

Abstract
The WNT pathway is a fundamental regulator of intestinal homeostasis and hyperactivation of WNT signaling is the major oncogenic driver in colorectal cancer (CRC). To date, there are no described mechanisms that bypass WNT dependence in intestinal tumors. Here, we show that while WNT suppression blocks tumor growth in most organoid and in vivo CRC models, the accumulation of CRC-associated genetic alterations enables drug resistance and WNT-independent growth. In intestinal epithelial cells harboring mutations in KRAS or BRAF, together with disruption of p53 and SMAD4, transient TGFβ exposure drives YAP/TAZ-dependent transcriptional reprogramming and lineage reversion. Acquisition of embryonic intestinal identity is accompanied by a permanent loss of adult intestinal lineages, and long-term WNT-independent growth. This work identifies genetic and microenvironmental factors that drive WNT inhibitor resistance, defines a new mechanism for WNT-independent CRC growth and reveals how integration of associated genetic alterations and extracellular signals can overcome lineage-dependent oncogenic programs.
Keywords: RSPO, CRC, WNT, TGFbeta YAP/TAZ, lineage plasticity, resistance
Introduction
The WNT signaling pathway is a key developmental regulator and essential for homeostasis of numerous adult cell types, including hematopoietic (1,2) and intestinal stem cells (3,4). Genetic alterations that hyperactivate the WNT pathway, including disruption of the APC tumor suppressor, mutational activation of CTNNB1 (β-catenin), or chromosomal translocations involving RSPO genes, occur in more than 90% of colorectal cancer (CRC) (5,6) and likely facilitate the initial growth of these tumors (7–9). We and others have shown that in many contexts, WNT hyperactivation is essential for tumor maintenance. For instance, restoration of endogenous APC expression in genetically engineered or tumor-engrafted mice leads to rapid and sustained tumor regression (10,11), while blocking WNT ligand secretion with porcupine (PORCN) inhibitors or blocking RSPO3 directly in human or murine tumors with RSPO3 fusions drives cell cycle arrest, differentiation, and tumor clearance (9,12,13). Thus, most normal and transformed cells of the intestine are thought to be WNT dependent.
Targeting the WNT pathway in cancer has clear therapeutic potential, and this has led to the recent initiation of multiple Phase I clinical trials (NCT02521844, NCT02649530, NCT03447470) for treatment of RSPO fusion cancers with PORCN inhibitors. However, due to the current lack of clinically approved WNT antagonists and scarcity of RSPO fusion model systems, it has not been possible to explore potential mechanism of therapy failure and resistance to WNT-targeted therapy. To further understand WNT dependence and the therapeutic potential of WNT inhibition, we engineered an array of organoid-based models capturing the most frequently observed genetic alterations in this molecular subtype. We found that while no individual oncogenic change influences sensitivity to WNT suppression, the combined alteration of multiple CRC-associated lesions enables rapid acquired resistance to WNT blockade. This occurs via a TGFβ-induced lineage conversion that reverts adult intestinal epithelium to a fetal intestinal state. The lineage switch is driven and maintained by YAP/TAZ signaling, and is not reversible. Consequently, WNT-independent cells become exquisitely and selectively sensitive to suppression of YAP/TAZ.
Results
Accumulation of CRC-associated oncogenic mutations leads to WNT independence
PTPRK-RSPO3 fusions drive the development of WNT-dependent murine intestinal adenomas that are extremely sensitive to treatment with the PORCN inhibitor WNT974 (9). Human RSPO fusion tumors carry an array of cooperating oncogenic lesions, including frequent mutational activation of KRAS or BRAF, as well as disruption of TP53 and/or SMAD4 (Figure 1a). In many cases, three or four of these oncogenic events occur within the same tumor, but how such co-occurring alterations contribute to WNT-dependence is not known. Unlike APC mutant CRC, there are very few human RSPO3 CRC cell lines or organoid models, and until recently (14), no engineered human cell systems with which to interrogate RSPO disease biology. To recapitulate some of the genetic complexity of human RSPO3 fusion tumors, we used CRISPR to develop a series of murine organoid models harboring common oncogenic genotypes (Figure 1b). We first generated multiple independent organoid lines, each carrying an endogenous Ptprk-Rspo3 (R) fusion (9), and an endogenous KrasG12D (K) mutation (from the KrasLSL-G12D allele) (15). We then used Cas9 and sgRNAs targeting Trp53 (P) and Smad4 (S) to create triple (KRP and KRS) and quadruple (KRPS) mutants (Figure 1b; see methods for details). As previously described (16,17), organoids containing loss of function alterations in p53 and SMAD4 were selected by treating for seven days with Nutlin-3 (5μM) or TGFβ (5ng/ml), respectively. We confirmed the presence of each mutation by sequencing or assessed protein disruption by western blot (Figure 1c-e). RSPO3 fusion (R), KR and KRP cultures resembled wildtype intestinal organoids, with proliferative crypt-like budding protrusions and a KRT20-positive differentiated core (Figure 1f). In contrast, KRS and KRPS were proliferative spheroid structures with very few KRT20-positive differentiated cells.
Figure 1. Engineering of mouse-derived intestinal tumor organoids.
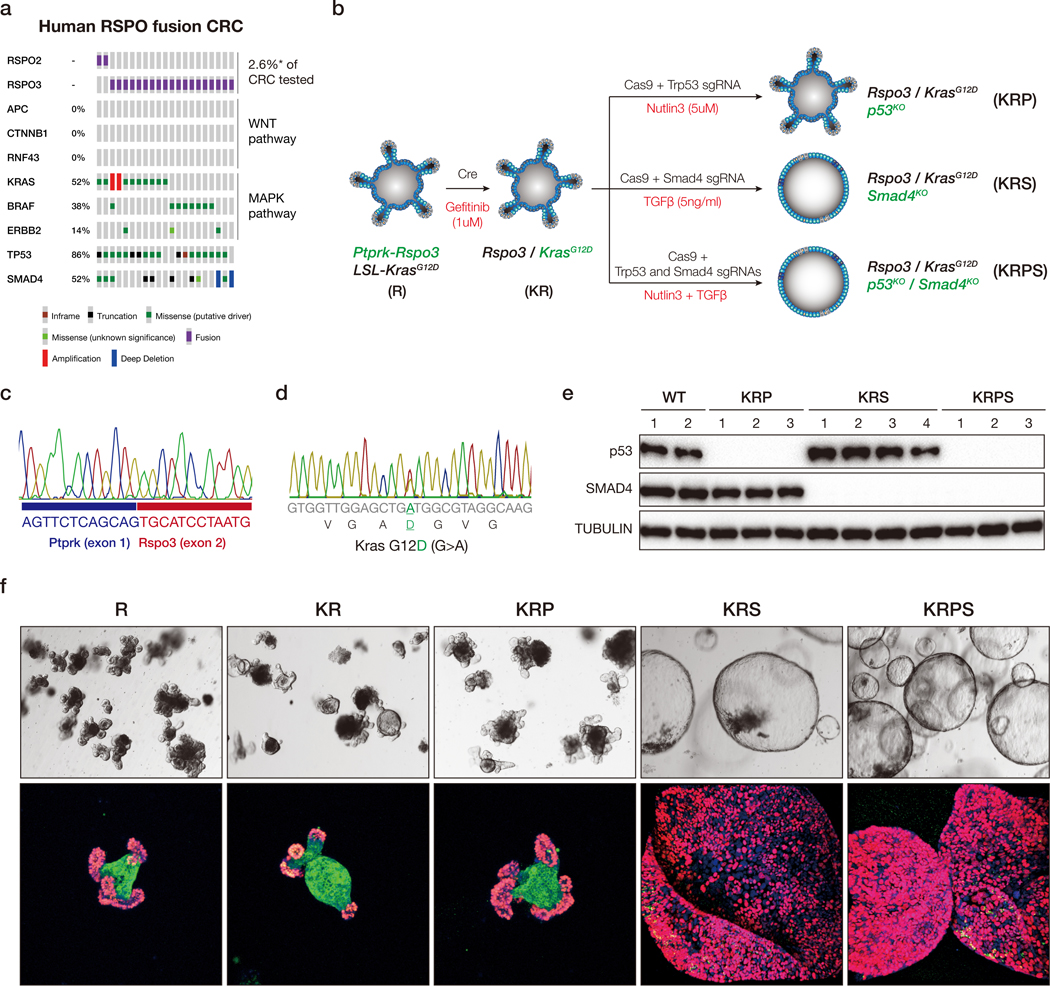
a. Oncoprint derived from MSK IMPACT sequencing and ArcherDx fusion testing, showing co-occurring mutations in WNT, MAPK, TP53 and SMAD4 genes in RSPO fusion-positive human CRCs. b. Schematic representation of the generation of sequentially mutated murine intestinal organoids. c. Representative sanger sequencing chromatogram confirming expected Ptprk-Rspo3 mRNA fusion junction following CRISPR-mediated intrachromosomal inversion. d. Representative Sanger sequencing chromatogram from cDNA, confirming expression of the KrasG12D (G>A) mutant transcript. e. Western blots showing loss of p53 and SMAD4 in independent biological replicates of edited and selected organoids. f. Bright field (upper) and immunofluorescent (lower) images of sequential mutants. Proliferating cells are marked by EdU (Red), and differentiated cells by KRT20 (green).
Similar to organoids carrying only Rspo3 fusions (9), KRP and KRS organoids showed rapid cell cycle arrest (loss of EdU incorporation) and differentiation (induction of KRT20) 4 days following PORCN inhibition with 500nM WNT974 (Figure 2a-b, Figure S1a). Quadruple KRPS mutants showed a marked decrease in EdU incorporation and increased KRT20 expression, however, unlike KRP and KRS, a subpopulation of KRPS cells (~4–5%) continued to proliferate and expand in the presence of drug (Figure 2a-b). Drug resistance was not specific to WNT974 as we observed an identical response with an independent PORCN inhibitor (ETC-159) (Figure 2b). Further, resistance to both drugs was long-lived as re-challenge with WNT974 or ETC159 2–4 weeks after drug withdrawal caused only a moderate reduction in proliferation (Figure 2b), and no change in cell morphology over continual culture with WNT974 (Figure S1b). Like KRPS cells, quadruple mutant organoids expressing an endogenous murine BrafV619E allele (18), equivalent to human BRAFV600E (“BRPS”), also showed rapid resistant outgrowth in the presence of WNT974 (Figure S1c).
Figure 2. Kras, p53 and Smad4 mutations enable Wnt independence.
