

Abstract
Endometrial cancer remains the most common gynecological malignancy in the United States. While the loss of the tumor suppressor, PTEN (phosphatase and tensin homolog), is well studied in endometrial cancer, recent studies suggest that DICER1, the endoribonuclease responsible for miRNA genesis, also plays a significant role in endometrial adenocarcinoma. Conditional uterine deletion of Dicer1 and Pten in mice resulted in poorly-differentiated endometrial adenocarcinomas, which expressed Napsin A and HNF1B (hepatocyte nuclear factor 1 homeobox B), markers of clear-cell adenocarcinoma. Adenocarcinomas were hormone-independent. Treatment with progesterone did not mitigate poorly-differentiated adenocarcinoma, nor did it affect adnexal metastasis. Transcriptomic analyses of DICER1 deleted uteri or Ishikawa cells revealed unique transcriptomic profiles and global miRNA downregulation. Computational integration of miRNA with mRNA targets revealed deregulated let-7 and miR-16 target genes, similar to published human DICER1-mutant endometrial cancers from TCGA (The Cancer Genome Atlas). Similar to human endometrial cancers, tumors exhibited dysregulation of ephrin-receptor signaling and transforming growth factor beta signaling pathways. LIM kinase 2 (LIMK2), an essential molecule in p21 signal transduction, was significantly upregulated and represents a novel mechanism for hormone-independent pathogenesis of endometrial adenocarcinoma. This preclinical mouse model represents the first genetically engineered mouse model of poorly-differentiated endometrial adenocarcinoma.
Introduction
Endometrial cancer will account for 65,620 new cases and 12,590 deaths in 2020 in the United States [1]. Well-differentiated [Fédération Internationale de Gynécologie Obstétrique (FIGO), grade 1] endometrioid endometrial adenocarcinoma represents the most common histology and has a 5-year survival for all stages that approaches 83% [2]. High-risk histologic endometrial adenocarcinoma, such as poorly-differentiated (FIGO grade 3) endometrioid, serous, and clear-cell adenocarcinoma, encompasses 15% of endometrial cancer cases but accounts for nearly 50% of deaths [3, 4]. High tumor grade is the most significant risk factor for disease recurrence and subsequent death [5, 6].
In endometrial cancer, DICER1 is a prominent cancer-driver gene [7]. Hotspot biallelic mutations in DICER1, the endoribonuclease responsible for miRNA genesis, are enriched in endometrial adenocarcinomas over other cancers profiled in both TCGA (The Cancer Genome Atlas) PanCancer and MSK-IMPACT (Memorial Sloan-Kettering Integrated Mutation Profiling and Actionable Cancer Targets) studies [8]. DICER1 dices the precursor stem-loop miRNA into two single-stranded mature miRNAs [9, 10]. Studies have shown that recurrent DICER1 hotspot mutations from ovarian and endometrial cancers resulted in altered miRNA processing [8, 11-15]. Further, downregulation of DICER1 expression was associated with features of more aggressive endometrial adenocarcinoma, including myometrial invasion, high FIGO grade tumors, and disease recurrence [16-18].
Uterine-specific deletion of Dicer1 using PgrCre/+; Dicer1flox/flox mice resulted in a uterine phenotype containing a single layer of luminal epithelium, lack of glandular epithelium, and thin to zero endometrial stroma, consistent with atrophic endometrium from postmenopausal women [19]. Loss or mutation of the tumor suppressor, phosphatase and tensin homolog (PTEN), occurs in more than 80% of endometrial adenocarcinomas [20]. Mice with conditional deletion of Pten in the uterus (PgrCre/+; Ptenflox/flox) have high-penetrance (88.9%) well-differentiated endometrial adenocarcinoma by 90 days [21]. To study the role of Dicer1 in endometrial cancer, Dicer1 was conditionally deleted in PgrCre/+; Ptenflox/flox mice. Because downregulation of DICER1 expression was clinically associated with high FIGO grade endometrial cancer in women [16], we hypothesized that deletion of Dicer1 with deletion of Pten in the mouse uterus would result in high FIGO grade endometrial adenocarcinomas.
