

Abstract
We generated eight multiple myeloma (MM) cell lines resistant to bortezomib (BTZ); five acquired PSMB5 mutations. In 1,500 patients such mutations were rare clinically. To better understand disruption of proteasomes on MM viability and drug sensitivity, we systematically deleted the major proteasome catalytic subunits. MM cells without PSMB5 were viable. Drug resistant, PSMB5 mutated, cell lines were re-sensitized to BTZ by PSMB5 deletion, implying PSMB5 mutation is activating in its drug resistance function. In contrast, PSMB6 knockout was lethal to MM cell lines. Depleting PSMB6 prevented splicing of the major catalytic subunits PSMB5, PSMB7, PSMB8 and PSMB10, however PSMB6 engineered without splicing function or catalytic activity, also restored viability, inferring the contribution of PSMB6 to proteasome structure to be more important than functional activity. Supporting this, BTZ sensitivity was restored in drug resistant MM cell lines by low level expression of mutated PSMB6 lacking splicing function. Loss of PSMB8 and PSMB9 was neither lethal nor restored BTZ sensitivity. Significant co-dependency of PSMB5, PSMB6 and PSMB7 expression was observed. We demonstrated elevated levels of PSMB6 and 7 but not 8 and 9 in some, but not all, serial patient samples exposed to proteasome inhibitors. In summary, we show PSMB6 and PSMB7 but not PSMB5 to be essential for MM cell survival, this dependency is structural and that up-regulation or activating mutation of PSMB5, 6 and 7 confers proteasome inhibitor resistance while depletion confers sensitivity. These findings support modulation of PSMB5, PSMB6 or PSMB7 expression as a new therapeutic strategy.
Keywords: Proteasome, PSMB5, PSMB6, PSMB7, PSMB8, PSMB9, multiple myeloma, Bortezomib resistance
Introduction
Multiple myeloma (MM) is a plasma cell malignancy characterized by the accumulation of plasma cells in the bone marrow (1). Treatment outcomes have improved significantly following the introduction of the proteasome inhibitors (PI), bortezomib (BTZ), carfilzomib and ixazomib, into routine clinical practice (2-7). PIs used in combination with other compounds, such as immunomodulatory drugs (IMiDs) and steroids achieve an initial response in 90-100% of patients (8-10). Despite the initial response, almost all patients eventually relapse and become refractory to multiple anti-MM therapies, including PIs (11).
The proteasome complex 26S consists of the 20S proteolytic core and one or two 19S regulatory complexes, which function as ubiquitin receptors to recognize substrates, deubiquitinate enzymes and facilitate substrate unfolding and translocation into the 20S proteasome. The 20S core has three β subunits that catalyze peptide hydrolysis, PSMB5 (β5), PSMB6 (β1) and PSMB7 (β2), with a chymotrypsin, caspase and trypsin-like activity, respectively (12). Upon exposure to interferon-γ these three subunits are replaced by their corresponding homologous, the immunoproteasome subunits PSMB8 (β5i), PSMB9 (β1i) and PSMB10 (β2i), respectively (13). These three constitutive catalytic β-subunits as well their immuno counterpart are expressed as a proprotein, whose catalytic functions are protected by a propeptide. These subunits are activated upon removing their propeptide by autocatalytic processing (14). Bortezomib directly inhibits the functional activity of PSMB5, PSMB6, PSMB8 and PSMB9 (15). It has long been believed that PSMB5, and not PSMB6 and PSMB7, plays an important role in cell viability, since a PSMB5 p.Thr1Ala mutation is lethal in yeast (16-19). However, subsequent publications demonstrated that significant inhibition of protein degradation can only be achieved when both, PSMB5 and either PSMB6 or PSMB7 sites are inhibited (20,21). Furthermore, proteasome inhibitor cytotoxicity does not correlate with inhibition of chymotrypsin-like sites alone, and the co-inhibition of either PSMB6 or PSMB7 sites are required for PSMB5-specific inhibitors to achieve maximum cytotoxicity (22).
Recently we employed CRISPR targeting of 19,052 human genes to identify unbiased targets that contribute to BTZ resistance(23). Of 20 targets conferring drug resistance, the proteasome regulatory subunit PSMC6 was the only validated gene to reproducibly confer BTZ resistance in our experimental model. We confirmed that inhibition of the chymotrypsin-like proteasome activity by BTZ was significantly reduced in cells lacking PSMC6. We investigated other members of the PSMC group individually (PSMC1 to 5) and found that deficiency in each of those subunits also imparts bortezomib resistance.
More recently, we have demonstrated an increased incidence of acquired proteasomal subunit mutations in relapsed MM compared to newly diagnosed disease, underpinning a potential role of point mutations in the clonal evolution of MM, including mutations in 19S proteasome subunits in 4% of patients (N=895, IA10 release of the CoMMpass study; https://themmrf.org).
Furthermore, we functionally characterized four somatic PSMB5 mutations (three of them affecting the PI-binding pocket) in primary MM cells identified in a patient under prolonged proteasome inhibition (24). Nevertheless, PSMB5 mutations are very uncommon even in relapsed MM. Since subunit mutations appears to explain only a fraction of proteasome inhibitor resistance seen in patients, we were interested in further exploring the role of proteasome subunit mutation and its role in BTZ sensitivity and resistance.
Materials and Methods
Cell lines
Eight human multiple myeloma cell lines expressing Cas9 were used in this study: RPMI-8226, FR4, U266 and JJN3(23), KMS12PE, XG-1, MM1.S and OCI-MY5, which were obtained from Dr. Leif Bergsagel’s lab (Mayo Clinic, Scottsdale, AZ) from 2014 to 2016. An initial genetic analysis of these lines (copy number variation analysis) established a baseline, identifying fingerprint (developed by Dr. Leif Bergsagel and Dr. Jonathan Keats, Translational Genomics Research Institute, Phoenix, AZ). The identity of cell lines was confirmed using copy number variation analysis each time samples are removed from liquid nitrogen storage for propagation. All cell lines taken from liquid nitrogen were thawed and maintained in RPMI1640 media, supplemented with 5% of sterile FCS and antibiotics. All cell lines were maintained for 4 to 8 weeks (less than 20 passages) before starting experiment and they were tested negative for mycoplasma at the beginning and during the experiments (using Mycoplasma Detection Kit from Lonza). BTZ resistance was developed by a stepwise increase in BTZ concentration.
Targeted sequencing
We performed DNA targeted sequencing in 8 multiple myeloma cell lines using semiconductor-sequencing technology (IonTorrent PGM) as per manufacturer’s protocol (25). All coding regions of the proteasome subunits were amplified in 200bp amplicons by multiplex PCR using customized oligos (Ion Ampliseq designer). Libraries were templated and enriched using IonOneTouch2 and IonOneTouch ES automated systems, respectively. Samples were sequenced using the 318™ chip (ThermoFisher). Raw data was aligned and indexed in BAM and BAI files using the IonTorrent suite. Variants were called using IonTorrent Somatic VariantCaller and low stringency settings (ThermoFisher). VCF files were annotated using a standalone application developed by the Mayo Clinic Bioinformatics Program called BioR (Biological Annotation Data Repository)(26). Somatic variants with a Mapping Quality <20, read depth <10X, or found in <1% of reads were removed. To remove germline mutations, common variants were eliminated based on the minor allele frequencies (>0.01%) available in one of the following germline variant databases: 1000 Genomes Project, ExAC and ESP6500, unless present in known CLL/MBL mutation hotspots or in COSMIC. Finally, variants of significant interest were visually inspected using Integrative Genomics Viewer (IGV)(27).
