions, and the best of the two values was chosen. This procedure was repeated 106 times to build the simulated response distribution. A number of responses equal to the number of observed combination responses was then selected at equally spaced time intervals from the full simulated response distribution. The final simulated response distribution and the observed combination response distribution were compared using a Cox proportional hazards model, with corresponding relative risk scores and p-values.
To determine statistical significance, we examined differences in responses to two PI3K inhibitors (BKM120 and CLR457) to estimate the magnitude of experimental error. A null distribution of differences in drug response was constructed by comparing tumor volume changes from BKM120 and CLR457 in 29 PDX models. For each combination therapy, volume changes expected by independent action were calculated by picking the better of the two constituent monotherapy volume changes for each PDX model. This resulting distribution of observed volume changes minus the expected independent action volume changes was compared to the null distribution (differences expected from experimental error). A Mann-Whitney test assessed whether the difference between the two volume difference distributions was significant and therefore whether the combination was significantly superior to independent action.
Discovery of drug-response biomarkers
Identification of genes whose expression levels are associated with drug sensitivity was performed separately for BKM120 plus binimetinib, LSU691, HKT288, and olaparib. Genes were selected that had highly variable in expression across the population of PDX tumors; specifically, those with variance in log2 (transcript abundance in TPM) > 2. For each such high-variance gene, two metrics were calculated: (1) the significance of association (−log10 P) between transcript abundance and volume change (best average response) by Spearman’s rank correlation, and (2) the significance (−log10 P) of the Hazard Ratio for disease progression between PDXs with less than median expression, and PDXs with greater or equal to median expression, by the Cox Proportional Hazards model. We elected not to explore different expression thresholds to avoid the risk of ‘p-hacking’. A dichotomous threshold for ‘biomarker positivity’ was implemented in accordance with clinical practice in oncology; for instance, companion diagnostics for immune checkpoint inhibitors assess whether histological samples surpass a pre-defined threshold for PD-L1 expression (26).
On a scatterplot of −log10 P (association with tumor volume change) versus −log10 P (association with hazard ratio for PFS), the 20 genes at the Pareto frontier were selected, that is, those most strongly associated with both volume change and PFS. For these genes, literature was searched for research articles describing the gene and the drug, or the gene and the drug’s target. Genes were only considered candidate biomarkers if literature described a mechanistically plausible interaction that might determine drug response (for example, an efflux pump with known activity against the drug). Finally, the procedure was repeated with scrambled drug response labels 100 times per drug, and the fraction of cases was counted in which any gene exhibited equal or better significance of association with tumor volume change and PFS. A gene only remained a candidate biomarker if this procedure indicated a False Discovery Rate ≤ 25% for the statistical association with drug response. Given that few candidate genes found literature support for interacting with a drug, the False Discovery Rate for both statistical association and literature support is estimated to be <5%.
RESULTS
Ovarian PDXs recapitulate the histopathology and genomics of human epithelial ovarian cancers
We studied 29 ovarian PDXs established from anonymously donated treatment-naïve patient tumors. The panel was representative of the histological and genetic diversity of human ovarian tumors and consisted of 18 serous, 5 mixed, two NS, one clear cell, one endometrioid subtypes as well as two additional gynecologic PDXs with unconfirmed origin. H&E staining showed that the PDXs closely recapitulated the histopathologic characteristics of the original patient tumors (Fig. 1A) except that human stroma were replaced by mouse stroma after two passages in mice, as previously reported for other ovarian PDX studies (27–29).
Figure 1. Histopathologic and genomic characterization of ovarian PDXs.
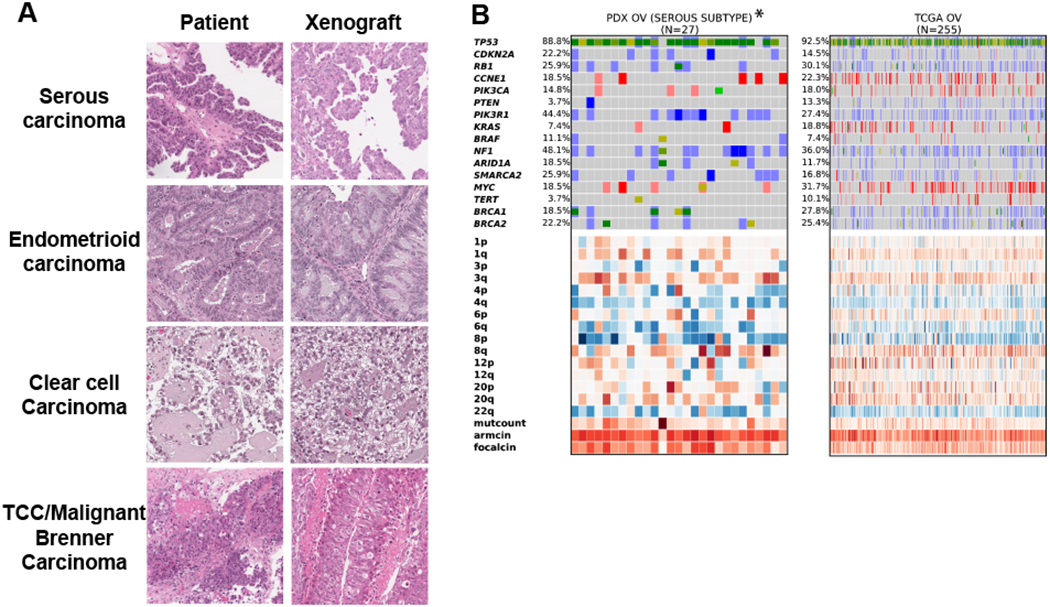
A, Representative histologic characteristics of the original patient tumors and corresponding xenografts (passage 2) by hematoxylin and eosin staining. B, Genomic landscape analysis of serous ovarian carcinoma of PDXs and patient tumors (TCGA). Parenthesis, number of models per indication; blue, homozygous deletions; light blue, heterozygous deletion; salmon, amplification > 5 copies; red, amplification > 8 copies; bright green, known COSMIC gain-of-function mutations; dark green, truncating mutations / frameshift or known COSMIC loss-of-function; mustard, novel mutation. Copy number heatmap scaled from blue (deletion) to white (average CN) to red (amplification); expression heatmap scaled from blue (3 standard deviations below mean) to red (3 standard deviations above mean).