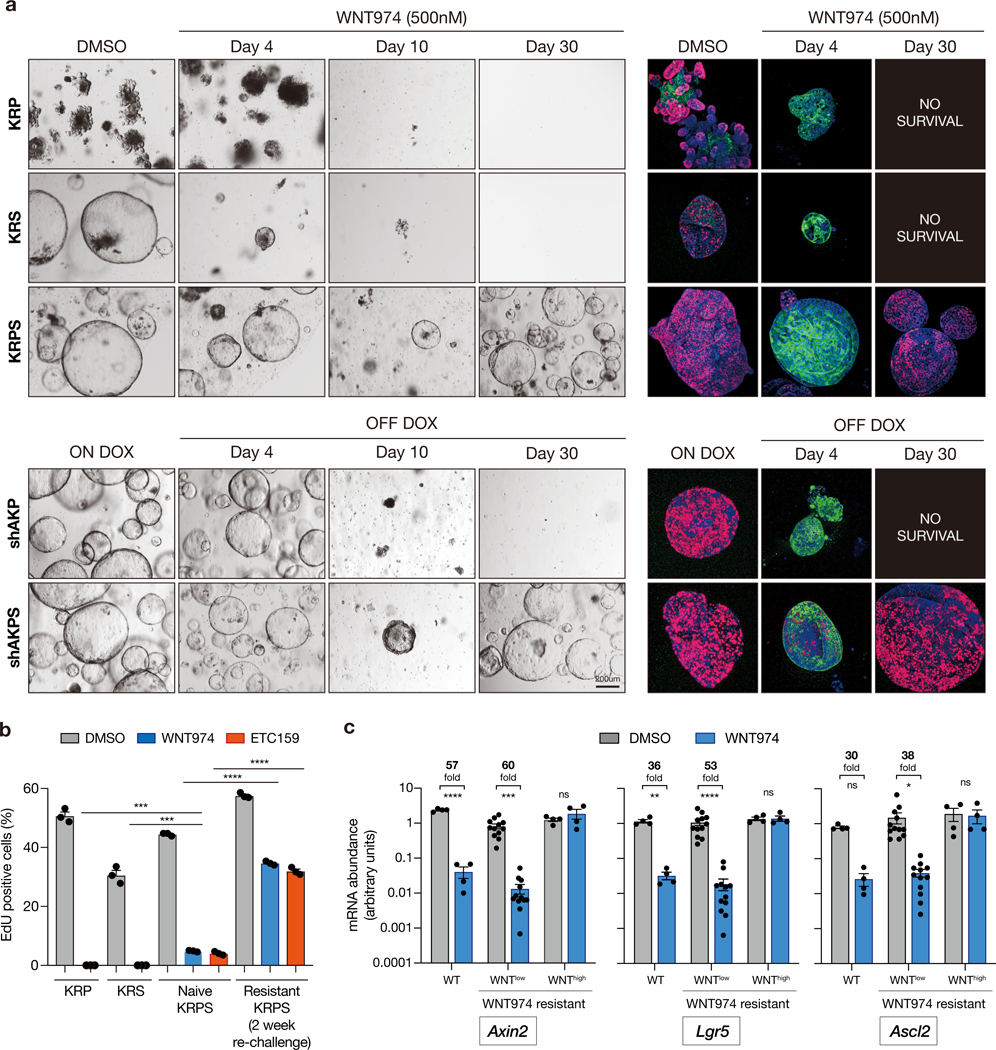
a. Bright field and immunofluorescent images of KRP, KRS and KRPS organoids treated with DMSO or WNT974 (upper panel). Bright field and immunofluorescent images of shAKP and shAKPS organoids before and following APC restoration (dox withdrawal) (lower panel). Proliferating cells are marked by EdU (Red), and differentiated cells by KRT20 (green). b. Percentage of EdU-positive cells measured by flow cytometry in KRP, KRS, naive KRPS and resistant KRPS organoids treated with DMSO or WNT974 (n=3 independent biological replicates per condition). During the initial establishment of resistant cells, organoids were treated with PORCN inhibitor continuously for 30 days. After that, resistant cultures were maintained without WNT974. EdU incorporation was assessed following re-treatment with WNT974 for 4 days, at least two weeks after cultures were removed from WNT974. c. qPCR of canonical WNT targets showing WNT signaling is well suppressed in the majority (16/20) of WNT974-resistant quadruple mutant organoid lines and is reactivated in a subset (4/20) of organoid lines. qPCR of canonical WNT targets from WT cells treated with WNT974 is used as a control of maximum WNT pathway suppression. All error bars represent the +/− standard error of the mean (SEM). All statistical tests are two-way ANOVA with Sidak’s correction for multiple comparisons; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To determine whether the (KRPS/BRPS) genotype-dependent response was specific to PORCN inhibition, or reflected a more general bypass of WNT-dependence, we generated organoids carrying KRASG12D and p53 mutations and an inducible Apc-targeted shRNA that enables doxycycline (dox)-regulated control of APC expression (shAKP). In this context, withdrawal of dox drives APC-mediated suppression of hyperactive WNT signaling and in the absence of exogenous RSPO1/WNT ligand, promotes cell cycle arrest and differentiation (10). Consistent with previously work (10), APC restoration in shAKP cells induced cell cycle arrest and differentiation, preventing long-term RSPO-independent growth (Figure 2a, lower panels). However, similar to KRPS cells, a subpopulation of SMAD4 mutant ‘shAKPS’ cells were able to expand in RSPO-free media following APC re-expression (Figure 2a). Further, these RSPO/WNT-independent shAKPS cells showed no acute response to treatment with WNT974 (Figure S1d), confirming they did not escape APC restoration by upregulating WNT ligand expression. Together, these data suggest that the combination of Kras/Braf, p53 and Smad4 mutations can enable the generation of WNT-independent intestinal organoids, following pharmacologic or genetic suppression of WNT signaling.
In total, we generated 20 WNT-independent organoid lines using both shRNA and CRISPR-based approaches (Table S1), which showed two distinct patterns of resistance. A subset of the lines (4/20) showed robust reactivation of the WNT pathway (“WNThigh”; Figure 2c Table S1). In one instance this was associated with a 25-fold genomic amplification of Ctnnb1 (Figure S2a), and in another, a hotspot S33C mutation in Ctnnb1 (Figure S2b). Using base editing, we confirmed that, as expected, organoids with mutational activation of Ctnnb1 (S33F) were dramatically enriched following treatment with WNT974 (Figure S2c). The majority of resistant cultures (16/20) maintained very low levels of WNT target gene expression while cultured in WNT974 (“WNTlow”; Figure 2c, Figure S3), suggesting they did not escape treatment by downstream activation of WNT signaling. We further saw no evidence of genomic WNT pathway alterations in those WNTlow resistant cases examined by WES (Table S1). Together, these data reveal the unexpected appearance of intestinal cells that remain responsive to WNT-suppressive stimuli, yet do not require WNT pathway activation for growth and proliferation. We defined organoids with the “WNTlow resistant” phenotype as KRPSR, BRPSR and shAKPSR.
Transient TGFβ stimulation is required for resistance to WNT inhibition
Transcriptome analysis of WNT974-naïve and resistant isogenic organoid pairs by RNAseq revealed a number of differentially regulated genes and pathways in both KRPSR and shAKPSR genotypes, including a series of inflammation-associated gene signatures (Figure 3a). We noted that during the generation of KRPS, BRPS, and shAKPS cultures, each sample was treated transiently with TGFβ to select for SMAD4 mutant populations (Figure 1b) and reasoned that TGFβ-treatment might be altering transcriptional networks and conditioning or ‘priming’ cells to become WNT-independent. To determine whether TGFβ-priming was important for WNT independence, we generated KRPshS organoids in which SMAD4 expression was silenced by an shRNA, enabling the enrichment of cells with SMAD4 depletion, without functional (TGFβ) selection (Figure 3b). In contrast to spheroid KRPS cells, KRPshS cultures retained a budding organoid morphology similar to R, KR, or KRP cells (Figure 3c). However, short-term treatment (3 days) with TGFβ (5ng/ml) induced a rapid morphological shift toward spheroid structures (Figure 3c-d) that was maintained 2-weeks following withdrawal of TGFβ (Figure 3d), up to at least six months in culture (not shown). The spheroid change was driven by signaling through the canonical TGFβ receptor complex as co-treatment with a TGFβR1 inhibitor (LY2157299) or CRISPR-mediated genetic disruption of Tgfbr2 completely abolished the morphological response (Figure 3c-d).
Figure 3. TGFβ is required for WNT independence.
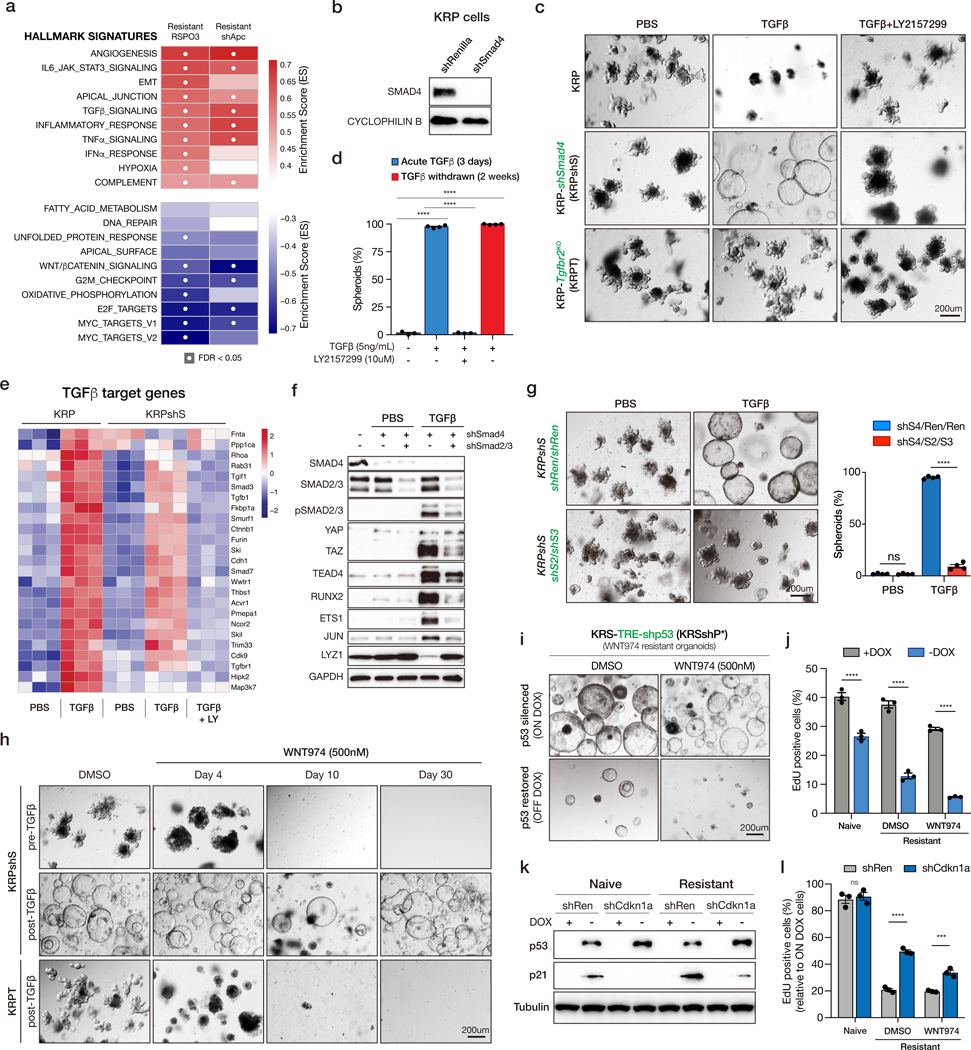
a. GSEAs show inflammation-related pathways are enriched in WNT independent lines. b. Western blot analysis showing Smad4 knockdown. KRPshS organoids are generated by transducing KRP organoids with SGEN-shSmad4, then selected by G418 at the concentration of 500ug/mL. c. Bright field images of KRP, KRPshS and KRPT organoids treated with PBS, 5ng/mL TGFβ or 5ng/mL TGFβ plus 10uM TGFBR1 inhibitor LY2157299 as indicated. d. Quantification of TGFβ-induced organoid morphology change. e. Heatmap of TGFβ target genes in KRP and KRPshS acutely treated with TGFβ, or TGFβ plus LY2157299. f. Western blots showing Smad2/3 depletion inhibits the induction of TGFβ downstream targets. g. Bright field images (left) and quantifications (right) showing that Smad2/3 depletion blocks TGFβ-induced spheroid formation. h. Bright field images of KRPshS (pre- and post-TGFβ) and KRPT (post-TGFβ) showing TGFβ priming is required for WNT974 resistance. i. Bright field images of p53 restoration in KRSshP* WNT974-resistant line showing p53 is required for the maintenance of WNT independence. j. EdU flow cytometry on p53 restored WNT974-naive KRSshP* and WNT974-resistant KRSshP* lines. All error bars represent +/− standard error of the mean (SEM). k. Western blot showing the induction of p21 after p53 restoration is inhibited by shCdkn1a in KRSshP*. l. EdU flow cytometry on p53 restored WNT974-naive KRSshP* and WNT974-resistant KRSshP* lines expressing either shRenilla or shCdkn1a. Percentage of relative EdU positive cells is calculated by normalizing the off-Dox EdU percentage to the on-Dox EdU percentage. All error bars represent +/− standard error of the mean (SEM). All statistical tests are two-way ANOVA with Sidak’s correction for multiple comparisons; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
In many contexts, SMAD4 association with SMAD2/3 is required for canonical TGFβ signaling (19). Indeed, the magnitude of TGFβ target gene expression following acute TGFβ stimulation was reduced in SMAD4-depleted cells (KRPshS) compared to those with wildtype SMAD4 (KRP) (Figure 3e). However, most downstream targets were still strongly induced compared to isogenic untreated cells, highlighting a widespread SMAD4-independent transcriptional response to TGFβ in these cells (Figure 3e, Figure S4a), as has been reported in other settings (20,21). In support of the notion that TGFβ drives SMAD4-independent canonical signaling, we saw a marked increase in SMAD2/3 phosphorylation, but no change in the activation of non-canonical TGFβ pathways (MAPK, AKT, p38, JNK) (Figure S4b). Further, TGFβ-mediated transcriptional changes in SMAD4-depleted cells were SMAD2/3-dependent, as simultaneous silencing of these SMADs suppressed target gene induction and spheroid transformation (Figure 3f-g). Most importantly, and consistent with results from CRISPR-derived KRPS cultures, TGFβ-treated KRPshS organoids developed resistance to WNT974, while TGFβ-naïve KRPshS organoids remained WNT-dependent and could not survive continuous treatment with the drug (Figure 3h). Similarly, disruption of Tgfbr2 prevented the emergence of WNT974 resistant cells (Figure 3h). Thus, TGFβ priming via canonical TGFβ receptors can initiate the induction of WNT-independence.