SgRNAs construction, lenti-vector packaging and infection
All SgRNAs were synthesized by IDT technology (Coralville, Iowa USA 52241). SgRNAs were cloned into lenti-vector pCDHPuroCr, as described before (23). SgRNA sequences are shown in supplemental Data. Packaging plasmids psPAX2 (Addgene plsmid # 12260) and pMD2.G (Addgene plasmid # 12259) were gifts from Dr. Didier Trono. A modified Graham’s calcium-phosphate method was used for packaging lentiviral vectors (28,29). Briefly, 293T cells were split into 100mm tissue culture dishes (15 ml total volume medium, no more than 70% confluent before transformation). Transformation was performed 24 hours later. The 2ml calcium phosphate-DNA co-precipitation solution was made with 100 ul 2.5M CaCl2 and plasmid DNA (18.34 ug for pSPAX2, 7.34 ug for pMD2.G and 24.34 ug for lenti vector), diluted with 1/10 TE, pH 8.0 to the final total volume of 1 ml. Next, one volume of 2xCa/DNA solution was added to one volume of 2x HEPES. After 1 minute, 1.5 ml of the mixed solution was added to the cell culture medium. The next morning the medium was changed with 12 ml fresh DMEM medium and 10% FBS. The viruses were harvested 20 hours after medium change. For infection of MM cell lines, one million cells were added into 6-well plates with 6 ul polybrene (5 mg/ml), 1.5 ml lenti virus and fresh medium to the total volume of 3 ml. Four hours later, 3 ml of fresh medium was added. Next morning the RPMI medium was changed.
Construction of the PSMB5 and PSMB6 mutants
The wild type PSMB5 was engineered to have a synonymous mutation in the SgRNA targeted region (protospacer adjacent motif sequence CCA was mutated to TCA, which is required for SgRNA PSMB5CrA CAAGTCCGAAAAACCCGCGC targeting); Two additional mutationsin PSMB% were introduced (PSMBM1 and PSMB5M2). The PSMB5 mutation 1 has G-1A, T1A, K32A and K33A mutations, resulting in no self-splicing function or catalytic activity; the PSMB5M2 mutation was similar to mutation 1 but with no propeptide, resulting in no catalytic activity. We also constructed a lenti-vector expressing wild type PSMB6 in which the protospacer adjacent motif sequence TGG was mutated to AGG (CCCGGAGCCTCCAATGGCAA) such that PSMB6 SgRNA 10 cannot target it. In addition, two versions of mutated PSMB6 (M1 and M2) were constructed. The PSMB6 M1 has a −1G and 1T, which were mutated to −1A and 1A, and a 33K, mutated to 33A. These mutations were directly synthesized by IDT (Coralville, IOWA 52241 USA) and cloned into a lentivector.
Western blot
We used a standard a Western Blot technique. Equal amounts of protein were subjected to SDS-PAGE gels followed by transfer to PVDF membranes. Membranes were probed with primary antibodies overnight and then washed and incubated with horseradish peroxidase-conjugated secondary antibodies. Detection was performed by the enhanced chemical luminescence method. When probing for multiple targets or validating equal amount of protein load, stripping and re-probing a single membrane were conducted. In the case of protein reprobed was absent, the same samples were usually re-run to validate the result. HA antibody (Cat. No. MMS-101P Clone 16B12) was from Covance (Emeryville California USA 94608). Antibodies against proteasome subunits PSMB5 (Cat. BML-PW8895), PSMB6 (Cat. BML-PW8140), PSMB7 (Cat. BML-PW8145), PSMB8 (Cat. BML-PW8840), PSMB9 (Cat. BML-PW8150), PSMB10 (Cat. BML-PW8350) and PSMA1 (α6) (Cat. BML-PW8100) were purchased from ENZO life science (Farmingdale, NY 11735 USA). PSMB2 (β4) was purchased from Proteintech (Rosemont, IL 60018, USA).
MTT assay
Cellular viability of the MM cell lines was determined by the MTT assay (Sigma-Aldrich, 3050 Spruce St.St. Louis, MO 63103) following the manufacturer’s instructions. Briefly, Seed cells at a concentration of 2 × 104 cells/ well in 90 μl culture medium and 10 ul medium with drug was added. Incubate cell cultures for 72 h at +37°C and 5% CO2. After the incubation period, add 10 μl of the MTT labeling reagent (final concentration 0.5 mg/ml) to each well. Incubate the microplate for 4 h in a humidified atmosphere (37°C, 5% CO2). Add 100 μl of the Solubilization solution into each well. Allow the plate to stand overnight in the incubator in a humidified atmosphere (37°C, 5% CO2). Check for complete solubilization of the purple formazan crystals and measure the absorbance of the samples using a microplate (ELISA) reader. The wavelength to measure absorbance of the formazan product is 550 nm by the ELISA reader. The reference wavelength is 650 nm. All MTT assay results were the average of triplicate wells on 96 well plates. STDEV was calculated by Excel. IC50 was calculated by PRISM software.
mRNA sequencing (mRNA-seq)
MM bone marrow CD138+ cells were harvested and total RNA was prepared using RNeasy plus using the protocols of the manufacturer (Qiagen, Valencia). mRNA-seq experiments were performed and analyzed as described previously(30) at the medical genome facility in Mayo Clinic, Rochester. RNAseq data was processed using Mayo RNA-Seq Bioinformatics workflow MAP-RSeq(31), including read alignment against human hg19 genome build (by TopHat v2.0.12) and read count quantification for genes (by featureCount v1.4.4). We analyzed differential expression of PSMB5, PSMB6, PSMB7, PSMB8, PSMB9 and PSMB10 between pre- and post BTZ treatment samples. Patient bone marrow samples were obtained by written informed consent from the patients, that the studies were conducted in accordance with Declaration of Helsinki and that the studies were approved by Mayo Clinic Institutional Review Board.
Results
Generation and sequencing of BTZ resistant cell lines
Eight MM cell lines (RPMI-8226, FR4, KMS11, XG-1, MM1S, U266, JJN3 and OCI-MY5) sensitive to BTZ (IC50 < 5 nM) were selected for relative clonal resistance through stepwise increases of BTZ concentration (post treatment IC50 ranging from 13 to 38 nM) (Fig. 1A). We defined BTZ resistance in cell lines as an IC50 of resistant cells more than five times higher than the parental cells. We screened all 49 proteasome subunits (Sup. Table 1) in these isogenic cell line pairs by targeted sequencing, and found PSMB5 mutations in five of the eight lines (RPMI-8226R, KMS11R, FR4R, XG-1R and MM1.SR) (Fig. 1A). Specifically, a p.Met104Val mutation was found in four cell lines (RPMI-8226R, FR4R, KMS11R and XG-1R), and a p.Thr80Ala in MM1.S. XG-1R also acquired a mutation in PSMD9 (p.Glu54Lys), a proteasome 19S subunit. No mutations were found in the remaining 3 BTZ resistant cell lines, but in two of them (U266 and JJN3) exhibited higher protein expression levels of PSMB5 and PSMB6 when compared to parental, drug sensitive, cell lines (Sup. Fig.1). Therefore, we identified a potential resistance mechanism secondary to proteasome subunit mutation or increased proteasome subunit expression in seven out of eight BTZ resistant cell lines.
Figure 1. Sensitivity to BTZ is restored by CRISPR depletion of PSMB5.

(A) Eight resistant MM cell lines were developed using a stepwise increase in BTZ concentration. Genome mutations were analyzed using a targeted sequencing panel, which included all 49 proteasome subunits. An IC50 was obtained for both parental and isogenic resistant cell lines (72 hour treatment with BTZ). (B) One of the BTZ resistant MM cell lines, RPMI-8226R, was depleted of PSMB5 using CRISPR guide RNA. Top: One to six are PSMB5 depleted clones as detected by Western blot with % reductions noted. C and control represent the RPMI-8226R cell line (BTZ resistant). Control 2 is the BTZ sensitive parental RPMI-8226 cells. Middle: MTT assay of the depleted and control cell lines for cell viability. Bottom: Drug response of PSMB5 depleted and controls following treatment with BTZ . (C) This represents a rescue experiment using lentiviral re-introduction of PSMB5 performed on PSMB5 depleted clones 1 and 5 from Fig. 1B. Labelling indicates B5: cells manipulated with a lenti-vector expressing wild type PSMB5. B5M: a lenti-vector expressing PSMB5 with mutation at Ala108Thr. NS: empty lenti-vector used as a non-specific control. C+: RPMI-8226R cells maintained with 20 nm BTZ. C-: RPMI-8226R cells maintained without BTZ. Re-introduction of wild type or mutant PSMB5 restored drug resistance. Expressed wild or mutated PSMB5 detected by Western blot (bottom). Protein expression was detected by western blot and analyzed using ImageJ software (the numbers below the blots from the Figure 1B and 1C are normalized to β-actin and untreated control).