To determine if the PDX cohort is representative of the genetic heterogeneity of human disease, we generated transcript and copy number profiles and compared the data to profiles from 255 patients with high-grade serous tumors found in The Cancer Genome Atlas (TCGA) (30) (Fig. 1B). Transcripts were mapped simultaneously to either human (tumor) or mouse (stroma). We observed that the frequency of genetic alterations across the two datasets was similar. For instance, TP53 was mutated and/or deleted in 89% of PDXs and 93% of patient tumors, and Cyclin E was amplified in 19% of PDXs and 22% of patient tumors. None of the 16 oncogenes or tumor suppressors frequently mutated in ovarian cancers demonstrated significant enrichment or depletion in high-grade serous PDX tumors relative to TCGA data. As is typical for high-grade serous ovarian cancer (and TP53 mutant cancers in general), most PDXs and TCGA tumors demonstrated high levels of chromosomal instability and relatively low mutation rates (Fig. 1B). Genetic data for all profiled ovarian PDXs (n = 37), cell lines (CCLE), and patient tumors (TCGA) is available in Supplementary Fig. S2 and Supplementary Table 2. We were not able to match PDX characteristics to patient data because the cohort was assembled from anonymous donors (as is common for large PDX panels) and the study protocol did not include clinical data collection subsequent to sample collection. We nonetheless conclude that the genetic profiles of PDX tumors used in this study are a representative sample of the genetic variability of human ovarian tumors.
Testing new therapies for ovarian cancer in PDX libraries
To identify new therapeutic strategies for advanced-stage epithelial ovarian cancer, we tested 21 therapeutic approaches involving 10 single agents and 11 drug combinations (Supplementary Table 3). Maximum tolerated dose was used for agents that have not entered the clinics, while clinically relevant doses were used for agents that are currently in use or have been evaluated in humans (Methods, Supplementary Table 1). As reported previously, good reproducibility is observed when the same PDX tumor is challenged with the same drug in different mice (10). In a data set comprising 440 examples of replicate treatment for a single type of PDX (2138 animals total) fewer than 10% of responses differed from the consensus RECIST response by more than one category (categories comprised: CR, complete response; PR, partial response; SD stable disease; or PD, progressive disease). Additionally, the distinction between any response (CR, PR, SD) and no response (PD) was consistent in 95% of individual animals. This finding justifies the use of one animal per tumor per treatment (a 1×1×1 design) to enable screening of a larger collection of treatments.
We used RECIST criteria to summarize response rates in a manner comparable to human trials, but for all analyses we used continuous data on changes in tumor volume and duration of progression-free survival (PFS; defined as time until tumor volume reached 200% of baseline (10)). Treatments were maintained for up to 150 days unless animal welfare considerations required euthanasia, in accordance with Novartis’ Global Animal Welfare Policy. Only 11 cases of treatment-related adverse events requiring euthanasia were observed for the 568 drug-treated murine cohort (2%); thus, euthanasia of on-treatment animals did not substantially impact our results. In cases in which animals experienced toxicities, their best response prior to removal from the trial was used for drug response analysis (Figure 2).
Figure 2. Tumor responses to novel treatments and standard chemotherapy in PDX-based clinical trials.
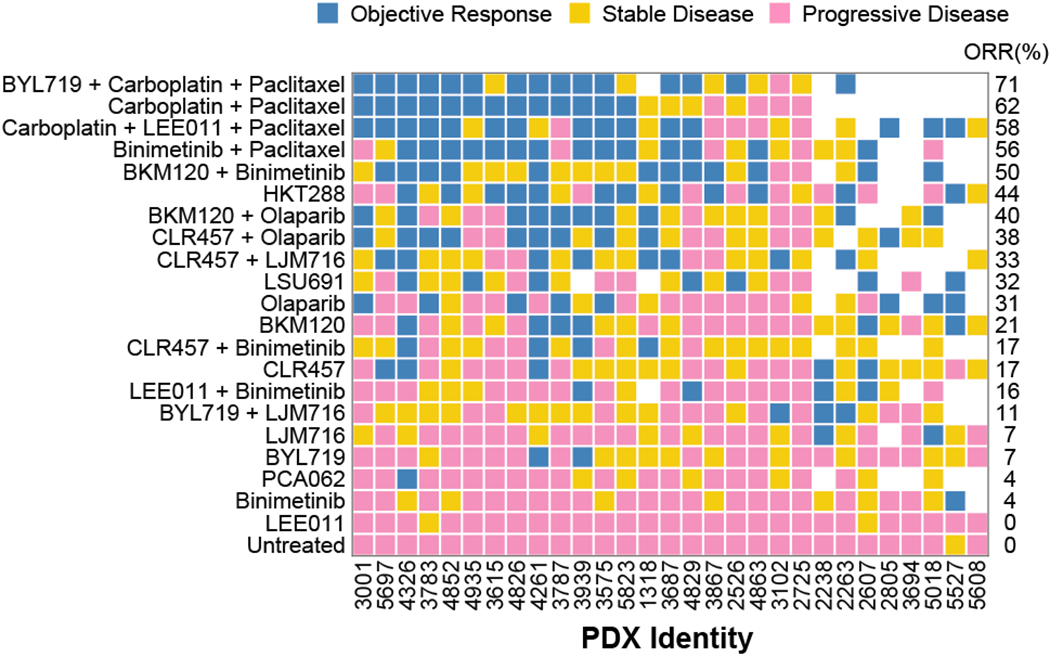
21 treatments were tested in 29 PDXs; each square represents a treated PDX. Objective response rate (ORR) is the percentage of PDXs with partial and complete responses to each therapy.
Combination chemotherapy (carboplatin/paclitaxel at clinically relevant doses) served as a standard-of-care comparator and elicited response rates comparable to those observed in patients with high-grade serous carcinoma (du Bois et al, 2003) (2): objective response rate (ORR, which is the sum of the CR and PR rates) was 62% in PDXs vs. 67% in humans, and disease control rate (the sum of CR, PR, and SD) was 81% in PDXs vs. 90% in humans. We found that all drugs and drug combinations tested were well-tolerated, with no significant drug-induced weight loss.