p53 loss, but not Smad4 loss, is essential for maintenance of WNT independence
As noted above, withdrawal of TGFβ from the culture media had no impact on TGFβ-induced spheroid morphology (Figure 3d). Likewise, treatment of WNT-independent organoids with a TGFβR1 inhibitor (LY2157299) had no effect on morphology or growth of organoids (Figure S4c-d). This observation raised the possibility that SMAD4 loss was important to enable TGFβ priming, but was dispensable beyond this event. To directly test the requirement for SMAD4 loss in regulating WNT-independence we generated KRPshS organoids in which the expression of the SMAD4 shRNA was controlled by a dox-regulated element (TRE) and a reverse tet-transactivator. We generated WNT974-resistant organoids in the presence of dox, exactly as described above, and restored SMAD4 expression by withdrawal of dox from the media (Figure S5a-b). As expected, re-expression of SMAD4 sensitized organoids to acute exposure to TGFβ (Figure S5c-d), however it had no impact on organoid morphology or the maintenance of WNT independence in the absence of exogenous TGFβ ligand (Figure S5e). Proliferation was slightly increased following dox withdrawal, perhaps owing to subtle effects of dox on organoid growth (Figure S5f).
We next used a similar approach to generate WNT-resistant organoids in which p53 expression was controlled by a dox-regulated shRNA (KRSshP*) (Figure S5a). In contrast to the effect of Smad4 re-expression, restoration of endogenous p53 significantly reduced cell proliferation in both drug-naïve and WNT974-resistant cells, though WNT-independent cells were 2–3 times more sensitive to p53 restoration (Figure 3i-j, Figure S6a). Notably, cell cycle arrest occurred both in the presence and absence of WNT974, suggesting that p53 was not simply restoring sensitivity to WNT974, but that WNT independent cells were hypersensitive to p53-mediated tumor suppression. Cell cycle arrest downstream of p53 is often mediated by p21 (22). Indeed, we noted that resistant cells showed increased chromatin accessibility at the Cdkn1a locus (Figure S6b), and both p21 transcript and protein was more dramatically induced in resistant cells compared to their drug-naïve counterparts (Figure 3k, Figure S6c). Silencing of p21 partially blocked cell cycle arrest following p53 restoration (Figure 3l), suggesting that bypassing p21-induced arrest contributes, but is not the only factor underlying the importance of p53 disruption in initiating and maintaining WNT independence.
Together, these data support the notion that oncogenic alterations play a critical role in the development of WNT independence, but that the requirements for initiation and maintenance of this program are different. Transient TGFβ-priming in KRPS/shAKPS intestinal cells drives SMAD2/3-dependent TGFβ pathway activation that avoids SMAD4-dependent cell death (21), but continual TGFβ signaling is not essential to maintain the WNT independent state. Conversely, p53 disruption is required for both initiation and maintenance of WNT pathway independence.
The in vivo tumor microenvironment is sufficient to prime cells for WNT independence
TGFβ is an abundant cytokine in the tumor microenvironment and modulation of TGFβ receptor has been shown to impact colorectal cancer progression in animal models by altering tumor-stroma signaling (23,24). We sought to determine whether exposure to tumor microenvironmental signals were sufficient to prime TGFβ-naïve organoids and induce WNT independence in vivo.
Long-term treatment with WNT974 can cause intestinal and bone damage in mice (25,26). To enable potent and sustained WNT suppression in tumor cells without effects on surrounding tissues, we used the regulatable shApc model, whereby withdrawal of dox from chow drives rapid APC restoration and potent cell-intrinsic WNT pathway suppression (10,11). We generated TGFβ-naïve shAKPS cells by simultaneous delivery of sgRNAs targeting both Trp53 and Smad4, and selected for p53 disruption by Nutlin-3 treatment. Surrogate selection for p53 mutation produced a polyclonal population of cells that were TGFβ-naïve, but contained greater than 95% Smad4 disruption (Figure S7a-b). As expected, TGFβ-naïve shAKP and shAKPS organoids, and TGFβ-treated shAKPT organoids were sensitive to WNT suppression by APC restoration, and never became WNT-independent in vitro (Figure S7c).
Each of the organoid lines were injected in mice pre-treated with dox (200mg/kg in chow). When tumors reached ~100mm3 (10–14 days post-injection), animals were either maintained on dox or returned to normal chow to restore APC. As previously described, shAKP organoids showed cell cycle arrest (loss of Ki67; Figure 4a-b), marked reduction of WNT target expression (SOX17; Figure 4a), and increased differentiation (KRT20; Figure S8) in response to APC restoration, and showed little-to-no tumor growth following the dox switch (Figure 4c). As expected, tumors derived from TGFβ-treated shAKPS organoids continued to grow following APC restoration (Figure 4a). At harvest, these tumors showed evidence of WNT pathway suppression (SOX17; Figure 4a), but in contrast to shAKP tumors, were highly proliferative (Figure 4a-b) and showed little evidence of differentiation (KRT20; Figure S8), similar to dox-treated tumors. These tumors also showed frequent phosphorylated Smad3, highlighting widespread TGFβ-mediated signaling throughout the tumor mass (pSMAD3; Figure S8). Tumors derived from TGFβ-naïve shAKPS organoids, that were entirely WNT-dependent in vitro (Figure S7c), showed a mixed response in vivo, with clusters of arrested differentiated cells interspersed with poorly differentiated and proliferative tumor cells (Figure 4a). This is consistent with the outgrowth of a subset of WNT independent tumor cells, as observed in TGFβ-primed ex vivo cultures (Figure 2a-b). Similar to TGFβ-primed shAKPS tumors, TGFβ-naïve tumors showed widespread pSMAD3 (Figure S8), indicating activated TGFβ signaling. As expected, genetic disruption of Tgfbr2 prevented Smad3 phosphorylation and WNT-independent tumor growth (Figure 4a-c; Figure S8). Similar effects were seen with KRP, KRPS, and KRPT-derived tumors, following 2 weeks of WNT974 treatment (Figure 4d; Figure S9a-b). In all, these data show that the tumor microenvironment is sufficient to prime cells for WNT independence, and that this is likely driven by canonical TGFβ signaling.
Figure 4. The in vivo tumor microenvironment is sufficient to prime cells for WNT independence.
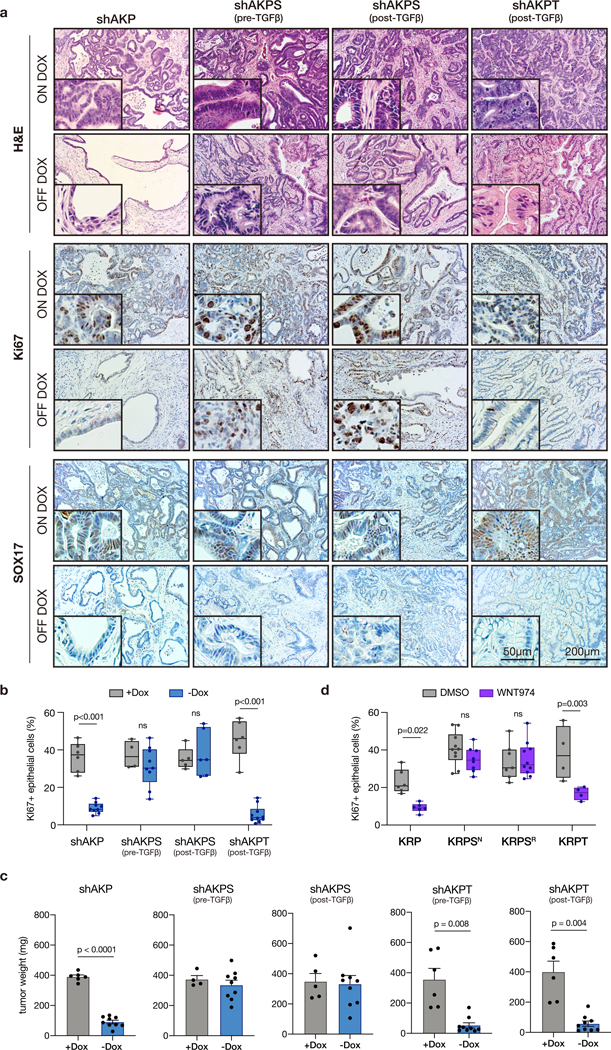
a. Immunohistochemical stains of organoid transplants on dox for 7 weeks (ON DOX), or on dox for 2 weeks then off dox for 5 weeks (OFF DOX). b. Quantification of Ki67 positive epithelial cells in individual tumors from Apc restoration experiments (n≥4 individual tumors). Box and whisker plots represent the full data range (min-max), p-values calculated using two-way ANOVA with Sidak’s correction for multiple comparisons. c. Tumor weight quantification following transplant of different organoid genotypes. Error bars represent +/− standard error of the mean (SEM), n≥4 individual tumors, p-values calculated using a two-sided t-test, with Welsh’s correction. d. Quantification of Ki67 positive epithelial cells in individual tumors from WNT974 treatment experiments (n≥4 individual tumors).
Lineage reversion underlies WNT independence
As WNT independence was initiated, but not maintained by TGFβ, we sought to identify downstream signaling pathway that were altered following TGFβ exposure and maintained following TGFβ withdrawal. To do this, we again compared pathway enrichment using the hallmark GSEA dataset (27), as well as a range of unique gene signatures that have recently been identified from single-cell and bulk transcriptome analyses of the mouse intestine (28–31). As expected, TGFβ signaling was the most enriched pathway following acute (3d) TGFβ treatment, but was lost after long-term (>30d) TGFβ−free culture (Figure 5a). Strikingly, the only molecular signature that was increased and robustly maintained in post-TGFβ organoids was a collection of genes upregulated in embryonic or fetal intestinal organoids, compared to adult-derived cultures (Figure 5a-b) (31). Accordingly, genes downregulated in fetal intestinal cultures were also strongly suppressed following TGFβ-priming (Figure 5a). Many of the genes downregulated in TGFβ-primed organoids specified differentiated intestinal lineages, including enterocytes, enteroendocrine, and Paneth cells (Figure 5a-b). These changes identified by transcriptome analysis were readily apparent in tumors in vivo, with KRPS and shAKPS tumors, but not KRP, shAKP, KRPT, or shAKPT tumors, showing loss of adult lineages (Paneth cells marked by LYZ1) and induction of fetal markers (SPP1 and ANXA1) (Figure 5c, Figure S10)
Figure 5. TGFβ drives fetal-like lineage reversion.