PSMB5 depleted cells are viable and confer Bortezomib sensitivity
Since PSMB5 mutations were present in five of eight BTZ resistant cell lines, but rare in primary patients (see introduction above) we began a deeper analysis by CRISPR depleting PSMB5 in the RPMI-8226R cell line (Fig. 1B top) using SgRNAs (Sup. Fig.2). Three subclones (clone 1.1, 1.2 and 1.3) had no detectable PSMB5 expression, both in denaturing and native gels (Sup. Fig.2). No significant effect on cell growth was observed in cells without PSMB5 (Fig. 1B middle). Depleting PSMB5 did however restore BTZ sensitivity comparable to the parental cell line (Fig.1B bottom of figure). Next, we introduced a new mutation (p.Ala108Thr)(32-35) deliberately restoring BTZ resistance (Fig. 1C) and confirming that each of two different identified PSMB5 mutations conferred proteasome inhibitor resistance. The six most efficient SgRNAs targeting PSMB5 (Sup.Fig.3) were used for further confirmatory studies in a second PSMB5 mutant, BTX resistant, cell line, KMS11R, which also remained viable after PSMB5 knock out. Five of the SgRNAs significantly restored KMS11R sensitivity to BTZ (Sup. Fig.4). PSMB5 was also successfully depleted in two BTZ sensitive cell lines RPMI-8226 (clone 11.2, 11.4, and 11.8 seen in Sup.Fig.2) and OCI-MY5 cells (Sup.Fig.5). Surprisingly then, and in contrast to published literature in yeast, removal of PSMB5 did not reduce viability in four different MM cell lines, including BTZ resistant and sensitive MM cell lines. Our in vitro data thus confirm that PSMB5 mutations confer BTZ resistance, that complete loss in PSMB5 expression is both tolerated and restores BTZ sensitivity and infer that induced PSMB5 mutations are a gain of function or confer stability of expression.
Absence of PSMB6 is lethal in MM
After confirming that PSMB5 knock out was not lethal in MM cell lines (while the mutation is lethal in yeast) we explored the role of other proteasome subunits on viability and drug sensitivity. After infection and western blot screening of RPMI-8226, six of 24 guides RNAs targeting PSMB6 were selected (Sup.Fig.6). All six SgRNAs efficiently depleted PSMB6 (Fig. 2A) and in contrast to PSMB5, all of them had a lethal effect (Fig. 2B). While still viable we then examined the expression of PSMB5, PSMB7, PSMB8, PSMB9 and PSMB10 in the PSMB6 deficient cells, and show that expression and splicing of each of these proteins, with the exception of PSMB9, was significantly inhibited (Fig. 2A). To exclude off-target CRISPR effects, we infected RPMI-8226R cells with a lentivirus expressing either the guide RNA resistant WT PSMB6, two functionally deficient PSMB6 mutants (M1 and M2) (Fig. 2C) or empty vector (control). After the establishment of stable cell lines expressing guide resistant PSMB6 WT, M1 and M2, or empty vector we reinfected these stable cell lines with SgRNA targeting PSMB6, and in this instance successfully rescued the lethal phenotype (Fig. 2D). Restoration of cell viability correlated with PSMB6 expression, including the mutated PSMB6 M1 and M2 lacking critical catalytic or splicing function (Fig. 2E). This experiment was repeated in a second BTZ resistant cell line, KMS11R, and a similar toxicity of PSMB6 knock out was observed (Sup. Fig.7). To exclude mutation in PSMB5 as a confounding factor the toxicity of PSMB6 knock out was also confirmed in a BTZ sensitive, unmutated OCI-MY5 cell line (Sup. Fig.8).These results demonstrate the importance of PSMB6, highlighting that these human MM cell lines depend on PSMB6 structurally, rather than functionally.
Figure 2. PSMB6 is essential for MM cell survival.
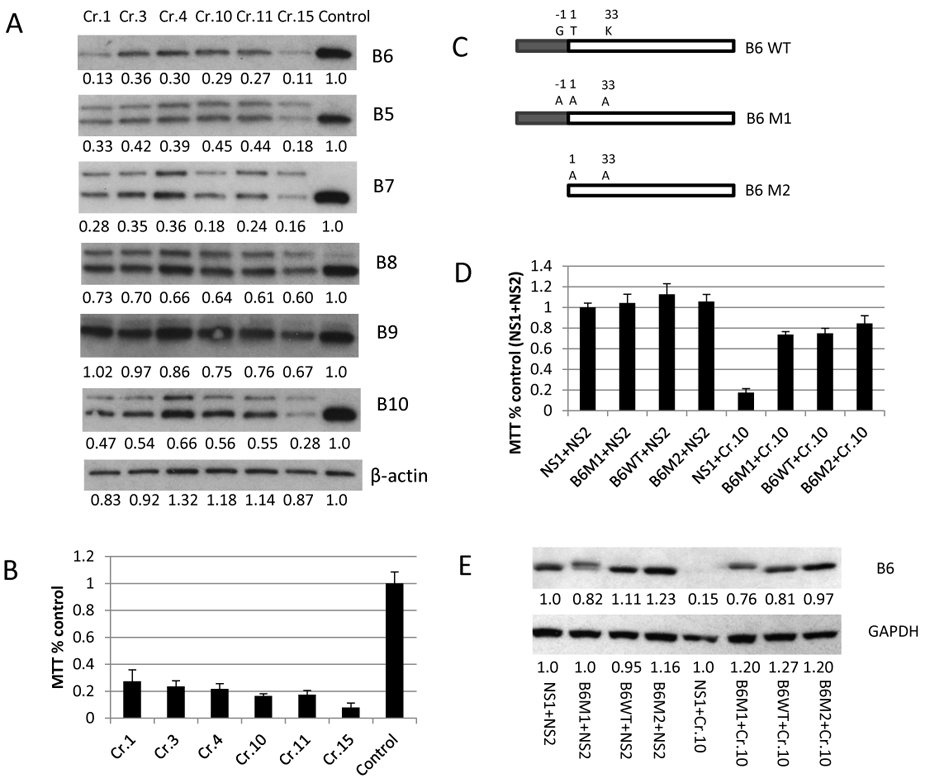
(A) Depletion of PSMB6 with six different SgRNAs (Cr.1, Cr.3, Cr.4, Cr.10, Cr.11 and Cr.15) demonstrates on day 4 that splicing for PSMB5, PSMB7, PSMB8 and PSMB10 was significantly inhibited (the top band is the proteasome subunit proprotein and the bottom band is the mature proteasome subunit). (B) The same panel of cell lines were analyzed using an MTT assay on day 8 post transfection (96 well plate with triplicate wells for each sample for 72 hours of BTZ treatment). (C) A schematic presentation of the mutations introduced in PSMB6. B6 WT: PSMB6 wild type protein. B6 M1: PSMB6 with mutations at G-1A, T1A and K33A. B6 M2: PSMB6 with mutations at T1A and K33A without the propeptide. The mature PSMB6 is characterized by an open box and the dark box marks the PSMB6 pro-sequence. B6WT, B6M1, and B6M2 were modified to resistant PSMB6 Cr10 targeting [protospacer adjacent motif sequence TGG was mutated to AGG for PSMB6 SgRNA 10 (CCCGGAGCCTCCAATGGCAA) resulting in SgRNA 10 cannot target it]. (D) The toxicity of PSMB6 depletion was rescued by expressing either PSMB6 wild type or PSMB6 mutants. RPMI-8226R BTZ resistant cell lines expressing wild type PSMB6 had re-introduced stable expression of PSMB6 WT, M1 and M2 described at Fig. 2C and was then infected with SgRNA targeting PSMB6 (Cr10). The infected cells were aliquoted into 96 well at day 4 and MTT reagent was added at day 8 after infection. Control NS1: RPMI-8226R cell line infected with an empty lenti-vector. Control NS2: A lenti-vector expressing non-specific SgRNA. B5 to B10 represents PSMB5 to PSMB10. (E) PSMB6 expression was measured on the cell lines from Fig. 2D at day 4 after infection. Protein expression was detected by western blot and analyzed by ImageJ software [numbers below the blots from the Figure 2A and 2E are normalized to β-actin (Figure 2A) and GAPDH (Figure 2E) and untreated control].