The drug sensitivity landscape was heterogeneous: no single therapy exhibited a better overall response rate than carboplatin/paclitaxel (62%), but 88% of tumors exhibited an objective response to at least one other treatment (Fig. 2, Supplementary Tables 2, 3). For all treatments, changes in tumor size were strongly correlated with duration of PFS (Spearman correlation −0.77, P = 10−109). Three drugs elicited objective responses in 25% or more of ovarian PDXs as single agents: (i) HKT288, an antibody-drug conjugate directed against Cadherin 6 (CDH6) (whose development we recently described (31)), (ii) LSU691, a small molecule inhibitor of Nicotinamide Phosphoribosyltransferase (NAMPT), and (iii) Olaparib, a small molecule inhibitor of Poly-ADP-Ribose Polymerases (PARP). PDX trials make it possible to compare the response of a single tumor to different drugs. This revealed that HKT288, LSU691, and BKM120 plus binimetinib induced responses in different subsets of tumors, including several that were resistant to standard chemotherapy (Fig. 2), resulting in an aggregate response rate of 88% as compared to 62% for carboplatin/paclitaxel.
A number of combination therapies exhibited promising activity in the PDX panel; in nine cases it was possible to compare the combination to its constituent drugs administered individually and test for independent or additive/synergistic activity (22). We found that, for eight of nine combinations, the response of each PDX to a combination was no better than the responses to individual drugs making up the combination, consistent with independent drug action (Supplementary Table 4). The single exception was the PI3K inhibitor BKM120 plus the MEK inhibitor binimetinib. Across all drugs tested as monotherapies, three PI3K inhibitors (BKM120, BYL719, and CLR457) resulted in modest and short-lived responses (by tumor volume and PFS respectively), recapitulating data in human ovarian cancers (32); binimetinib was even less active as a monotherapy. In contrast, BKM120 plus binimetinib (Fig. 3A) was superior to either drug alone and to a model of independent action with respect to both duration of progression-free survival (Fig. 3B) and changes in tumor volume (Fig. 3C). The combination achieved 50% ORR (as compared to 21% for BKM120 and 4% for binimetinib) and a median PFS >100 days as compared to 43 or 14 days respectively. For the combination vs. BKM120 alone, the Hazard Ratio (HR) was 0.24 (P=0.01 log-rank test) and for the combination vs. binimetinib alone HR=0.09 (P=10−5); relative to an independent action model, the combination achieved an HR=0.27 (P=0.01) demonstrating highly superior activity (Supplementary Table 3). We conclude that, among combinations tested, BKM120 plus binimetinib is unique in exhibiting strong positive drug-drug interaction; this observation is consistent with extensive pre-clinical evidence that the two drugs are synergistic in cell lines and that that MAPK activation can mediate resistance to PI3K inhibitors (32).
Figure 3: BKM 120 plus Binimetinib combination is superior to response expected by independent action.
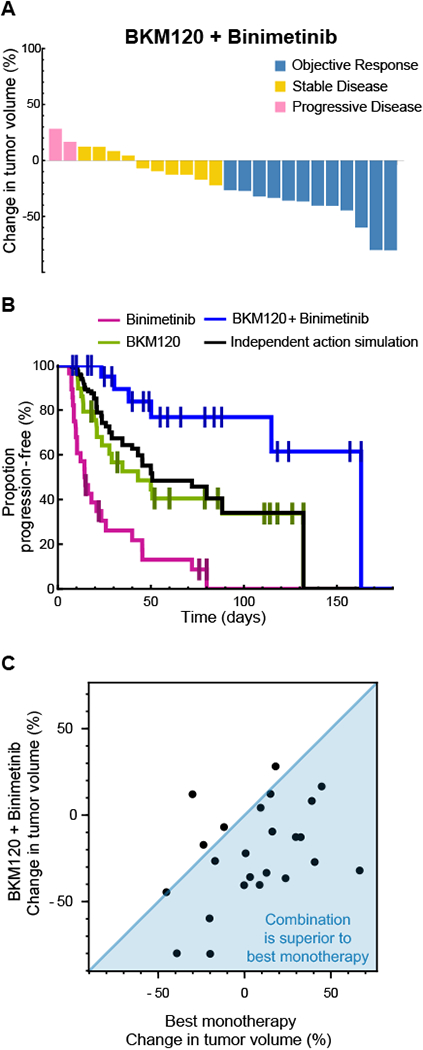
A, Tumor volume changes in ovarian PDXs (n=24) treated with BKM120 + Binimetinib. Color indicates RECIST response. B, Kaplan-Meier PFS curve of PDXs treated with: BKM120 (n=29), Binimetinib (n=28), and BKM120 + Binimetinib (n=24). Responses from each monotherapy arm were randomly sampled, and the best of the two responses was used to simulate the benefit of BKM120 + Binimetinib expected due to independent action alone. C, BKM120+Binimetinib tumor volume change per PDX plotted against best monotherapy volume shrinkage (BKM120 or Binimetinib response) for the same tumor (n=24).
Identifying biomarkers for targeted therapies using PDX libraries
Biomarkers associated with drug response are required to for stratified therapeutic approaches. To identify potential biomarkers we compared treatment data for PDX models with RNAseq profiles. We limited the search to ~5,000 protein-coding genes having a high variance in transcript abundance among PDX models. For each gene we calculated the significance of the correlation between transcript expression and treatment-induced changes in tumor volume change, and the significance of response duration (as assessed by hazard ratio) between tumors with higher or lower than median expression (Methods). We considered a gene a candidate response biomarker if it satisfied three criteria: (1) the gene was at the ‘Pareto front’ of significance - that is, among the 20 most significant in correlating with volume change and response duration; (2) the statistical association had a false discovery rate ≤ 25%, based on simulations in which drug responses were scrambled among tumors; (3) a literature search revealed a mechanistic relationship between the gene product and the drug or its target (Methods). The resulting rigorous quantification of false discovery rate, and integrating this feature into the process of biomarker discovery, are methodological advances of this study.