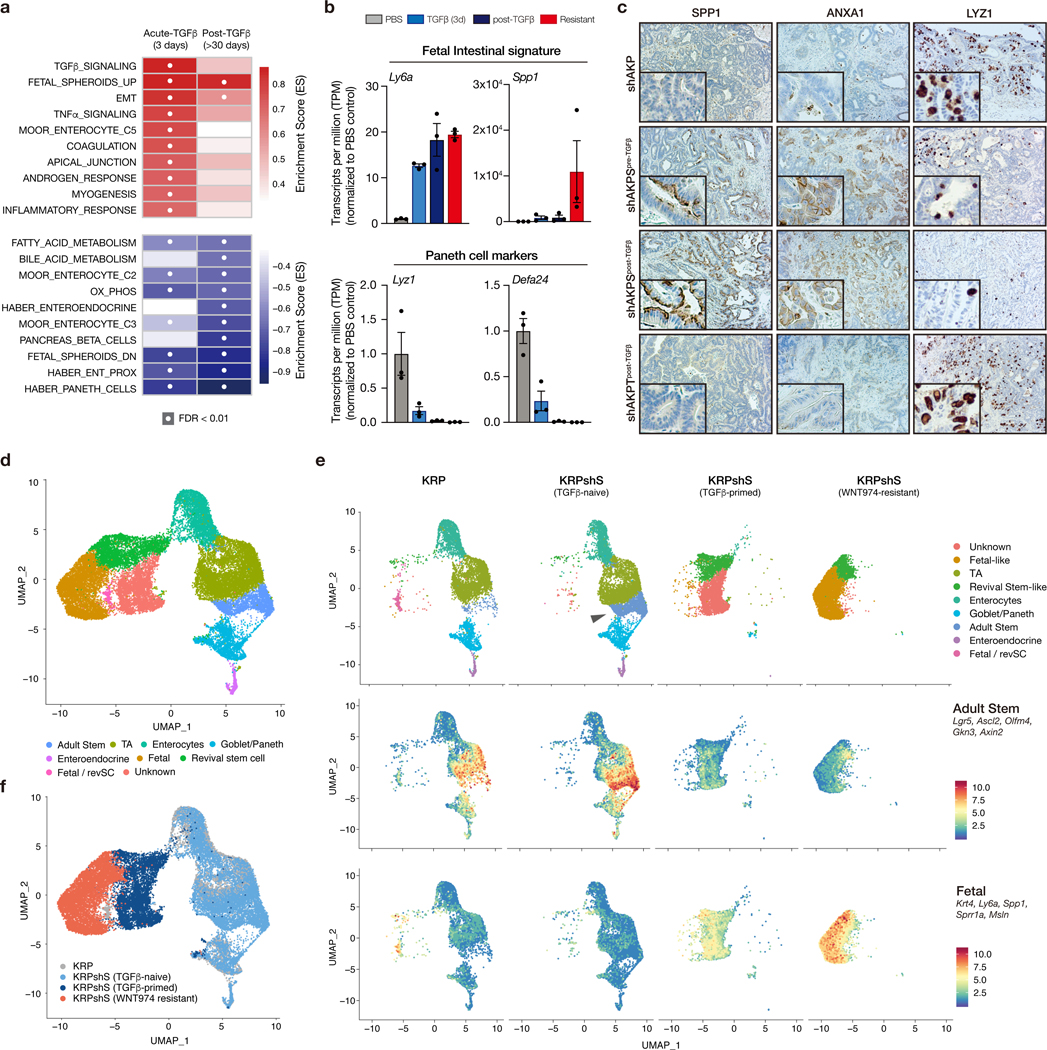
a. GSEA summary showing pathway enrichment scores for KRPshSmad4 organoids treated for 3 days with TGFβ (acute) and the same organoids one month after TGFβ withdrawal. White dots represent significantly enriched gene sets (FDR<0.01). b. mRNA expression (transcript per million estimates) of fetal intestinal markers (upper) and Paneth cell markers (lower) in KRPshS organoids. Error bars are +/− SEM. c. Immunohistochemical staining of SPP1, ANXA1, and LYZ1 on organoid-derived tumors, as labelled. d. UMAP plot of merged scRNAseq from KRP, KRPshS (pre-TGFβ), KRPshS (post-TGFβ), and KRPshS (WNT974-resistant) organoids, showing identified and putatively identified cell populations. e. UMAP plot (upper) of individual scRNAseq samples shows expansion of Lgr5high adult stem cell population following Smad4 silencing (arrow), and transition of cell lineage following TGFβ-priming and development of WNT-independence. Feature plots (middle and lower) highlight the mean expression of adult stem cell and fetal intestinal markers, as indicated. f. UMAP plot of merged scRNAseq data highlighting specific samples. All error bars represent +/− standard error of the mean (SEM).
To more closely interrogate the dynamics of cell fate change following TGFβ priming and the development of WNT independence, we performed single cell RNA sequencing (scRNAseq) in KRP, and KRPshS TGFβ-naïve, TGFβ-primed, and WNT974-resistant organoids. Mapping of marker gene expression across 12 identified cell clusters revealed multiple distinct populations (Figure S11a-c), including Lgr5-positive adult stem cells, enterocytes, goblet, Paneth, and enteroendocrine cells (Figure 5d; Figure S12). Depletion of SMAD4 (in KRPshS organoids) induced expansion of a WNT-high and Lgr5-high stem cell compartment compared to KRP (Figure 5e, Figure S12), as reported in other intestinal organoid and in vivo models (32,33). Despite the expansion of the stem cell pool, SMAD4 loss alone did not alter lineage differentiation (Figure 5e, upper; Figure S12). In contrast, TGFβ priming of KRPshS cells caused a dramatic shift in transcriptional identity, depleting multiple differentiated intestinal lineages (Figure 5e-f). In addition, we noted the expansion of a cell population with marker gene expression reminiscent of recently identified ‘revival stem cells’ (expressing Clu, Anxa1, Cxadr, and Basp1; hereafter “RevSCs”) that are activated in vivo following intestinal injury (34) (Figure 5d, Figure S12, Figure S13a). WNT974-resistant KRPshS cells were transcriptionally distinct from TGFβ-primed cultures, with some overlap in the RevSC-like cluster. They showed further depletion of differentiated intestinal lineages (Figure S13b) as well as low Wnt and adult stem cell signatures (Figure 5e, middle; Figures S12, Figure S13a-b). Most notably, WNT974-resistant organoids showed marked elevation of the fetal intestinal signature (Figure 5e, lower), consistent with a role for this transition in the development of WNT-independence. Importantly, all scRNAseq profiling was performed more than 6 weeks after the withdrawal of TGFβ and 30 days after withdrawal of WNT974. Thus, transcriptional reprogramming and the associated lineage switch is maintained over long-term culture and does not require sustained TGFβ signaling or active WNT suppression.
YAP/TAZ signaling is necessary and sufficient to drive lineage reversion and WNT independence
YAP1 and TAZ (WWTR1) are closely related transcriptional co-activators in the Hippo signaling pathway that have been linked to the acquisition of stem and progenitor cell properties in breast, pancreas, and neuronal tissue (35), and for tissue regeneration in the colonic epithelium (36). YAP1 and TAZ are direct transcriptional targets of SMAD2/3 (37,38) (Figure S14a), and were induced following TGFβ treatment (Figure 6a). YAP transcriptional signatures were strongly induced by TGFβ treatment and remained high following TGFβ-withdrawal (Figure 6a-b). Remarkably, the expression of a small set of recently reported canonical YAP target genes (39) was sufficient to accurately cluster the organoid genotypes into pre-TGFβ, post-TGFβ, and WNT-independent subtypes (Figure S14b). Moreover, this comparison, along with single-cell analysis showed that the relative amplitude of the YAP signature was increased in WNT-independent organoids relative to TGFβ-primed cells (Figure 6c, Figure S14b). Finally, analysis of chromatin accessibility TGFβ-primed and WNT974 resistant organoids by ATACseq revealed the consensus TEAD binding motif as the most significantly enriched of all known transcription factor binding motifs in WNT-independent cells (Figure 6d); the TEAD family are transcriptional co-factors for YAP and TAZ. Together, these molecular data suggest that transcriptional reprogramming associated with WNT independence is likely driven by direct YAP/TAZ/TEAD DNA-binding, and that robust YAP/TAZ activity may be critical for the establishment of WNT independence. YAP/TAZ induction was also clearly apparent in vivo, with KRPS tumors showing markedly elevated nuclear YAP/TAZ staining, compared to KRP and KRPT genotypes (Figure S14c). We further observed prominent nuclear (non-phosphorylated) active-YAP in 7/8 human RSPO2/RSPO3 fusion CRCs examined, all of which contained concomitant mutations in KRAS or BRAF (Figure S15). A subset of these tumors also showed weak-to-moderate expression of two markers associated with fetal intestinal identity, ANXA1 and MSLN (Figure S15a-b). Interestingly, 2/3 tumors that showed expression of all three markers carried mutations in KRAS (or BRAF), TP53 and SMAD4 (Figure S15b).
Figure 6. Yap/Taz is required for lineage reversion and WNT independence.
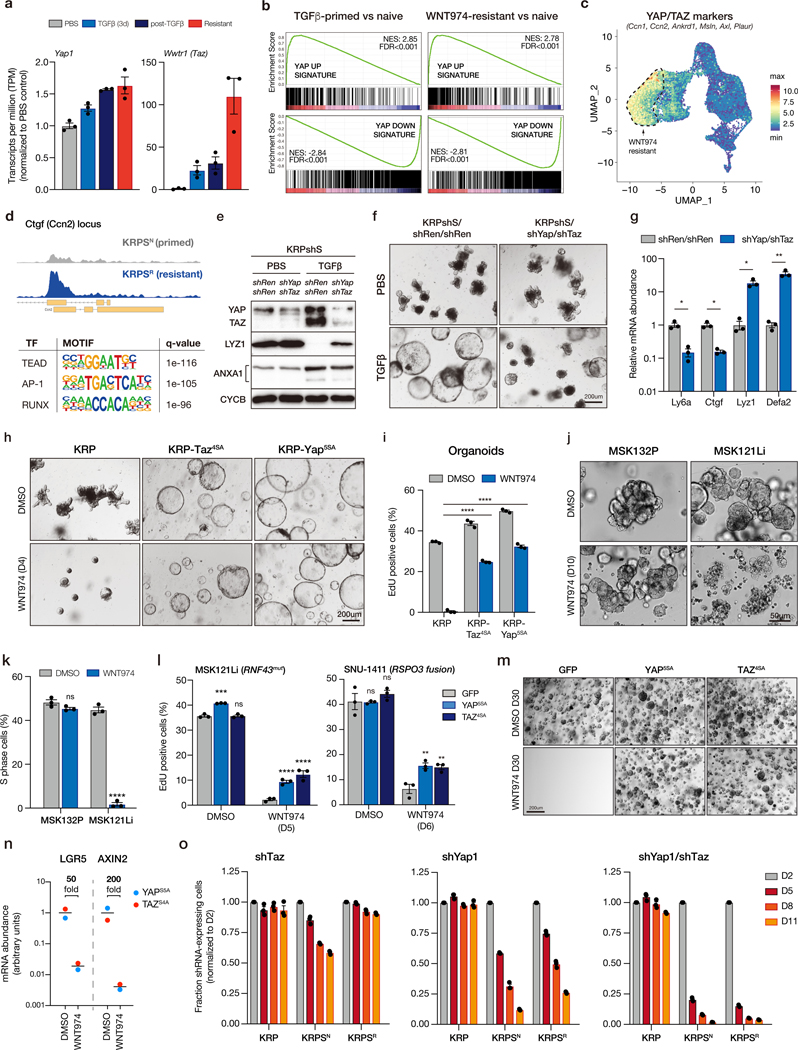
a. Normalized transcript per million (TPM) shows upregulation of Yap1 and Taz (Wwtr1) transcripts following TGFβ-priming and development of WNT-independence. b. Gene set enrichment analysis (GSEA) plots showing YAP signature is enriched in post-TGFβ KRPshS cells, compared to PBS-treated isogenic organoids, and in WNT974-resistant cells, compared to isogenic WNT974 naïve cells. c. UMAP plot of scRNAseq data showing relative mean gene expression of known YAP/TAZ transcriptional targets. Dotted outline indicates WNT974-resistant population. d. ATACseq profile showing increased accessibility of Ctgf (Ccn2) locus in WNT974-resistant KRPS compared to isogenic WNT974-naive KRPS. Lower panel shows HOMER motif analysis from ATACseq showing top 3 known transcription factor binding sites enriched in WNT974-resistant KRPS organoids compared to WNT974-naive cells. e. Western blot of whole cell lysates from KRPshS/shRen/shRen and KRPshS/shYap/shTaz organoids treated with PBS or 5ng/mL TGFβ as indicated. f. Bright field images of KRPshS/shRen/shRen and KRPshS/shYap/shTaz organoids treated with PBS or 5ng/mL TGFβ as indicated. g. qRT-PCR analysis of gene expression in KRPshS/shRen/shRen and KRPshS/shYap/shTaz organoids treated with 5ng/mL TGFβ. h. Bright field images of KRP, KRP-TAZ4SA and KRP-YAP5SA organoids treated with DMSO or 500nM WNT974 for 4 days. i. Percentage of EdU-positive cells measured by flow cytometry on KRP, KRP-TAZ4SA and KRP-YAP5SA organoids treated with DMSO or 500nM WNT974 for 4 days. j. Bright field images of patient-derived organoid line MSK132P (APC mutant) and MSK121Li (RNF43 mutant) treated with DMSO or 500nM WNT974 for 10 days. k. Percentage of EdU-positive cells measured by flow cytometry on MSK132P and MSK121Li organoids treated with DMSO or 500nM WNT974 for 5 days. l. Percentage of EdU-positive cells measured by flow cytometry on MSK121Li organoids (left) and SNU1411 human RSPO3 fusion CRC cells (right) expressing control vector (GFP) or YAP5SA or TAZ4SA treated with WNT974 in 3D Matrigel culture for 6 days, as indicated. Data represent treatment of 3 independent transductions (n=3, *p<0.05, one-way ANOVA, with Tukey’s correction). m. Bright field images of MSK121Li-GFP, MSK121Li- YAP5SA and MSK121Li-TAZ4SA organoids treated with DMSO or 500nM WNT974 for 30 days. n. qRT-PCR of WNT targets (LGR5 and AXIN2) from WNT-independent MSK121Li organoids treated with DMSO or 500nM WNT974, showing that WNT signaling is not reactivated in these WNT-independent lines. o. Cell competition assays on KRP, KRPS WNT974-naive (KRPSN) and KRPS WNT974-resistant (KRPSR) lines transduced with shTaz, shYap1 and shYap1/shTaz vectors. All error bars represent +/− standard error of the mean (SEM). All statistical tests are two-way ANOVA with Sidak’s correction for multiple comparisons; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To directly assess whether YAP/TAZ signaling was the key driver of WNT independence downstream of TGFβ, we silenced YAP and TAZ in TGFβ-naïve KRPshS organoids using tandem shRNAs. YAP/TAZ silencing in these cells had no obvious impact on morphology or organoid growth under basal conditions, but like SMAD2/3 knockdown, partially inhibited the transition to spheroid morphology (Figure S16a), and blocked key molecular and transcriptional changes associated with lineage reversion (Figure 6e-g). YAP/TAZ activity was not only required for spheroid formation and transcriptional changes downstream of TGFβ, but was sufficient to induce WNT independence. Expression of either stabilized YAP (YAPS5A) or TAZ (TAZS4A) in WNT-dependent KRP organoids was sufficient to downregulate adult intestinal lineage markers, upregulate fetal intestinal signature genes and drive spheroid transformation in the absence of TGFβ priming (Figure 6h, Figure S16b). Moreover, KRP-YAPS5A and KRP-TAZS4A organoids were refractory to WNT inhibition with WNT974, showing only a moderate decrease in EdU incorporation (Figure 6i), comparable to KRPSR organoids (Figure 2b).