Sensitivity to BTZ is restored by functionally deficient PSMB6 expression
In an attempt to develop MM cell lines that survive after PSMB6 deletion, we chose BTZ resistant KMS11R, which has a PSMB5 mutation (p.Met104Val). We selected two SgRNAs (Cr10 and Cr15) out of six SgRNAs targeting PSMB6 for further cloning (Fig. 3A) and identified two viable clones (10.3 and 15.2), which expressed significantly lower PSMB6 than the control cells, but sufficient to maintain cell viability. A PSMB6 loss of splicing function was observed in clone 10.3 (Fig. 3B). Sequencing data revealed that clone 10.3 had acquired an in-frame deletion with amino acid change in PSMB6, leading to the replacement of amino acids (160Ala and 161Ile) by 160Val (Fig. 3C). Furthermore, clone 15.2 had a deletion of PSMB6 181Met (Fig. 3C). The amino deletion and low expression of PSMB6 resulted in no significant decline in cell growth potential for clone 15.2, while clone 10.3 had a slower growth rate (Fig. 3D). However, both “PSMB6 low” clones exhibited restored sensitivity to BTZ (Fig. 3E). This demonstrates that a PSMB5 mutation causing BTZ resistance is dependent on the natural expression of PSMB6 and that deletion of certain amino acids resulting in lowering of PSMB6 levels can reverse resistance. This also highlights that PSMB6 amino acids 160Ala and 161Ile play an important role for appropriate splicing.
Figure 3. Sensitivity to BTZ is restored by even low level PSMB6 expression.

(A) The KMS11R BTZ resistant cell line was targeted by SgRNAs (Cr 1, 3, 4, 10, 11, and 15) against PSMB6 (B6). (B) Further subcloning was performed (Clone 10.1, 10.2, 10.3 and 10.4 were from KMS11R cells targeted by SgRNA Cr.10 and clone 15.1, 15.2 and 15.3 were from KMS11R cells targeted by SgRNA Cr.15). This identified Clone 10.3 which expressed low level un-spliced PSMB6 and clone 15.2 which expressed low levels of PSMB6. (C) Sanger sequencing revealed that clone 10.3 had acquired an Ala (A)160 and Ile (I)161 replaced by Val (V)160. Clone 15.2 had a Met (M) 181 deletion identified. (D) the MM cell lines expressing residual PSMB6 grow normally (Clone 15.2) while clone 10.3 grows more slowly than parental cells but both remained viable in contrast to cells completely deficient in PSMB6. (E) Clone 10.3 and 15.2 were sensitive to BTZ treatment. Controls included KMS11R (BTZ resistant) and Control 2 KMS11 (BTZ sensitive). Protein expression was detected by western blot and analyzed by ImageJ software (the numbers below the blots Figure 3 A and B are normalized to β-actin and untreated control).
Stability of the PSMB5 and PSMB6 proteasome subunits is co-dependent
Since MM cell lines survive without PSMB5, this raised the question of which proteasome subunit compensates for the loss of PSMB5. Consequently, we checked the expression of five other proteasome catalytic subunits (PSMB6, PSMB7, PSMB8, PSMB9 and PSMB10) and one non-catalytic subunit (PSMB2 as a control) on MM cell lines lacking PSMB5 (Fig. 4A). After PSMB5 deletion, PSMB6 and PSMB7 expression levels were significantly lower than in control cells. No significant change in PSMB8 and PSMB9 expression was observed in different RPMI-8226 cell subclones, even though PSMB8 was expected to significantly compensate for the loss of PSMB5, since PSMB8 replaces PSMB5 upon stimulation by interferon γ. PSMB9 was the only protein with a significant influence in maturation (significant pro-PSMB9 band was observed) by removing pro sequence following PSMB5 deletion. PSMB10 was expressed at higher levels than the control cells.
Figure 4. High inter-dependence was observed between expression of PSMB5 and PSMB6.
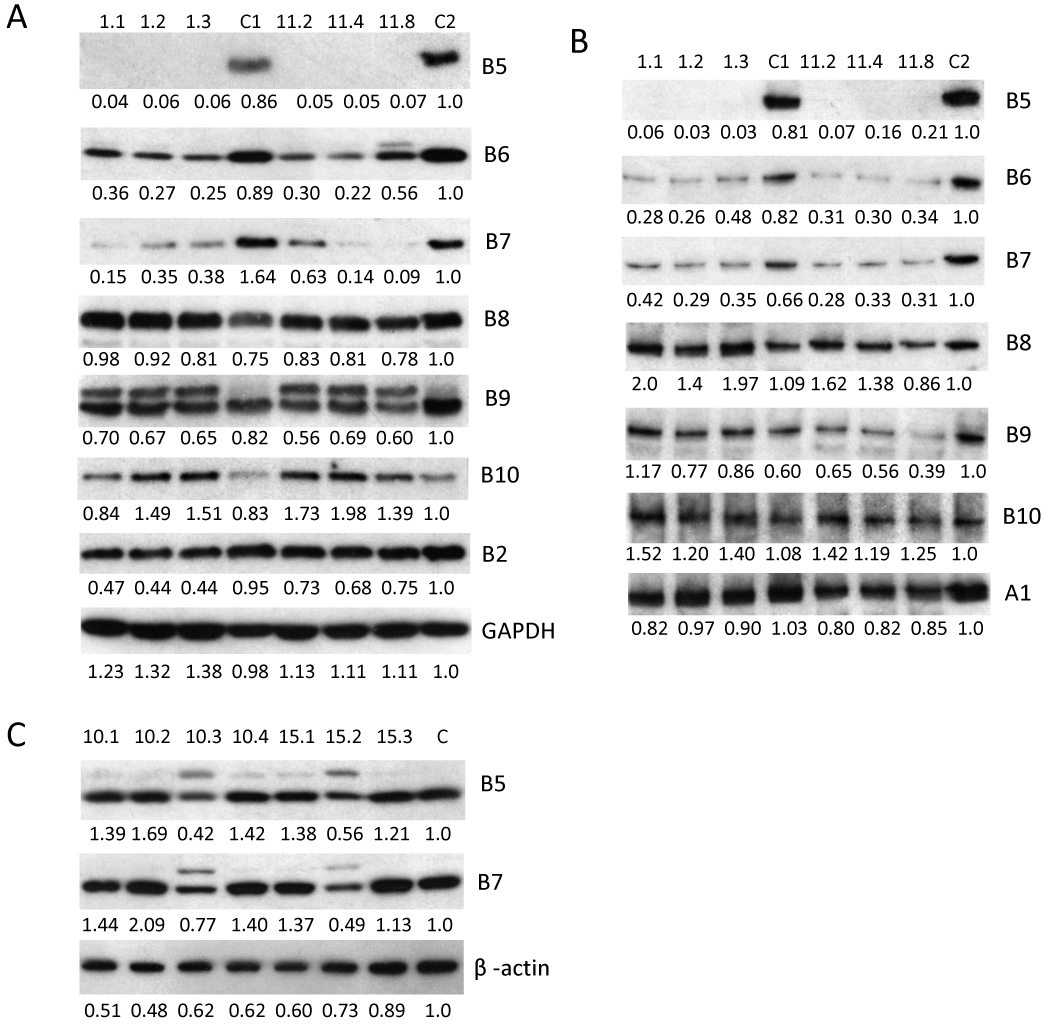
(A) Expression of PSMB6 and PSMB7 was significantly reduced in MM cell lines lacking the PSMB5 gene. Clones 1.1, 1.2 and 1.3 are subclones derived from clone 1 of BTZ resistant RPMI-8226R CRISPR depleted of PSMB5 and clones 11.2, 11.4 and 11.8 are subclones from clone 11 of BTZ sensitive RPMI-8226 depleted of PSMB5. C1:- is parental BTZ sensitive RPMI-8226. C2: is parental BTZ resistant RPMI-8226R. (B) Incorporation of PSMB6 and PSMB7 was significantly reduced in MM cell lines lacking the PSMB5 gene. The same panel of lysates from Fig. 4A was examined for protein expression using a native gel. (C) Expression of PSMB5 and PSMB7 was significantly reduced when MM cell lines expressed low levels of mutated PSMB6. The panel of lysates from Fig. 3B was used for western blot detection. B2 represents PSMB2. B5 to B10 represents PMBB5 to PSMB10. Protein expression was detected by western blot and analyzed by ImageJ software (numbers below the blots Figure 4 A, B and C are normalized to GAPDH (Figure 4A), PSMA1 (Figure 4B) and β-actin (Figure 4C) and untreated control].