This procedure revealed candidate biomarkers for all four targeted therapies. Resistance to BKM120 plus binimetinib was associated with high expression of the multi-drug efflux pump ABCG2 (Fig. 4A). Binimetinib and related MEK inhibitors are known to be substrates of ABCG2 (33) and we found that the association with resistance was independently supported by binimetinib responsiveness in a panel of breast cancer PDXs (10) (correlation of ABCG2 expression with volume change ρ=0.33, P=0.046, n=37; the proportional hazard of low vs high ABCG2 groups was HR=0.50, P=0.08) (Fig. 4B). Sensitivity to the CDH6-targeting antibody HKT288 is associated with high expression of CDH6 itself, as was previously reported based on CDH6 immunohistochemistry (31).
Figure 4: Discovery of novel biomarkers for targeted therapies in ovarian cancer.
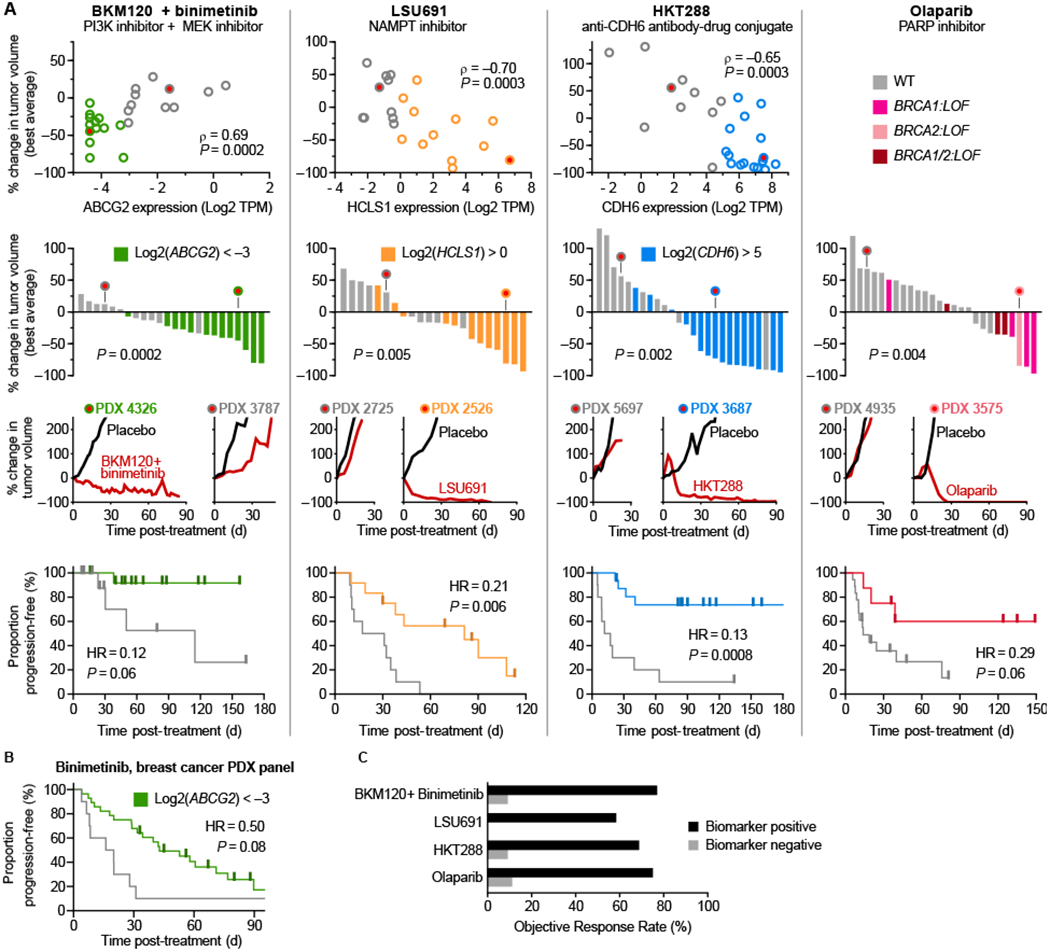
A, PDX treatment volume changes plotted against that model’s biomarker expression (TPM= transcripts per million) and as waterfall plots. Kaplan-Meier PFS curves are stratified into biomarker negative (gray) or positive (colored) groups (total n=24, 22, 26, and 26 respectively). B, Validation of ABCG2 biomarker in a separate data set measuring duration of binimetinib response in breast cancer PDXs (n=39). C, Objective response rate among biomarker positive and negative indications for each therapy.
Loss of BRCA1 and/or BRCA2 function (defined here as mutation of BRCA1 or BRCA2, or silencing of the BRCA1 transcript (34)) was the biomarker most strongly correlated with responsiveness to olaparib (Fig. 4). BRCA1/2 loss is a clinically validated biomarker of olaparib response (35) and reproducing this association serves as a validation of our PDX-based approach. In contrast, we find that data from cancer cell lines reveals no association between olaparib sensitivity and BRCA1/2 loss similarly defined (Supplementary Fig. S1). Our search for potential biomarkers identified USP51 expression as being similarly associated with olaparib response as BRCA loss-of-function; USP51 is recruited to double-strand breaks and regulates responses to DNA damage, including the assembly and disassembly of BRCA1 foci (36). These data suggest that a simple test involving measuring the levels of four transcripts might be sufficient to identify tumors with a higher than average rates of response to four targeted therapies. Moreover, when PDX tumors were divided into ‘treatment-eligible’ or ‘treatment ineligible’ groups based on these biomarkers (BRCA1/2 loss-of-function was used to score for olaparib), response rates were high in eligible PDX tumors (ORR 58% to 77%) and low in ineligible tumors (ORR 0% to 11%) (Fig. 4C).