To determine whether YAP (or TAZ) activation could induce WNT independence in human CRC, we identified a BRAF / TP53 / SMAD4 mutant patient-derived organoid line (“MSK121Li”) with a homozygous disruptive mutation in RNF43 (40). RNF43 is an E3 ligase that modulates WNT signaling by controlling the abundance of the Frizzled/LRP WNT receptors at the cell surface. Like RSPO fusions, RNF43 mutant cells are predicted to be sensitive to PORCN inhibitors. Indeed, unlike APC mutant cells (“MSK132P”) that were completely insensitive to WNT974 (Figure 6j-k), MSK121Li cultures showed morphological changes reminiscent of mucinous-like differentiation (Figure 6j), rapid cell cycle arrest (Figure 6k), and could not be maintained for more than 2 passages (~10 days) in the presence of WNT974. Similar to murine organoids, expression of YAPS5A or TAZS4A blunted the acute WNT974-induced cell cycle arrest (Figure 6l, left), and enabled the outgrowth of WNT974 resistant organoids when maintained in drug over 30 days (Figure 6m). As expected, YAP/TAZ expressing cells maintained very low levels of WNT target gene expression in the presence of WNT974 (Figure 6n), suggesting they had bypassed WNT dependence. Similarly, expression of YAPS5A or TAZS4A in the RSPO3 fusion-positive human CRC cell line SNU1411, led to a 2.5-fold increase in proliferation following treatment with WNT974 (Figure 6l, right).
To determine whether YAP/TAZ-mediated signaling was required for maintenance of WNT-independence, we silenced YAP, TAZ, or both YAP and TAZ in KRP, KRPSN and KRPSR cells and measured proliferation by fluorescence-based competition assay. TAZ knockdown alone had very little impact on the proliferation of organoids relative to untransduced cells, while YAP silencing had a more dramatic effect on KRPSR cells (Figure 6o). Combined YAP/TAZ suppression induced a rapid and near-complete depletion of shRNA-expressing cells by 8 days (Figure 6o). In contrast, YAP/TAZ knockdown caused only a minor (~20%) reduction in EdU incorporation in KRP cells (Figure S16c), highlighting the switch from WNT to YAP dependency following TGFβ-primed transcriptional rewiring and WNT independence. Interestingly, TGFβ-primed, WNT974-naïve (KRPSN) organoids were acutely sensitive to inhibition of both WNT (Figure 2b) and YAP (Figure 6o), suggesting they represent a dual-dependent lineage intermediate.
Together, these data suggest that YAP/TAZ transcriptional activity is the central requirement for TGFβ-primed lineage reversion, and that following the transition to WNT independence, YAP/TAZ signaling becomes essential for survival and proliferation.
Discussion
WNT pathway hyperactivation is the most frequent initiating in human colorectal cancer and thought to be one of the most important molecular drivers in this disease. Here, we show that intestinal tumor cells carrying common cancer-associated genetic alterations can become primed to evade targeted WNT inhibition and rapidly evolve to become completely WNT independent. WNT independence is initiated by canonical TGFβ signaling, which drives YAP/TAZ-dependent transcriptional reprogramming and lineage reversion. This lineage switch is irreversible, and consequently, WNT independent cells become reliant on YAP/TAZ signaling and can be selectively depleted by suppressing YAP/TAZ.
The role of YAP in intestinal biology is complex. YAP is required for early phases of tumor initiation in APC mutant cells (41,42), but in normal intestinal epithelium, YAP induction leads to crypt loss and intestinal dysfunction (43). Likewise, activating genetic alterations in YAP, TAZ and TEAD are rare in colorectal cancer, although YAP signaling can be important for proliferation and metastasis in WNT-driven CRC xenografts (44). Our data show that YAP/TAZ signaling can be potent driver of survival and proliferation in intestinal cancer cells, independent of WNT activation. This mirrors a recently described role for YAP/TAZ in normal tissue regeneration following colitis (36) and irradiation-induced mucosal damage (34). In both cases, YAP-mediated repair is associated with upregulation of fetal markers, though it is unclear whether the regenerating epithelium, like fetal organoids (31), are WNT-independent. Taken together, it seems likely that TGFβ-priming in cancer cells can promote WNT-independence by hijacking a normal physiological wound-repair process to drive lineage reversion.
Despite the similarities, resistance to WNT blockade in cancer cells has two critical and fundamental differences to the wound-repair program. First, TGFβ-priming, unlike injury-induced YAP activation, is dependent on the presence of multiple oncogenic mutations (e.g. KRAS/BRAF, p53 and SMAD4), indicating that this is mode of reprogramming is likely cancer-specific. Second, YAP-mediated wound repair is a tightly controlled and transient process (34,36), while lineage reversion of KRPS/AKPS cells is irreversible. Even following months of culture in the absence of WNT suppression, WNT-independent organoid cultures remain resistant to WNT974 and reliant on YAP/TAZ for survival. Understanding the factors that dictate the differences between reversible YAP activation in injury repair and permanent YAP-reprogramming in WNT inhibitor resistance may identify key cellular switches that could be harnessed to prevent or reverse WNT-independence in therapy refractory tumors. For instance, while blocking the TGFβ receptor or restoring SMAD4 has no impact on the maintenance of WNT-independence, it is possible that cytokine-dependent YAP induction guides reprogramming on a different course to transient wound repair.
Unlike SMAD4, we show that disruption of p53, which impacts cellular reprogramming and lineage plasticity in multiple settings (45–48), is critical for the initiation and maintenance of lineage reversion. Consistent with this, mutations in TP53 have been associated with the development of WNT/RSPO niche independence in human gastric and pancreatic organoids (49,50), although the precise mechanisms that drive pathway independence in each tissue-type may be distinct. Interestingly, TP53 disruption is extremely common in patients with colitis-associated CRC, whose tumors show relatively infrequent WNT activating mutations (51). It is tempting to speculate that induction of an inflammation-induced YAP regenerative program in p53 mutant cells would enable WNT-independent cancer growth. It is notable that TP53 (and KRAS/BRAF) mutations were present in each human RSPO3 fusion CRC case we identified co-incident active-YAP1, ANXA1 and MSLN expression, while it did not correlate precisely with SMAD4 status. Though the number of samples is limited, it is likely that SMAD4 loss is not an absolute requirement for YAP/TAZ activation in CRC, which can be induced by factors other than TGFβ (44). Hence, while we describe one mechanism of lineage reversion and WNT-independence, we suspect that there will be other genetic combinations, contexts, and/or extracellular stimuli that induce a similar WNT-independent pro-tumorigenic outcome. Similar to our work, previous studies have identified elevated expression of a variety of YAP and fetal-like markers in human CRC, including ANXA1 (52), SPP1 (53), and MSLN (54,55). Whether these markers alone can distinguish WNT independent or ‘primed’ human tumors is unknown.
Our functional studies indicate that both YAP and TAZ, individually, are capable of inducing lineage reversion and WNT independence, while knockdown experiments suggest YAP may be a more dominant driver of this process. Both YAP and TAZ are controlled post-translationally by phosphorylation and degradation (56), but it remains unclear exactly how these processes support ongoing pathway activation to maintain WNT independence. One plausible mechanism is through engagement with the extracellular matrix and activation of YAP/TAZ via FAK and SRC, as has been described in wound repair (34,36). Interestingly, while YAP and TAZ were sufficient to drive WNT independence in our patient-derived organoid model, TGFβ was not sufficient to induce this response in the same way as murine organoids, at least in the one patient-derived organoid sample we assessed. We do not yet know the reason for this, though it is possible that culture in mouse-derived ECM (Matrigel) does not provide the necessary extracellular cues to trigger appropriate intracellular signaling to YAP, as was observed in the context of wound repair (36). Such a mechanism would also provide a reasonable explanation for the lack of PORCN inhibitor resistance reported in previously xenograft studies (12,13). It is also likely that there are other unidentified factors that influence the activation of YAP/TAZ and WNT inhibitor resistance. Identifying those genes and pathways required to maintain WNT independence will help reveal these signaling networks and may highlight new therapeutic opportunities. Regardless of the mechanism of activation, the dependence on YAP/TAZ for survival of WNT-independent cells suggests that therapies directly targeting these effectors would be an attractive approach to prevent or reverse WNT inhibitor resistance. As potent and selective YAP/TAZ-targeted agents become available it will be important to determine whether they can be used safely in combination, or sequentially, with WNT inhibitors.
The work described here was guided by the clinical genetics of CRC, and in particular, RSPO fusion CRC. Due to the paucity of pre- and post-treatment clinical samples, we exploited engineered murine organoids to interrogate the mechanisms of WNT dependence, but this work has obvious implications for clinical application of WNT therapies. There is evidence that lineage changes during colonic wound repair are similar in mouse and human (36), but owing to lack of clinical treatment data, we don’t yet know the exact conditions or frequency with which lineage reversion occurs in human CRC or how it will impact clinical treatment response. To date, WNT-targeted drugs have shown a range of on-target dose-limiting toxicity, including bone fractures (57), though recent work has demonstrated that a combination of RANKL and PORCN inhibitors provide potent WNT pathway suppression while avoiding bone-related adverse events (26). Based on this data, new combination trials have been launched, specifically targeting RSPO3 fusion CRC. It is too early to know the outcome of these new trials, but given the prevalence of p53, SMAD4, and RAS/RAF alterations in RSPO fusion cancers, it will be critical to pay close attention to how genotype influences clinical tumor behavior, and to assess features of lineage reversion in emergent resistant disease. Ultimately, such genetic biomarkers may serve to further stratify patients to improve clinical outcomes.