We ran a native gel to check 20S proteasome protein expression levels (Fig. 4B). PSMB10 was the only protein expressed at higher levels than the control cells. Expression of both PSMB6 and PSMB7 was lower, while there was no significant change in PSMA1 expression (one of the α subunits used as a control).
The dependence of PSMB6 and PSMB7 expression on PSMB5 prompted us to query the corollary: is the dependence of PSMB5 and PSMB7 expression based on PSMB6 presence. The same two clones (10.3 and 15.2) from Fig. 3B expressing a lower level of mutated PSMB6 were employed. We detected expression of PSMB5 and PSMB7 in these two clones but at significantly lower expression than the control cells, with significant amounts of the proprotein of PSMB5 and PSMB7 appearing, indicating that splicing of these two proteins was also significantly inhibited (Fig.4C). Here, we therefore demonstrate that expression of PSMB5 and PSMB6 is co-dependent and that expression of PSMB7 is highly dependent on both PSMB5 and PSMB6.
Stability of PSMB5 and PSMB6 does not require normal functional activity
Next, we addressed if this mutual dependence is based on the proteasome’s catalytic function or structure. We constructed lenti-vectors expressing PSMB5 WT, M1 and M2 (Fig. 5A). We selected two subclones from SgRNA, PSMB5 depleted, RPMI-8226 and re-infected with guide resistant PSMB5 WT, M1 and M2. Unsurprisingly, re-expression of wild type PSMB5 restored the expression of PSMB6, PSMB7, PSMB8, PSMB9 and PSMB10 to the parental cell level (Fig. 5B). PSMB5 M1 was expressed as an un-spliced form, as expected (Fig. 5B). However, the un-spliced PSMB5 M1 was still able to restore PSMB6, PSMB7 and PSMB8 expression to parental cell levels. PSMB9 levels were partially restored, although remaining lower than the control and in its un-spliced form (Fig. 5B). Expression levels of PSMB5 M2 (without propeptide and lacking catalytic function) were lower than the WT and M1, and the PSMB5 deletion phenotype was not rescued (Fig. 5B). Native gel detection indicated that only a low level of PSMB5 was incorporated into 20S proteasome (Fig. 5C). This result shows that propeptide of PSMB5 is important for expression and incorporation into 20S proteasome. We repeated this experiment infecting KMS11R cells with WT PSMB6 and functionally deficient mutants. In this case, all PSMB6 variants restored the expression of PSMB5 to parental cell levels and partial rescue was seen for PSMB7 (Fig. 5D). We found that RPMI-8226 cells lacking PSMB5 had relatively higher ubiquitinated protein accumulation, but no significant difference was seen between RPMI-8226 cells without PSMB5 and the cells rescued by PSMB5 WT, M1 and M2 (Fig. 5E), which indicate that it is no significant influence to the function of human 20S proteasome without PSMB5. These results indicate that PSMB5 and PSMB6 expression is co- dependent but does not require catalytic or splicing functionally of the protein.
Figure 5. Expression of PSMB5 and PSMB6 is mutually dependent on structure, but not function.
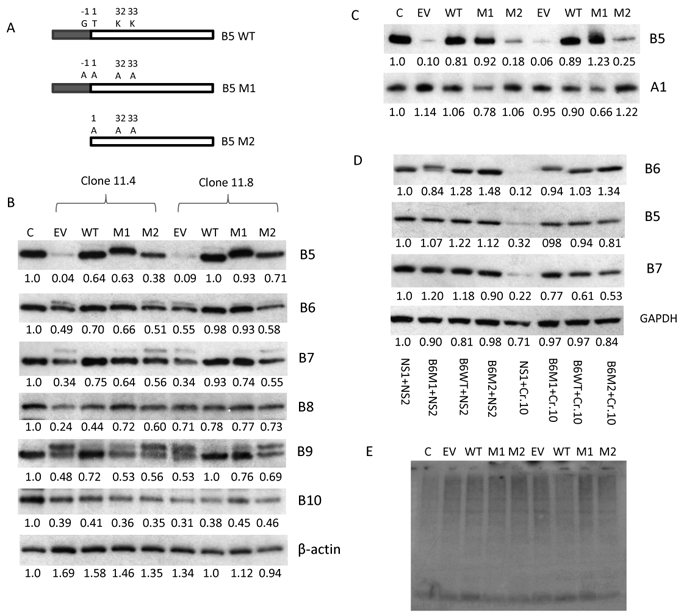
(A) Schematic presentation of mutations introduced into PSMB5. In the figure B5 WT is PSMB5 wild type protein. B5 M1 is PSMB5 with mutations at G-1A, T1A, K32A and K33A. The PAM sequence is mutated in the SgRNA target region. B5 M2 is PSMB5 with mutations at T1A K32A and K33A without expression of the propeptide. The mature part of PSMB5 is characterized by an open box and the dark box marks the PSMB5 pro-sequence. (B) Expression of PSMB6 and PSMB7 is restored by the expression of wild type or guide resistant mutated B5 M1 but not B5 M2. Cis for RPMI-8226R control cells. EV is empty lenti-vector. (C) Mutated PSMB5 with un-spliced prosequenence (PSMB5 M1) can be efficiently incorporated into the 20S proteasome. The panel of lysates from Fig. 5B was used for native gel detection. (D) Expression of PSMB5 and PSMB7 is restored by the expression of wild type or mutated PSMB6 (B6 M1 and B6 M2). The cell lysate from Fig. 2E was used for western blot assay. E. Ubiquitination of MM cell lines lacking PSMB5 by re-introducing expression of wild type and mutated PSMB5. The same panel of cell lysate as Fig. 5B was detected expression of ubiquitination. The proteins expression was detected by western blot and analyzed by ImageJ software [numbers below the blots from the Figure 5B, C and D are normalized to β-actin (Figure 5B), PSMA1 (Figure 5C) and GAPDH (Figure 5D) and untreated control].
PSMB7 depletion is lethal to MM cell lines
Since PSMB7 expression is highly dependent on the expression of PSMB5 and PSMB6, we proposed that expression of PSMB5 and PSMB6 would also be highly dependent on PSMB7. Nine of 12 PSMB7 SgRNAs (Sup. Table 2) were significantly toxic to the RPMI-8226R cell line (Fig. 6A), and with one exception correlated to the corresponding PSMB7 knock out efficiency (Fig. 6B). As expected, expression of both PSMB5 and especially PSMB6 were reduced significantly following PSMB7 depletion (Fig. 6C). PSMB8 was the only protein among the immunoproteasome subunits with a significant reduction in its expression. Unlike the PSMB6 knock out, constitutive and immunoproteasome subunit splicing was not influenced after PSMB7 knockout.
Figure 6. PSMB7 depletion is lethal to MM cells.