Sensitivity to the NAMPT inhibitor LSU691 was associated with high expression of HCLS1, an anti-apoptotic protein that is activated by the NAMPT/NAD+/SIRT1 pathway. This interaction has previously been proposed as a therapeutic target in leukemia (37) and our data suggests that it also affects responsiveness to NAMPT inhibition in ovarian tumors. HCLS1 has not been independently validated as drug response biomarker and we therefore searched for associations between sensitivity to NAMPT inhibitors and basal gene expression in a panel of ~800 cancer cell lines in the Cancer Therapeutics Response Portal v2 (19). For three of four NAMPT inhibitors examined, we observe a correlation between HCLS1 levels and drug sensitivity (correlation z-score for: daporinad, −6.8; CAY10618, −6.9; STF-31, −8.4).
A possible confounder in this analysis as that PDX-nominated biomarkers might be associated with greater survival in general rather than higher drug response. To address this issue we used TCGA primary tumor transcriptional and survival data to look for associations between biomarkers and rates of disease progression irrespective of therapy. We found that no biomarker was associated with disease-free duration when the proportions of disease-free patients in the biomarker positive and negative groups were scored for ABCG2, HCLS1, CDH6, and BRCA1/2 status (Supplementary Fig. S3A, S3B, S3C, and S3D). Thus, PDX-nominated biomarkers appear to be associated with responsiveness to therapy, not the rapidity of disease progression.
Most ovarian PDXs are candidates for biomarker-guided treatments
We next assessed the proportion of PDX models potentially benefiting from biomarker-guided treatment strategy. The least established biomarker in our panel is HCLS1 as a predictor of LSU691 and we therefore performed the analysis with and without this biomarker. We found that a large majority of PDX tumors (89%) were biomarker-positive candidates for one or more of four therapies: BKM120 plus binimetinib, HKT288, olaparib or LSU691; this fell to 85% when LSU691 was excluded (Fig. 5A, Supplementary Fig. S4A). Analysis of the same biomarkers in human primary epithelial ovarian cancers (n= 232) in The Cancer Genome Atlas (TCGA) revealed that a similar fraction of human tumors (93% with or without HCLS1) are positive for one or more of the same biomarkers. Furthermore, the proportion of biomarker positive tumors in TCGA does not significantly differ between rapidly progressing disease (less than 12 months disease-free) and better controlled disease (greater than 12 months disease-free). Thus, biomarker-guided targeted therapy may be an option in tumors responsive to chemotherapy as well as primary progressive disease (Fig. 5A).
Figure 5: Most ovarian tumors respond to one of three biomarker-guided treatments.
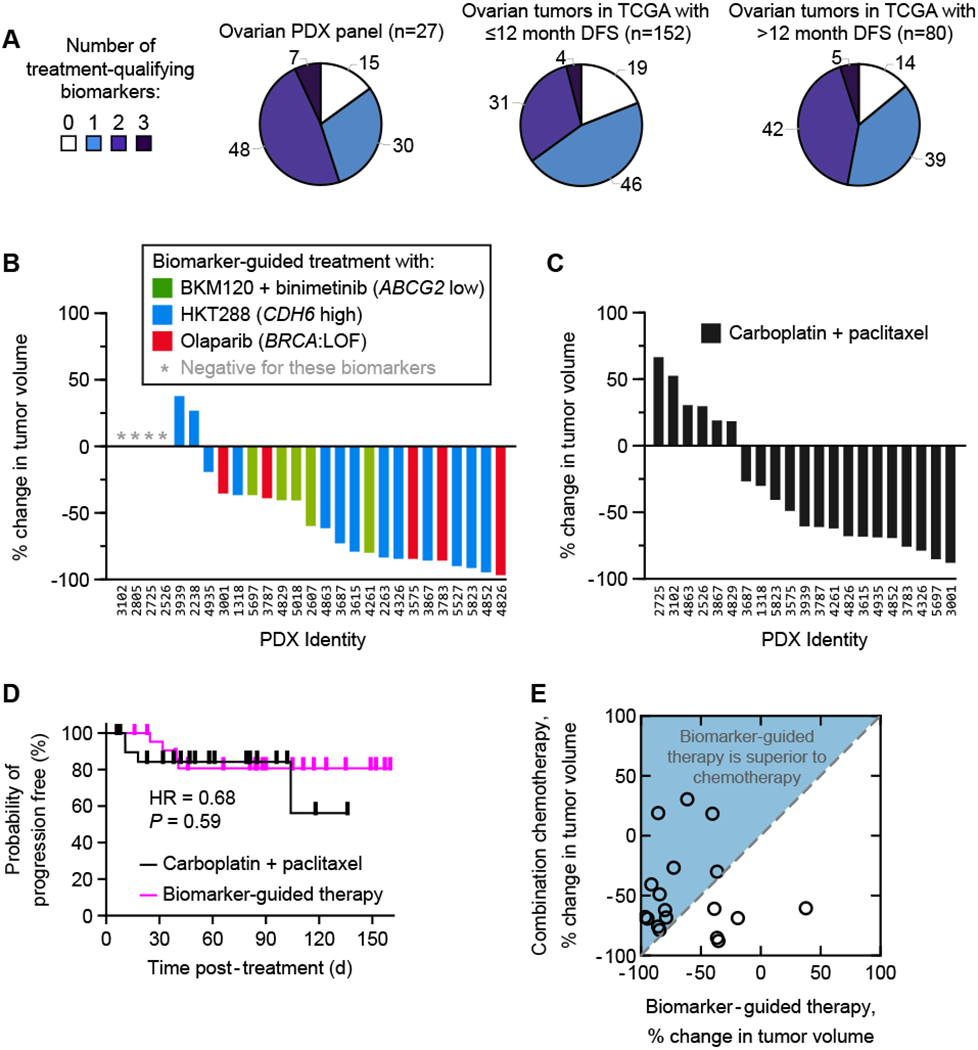
A, Proportion of ovarian tumors possessing one or more treatment-qualifying biomarkers (for BKM120 + binimetinib, HKT288 or Olaparib). B, Tumor volume changes in ovarian PDXs each treated with their best biomarker-guided therapy. C, Waterfall plots of PDX responses to standard of care chemotherapy (carboplatin + paclitaxel). D, Kaplan-Meier PFS curves of PDXs treated with biomarker-guided therapy (n= 23) as compared to standard of care paclitaxel/carboplatin (n=21). E, Biomarker-guided therapy tumor volume change per PDX plotted against combination chemotherapy volume shrinkage (for the same animal; n=18).