Methods
Cloning
Sequences encoding guide RNAs (Table S2) were cloned into the BsmBI site of a Cas9-P2A-Cre (LCC) lentiviral vector. For tandem guide RNA cloning, each U6-sgRNA cassette was amplified from px458 vector and ligated into the EcoRI site of LCC. For shRNA cloning, 97mer oligonucleotides (Table S2) were PCR amplified as previously described (58,59) using specific primers (Table S2) and cloned into the XhoI/EcoRI sites of SGEN (59) (Addgene #111171). For tandem shRNA cloning, shRNAs were cloned sequentially into the EcoRI site downstream of the existing shRNA in SGEN, as previously described (60). For cDNA cloning, cDNAs were PCR amplified using specific primers from gBlocks (Table S2) synthesized at Integrated DNA Technologies, and ligated into the BglII/MluI sites of LMN (61) using Gibson assembly.
Animal studies
All studies involving animals were approved by the Institutional Animal Care and Use Committee (IACUC) at Weill Cornell Medicine (NY), under protocol number 2014–0038. For Apc restoration studies, animals were fed with Doxycycline chow (200mg/kg) (Harlan Teklad) 1 day before transplantation. Organoids (~100,000 cells for each flank) were implanted subcutaneously into athymic nude mice. Animals were distributed randomly into “ON DOX” and “OFF DOX” groups when tumors reached a volume of approximately 100mm3. For WNT974 treatment studies, organoids (~100,000 cells for each flank) were implanted subcutaneously into athymic nude mice. After 2 weeks, animals were randomized for WNT974 treatment. WNT974 (5mg/kg, Selleckchem #S7143) was mixed with 0.5% methylcellulose and 0.1% Tween 80 and then administrated by daily oral gavage for 2 weeks. All animals were humanely euthanized after 5 weeks. Macrodissected tumors were weighed, fixed in fresh 4% paraformaldehyde (PFA), and processed for histology.
Immunohistochemistry and immunofluorescence
Tissue, fixed in freshly prepared 4% paraformaldehyde for 24 hours, was embedded in paraffin and sectioned by IDEXX RADIL (Columbia, MO). Sections were rehydrated and unmasked (antigen retrieval) by either: (i) Heat treatment for 5 mins in a pressure cooker in 10mM Tris / 1mM EDTA buffer (pH 9) containing 0.05% Tween 20 or (ii) (for Lysozyme staining only) Proteinase K treatment (200ug/ml) for 10 mins at 37ºC. For immunohistochemistry, sections were treated with 3% H2O2 for 10 mins and blocked in TBS / 0.1% Triton X-100 containing 1% BSA. MSLN staining was performed in a Leica Bond automated platform, with pre-treatment (ER2) for 30 mins. For immunofluorescence, sections were not treated with peroxidase. Primary antibodies, incubated at 4C overnight in blocking buffer, were: rabbit anti-ki67 (1:100, Sp6 clone, Abcam #ab16667), rabbit anti-KRT20 (1:200, Cell Signaling Technologies, #13063), rabbit anti-Lysozyme (1:1000, Dako, #EC 3.2.1.17), rat anti-BrdU (1:200, Abcam #ab6326), rabbit anti-YAP/TAZ (1:200, Cell Signaling Technologies, #8418), mouse anti-Spp1 (1:200, Santa Cruz Biotechnology, #sc-21742), anti-pSmad3 (1:200, Abcam #ab52903), anti-ANXA1 (1:200, Invitrogen, #PA5–27315, anti-MSLN (1:100 Vector Labs, clone 5B2, # VP-M649), anti-active-YAP (1:2000, Abcam #205270),. For immunohistochemistry, sections were incubated with anti-rabbit or anti-rat ImmPRESS HRP-conjugated secondary antibodies (Vector Laboratories, #MP7401) and chromagen development performed using ImmPact DAB (Vector Laboratories, #SK4105). Stained slides were counterstained with Harris’ hematoxylin. For immunofluorescent stains, secondary antibodies were applied in TBS for 1 hour at room temp in the dark, washed twice with TBS, counterstained for 5 mins with DAPI and mounted in ProLong Gold (Life Technologies, #P36930). Secondary antibodies used were: anti-rabbit 568 (1:500, Molecular Probes, #a11036). Images of fluorescent and IHC stained sections were acquired on a Zeiss Axioscope Imager (chromogenic stains), Nikon Eclipse T1 microscope (IF stains). Raw .tif files were processed using FIJI (Image J) and/or Photoshop (Adobe Systems Inc., San Jose, CA) to create stacks, adjust levels and/or apply false coloring.
Isolation and culture of intestinal organoids
Isolation, maintenance and staining of mouse intestinal organoids has been described previously (62,63). Briefly, for isolation, 15 cm of the proximal small intestine was removed and flushed with cold PBS. The intestine was then cut into 5 mm pieces, vigorously resuspended in 5mM EDTA-PBS using a 10ml pipette, and placed at 4ºC on a benchtop roller for 10 minutes. This was then repeated for a second time for 30 minutes. After repeated mechanical disruption by pipette, released crypts were mixed with 10ml DMEM Basal Media (Advanced DMEM F/12 containing Pen/Strep, Glutamine, 1mM N-Acetylcysteine (Sigma Aldrich A9165-SG)) containing 10 U/ml DNAse I (Roche, 04716728001), and filtered sequentially through 100μm and 70μm filters. 1ml FBS (final 5%) was added to the filtrate and spun at 125g for 4 minutes. The purified crypts were resuspended in basal media and mixed 1:10 with Growth Factor Reduced Matrigel (BD, 354230). 40μl of the resuspension was plated per well in a 48 well plate and placed in a 37ºC incubator to polymerize for 10 minutes. 250μl of small intestinal organoid growth media (Basal Media containing 50 ng/mL EGF (Invitrogen PMG8043), 100ng/ml Noggin (Peprotech 250–38), and 500 ng/mL R-spondin (R&D Systems, 3474-RS-050, or from conditioned media) was then laid on top of the Matrigel. Where indicated, dox was added to experiments at 500 ng/ml.
For sub-culture and maintenance, media was changed on organoids every two days and they were passaged 1:4 every 3–5 days. To passage, the growth media was removed and the Matrigel was resuspended in cold PBS and transferred to a 15ml falcon tube. The organoids were mechanically disassociated using a p1000 or a p200 pipette and pipetting 50–100 times. 7 ml of cold PBS was added to the tube and pipetted 20 times to fully wash the cells. The cells were then centrifuged at 125g for 5 minutes and the supernatant was aspirated. They were then resuspended in GFR Matrigel and replated as above. For freezing, after spinning the cells were resuspended in Basal Media containing 10% FBS and 10% DMSO and stored in liquid nitrogen indefinitely.
Organoid Imaging and Counting
For fixed staining, organoids were grown in 40μl of Matrigel plated into an 8-well chamber slide (Lab-Tek II, 154534). Where indicated, 10μM EdU was added to the growth media for 6 hours before fixing. The growth media was removed and the cells were fixed in 4% PFA-PME (50mM PIPES, 2.5 mM MgCl2, 5mM EDTA) for 20 minutes. They were then permeabilized in .5% Triton for 20 minutes and blocked in IF Buffer (PBS, .2% Triton, .05% Tween, 1% BSA) for 1 hour or immediately processed for EdU staining performed according to directions provided with the Click-iT Edu Alexa Fluor 647 Imaging Kit (Invitrogen C10340). For alkaline phosphatase staining, fixed cells were washed twice with TBS and then incubated with the BCIP/NBT Substrate Kit (Vector Laboratories, SK-5400) for 15 minutes in the dark. The chambers were then washed twice with TBS and then imaged using bright field microscopy. For immunofluorescent staining, cells were incubated in primary antibodies overnight in IF buffer: rabbit anti-KRT20 (1:200, Cell Signaling Technologies, #13063), rabbit anti-Lysozyme (1:200, Dako, #EC 3.2.1.17). They were then washed 3 times with TBS .1% Tween. Secondary antibodies (1:1000, same reagents as above) were incubated with or without Alexa-647 Phalloidin (Molecular Probes, #A22287) for 1 hour. The solution was removed and DAPI in PBS was added for 5 minutes and washed twice with TBS .1% Tween. The chambers were then removed and cover slips were mounted using Prolong Gold antifade medium (Invitrogen P36930). Images were acquired using Zeiss LSM 880 laser scanning confocal microscope, and Zeiss image acquiring and processing software. Images were processed using FIJI (Image J) and Photoshop CS software (Adobe Systems Inc., San Jose, CA). For determining the proportion of spheroids to organoids, both total number of live organoids and spheroids were counted in representative microscopy fields using 4X objective lens. At least three different fields for each sample were counted. The percentage of spheroids is calculated by dividing the number of spheroids to total number of organoids.
Organoid transfection, transduction, and selection
Murine small intestinal organoids were cultured in transfection medium containing CHIR99021 (5μΜ) and Y-27632 (10μΜ) for 2 days prior to transfection. Single cells suspensions were produced by dissociating organoids with TrypLE™ express (Invitrogen #12604) for 5 min at 37°C. After trypsinization, cell clusters in 300μl transfection medium were combined with 100μl DMEM/F12-lipofectamine2000 (Invitrogen #11668)-DNA mixture (97ul-2ul-1ug), and transferred into a 48-well culture plate. The plate was centrifuged at 600g at 32°C for 60 min, followed by another 6h incubation at 37°C. The cell clusters were spun down and plated in Matrigel. For transduction, organoids were pre-treated and dissociated as for transfection, then mixed with viral supernatant and transferred to a 48-well plate for spinocculation. The plate was centrifuged at 600g at 32°C for 60 min, followed by another 4–6h incubation at 37°C. The cell clusters were spun down and plated in Matrigel. Functional selection: To select for organoids with Ptprk-Rspo3 rearrangements, exogenous RSPO1 was withdrawn from the culture media 1 week after transfection; For selection of organoids with Kras or Braf mutations, organoids were treated with 1μM Gefitinib for 2 weeks; For selection of organoids with loss-of-function Trp53 mutations, organoids were cultured in medium containing Nutlin3 (5μM); For selection of organoids with loss-of-function Smad4 or Tgfbr2 alterations, cell were cultured in medium containing TGFβ1 (5ng/mL) for 1 week. To generate TGFβ-naïve Smad4/Tgfbr2 organoids, cells transduced with both Trp53 and Smad4/Tgfbr2 sgRNAs were selected using Nutlin3 only.
Human RSPO fusion identification
Human cancer samples were collected and analyzed under appropriate Institutional Review Board protocols and waivers (#06–107, 12–245, 16–1261). DNA sequencing was performed with the MSK-IMPACT assay, a targeted exome capture assay with deep sequencing coverage (64). RSPO2/3 fusions were detected with a custom Archer-targeted RNA-seq-based next-generation sequencing assay (65). Human cancer samples were collected and analyzed under appropriate Institutional Review Board protocols and waivers (#06–107, 12–245, 16–1261). DNA sequencing was performed with the MSK-IMPACT assay, a targeted exome capture assay with deep sequencing coverage (61). RSPO2/3 fusions were detected with a custom Archer-targeted RNA-seq-based next-generation sequencing assay (62). Cases with both APC / CTNNB1 / RNF43 and RAS hotspot/ BRAF p. V600E mutations were not analyzed by Archer for RNA level fusions. Therefore, some selection bias is present. All studies were conducted in accordance with the Declaration of Helsinki. Classification of tumor immunohistochemistry was performed while blinded to sample genotype.
Patient-derived organoids
Patient-derived organoids were derived under Memorial Sloan Kettering Institutional Review Board biospecimen research protocols 14–244. All studies were conducted in accordance with the Declaration of Helsinki, and all patients provided pre-procedure written informed consent. Tissue was processed within 1 h of surgical resection and organoids derived as described (40). Conservation of truncal driver mutations was verified by MSK-IMPACT of FFPE tissue block and cognate organoids.