In this study, we present data that underscores the impact of appropriate Dicer1 function in endometrial adenocarcinoma. Importantly, we describe the development of hormone-independent, poorly-differentiated adenocarcinoma with adnexal metastasis that arises from an atrophic endometrium. High-fidelity mouse models of poorly-differentiated endometrial adenocarcinomas have not been described. Further, xenograft models have shown limited success for high-risk histologic endometrial adenocarcinoma types [22]. Our findings indicate a relationship between appropriate miRNA expression and molecular signaling pathways in the development of poorly-differentiated endometrial adenocarcinoma.
Materials and Methods
Breeding
All animals were bred and kept under standard conditions. Pgrcre/+; Dicer1flox/flox [19] mice were crossbred to Pgr+/+; Ptenflox/flox mice [21] and maintained on a C57BL/6J;129S5/Brd mixed hybrid background. Crossbreeding and genotyping was as described (Supplementary Methods and Figure S1).
Survival studies, histological analyses, immunohistochemistry, and immunofluorescence staining
For survival studies, mice were caged and examined twice weekly. Mice were euthanized at humane endpoints of loss of normal behavior or ambulation, obvious distress, not eating or drinking, loss of 20% of body weight, tumor size greater than 10% of body weight, palpable tumor >1.5 cm in any diameter, ulcerations, roughened hair coat or hunchback, or poor body conditioning score [23]. At the time points listed, mice were sacrificed; body and uterine weight were recorded. One uterine horn, oviduct, and ovary were snap-frozen, and the other, fixed, and processing, paraffin embedding, and histological and immunohistochemistry staining were as described [19, 24]. All histology was interpreted by clinical pathologists (R.E.E. and R.R.B.). FIGO grading was used. A binary system of low-grade (FIGO grade 1 or 2) and high-grade (FIGO grade 3) was used as clinically recommended [25]. Primary antibodies and conditions are listed (Supplementary Table S1). Immunohistochemistry staining was imaged on a Zeiss Axio Lab.A1 microscope (Oberkochen, Germany) and scored based on the frequency of staining as the percentage of positive staining cells and confirmed with ImageJ using FIJI [26, 27]. Immunofluorescence staining was imaged on an EVOS FL Cell Imaging System (Thermo Fisher Scientific, Waltham, MA). Due to the extreme gross and histological differences noted during dissection, investigators were not blinded to genotype. Sample size, statistical analysis, and randomization are described in Supplementary Methods.
Removal of ovaries and steroid hormone treatment
Mice underwent ovariectomy (ovex) at six weeks. Mice were divided into treatment groups: progesterone pellet (Innovative Research of America, Sarasota, Florida, 25mg/60-day release pellet) or placebo pellet, for 60 days.
CRISPR-Cas9 DICER1 deletion and cell culture
Ishikawa cells were obtained from the Cytogenetics and Cell Authentication Core at MD Anderson Cancer Center (Houston, TX) and maintained in RPMI1640 (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Minneapolis, MN) and 1% penicillin/streptomycin (Thermo Fisher Scientific). Cell line authentication was confirmed using a short tandem repeat (STR) marker profile (IDEXX BioAnalytics, Westbrook, ME) within six months of experiments and tested for mycoplasma contamination monthly (MycoAlert Plus Mycoplasma Detection Kit, Lonza, Switzerland). DICER1 was deleted in Ishikawa cells using CRISPR-Cas9 (Supplementary Methods and Figure S2). Western blot, proliferation, immunocytochemistry, and colony formation assay are described in Supplementary Methods.
RNA studies
Total RNA was extracted from uteri and Ishikawa cells, DNase treated, and assessed for quality [19]. Details of next-generation sequencing are provided in Supplementary Methods. The mRNA reads were mapped against GRCm38 and GRCh38.p5. Initial mRNA differential expression analysis was carried out using the DESeq2 package (version 1.10.1) in R/Bioconductor (R version 3.2.1) [28]. MiRNA reads were mapped to miRBase V21. Initial miRNA differential expression analysis was carried out as described [29]. Dysregulated canonical pathways and gene ontology enrichment analysis were determined from Web Gestalt (WEB-based Gene SeT AnaLysis Toolkit) [30]. Integrated miRNA:mRNA analyses were carried out using miRTarBase [31]. Reverse transcription and real-time qPCR were performed on a QuantStudio3 Real-Time PCR System (Thermo Fisher Scientific) and analyzed [19]. Supplementary Table S1 lists TaqMan assays and Sybr primer sequences. Transcriptomic data have been deposited into the Gene Expression Omnibus (accession pending).