Among PDX tumors, 26% were ineligible or did not respond to one of three biomarker-indicated treatments, 37% experienced disease control from only one indicated treatment, and 37% had disease control from two indicated treatments (19%, 33%, and 48% respectively when the analysis was performed for four therapies, Supplementary Fig. S4A). When each PDX tumor was assigned to its single best biomarker-indicated treatment (as measured by tumor volume change), the resulting set of responses was comparable to the best responses observed for carboplatin/paclitaxel, both by volume change (Fig. 5B, 5C: no significant difference by Mann-Whitney test, P=0.47; Supplementary Fig. S4B, S4C: also not significant by analysis with four therapies P=0.17) and by response duration (Fig. 5D: no significant difference in hazard ratio, P= 0.59; Supplementary Fig. S4D: not significant with four therapies, P=0.44). Thus, a majority of ovarian PDX tumors respond strongly to a biomarker-guided treatment (ORR 74% in eligible tumors; 88% with a four-therapy strategy). Moreover, among the 38% of tumors that did not respond to paclitaxel/carboplatin, a majority (5 of 8) had an objective response to a biomarker-indicated targeted therapy (Fig. 5E; Supplementary Fig. S4E). Biomarker-guided therapy may therefore be an effective option for chemotherapy-resistant ovarian cancers, the most realistic setting in which to commence human testing.
DISCUSSION
Approval of the angiogenesis inhibitor bevacizumab and of PARP inhibitors has demonstrated the potential utility of targeted therapy in ovarian cancer, which is currently treated primarily with surgery and chemotherapy. The path to identifying additional therapies is complicated by a difficult-to-target mutational landscape (38). Moreover, standard combination chemotherapy is sufficiently effective to provide a high bar to a new therapy while nonetheless being ineffective for some patients and ultimately inadequate with respect to lasting tumor control. The panel of patient-derived ovarian tumor xenografts described in this paper was established to enable empirical comparison of multiple drug and drug combinations against standard chemotherapy, both across and within individual human tumors. The overall goal was to determine if a stratified approach to treating ovarian cancer involving multiple biomarkers and drugs was feasible in principle, while also identifying drug-gene associations with a sufficient mechanistic basis that follow-on analysis in human patients might be warranted.
In our study, strong anti-tumor activity (greater than 25% ORR) was observed for three single agents and one additive/synergistic drug combination, but as is common for targeted therapies, each drug or combination was active in only a subset of tumors. We identified genes whose expression was significantly associated with drug sensitivity for each of the four therapies (for olaparib, BRCA status is already an established biomarker (35) that was recapitulated in the PDX cohort), making it possible to stratify tumors into likely responders and non-responders. For example, a combination of the PI3K inhibitor BKM120 plus the MEK inhibitor binimetinib was most active in PDX tumors with low expression of the ABCG2 efflux pump, which has previously been shown to export binimetinib (33); we were able to validate the relationship between ABCG2 and binimetinib sensitivity in a breast cancer xenograft panel that is part of a previously described study (Fig. 4B). The correlation of ABCG2 with drug response was similar to that of other biomarkers in this study including BRCA loss-of-function and olaparib sensitivity; this is important because BRCA mutation is an approved clinical test in breast and ovarian cancers (35,39).
Encouragingly, recent work in triple-negative breast cancer has demonstrated a concordance between a de novo bio-marker discovered in PDX models (e.g. low SLFN11 expression) and prognostic power in patients (low survival in a 250 patient population) (40). In our study, four treatment options exhibited unstratified response rates between 31% and 48% and choosing among these using biomarkers achieved an aggregate objective response rate of 88% (a more conservative choice omitting the least validated drug and biomarker, namely LSU691 and HCLS1, achieved an aggregate ORR of 74%). Thus, most PDX tumors tested had the potential to benefit from a biomarker-guided precision therapy approach. We note that, even if the novel therapies described in this paper are not ultimately successful in humans (due to excess toxicity, for example), the heterogeneous drug response landscape (Fig. 2) and the capacity of PDX studies to identify responsive subpopulations (Fig. 4) constitutes a proof of concept for the wider use of precision therapy in ovarian cancer.
A more general result from our analysis is that biomarkers other than oncogenic mutations or amplifications can be used to nominate targeted therapies including antibody-drug conjugate, kinase inhibitors, and inhibitors of a metabolic enzyme. From the perspective of future translation, we believe that it is appropriate to search for biomarkers with a logical or mechanistic connection to drug response. In the case of ovarian cancer and the therapies studied here, analysis of biomarker prevalence in TCGA suggests that a precision medicine approach might be applicable to > 90% of patients. We have described independent validation for two of those biomarkers (from PDX or clinical data) and a third biomarker is self-evident. In particular, the relationship between the ABCG2 efflux pump and binimetinib could be confirmed in breast cancer PDXs (Figure 4B) and has previously been demonstrated in a mechanistic in vivo study of pharmacokinetics (33). Second, the association between BRCA loss-of-function and olaparib response is clinically validated, being the subject of an FDA-approved companion diagnostic. Third, Cadherin-6 is a logically compelling biomarker for an anti-cadherin-6 antibody and its strong association is unlikely to be a false discovery (P = 0.0008).
Limitations of the study
PDX drug trials have important limitations but they nonetheless represent a unique setting in which to answer scientific questions that cannot be addressed in humans. Among the limitations of PDXs are potential biases introduced by low engraftment rates and the inevitable selection of faster growing tumors, the replacement of human stroma with mouse stroma, and the small size of tumors in mice as compared to humans (particularly for ovarian cancers, which can be very large) and attendant limits on intra-tumor heterogeneity. The ovarian cancer PDX panel used in this study was generated with the goal of capturing the clinically observed histological heterogeneity and mutational spectrum of human ovarian cancers at a population level. Reassuringly, the panel exhibits the same rate of response to first-line chemotherapy as patients with ovarian cancer, reproduces the correlation between BRCA loss-of-function and olaparib response and has histology, genomics, and bulk transcriptomics (RNAseq) concordant with human cancers (as documented at PRoXe). Responses to systemic chemotherapy for epithelial ovarian cancer are similar across multiple histological subtypes, making it appropriate to study these subtypes together (4).