Protein analysis
Small intestine organoids were grown in 300μl of Matrigel in 1 well of 6-well plate for 4 days post-passage. Organoids were then recovered from the Matrigel using cell recovery solution (Corning #354253). Organoid pellets were lysed with RIPA buffer. Antibodies used for Western blot were: mouse anti-p53 (1:1000, Cell Signaling Technologies, #2524), rabbit anti-Smad4 (1:1000, Cell Signaling Technologies, #46535), rabbit anti-Smad2/3 (1:1000, Cell Signaling Technologies, #8685), rabbit anti-YAP (1:1000, Cell Signaling Technologies, #14074), rabbit anti-YAP/TAZ (1:1000, Cell Signaling Technologies, #8418), rabbit anti-Runx2 (1:1000, Cell Signaling Technologies, #12556), mouse anti-E-Cadherin (1:1000, Cell Signaling Technologies, #14472), rabbit anti-phospho-Akt (Ser473) (1:1000, Cell Signaling Technologies, #4060), rabbit anti-N-Myc (1:1000, Cell Signaling Technologies, #84406), rabbit anti-c-Jun (1:1000, Cell Signaling Technologies, #9165), rabbit anti-Junb (1:1000, Cell Signaling Technologies, #3753), rabbit anti-ETS-1 (1:1000, Cell Signaling Technologies, #14069), rabbit anti-GAPDH (1:1000, Cell Signaling Technologies, #5174), rabbit anti-Cyclophilin B (1:1000, Cell Signaling Technologies, #43603), rabbit anti-pan-TEAD (1:1000, Cell Signaling Technologies, #13295), rabbit anti-phospho-Smad2 (Ser465/467) (1:1000, Cell Signaling Technologies, #3108), mouse anti-phospho-SAPK/JNK (Thr183/Tyr185) (1:1000, Cell Signaling Technologies, #9255), rabbit anti-phospho-p38 MAPK (Thr180/Tyr182) (1:1000, Cell Signaling Technologies, #4511), mouse anti-TEAD4 (1:1000, Abcam, #ab58310), rabbit anti-phospho-Smad3 (Ser423/425) (1:1000, Abcam, #ab52903).
Flow Cytometry
Organoids were pulsed with 10uM EdU for 3 hours in regular tissue culture incubator. Cells were pelleted and trypsinized using 0.25% Trypsin-EDTA (ThermoFisher scientific #25200056) at 37°C for 5 mins, re-suspended using FACS buffer (PBS+2%FBS) then filtered through cell strainer (Corning #352235). EdU was stained using Click-iT™ EdU Alexa Fluor™ 647 flow cytometry assay kit (C104424) following the manufacturer instructions. Flow cytometry was conducted on Invitrogen Attune NxT following the manufacturer instructions. For fluorescence-based competition assay, cells were transduced with lentiviral vectors containing GFP-linked shRNA/s or cDNAs. Two days after viral transduction, the frequency of GFP was measured by flow cytometry two days post-transduction (D2) and remaining cells were replated in Matrigel culture. Every three days cells were split for flow cytometry analysis and replating. The percentage of GFP positive cells at each passage is normalized to D2.
RNA isolation, cDNA synthesis and QPCR
RNA was extracted using TRIzol according to the manufacturer’s instructions and contaminating DNA was removed by DNase treatment for 10 mins and column purification (Qiagen RNAeasy). cDNA was prepared from 1μg total RNA using qScript reverse transcription kit (Quantabio, #95047). Quantitative PCR detection was performed using SYBR green reagents (Quantabio #101414–288) and specific primers listed in Table S2.
Whole exome sequencing
Each gDNA sample based on Qubit quantification are mechanically fragmented on an Covaris E220 focused ultrasonicator (Covaris, Woburn, MA, USA). Two hundred ng of sheared gDNA were used to perform end repair, A-tailing and adapter ligation with Agilent SureSelect XT (Agilent Technologies, Santa Clara, CA) library preparation kit following the manufacturer instructions. Then, the libraries are captured using Agilent SureSelectXT Mouse All Exon probes, and amplified. The quality and quantities of the final libraries were checked by Agilent 2100 Bioanalyzer and Invitrogen Qubit 4.0 Fluorometer (Thermo Fisher, Waltham, MA), libraries are pooled at 8 samples per lane and sequenced on an Illumina HiSeq 4000 sequencer (Illumina Inc, San Diego, CA) at PE 2×100 cycles. Copy number alterations were identified and plotted using cnvkit (v0.9.6) and single nucleotide varaiant called using MuTect2.
RNA sequencing
Total RNA was isolated using Trizol, DNAse treated and purified using the RNeasy mini kit (Qiagen, Hilden, Germany). Following RNA isolation, total RNA integrity was checked using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA concentrations were measured using the NanoDrop system (Thermo Fisher Scientific, Inc., Waltham, MA). Preparation of RNA sample library and RNAseq were performed by the Genomics Core Laboratory at Weill Cornell Medicine. Messenger RNA was prepared using TruSeq Stranded mRNA Sample Library Preparation kit (Illumina, San Diego, CA), according to the manufacturer’s instructions. The normalized cDNA libraries were pooled and sequenced on Illumina NextSeq500 sequencer with single-end 75 cycles.
RNAseq analysis
The quality of raw FASTQ files were mapped to mouse reference GRCm38 using STAR two-pass alignment (v2.4.1d; default parameters) (66), and transcript abundance estimates were performed using Kallisto (67), aligned to the same (GRCm38) reference genome. Kallisto transcript count data for each sample was concatenated, and transcript per million (TPM) data was reported for each gene after mapping gene symbols to ensemble IDs using R packages, “tximport”, tximportData”, “ensembldb”, and “EnsDb.Mmusculus.v79”. Differential gene expression was estimated using DESeq2 (68). For data visualization and gene ranking, log fold changes were adjusted using lfcShrink in DESeq2, to minimize the effect size of poorly expressed genes. GSEA analysis (v3.0) was performed on pre-ranked gene sets from differential expression between control and treated groups. WE used a custom curated geneset including the Broad “HALLMARK” collection and published bulk and single cell RNAseq from murine intestine (https://github.com/lukedow/Genesets.git). We used R (v3.6.1) and R Studio (v1.2.1335) to create all visualizations, perform hierarchical clustering and principal component analysis. Volcano plots, heatmaps and other visualizations were produced using the software packages:
Enhanced Volcano (https://bioconductor.org/packages/devel/bioc/html/EnhancedVolcano.html)
pheatmap (https://cran.r-project.org/web/packages/pheatmap/index.html)
ggplot2 (https://cran.r-project.org/web/packages/ggplot2/index.html)
ATAC sequencing
ATAC-seq library preparation, sequencing and post-processing of the raw data was performed at the Epigenomics Core at Weill Cornell Medicine by using the OMNI-ATAC-seq method previously described (69). Briefly, organoids were dissociated from Matrigel using cell recovery solution (Corning #354253), and 50,000 live cells were spun down and incubated 3 min at 4oC in 25μl of a detergent buffer containing 0.2% Igepal CA-630 (Sigma-Aldrich, St.Louis, MA), 0.2% Tween 20 (Sigma-Aldrich, St.Louis, MA) and 0.02% Digitonin (Promega Corporation, Madison, WI). Nuclei were centrifuged at 500 g for 10 min and immediately resuspended in 25 μl of buffer containing 2.5 μl of Tn5 transposase (Illumina, Inc., San Diego, CA cat # 15027865) for a 30 min incubation at 37oC. Fragments generated by the Tn5 transposase were purified using the DNA Clean and Concentrate kit from Zymo Research (Zymo Research, Irvine, CA cat #D4014). Uniquely indexed libraries were obtained by amplification of the purified fragments with indexed primers using 9 cycles of PCR (5 min x 72 oC, 5 cycles each 10 sec x 98 oC, 30 sec x 63 oC, 1 min x 72 oC). Resulting libraries were subjected to a two-sided size clean up using SPRI beads (Beckman Coulter, Brea, CA) to obtain sizes between 200–1000bp, and pooled for sequencing. The pool was clustered at 9 pM on a pair end read flow cell and sequenced for 50 cycles on an Illumina HiSeq 2500 to obtain ~40M reads per sample. Primary processing of sequencing images was done using Illumina’s Real Time Analysis software (RTA) as suggested by the Illumina. CASAVA 2.17 software was used to perform image capture, base calling and demultiplexing of the raw reads. FASTQ files generated were aligned to the mouse mm10 genome build using the BWA aligner.
ATACseq analysis
Raw ATACseq reads were processed using ENCODE ATACseq-pipeline. This includes Bowtie2(70) alignment against mm10 and MACS2(71) as peak caller. Duplicated reads and reads from mitochondria were filtered out. Peaks for all samples were merged together into a union of peaks, and the ones that existed in at least two samples were kept for further analysis. Peaks were annotated using the ChIPseeker (72) “annotatePeak” function with “TxDb.Mmusculus.UCSC.mm10.knownGene”. Numbers of reads mapped to each peak were counted with featureCounts (73), and raw read counts were normalized using rlog in DESeq2(68). Differentially accessible peaks were defined as peaks with p.adj smaller than 0.05 and absolute value of log2(FC) >1. Known and de novo motifs were idetified using findMotifsGenome.pl from Homer (74) with ‘-size given -mask’ was applied on the differentially accessible peaks, with non-differentially accessible peaks as background.
Single cell RNA sequencing
Organoids were dissociated from Matrigel using cell recovery solution (Corning #354253), then trypsinized using 10X trypsin (ThermoFisher Scientific #15090046) for 15 minutes at 37C. Single cells were resuspended using organoid culture medium and stained with DAPI. DAPI negative single cells were sorted using BD Aria II FACS machine with 130um nozzle. Sorted single cell suspensions were transferred to the Genomics Core Facility at Weill Cornell Medicine to proceed with the Chromium Single Cell 3’ Reagent Kit v3 (10x Genomics, product code # 1000075) using 10X Genomics’ Chromium Controller. A total of 10,000 cells were loaded into each channel of the Single-Cell A Chip to target 5000 cells in the end. Briefly, according to manufacturer’s instruction, the sorted cells were washed with 1x PBS + 0.04% BSA, counted by Bio-Rad TC20 Cell Counter, and cell viability was assessed and visualized. A total of 10,000 cells and Master Mixes were loaded into each channel of the cartridge to generate the droplets on Chromium Controller. Beads-in-Emulsion (GEMs) were transferred and GEMs-RT was undertaken in droplets by PCR incubation. GEMs were then broken and pooled fractions are recovered. After purification of the first-strand cDNA from the post GEM-RT reaction mixture, barcoded, full-length cDNA was amplified via PCR to generate sufficient mass for library construction. Enzymatic fragmentation and size selection were used to optimize the cDNA amplicon size. TruSeq Read 1 (read 1 primer sequence) was added to the molecules during GEM incubation. P5, P7, a sample index, and TruSeq Read 2 (read 2 primer sequence) were added via End Repair, A-tailing, Adaptor Ligation, and PCR. The final libraries were assessed by Agilent Technology 2100 Bioanalyzer and sequenced on Illumina NovaSeq sequencer with pair-end 100 cycle kit.
scRNAseq analysis
Reads were pseudoaligned and transcript abundances estimated using kallisto (v0.46.0), and gene count matrices were produced using bustools (v0.39.3) (75) and BUSpaRse. Count matrices were processed and analysed using the Seurat package (v3.1.1) in R (v3.6.1) and R Studio (v1.2.1335). Low quality cells (Less than 1000 identified genes and greater than 10% mitochondrial reads) were removed from the dataset after normalization. Individual scRNAseq datasets were merged function and clusters were resolved on the combined data. Expression of cell type markers were plotted by calculating the mean expression of the genes indicated for each individual cell, using a custom function (“FeaturePlot.1”; https://github.com/nyuhuyang/SeuratExtra”).
Data availability
Raw exomeSeq, RNAseq, ATACseq and scRNAseq data have been deposited in the sequence read archive (SRA) under accession PRJNA578488.
Supplementary Material
Statement of Significance.
Colorectal and intestinal cancers are driven by mutations in the WNT pathway and drugs aimed at suppressing WNT signaling are in active clinical development. Our study identifies a mechanism of acquired resistance to WNT inhibition and highlights a potential strategy to target those drug-resistant cells.
Acknowledgements
We thank Wouter Karthaus, Charles Sawyers, and Darjus Tschaharganeh for technical and experimental advice. We thank Marie Parsons, Francisco Barriga, Michal Nagiec, Ashley Laughney, and J. Joshua Smith for advice and comments on preparation of the manuscript. This work was supported by a Research Scholar Award from the American Cancer Society (RSG-17–202-01-TBG), project grant from the NIH/NCI (1R01CA222517–01A1), project grants from the Starr Cancer Consortium (#I10–0095 and #I11–0040) and a Stand Up to Cancer Colorectal Cancer Dream Team Translational Research Grant (SU2C-AACR-DT22–17). Stand Up to Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, a scientific partner of SU2C. We thank the Weill Cornell Genomics Resource Core Facility who performed library preparation and sequencing for WES, RNAseq, and scRNAseq, and the Weill Cornell Epigenomics Core Facility who performed library preparation and ATACseq. MPZ is supported in part by National Cancer Institute (NCI) Grant NIH T32 CA203702. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest Statement
LED is a scientific advisor for Mirimus Inc.