Statistical analysis
Statistical analysis was performed with one of the following: Student’s t-test, Fisher’s exact test, Chi-squared test, multiple t-test, 2-way ANOVA, or log-rank (Mantel-Cox) pairwise comparison with Bonferroni correction. Differences between groups were determined by P<0.05. Data presented are mean and standard error of the mean (SEM) or 95% confidence interval (CI). Statistical analyses were conducted using the InStat package for Prism8 (GraphPad, San Diego, CA).
Results
Bulky endometrial tumors
Pgr+/+ (control), Pten cKO [Pten conditional knockout (Pgrcre/+; Ptenflox/flox; Dicer1+/+)], and dcKO [double conditional knockout (Pgrcre/+; Ptenflox/flox; Dicer1flox/flox)] were generated. Pten-Dicer het [Pten-Dicer heterozygous (Pgrcre/+; Ptenflox/flox; Dicer1flox/+)] were studied in parallel, but they were similar to Pten cKO (Supplementary Figures S3 and Table S2). Examination of the reproductive tract of 6-month-old female mice revealed bulky endometrial tumors in both Pten cKO and dcKO mice (Figure 1A). Uteri from Pten cKO mice revealed large tortuous uteri with bulky nodules of solid tumors protruding onto the surface, but homogeneous in gross morphology along both uterine horns. Uteri from dcKO mice also contained solid, bulky tumors. Histological analyses revealed adenocarcinoma invading through the myometrium of the uterus and into the adnexa, but without gross metastatic disease outside the female reproductive tract (Supplementary Figure S4-S5). Poorly-differentiated adenocarcinoma was found in both the uterus and adnexa in 6-month-old dcKO mice (Supplementary Figure S6 and Supplementary Table S3). Due to morbidity, only a small number of mice at this time point were examined (Supplementary Table S3).
Figure 1.

Unique morphology and poor survival with deletion of Dicer1. (A) Postnatal mice at 6 months were dissected, and female reproductive tracts were examined grossly. Tumor burden was seen in both Pten cKO and dcKO mice. Ov, ovary; Ut, uterus; Vag, vagina. (Ruler, tick mark indicates 1mm, but dcKO photo is further zoomed.) (B) Kaplan-Meier survival curves were analyzed by log-rank (Mantel-Cox) pairwise comparison with Bonferroni correction. Both Pten cKO and dcKO mice have decreased survival compared to control mice. ***, P < 0.001. (C) At 12 weeks, uteri were weighed and normalized to body weight. Both Pten cKO and dcKO mice had larger uteri than Pgr+/+. Mean ± SEM; *, P < 0.05; ***, P < 0.001; multiple t-test; n > 8 per group.
Based on criteria for humane endpoints, Pten cKO and dcKO mice showed decreased survival compared to Pgr+/+ (Figure 1B and Supplementary Figure S7). Pten cKO had a median survival of 240 days, similar to published results [21]. The median survival for dcKO mice was 327 days. Many survival mice had necrotic tumors throughout the reproductive tract, making an accurate assessment of tumor histology impossible (Supplementary Table S3).