Our study is unable to compare outcomes between PDX models and human participants since tissue was donated anonymously for engraftment in mice. Such concordance has been established by prior studies (in what are often called patient avatar experiments (10,41–45)), which support the validity of PDXs as a model system to assess human tumor responses to therapy. The strength of our study is that it PDXs from anonymous donors that can be readily accessed by other investigators via the Public Repository of Xenografts (PRoXe; www.proxe.org) (46) facilitating additional research on new therapies ovarian cancer.
Intra-patient heterogeneity complicates the assessment of drug response biomarkers, particularly in the case of ovarian cancers, which are often quite large. This is not an issue we are able to address here. We believe that studies of inter-lesion variability in response within a single patient, and of cellular heterogeneity within a single tumor, are potentially best performed in a human setting because xenografts typically involve only a small amount of tissue from a single tumor site. We note that translational and clinical studies of this type are currently underway.
Clinical Translation
A second PDX trial with an independently derived set of tumors is a potential avenue to further testing the biomarkers proposed here, but these pre-clinical results would still need testing in a clinical trial. We therefore propose, as have others (40), that an efficient path to discovery and validation of drug-response biomarkers involves using PDX panels for discovery followed by incorporating a pre-planned subgroup analysis to evaluate biomarker performance in humans during the phase 1b or 2 trials, which take place as a standard part of clinical development.
Possible clinical uses of biomarker-guided treatments for ovarian cancer include single-agent treatments for relapsed or chemotherapy-resistant disease, maintenance therapies following an initial course of chemotherapy (as PARP inhibitors are currently used (6,7)), or in combination with chemotherapy. The current findings in treatment-naive ovarian tumors are most directly relevant to the use of novel agents at first-line (as maintenance following, or combined with, chemotherapy), whereas chemoresistant PDX models would best inform the design of salvage therapies. We show that response to olaparib and chemotherapy is correlated, suggesting high cross-resistance between these two treatment modalities. In contrast, targeted therapies used in the study (including HKT288, BKM120/binimetinib, and LSU691) show little cross-resistance with chemotherapy, suggesting their potential as combination strategies with current front-line standard of care. Additionally, our work identified two novel single agents with greater than 25% ORR: HKT288 (an antibody-drug conjugate directed against Cadherin 6) and LSU691 (a small molecule inhibitor of Nicotinamide Phosphoribosyltransferase (NAMPT)), demonstrating the potential use for these agents in the neo-adjuvant setting. Clinical testing, which is not limited to the therapies studied here, would most efficiently be conducted using a master protocol in which multiple biomarker-guided therapies are evaluated, as in the ongoing Lung-MAP trial (47).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
STATEMENT OF SIGNIFICANCE.
This study exploits a panel of patient-derived xenografts to demonstrate that most ovarian tumors can be matched to effective biomarker-guided treatments.
ACKNOWLEDGEMENTS
We thank A. Färkkilä for helpful discussions and comments on this manuscript. This work was funded in part by Novartis, Inc. and by NCI Grant U54-CA225088 to P.K. Sorger. D. Plana is also supported by NIGMS grant T32-GM007753.
Footnotes
ADDITIONAL INFORMATION
Competing interests
With the exception of ACP, DP and PKS, authors were employees of Novartis, Inc. at the time this study was performed, PKS is a member of the SAB or Board of Directors of Glencoe Software, Applied Biomath and RareCyte Inc and has equity in these companies. PKS declares that none of these relationships are directly or indirectly related to the content of this manuscript.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015. February;65(1):5–29. [DOI] [PubMed] [Google Scholar]
- 2.du Bois A, Lück H-J, Meier W, Adams H-P, Möbus V, Costa S, et al. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J Natl Cancer Inst. 2003. September 3;95(17):1320–9. [DOI] [PubMed] [Google Scholar]
- 3.Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003. September 1;21(17):3194–200. [DOI] [PubMed] [Google Scholar]
- 4.Coward JI, Middleton K, Murphy F. New perspectives on targeted therapy in ovarian cancer. Int J Womens Health. 2015. February 4;7:189–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cook SA, Tinker AV. PARP Inhibitors and the Evolving Landscape of Ovarian Cancer Management: A Review. BioDrugs. 2019. June;33(3):255–73. [DOI] [PubMed] [Google Scholar]
- 6.Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N Engl J Med. 2019. December 19;381(25):2416–28. [DOI] [PubMed] [Google Scholar]
- 7.FDA approves olaparib plus bevacizumab as maintenance treatment for ovarian, fallopian tube, or primary peritoneal cancers [Internet]. FDA. FDA; 2020. [cited 2020 May 26]. Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-olaparib-plus-bevacizumab-maintenance-treatment-ovarian-fallopian-tube-or-primary [Google Scholar]
- 8.Siolas D, Hannon GJ. Patient-Derived Tumor Xenografts: Transforming Clinical Samples into Mouse Models. Cancer Res. 2013. September 1;73(17):5315–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discovery. 2014. September;4(9):998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015. November;21(11):1318–25. [DOI] [PubMed] [Google Scholar]