REFERENCES
- 1.Luis TC, Weerkamp F, Naber BA, Baert MR, de Haas EF, Nikolic T, et al. Wnt3a deficiency irreversibly impairs hematopoietic stem cell self-renewal and leads to defects in progenitor cell differentiation. Blood 2009;113(3):546–54 doi 10.1182/blood-2008-06-163774. [DOI] [PubMed] [Google Scholar]
- 2.Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007;12(6):528–41 doi 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev 2003;17(14):1709–13 doi 10.1101/gad.267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fevr T, Robine S, Louvard D, Huelsken J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol 2007;27(21):7551–9 doi 10.1128/MCB.01034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7 doi 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature 2012;488(7413):660–4 doi 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moser AR, Shoemaker AR, Connelly CS, Clipson L, Gould KA, Luongo C, et al. Homozygosity for the Min allele of Apc results in disruption of mouse development prior to gastrulation. Dev Dyn 1995;203(4):422–33. [DOI] [PubMed] [Google Scholar]
- 8.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J 1999;18(21):5931–42 doi 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han T, Schatoff EM, Murphy C, Zafra MP, Wilkinson JE, Elemento O, et al. R-Spondin chromosome rearrangements drive Wnt-dependent tumour initiation and maintenance in the intestine. Nat Commun 2017;8:15945 doi 10.1038/ncomms15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dow LE, O’Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, et al. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 2015;161(7):1539–52 doi 10.1016/j.cell.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Rourke KP, Loizou E, Livshits G, Schatoff EM, Baslan T, Manchado E, et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat Biotechnol 2017;35(6):577–82 doi 10.1038/nbt.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madan B, Ke Z, Harmston N, Ho SY, Frois AO, Alam J, et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016;35(17):2197–207 doi 10.1038/onc.2015.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storm EE, Durinck S, de Sousa e Melo F, Tremayne J, Kljavin N, Tan C, et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature 2016;529(7584):97–100 doi 10.1038/nature16466. [DOI] [PubMed] [Google Scholar]
- 14.Kawasaki K, Fujii M, Sugimoto S, Ishikawa K, Matano M, Ohta Y, et al. Chromosome Engineering of Human Colon-Derived Organoids to Develop a Model of Traditional Serrated Adenoma. Gastroenterology 2020;158(3):638–51 e8 doi 10.1053/j.gastro.2019.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001;15(24):3243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015;521(7550):43–7 doi 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- 17.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nature medicine 2015;21(3):256–62 doi 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 18.Karreth FA, Tay Y, Perna D, Ala U, Tan SM, Rust AG, et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 2011;147(2):382–95 doi 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Batlle E, Massague J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019;50(4):924–40 doi 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol 2005;25(18):8108–25 doi 10.1128/MCB.25.18.8108-8125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, et al. TGF-beta Tumor Suppression through a Lethal EMT. Cell 2016;164(5):1015–30 doi 10.1016/j.cell.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell 2017;170(6):1062–78 doi 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554(7693):538–43 doi 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 24.Jackstadt R, van Hooff SR, Leach JD, Cortes-Lavaud X, Lohuis JO, Ridgway RA, et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell 2019;36(3):319–36 e7 doi 10.1016/j.ccell.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guimaraes PPG, Tan M, Tammela T, Wu K, Chung A, Oberli M, et al. Potent in vivo lung cancer Wnt signaling inhibition via cyclodextrin-LGK974 inclusion complexes. J Control Release 2018;290:75–87 doi 10.1016/j.jconrel.2018.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madan B, McDonald MJ, Foxa GE, Diegel CR, Williams BO, Virshup DM. Bone loss from Wnt inhibition mitigated by concurrent alendronate therapy. Bone Res 2018;6:17 doi 10.1038/s41413-018-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1(6):417–25 doi 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, et al. A single-cell survey of the small intestinal epithelium. Nature 2017;551(7680):333–9 doi 10.1038/nature24489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim TH, Li F, Ferreiro-Neira I, Ho LL, Luyten A, Nalapareddy K, et al. Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature 2014;506(7489):511–5 doi 10.1038/nature12903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moor AE, Harnik Y, Ben-Moshe S, Massasa EE, Rozenberg M, Eilam R, et al. Spatial Reconstruction of Single Enterocytes Uncovers Broad Zonation along the Intestinal Villus Axis. Cell 2018;175(4):1156–67 e15 doi 10.1016/j.cell.2018.08.063. [DOI] [PubMed] [Google Scholar]
- 31.Mustata RC, Vasile G, Fernandez-Vallone V, Strollo S, Lefort A, Libert F, et al. Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep 2013;5(2):421–32 doi 10.1016/j.celrep.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Qi Z, Li Y, Zhao B, Xu C, Liu Y, Li H, et al. BMP restricts stemness of intestinal Lgr5(+) stem cells by directly suppressing their signature genes. Nat Commun 2017;8:13824 doi 10.1038/ncomms13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perekatt AO, Shah PP, Cheung S, Jariwala N, Wu A, Gandhi V, et al. SMAD4 Suppresses WNT-Driven Dedifferentiation and Oncogenesis in the Differentiated Gut Epithelium. Cancer Res 2018;78(17):4878–90 doi 10.1158/0008-5472.CAN-18-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, et al. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 2019;569(7754):121–5 doi 10.1038/s41586-019-1154-y. [DOI] [PubMed] [Google Scholar]
- 35.Panciera T, Azzolin L, Fujimura A, Di Biagio D, Frasson C, Bresolin S, et al. Induction of Expandable Tissue-Specific Stem/Progenitor Cells through Transient Expression of YAP/TAZ. Cell Stem Cell 2016;19(6):725–37 doi 10.1016/j.stem.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yui S, Azzolin L, Maimets M, Pedersen MT, Fordham RP, Hansen SL, et al. YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell 2018;22(1):35–49 e7 doi 10.1016/j.stem.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koinuma D, Tsutsumi S, Kamimura N, Taniguchi H, Miyazawa K, Sunamura M, et al. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor beta signaling. Mol Cell Biol 2009;29(1):172–86 doi 10.1128/MCB.01038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li QV, Dixon G, Verma N, Rosen BP, Gordillo M, Luo R, et al. Genome-scale screens identify JNK-JUN signaling as a barrier for pluripotency exit and endoderm differentiation. Nat Genet 2019;51(6):999–1010 doi 10.1038/s41588-019-0408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK, et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep 2018;25(5):1304–17 e5 doi 10.1016/j.celrep.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ganesh K, Basnet H, Kaygusuz Y, Laughney AM, He L, Sharma R, et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nature Cancer 2020;1(1):28–45 doi 10.1038/s43018-019-0006-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014;158(1):157–70 doi 10.1016/j.cell.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 42.Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 2015;526(7575):715–8 doi 10.1038/nature15382. [DOI] [PubMed] [Google Scholar]
- 43.Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2013;493(7430):106–10 doi 10.1038/nature11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016;29(6):783–803 doi 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014;158(3):579–92 doi 10.1016/j.cell.2014.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355(6320):84–8 doi 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355(6320):78–83 doi 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loizou E, Banito A, Livshits G, Ho YJ, Koche RP, Sanchez-Rivera FJ, et al. A Gain-of-Function p53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia through the Pluripotency Factor FOXH1. Cancer Discov 2019;9(7):962–79 doi 10.1158/2159-8290.CD-18-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nanki K, Toshimitsu K, Takano A, Fujii M, Shimokawa M, Ohta Y, et al. Divergent Routes toward Wnt and R-spondin Niche Independency during Human Gastric Carcinogenesis. Cell 2018;174(4):856–69 e17 doi 10.1016/j.cell.2018.07.027. [DOI] [PubMed] [Google Scholar]
- 50.Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M, et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018;22(3):454–67 e6 doi 10.1016/j.stem.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 51.Yaeger R, Shah MA, Miller VA, Kelsen JR, Wang K, Heins ZJ, et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2016;151(2):278–87 e6 doi 10.1053/j.gastro.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaspar C, Cardoso J, Franken P, Molenaar L, Morreau H, Moslein G, et al. Cross-species comparison of human and mouse intestinal polyps reveals conserved mechanisms in adenomatous polyposis coli (APC)-driven tumorigenesis. Am J Pathol 2008;172(5):1363–80 doi 10.2353/ajpath.2008.070851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin AY, Chua MS, Choi YL, Yeh W, Kim YH, Azzi R, et al. Comparative profiling of primary colorectal carcinomas and liver metastases identifies LEF1 as a prognostic biomarker. PLoS One 2011;6(2):e16636 doi 10.1371/journal.pone.0016636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Serna G, Ruiz-Pace F, Cecchi F, Fasani R, Jimenez J, Thyparambil S, et al. Targeted multiplex proteomics for molecular prescreening and biomarker discovery in metastatic colorectal cancer. Sci Rep 2019;9(1):13568 doi 10.1038/s41598-019-49867-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shiraishi T, Shinto E, Mochizuki S, Tsuda H, Kajiwara Y, Okamoto K, et al. Mesothelin expression has prognostic value in stage IotaIota/IotaIotaIota colorectal cancer. Virchows Arch 2019;474(3):297–307 doi 10.1007/s00428-018-02514-4. [DOI] [PubMed] [Google Scholar]
- 56.Moya IM, Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol 2019;20(4):211–26 doi 10.1038/s41580-018-0086-y. [DOI] [PubMed] [Google Scholar]
- 57.Tan D, Ng M, Subbiah V, Messersmith W, Teneggi V, Diermayr V, et al. 71OPhase I extension study of ETC-159 an oral PORCN inhibitor administered with bone protective treatment, in patients with advanced solid tumours. Annals of Oncology 2018;29(suppl_9) doi 10.1093/annonc/mdy430.002. [DOI] [Google Scholar]
- 58.Dow LE, Premsrirut PK, Zuber J, Fellmann C, McJunkin K, Miething C, et al. A pipeline for the generation of shRNA transgenic mice. Nat Protoc 2012;7(2):374–93 doi 10.1038/nprot.2011.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep 2013;5(6):1704–13 doi 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 60.Schatoff EM, Goswami S, Zafra MP, Foronda M, Shusterman M, Leach BI, et al. Distinct Colorectal Cancer-Associated APC Mutations Dictate Response to Tankyrase Inhibition. Cancer Discov 2019;9(10):1358–71 doi 10.1158/2159-8290.CD-19-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol 2011;29(1):79–83 doi 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Rourke KP, Ackerman S, Dow LE, Lowe SW. Isolation, Culture, and Maintenance of Mouse Intestinal Stem Cells. Bio Protoc 2016;6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Rourke KP, Dow LE, Lowe SW. Immunofluorescent Staining of Mouse Intestinal Stem Cells. Bio Protoc 2016;6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17(3):251–64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu G, Benayed R, Ho C, Mullaney K, Sukhadia P, Rios K, et al. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod Pathol 2019;32(5):609–20 doi 10.1038/s41379-018-0175-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016;34(5):525–7 doi 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 68.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 2017;14(10):959–62 doi 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods 2012;9(4):357 doi doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9(9):R137 doi 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu G, Key Laboratory of Functional Protein Research of Guangdong Higher Education Institutes CoLSaT, Jinan University, Guangzhou 510632, China,, State Key Laboratory of Emerging Infectious Diseases SoPH, The University of Hong Kong, Hong Kong SAR, China and, Wang L-G, Guangdong Information Center G, China, He Q-Y, et al. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2018;31(14):2382–3 doi 10.1093/bioinformatics/btv145. [DOI] [Google Scholar]
- 73.Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 2019;47(8):e47 doi 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38(4):576–89 doi 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Melsted P, Booeshaghi AS, Gao F, Beltrame E, Lu L, Hjorleifsson KE, et al. Modular and efficient pre-processing of single-cell RNA-seq. bioRxiv 2019. doi 10.1101/673285. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw exomeSeq, RNAseq, ATACseq and scRNAseq data have been deposited in the sequence read archive (SRA) under accession PRJNA578488.