Poorly-differentiated adenocarcinoma in dcKO mice
By 12 weeks, dcKO uteri were nearly twice as large as Pgr+/+ but remained significantly smaller than Pten cKO (Figure 1C). Uterine morphology, survival studies, and uterine weight suggested distinct phenotypes between Pten cKO and dcKO uteri. Similar to published [21], a majority of the Pten cKO uteri contained well-differentiated adenocarcinoma, with glands in columnar architecture and nuclear pseudostratification but with retained polarity in many of the nuclei, mild nuclear atypia, and little or no solid pattern (Figure 2A). Endometrial adenocarcinoma was defined as confluent growth of the endometrium, with cribriform pattern and/or expansion of the endometrium into the endometrial stroma, or invasion into the surrounding myometrium [32]. With deletion of Dicer1, the frequency of poorly-differentiated adenocarcinoma increased significantly (χ2 = 12.8; P = 0.00035). Most of the dcKO mice exhibited poorly-differentiated adenocarcinoma (Supplementary Table S3), as evidenced by poorly formed malignant glands, solid microscopic architecture, or tubulocystic architecture (Figure 2A). Epithelial cells were stained with cytokeratin-8 and myometrium with smooth muscle actin (Supplementary Figure S8). Myometrial invasion was defined as tumor between the two layers of the myometrium. Similar to published results [21], 85% of tumors from Pten cKO uteri exhibited invasion through the myometrium. All dcKO adenocarcinomas at 12 weeks showed myometrial invasion (Supplementary Table S3).
Figure 2.

Invasive adenocarcinoma with metastasis to the adnexa. (A) Pgr+/+ uteri showed normal luminal epithelium (LE) surrounding a lumen (L), with glandular epithelium (GE) and endometrial stroma (S), and two layers of smooth muscle in the myometrium (M). Pten cKO uteri exhibited well-differentiated adenocarcinoma. Uteri from dcKO mice exhibited poorly-differentiated adenocarcinoma (black dashed lines) with pale cytoplasm (black arrow). H&E. (Scale bars, low-power, 200μm; high-power, 20 μm.) (B) Pgr+/+ oviducts contained a single layer of columnar epithelium (CE, arrow) surrounding a lumen (L), surrounded by a layer of smooth muscle. Pten cKO oviduct showed atypical epithelium as evidenced by >1 cell thickness epithelium (line). The dcKO oviduct showed adenocarcinoma within the oviduct as well as next to the oviductal lumen (grey dashed lines). (Scale bars, 50 μm.) (C) Pgr+/+ ovary showed normal histology with follicles in each stage of follicular development. There were primary (PF), secondary (SF), and antral (A) follicles and corpus luteum (CL). Pten cKO ovaries had normal appearing follicles and well-differentiated adenocarcinoma (black dashed lines) that appeared to be invading from outside the ovary. Ovaries from dcKO mice did not frequently show adenocarcinoma close to the ovary. (Scale bars, 200 μm.) (D) Adult dcKO female reproductive tract showed direct invasion from moderately-differentiated adenocarcinoma in the uterus (UT) to the oviduct (OVI). Inset shows normal single layer of columnar epithelium (CE) in the oviduct next to adenocarcinoma (black arrow). Distal fimbria (white arrow) was normal. Ovary showed normal follicles, including primary (PF), secondary (SF), and antral (A) follicles. (Scale bars, low-power, 200 μm; high-power, 50 μm.)
Malignant metastatic adnexal pathology
PgrCre/+ mice have homologous recombination of floxed alleles within the columnar epithelial cells of the oviduct and the corpus luteum of the ovary when stimulated with gonadotropins [33]. Older mice of all genotypes (i.e., survival and 6-month old) exhibited adenocarcinoma of the uterus, oviduct, and ovaries. However, the origin of the tumor, driven by Cre-recombination events in the oviduct and/or ovary versus metastatic disease from the uterus, was unable to be discerned. The oviducts and ovaries of 12-week-old mice were carefully examined for any signs of cancer-associated histological changes. Pten cKO oviducts frequently showed epithelial atypia in the oviducts defined as nuclear stratification (>1 cell thickness epithelium, loss of nuclear polarity) and nuclear atypia in the form of enlarged, rounded nuclei (Figure 2B). Adenocarcinoma was infrequently discovered in the oviducts of Pten cKO mice. Adenocarcinoma was found in nearly 50% of the oviducts of dcKO mice. The histology was similar to uterine histology and adenocarcinoma was located on the inside and outside of normal oviducts (Figure 2B), consistent with metastatic spread. Pten cKO ovaries showed evidence of both normal follicular development and metastatic disease, as cancer appeared to have invaded at the ovarian hilum (Figure 2C). Ovaries of dcKO mice were most frequently normal (Figure 2C-D). Examination of the reproductive tracts of dcKO mice showed evidence of direct extension from the uterus to the oviduct with normal, cancer-free, distal fimbria and ovary (Figure 2D). Evidence of metastatic disease to the adnexa was observed in approximately 50% of Pten cKO and dcKO mice. Histology was largely similar between the uterus and adnexa in many dcKO mice (Supplementary Table S3 and Supplementary Figure S9).