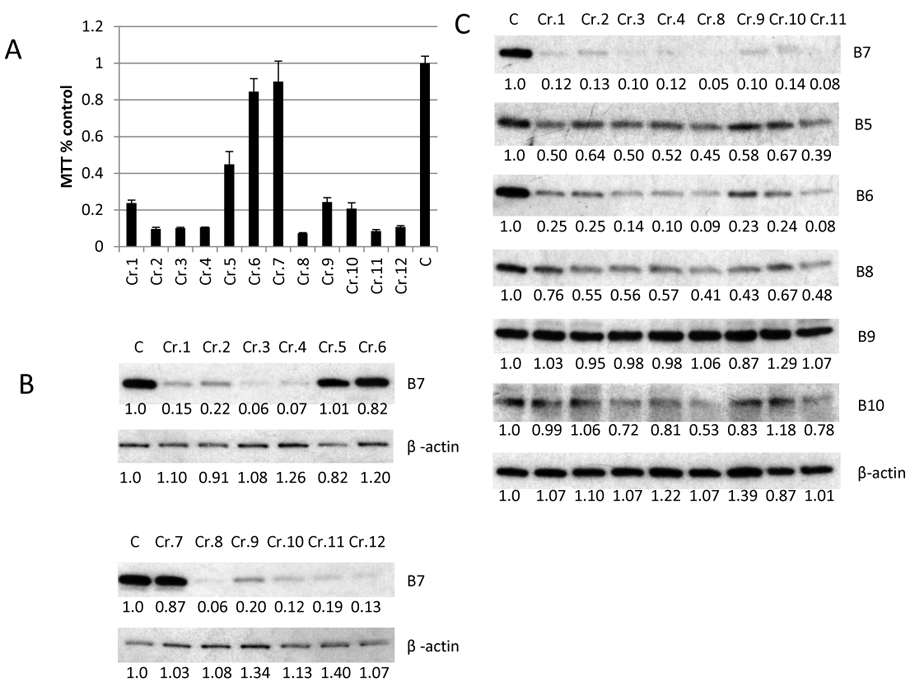
(A) MTT assay for RPMI-8226R cells infected with lenti-vector expressing SgRNAs (Cr.1 to Cr.12) targeting PSMB7. On day 4 after infection the infected cells were aliquoted for an MTT assay (triplicate wells), which was read on day 8. C is control cell line RPMI-8226R. (B) The panel from Fig. 6A were examined for the expression of PSMB7 by western blot. (C) The expression of PSMB5 to PSMB10 (B5 to B10) was detected by western blot from RPMI-8226R cells targeted by eight SgRNAs against PSMB7. The proteins expression was detected by western blot and analyzed by ImageJ software (numbers below the blots from the Figure 6B and C are normalized to β-actin and untreated control).
Increased expression levels of PSMB5, 6 and 7 in patients after BTZ treatment
Recently we finished RNA sequencing for 450 MM patients (unpublished data). Among these, ten patients with paired samples were collected before and after multiple rounds of chemotherapy treatment (Sup.data). Eight of these had received proteasome inhibitors. mRNA sequencing results indicated that two (patient one ID: 6496153 and patient two ID: 5493955) of the eight patients has notable and significant increased expression levels of PSMB5, 6 and 7, but not PSMB8, 9 and 10 (Fig. 7). PSMB5, 6 and 7 from one patient increased 1.5, 2.0 and 2.0 fold correspondingly in late stage disease compared to pre-treatment. PSMB5, 6 and 7 from patient two increased 5.3, 1.9 and 1.7 fold correspondingly in late stage disease compared to pre-treatment. On the other hand in 6 of the patients an increase was not observed.
Figure 7. Significant increase in expression levels of PSMB5, 6 and 7 in late disease stage patients.
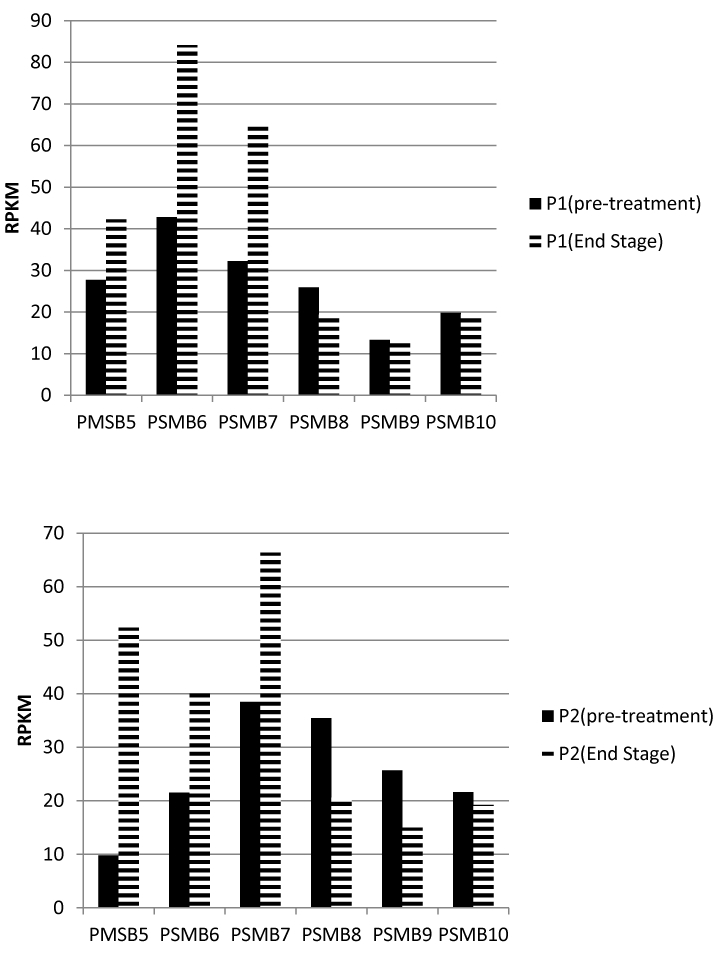
mRNA sequencing results revealed significant increased PSMB5, 6 and 7 levels when early and late stages of disease were examined., while levels of PSMB8, 9 and 10 did not change. P1=patient one. P2=patient two.
Absence of the immunoproteasome subunits PSMB8 and PSMB9 does not restore sensitivity to BTZ in MM cell lines
PSMB8 and PSMB9 are also targets of proteasome inhibitors BTZ and carfilzomib. We designed three SgRNAs for PSMB8 or PSMB9 (Sup.Table 3). We did not observe any significant correlation in BTZ sensitivity with or without the PSMB8 and PSMB9 subunits in human MM cell lines (Sup.Fig.9), indicating that inhibition of the activity of these immunoproteasome subunits may not overcome BTZ resistance.
Discussion
Our results indicate that alterations in PSMB5 (via mutation or over expression) are the predominant mechanism for in vitro induced BTZ resistance, consistent with previous reports (32,33,35-37). Depleting PSMB5 in PSMB5 mutant cell lines restored sensitivity to BTZ and introducing the frequently reported PSMB5 mutation (p.Ala108Thr) into PSMB5 depleted lines restored resistance to BTZ, further establishing PSMB5 mutations as a major contributor to BTZ resistance in MM cell lines, and consistent with the report by Hess et al. wherein PSMB5 of K562 cells mutated by CRISPR technology (p.Arg78Asn, p.Ala79Gly, p.Ala79Thr and p.Ala108Val) but expressing physiological level of PSMB5 developed resistance to BTZ(38). Different PI-resistant mechanisms may well exist in MM cell lines, as in a report by Soriano et al (39), which concluded that PI-adapted cells were largely independent of proteasome activity and that resistance was due to metabolic adaptions.
With respect to primary patient samples, in Barrio et al. (24), we reported the first example of a drug resistant patient with multiple PSMB5 mutations. The same mutation (p. Met104Val) that appeared in the four cell lines in our analysis was also noted in Barrio’s report, although mutated to a different amino acid (p.Met104Ile, position with pro-sequence = p.Met45Ile without pro-sequence). Two closely located PSMB5 amino acids mutations (p.Thr80Ala in our study VS p.Ala86Pro and p.Ala79Thr in Barrio’s study) were also reported by Barrio’s study. The patient in this instance had been treated with multiple regimens containing BTZ. We would predict that in future deep sequencing studies (as opposed to exome) of relapsed patients substantially exposed to PIs, a higher frequency of PSMB5 mutation will be discovered.