- 11.Drapkin BJ, George J, Christensen CL, Mino-Kenudson M, Dries R, Sundaresan T, et al. Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discov. 2018. May;8(5):600–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung J, Seol HS, Chang S. The Generation and Application of Patient-Derived Xenograft Model for Cancer Research. Cancer Res Treat. 2018. January 15;50(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whittle JR, Lewis MT, Lindeman GJ, Visvader JE. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015. February 10;17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeRose YS, Wang G, Lin Y-C, Bernard PS, Buys SS, Ebbert MTW, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011. November;17(11):1514–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-David U, Ha G, Tseng Y-Y, Greenwald NF, Oh C, Shih J, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017. November;49(11):1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015. October 8;526(7572):263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy B, Yin H, Maris JM, Kolb EA, Gorlick R, Reynolds CP, et al. Evaluation of Alternative In Vivo Drug Screening Methodology: A Single Mouse Analysis. Cancer Res. 2016. 01;76(19):5798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013. January 1;41(D1):D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016. February;12(2):109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu C, Mannan AM, Yvone GM, Ross KN, Zhang Y-L, Marton MA, et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol. 2016. April;34(4):419–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Politi K, Herbst RS. Lung Cancer in the Era of Precision Medicine. Clin Cancer Res. 2015. May 15;21(10):2213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer AC, Sorger PK. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell. 2017. December 14;171(7):1678–1691.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer AC, Izar B, Sorger PK. Combinatorial benefit without synergy in recent clinical trials of immune checkpoint inhibitors. medRxiv. 2020. January 1;2020.01.31.20019604. [Google Scholar]
- 24.Berenbaum MC. What is synergy? Pharmacol Rev. 1989. June;41(2):93–141. [PubMed] [Google Scholar]
- 25.Loewe S The problem of synergism and antagonism of combined drugs. Arzneimittelforschung. 1953. June;3(6):285–90. [PubMed] [Google Scholar]
- 26.Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. j immunotherapy cancer. 2019. December;7(1):278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Topp MD, Hartley L, Cook M, Heong V, Boehm E, McShane L, et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Molecular Oncology. 2014. May;8(3):656–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.George E, Kim H, Krepler C, Wenz B, Makvandi M, Tanyi JL, et al. A patient-derived-xenograft platform to study BRCA-deficient ovarian cancers. JCI Insight [Internet]. 2017. January 12 [cited 2019 Sep 9];2(1). Available from: https://insight.jci.org/articles/view/89760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong R, Qiang W, Guo H, Xu X, Kim JJ, Mazar A, et al. Histologic and molecular analysis of patient derived xenografts of high-grade serous ovarian carcinoma. J Hematol Oncol. 2016. December;9(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011. June 29;474(7353):609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bialucha CU, Collins SD, Li X, Saxena P, Zhang X, Dürr C, et al. Discovery and Optimization of HKT288, a Cadherin-6-Targeting ADC for the Treatment of Ovarian and Renal Cancers. Cancer Discov. 2017;7(9):1030–45. [DOI] [PubMed] [Google Scholar]
- 32.Cheaib B, Auguste A, Leary A. The PI3K/Akt/mTOR pathway in ovarian cancer: therapeutic opportunities and challenges. Chin J Cancer. 2015. January;34(1):4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gooijer MC de, Zhang P, Weijer R, Buil LCM, Beijnen JH, Tellingen O van. The impact of P-glycoprotein and breast cancer resistance protein on the brain pharmacokinetics and pharmacodynamics of a panel of MEK inhibitors. International Journal of Cancer. 2018;142(2):381–91. [DOI] [PubMed] [Google Scholar]
- 34.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, et al. Promoter Hypermethylation and BRCA1 Inactivation in Sporadic Breast and Ovarian Tumors. J Natl Cancer Inst. 2000. April 5;92(7):564–9. [DOI] [PubMed] [Google Scholar]
- 35.George A, Kaye S, Banerjee S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nat Rev Clin Oncol. 2017;14(5):284–96. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Zhang H, Liu J, Cheruiyot A, Lee J-H, Ordog T, et al. USP51 deubiquitylates H2AK13,15ub and regulates DNA damage response. Genes Dev. 2016. April 15;30(8):946–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samareh B, Zimmer I, Zikic A, Klimenkova O, Loghmani-Khouzani H, Suttorp M, et al. Inhibition of the NAMPT-Triggered Deacetylation of HCLS1 Protein: A New Therapeutic Option in Chronic Myeloid Leukemia. Blood. 2015;126(23):1001–1001. [Google Scholar]
- 38.Tsibulak I, Zeimet AG, Marth C. Hopes and failures in front-line ovarian cancer therapy. Critical Reviews in Oncology/Hematology. 2019. November 1;143:14–9. [DOI] [PubMed] [Google Scholar]
- 39.Tung NM, Garber JE. BRCA 1/2 testing: therapeutic implications for breast cancer management. British Journal of Cancer. 2018. July;119(2):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coussy F, El-Botty R, Château-Joubert S, Dahmani A, Montaudon E, Leboucher S, et al. BRCAness, SLFN11, and RB1 loss predict response to topoisomerase I inhibitors in triple-negative breast cancers. Sci Transl Med. 2020. February 19;12(531):eaax2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vargas R, Gopal P, Kuzmishin GB, DeBernardo R, Koyfman SA, Jha BK, et al. Case study: patient-derived clear cell adenocarcinoma xenograft model longitudinally predicts treatment response. npj Precision Onc. 2018. December;2(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, et al. A Renewable Tissue Resource of Phenotypically Stable, Biologically and Ethnically Diverse, Patient-Derived Human Breast Cancer Xenograft Models. Cancer Research. 2013. August 1;73(15):4885–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart EL, Mascaux C, Pham N-A, Sakashita S, Sykes J, Kim L, et al. Clinical Utility of Patient-Derived Xenografts to Determine Biomarkers of Prognosis and Map Resistance Pathways in EGFR -Mutant Lung Adenocarcinoma. JCO. 2015. August 1;33(22):2472–80. [DOI] [PubMed] [Google Scholar]
- 44.Isler J, Katz A, Sorogin A, Scoppetuolo M, Nechushtan H, Pass H, et al. The Use of Patient-Derived Xenograft (PDX) Models to Predict Patient Response in Non-Small Cell Lung Cancer (NSCLC). International Journal of Radiation Oncology*Biology*Physics. 2014. November;90(5):S42–3. [Google Scholar]
- 45.Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Annals of Oncology. 2017. October;28(10):2595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Townsend EC, Murakami MA, Christodoulou A, Christie AL, Köster J, DeSouza TA, et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell. 2016. 11;29(4):574–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferrarotto R, Redman MW, Gandara DR, Herbst RS, Papadimitrakopoulou VA. Lung-MAP--framework, overview, and design principles. Chin Clin Oncol. 2015. September;4(3):36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.