Nevertheless, , since PSMB5 mutations are generally uncommon in primary patients analyzed to date in large sequencing efforts we took a deeper dive into proteasome subunit deletion employing CRISPR. Our results demonstrate that deletion of PSMB6 and PSMB7, but not PSMB5, is lethal for human MM cell lines. Both BTZ resistant and sensitive human MM cell lines are able to survive in the absence of PSMB5. This result changes our knowledge about the role PSMB5 on human cell survival, at least in the context of malignant transformation. The yeast PSMB5 homologous gene, pre2, has been reported as an essential gene. By mutating pre2, but not pre3 (homologous to the human PSMB6) or pup1 (homologous to PSMB7), yeast growth defects are found (16,17,19,40). This might be explained by the immunoproteasome subunit PSMB8 (absent in yeast), compensating for the loss of PSMB5 in human cells. Our result in MM cells is in contrast to the findings reported by Hirano et al (41), in which depleting PSMB5 led to cytotoxicity in 293 cell lines, which could reflect off-target activity of shRNA or alternatively that non transformed 293 cells are, in contrast to transformed malignant cells dependent on PSMB5.
Our results demonstrate that expression of the three constitutive proteasome subunits PSMB5, PSMB6 and PSMB7 is highly co-dependent. This dependence is reliant on the structure, but not the function, of PSMB5 and PSMB6. PSMB6 plays an essential role in the 20S proteasome biogenesis. In the absence of PSMB6, the splicing of all other proteasome catalytic subunits, with the exception of PSMB9, is inhibited, indicating that these subunits cannot be incorporated into the 20S core without PSMB6 expression. This phenotype was not observed by deletion of PSMB5 and PSMB7. Our current knowledge of the incorporation of β-subunits is mostly derived from ShRNA knock down in a single β-subunit experiment (41). In that study, the authors proposed that addition of β subunits to the α ring occurred in an orderly manner, starting from β2 (PSMB7), followed by β3, β4, β5 (PSMB5), β6, β1 (PSMB6), and finally β7. However, our results highlight that PSMB6 plays a central role in the incorporation of β-subunits. Therefore, we propose a new model for constitutive proteasome assembly (Sup. Fig. 10) wherein PSMB6 is the first subunit to add to α ring, PSMB7 is added to the α ring immediately after PSMB6 (or same time with PSMB6), PSMB5 and other proteasome subunits are incorporated into the 20S proteasome later (we did not delete non-catalytic β subunit in this study, so the order of addition of non-catalytic β needs future study), since MM cell lines cannot survive without PSMB6 and PSMB7, but can survive without PSMB5.
Immunoproteasome subunits PSMB9 and PSMB10 have been reported as mutually required for incorporation into the 20S proteasome core, independently of PSMB8 (42). In a separate study, Griffin et al (43) reported that PSMB10 requires PSMB9 for efficient incorporation into preproteasomes and preproteasomes containing PSMB9 and 10 require PSMB8 for efficient maturation. The authors proposed a cooperative model for proteasome assembly, with one major pathway leading to immunoproteasomes containing all three IFN-γ-inducible catalytic β subunits (PSMB8, PSMB9 and PSMB10), another major pathway leading to constitutive proteasomes containing all three constitutive catalytic β subunits (PSMB5, PSMB6 and PSMB7), and a minor pathway possibly leading to mixed catalytic subunits. Our results demonstrate the existence of the pathway leading to the constitutive proteasome assembly. Our results also indicate a crosstalk between the two main pathways, since splicing of the PSMB9 was inhibited without expression of PSMB5 and splicing of PSMB8 and PSMB10 was inhibited by depletion of PSMB6.
Both, BTZ and carfilzomib target the constitutive proteasome and the immunoproteasome indiscriminately. It has been speculated that adverse events attributed to PIs, such as peripheral neuropathy and gastrointestinal effects, might be caused by targeting of the constitutive proteasome. Thus, developing immunoproteasome specific drugs was proposed as a new strategy for MM therapy (44-46). However, our results show that depletion of the immunoproteasome subunits PSMB8 and PSMB9 was non-relevant to BTZ resistance, showing no influence on the growth of multiple MM cell lines. Mice completely lacking immunoproteasome subunits (PSMB8, PSMB9 and PSMB10) are viable, fertile and appeared healthy, only displaying major alterations in antigen presentation (47). However despite this promising therapeutic index, our studies suggest that targeting the immunoproteasome in MM might not represent an ideal therapeutic strategy.
We analyzed ten patients with paired bone marrow samples collected before treatment and in later stage post treatment disease by mRNA sequencing. Eight of these patients were treated with multiple drugs including BTZ and developed multiple drug resistant diseases including to BTZ. Two of these patients had elevated expression levels of PSMB5, PSMB6 and PSMB7, but not PSMB8, PSMB9 and PSMB10. Since mutation of PSMB5 in vivo is rare, elevated expression levels of PSMB5, PSMB6 and PSMB7 might contribute significantly to BTZ resistance in the clinic. This rather preliminary observation requires larger data sets.
Since expression of PSMB5, PSMB6 and PSMB7 is co-dependent and because expression of low level of PSMB6 and knock out of PSMB5 can overcome BTZ resistance caused by PSMB5 mutation, we propose that inducing the down regulation or otherwise inhibiting expression of either PSMB5, PSMB6 or PSMB7 might be a novel strategy to overcome human MM resistance to proteasome inhibitors.
In summary, our current and previous studies demonstrated that PSMB5 mutation is a dominant mechanism in MM cell lines induced to be resistant to BTZ in vitro. However, such mutations are rare in vivo. Alterations in PSMB expression is perhaps an alternate and rational explanation for induced resistance in vivo.
In that regard our studies show that, PSMB5 and PSMB6 and PSMB7, but not PSMB8 and PSMB9, are highly relevant for BTZ sensitivity in MM. Absence of PSMB6 or PSMB7, but not PSMB5, was lethal in MM cell lines. Expression of PSMB5, PSMB6 and PSMB7 was highly co-dependent. Importantly, the physical structure, but not the catalytic function, is required for this dependence.
Supplementary Material
Acknowledgement
We thank Dr. Leif Bergsagel for providing human MM parent cell lines. We thank Dr. Didier Trono for providing packaging plasmids psPAX2 (Addgene plsmid # 12260) and pMD2.G (Addgene plasmid # 12259). We thank Mayo Clinic Medical Genome Facility for providing targeting sequencing. This study was supported by U54 grant 4U54CA224018, Mayo Clinic Multiple Myeloma Spore SPORE and Multiple Myeloma Foundation.
A.K.S is supported by National Institute of Health (1U54CA114018)
Footnotes
Conflict-of-interest disclosure
The authors declare no competing financial interests.
References:
- 1.Rajkumar SV, Kumar S. Multiple Myeloma: Diagnosis and Treatment. Mayo Clin Proc 2016;91(1):101–19 doi 10.1016/j.mayocp.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia 2009;23(11):1964–79 doi 10.1038/leu.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A, et al. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012;120(5):947–59 doi 10.1182/blood-2012-04-403733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laubach JP, Schlossman RL, Mitsiades CS, Anderson KC, Richardson PG. Thalidomide, lenalidomide and bortezomib in the management of newly diagnosed multiple myeloma. Expert Rev Hematol 2011;4(1):51–60 doi 10.1586/ehm.10.83. [DOI] [PubMed] [Google Scholar]
- 5.Rosenthal A, Kumar S, Hofmeister C, Laubach J, Vij R, Dueck A, et al. A Phase Ib Study of the combination of the Aurora Kinase Inhibitor Alisertib (MLN8237) and Bortezomib in Relapsed Multiple Myeloma. Br J Haematol 2016;174(2):323–5 doi 10.1111/bjh.13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar SK, Berdeja JG, Niesvizky R, Lonial S, Laubach JP, Hamadani M, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol 2014;15(13):1503–12 doi 10.1016/S1470-2045(14)71125-8. [DOI] [PubMed] [Google Scholar]
- 7.Reeder CB, Reece DE, Kukreti V, Mikhael JR, Chen C, Trudel S, et al. Long-term survival with cyclophosphamide, bortezomib and dexamethasone induction therapy in patients with newly diagnosed multiple myeloma. Br J Haematol 2014;167(4):563–5 doi 10.1111/bjh.13004. [DOI] [PubMed] [Google Scholar]
- 8.Stewart AK, Richardson PG, San-Miguel JF. How I treat multiple myeloma in younger patients. Blood 2009;114(27):5436–43 doi 10.1182/blood-2009-07-204651. [DOI] [PubMed] [Google Scholar]
- 9.Kouroukis TC, Baldassarre FG, Haynes AE, Imrie K, Reece DE, Cheung MC. Bortezomib in multiple myeloma: systematic review and clinical considerations. Curr Oncol 2014;21(4):e573–603 doi 10.3747/co.21.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guerrero-Garcia TA, Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Mitsiades C, et al. The power of proteasome inhibition in multiple myeloma. Expert Rev Proteomics 2018;15(12):1033–52 doi 10.1080/14789450.2018.1543595. [DOI] [PubMed] [Google Scholar]
- 11.Laubach J, Garderet L, Mahindra A, Gahrton G, Caers J, Sezer O, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia 2016;30(5):1005–17 doi 10.1038/leu.2015.356. [DOI] [PubMed] [Google Scholar]
- 12.Finley D Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 2009;78:477–513 doi 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol 1999;17:739–79 doi 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- 14.Budenholzer L, Cheng CL, Li Y, Hochstrasser M. Proteasome Structure and Assembly. J Mol Biol 2017;429(22):3500–24 doi 10.1016/j.jmb.2017.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berkers CR, Verdoes M, Lichtman E, Fiebiger E, Kessler BM, Anderson KC, et al. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat Methods 2005;2(5):357–62 doi 10.1038/nmeth759. [DOI] [PubMed] [Google Scholar]
- 16.Friedman H, Snyder M. Mutations in PRG1, a yeast proteasome-related gene, cause defects in nuclear division and are suppressed by deletion of a mitotic cyclin gene. Proc Natl Acad Sci U S A 1994;91(6):2031–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen P, Hochstrasser M. Biogenesis, structure and function of the yeast 20S proteasome. EMBO J 1995;14(11):2620–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem 1997;272(40):25200–9. [DOI] [PubMed] [Google Scholar]
- 19.Groll M, Heinemeyer W, Jager S, Ullrich T, Bochtler M, Wolf DH, et al. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci U S A 1999;96(20):10976–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kisselev AF, Callard A, Goldberg AL. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem 2006;281(13):8582–90 doi 10.1074/jbc.M509043200. [DOI] [PubMed] [Google Scholar]
- 21.Oberdorf J, Carlson EJ, Skach WR. Redundancy of mammalian proteasome beta subunit function during endoplasmic reticulum associated degradation. Biochemistry 2001;40(44):13397–405. [DOI] [PubMed] [Google Scholar]
- 22.Britton M, Lucas MM, Downey SL, Screen M, Pletnev AA, Verdoes M, et al. Selective inhibitor of proteasome's caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem Biol 2009;16(12):1278–89 doi 10.1016/j.chembiol.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi CX, Kortum KM, Zhu YX, Bruins LA, Jedlowski P, Votruba PG, et al. CRISPR Genome-Wide Screening Identifies Dependence on the Proteasome Subunit PSMC6 for Bortezomib Sensitivity in Multiple Myeloma. Mol Cancer Ther 2017;16(12):2862–70 doi 10.1158/1535-7163.MCT-17-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrio S, Stuhmer T, Da-Via M, Barrio-Garcia C, Lehners N, Besse A, et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019;33(2):447–56 doi 10.1038/s41375-018-0216-8. [DOI] [PubMed] [Google Scholar]
- 25.Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011;475(7356):348–52 doi 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 26.Kocher JP, Quest DJ, Duffy P, Meiners MA, Moore RM, Rider D, et al. The Biological Reference Repository (BioR): a rapid and flexible system for genomics annotation. Bioinformatics 2014;30(13):1920–2 doi 10.1093/bioinformatics/btu137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol 2011;29(1):24–6 doi 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 1973;52(2):456–67. [DOI] [PubMed] [Google Scholar]
- 29.Jordan M, Schallhorn A, Wurm FM. Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res 1996;24(4):596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu YX, Shi CX, Bruins LA, Jedlowski P, Wang X, Kortum KM, et al. Loss of FAM46C Promotes Cell Survival in Myeloma. Cancer Res 2017;77(16):4317–27 doi 10.1158/0008-5472.CAN-16-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalari KR, Nair AA, Bhavsar JD, O'Brien DR, Davila JI, Bockol MA, et al. MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinformatics 2014;15:224 doi 10.1186/1471-2105-15-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008;112(6):2489–99 doi 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 33.Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH, et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012;26(4):757–68 doi 10.1038/leu.2011.256. [DOI] [PubMed] [Google Scholar]
- 34.Lu S, Yang J, Song X, Gong S, Zhou H, Guo L, et al. Point mutation of the proteasome beta5 subunit gene is an important mechanism of bortezomib resistance in bortezomib-selected variants of Jurkat T cell lymphoblastic lymphoma/leukemia line. J Pharmacol Exp Ther 2008;326(2):423–31 doi 10.1124/jpet.108.138131. [DOI] [PubMed] [Google Scholar]
- 35.Ri M, Iida S, Nakashima T, Miyazaki H, Mori F, Ito A, et al. Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010;24(8):1506–12 doi 10.1038/leu.2010.137. [DOI] [PubMed] [Google Scholar]
- 36.Balsas P, Galan-Malo P, Marzo I, Naval J. Bortezomib resistance in a myeloma cell line is associated to PSMbeta5 overexpression and polyploidy. Leuk Res 2012;36(2):212–8 doi 10.1016/j.leukres.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 37.Ruckrich T, Kraus M, Gogel J, Beck A, Ovaa H, Verdoes M, et al. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia 2009;23(6):1098–105 doi 10.1038/leu.2009.8. [DOI] [PubMed] [Google Scholar]
- 38.Hess GT, Fresard L, Han K, Lee CH, Li A, Cimprich KA, et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat Methods 2016;13(12):1036–42 doi 10.1038/nmeth.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soriano GP, Besse L, Li N, Kraus M, Besse A, Meeuwenoord N, et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016;30(11):2198–207 doi 10.1038/leu.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinemeyer W, Gruhler A, Mohrle V, Mahe Y, Wolf DH. PRE2, highly homologous to the human major histocompatibility complex-linked RING10 gene, codes for a yeast proteasome subunit necessary for chrymotryptic activity and degradation of ubiquitinated proteins. J Biol Chem 1993;268(7):5115–20. [PubMed] [Google Scholar]
- 41.Hirano Y, Kaneko T, Okamoto K, Bai M, Yashiroda H, Furuyama K, et al. Dissecting beta-ring assembly pathway of the mammalian 20S proteasome. EMBO J 2008;27(16):2204–13 doi 10.1038/emboj.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Groettrup M, Standera S, Stohwasser R, Kloetzel PM. The subunits MECL-1 and LMP2 are mutually required for incorporation into the 20S proteasome. Proc Natl Acad Sci U S A 1997;94(17):8970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griffin TA, Nandi D, Cruz M, Fehling HJ, Kaer LV, Monaco JJ, et al. Immunoproteasome assembly: cooperative incorporation of interferon gamma (IFN-gamma)-inducible subunits. J Exp Med 1998;187(1):97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ettari R, Zappala M, Grasso S, Musolino C, Innao V, Allegra A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol Ther 2018;182:176–92 doi 10.1016/j.pharmthera.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Santos RLA, Bai L, Singh PK, Murakami N, Fan H, Zhan W, et al. Structure of human immunoproteasome with a reversible and noncompetitive inhibitor that selectively inhibits activated lymphocytes. Nat Commun 2017;8(1):1692 doi 10.1038/s41467-017-01760-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 2009;113(19):4667–76 doi 10.1182/blood-2008-07-171637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kincaid EZ, Che JW, York I, Escobar H, Reyes-Vargas E, Delgado JC, et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol 2011;13(2):129–35 doi 10.1038/ni.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.