Expressed clear-cell adenocarcinoma markers
Epithelial cells from dcKO uterine tumors exhibited large, often rounded, and hyperchromatic nuclei and pale-staining to clear cytoplasm (Figure 2A and Supplementary Figure S4). To better describe the poorly-differentiated adenocarcinomas with clear cytoplasm in dcKO mice, immunohistochemical markers of clear-cell adenocarcinoma, Napsin A and hepatocyte nuclear factor 1 homeobox B (HNF1B) [34, 35], were evaluated. Poorly-differentiated adenocarcinomas from dcKO mice exhibited malignant epithelial cells with high-frequency, high-intensity cytoplasmic Napsin A staining. Malignant epithelial cells from dcKO mice more frequently stained positive for nuclear HNF1B compared to Pten cKO tumors (Figure 3 and Supplementary Figure S10).
Figure 3.

Poorly-differentiated adenocarcinomas from 12-week-old dcKO mice expressed clear-cell adenocarcinoma markers. Napsin A staining was cytoplasmic, and HNF1B staining was nuclear. Quantification of the frequency of positive epithelial cells. Kidney is shown as positive control. Mean expression scores were compared using 2-tailed Student’s t-test. Average scoring ± SEM is shown; ***; P<0.001; n = 6. (Scale bars, low-power, 50 μm; high-power, 10 μm.)
No effect on the frequency of poorly-differentiated adenocarcinoma histology with steroid hormone depletion and/or progesterone treatment
To determine the effects of ovarian insufficiency in dcKO mice, both ovaries were removed, and uterine weight and histology were assessed. Gross uterine weight was lower in ovariectomized (ovex) mice compared to intact mice (Supplementary Figure S11A). However, the frequency of poorly-differentiated adenocarcinoma was not decreased in dcKO mice (Supplementary Table S3). To examine the effects of long-term progesterone therapy, mice underwent removal of ovaries at six weeks and were randomized to either placebo or progesterone pellets at eight weeks. Treatment lasted 60 days. Treatment with progesterone did not alter the frequency of poorly-differentiated adenocarcinomas in ovex dcKO mice (Supplementary Table S3). Carefully selected women with well-differentiated, early-stage endometrioid adenocarcinoma can be treated with progesterone to preserve fertility options [22]. Intact mice were randomized at six weeks to either placebo or progesterone pellet for 60 days. Treatment with progesterone did not significantly alter the frequency of poorly-differentiated endometrial adenocarcinomas in intact mice (Supplementary Table S3). Treatment of intact mice with long-term progesterone decreased the rate of metastatic adenocarcinoma to the adnexa, but it was only statistically significantly decreased for Pten cKO mice (Supplementary Figure S11B and Table S3).
Unique transcriptomic profile
To create an in vitro human model, CRISPR-Cas9 was used to delete DICER1. Ishikawa cells were chosen because they are PTEN mutant, derived from a well-differentiated adenocarcinoma, and exhibit high expression of E-cadherin (CDH1) consistent with epithelial cell differentiation [22, 36]. Western blot confirmed knockdown or knockout of DICER1 (Figure 4A). DICER1 deletion in Ishikawa cells led to decreased cellular proliferation and reduced colony formation in soft agar (Figure 4B-C), consistent with the smaller-sized uterine tumors in dcKO mice (Figure 1C). Examination of CDH1 confirmed high expression at the RNA level by qPCR and the protein level by immunocytochemistry in DICER1+/+ Ishikawa cells as previously reported [36]. DICER1−/− Ishikawa cells exhibited a 4-fold decrease in CDH1 RNA expression (Figure 4D) and a significant decline in CDH1 expression by immunocytochemistry (Figure 4E and Supplementary Figure S12), consistent with a more poorly-differentiated phenotype. DICER1−/− cells exhibited increased expression of the clear cell markers, Napsin A and HNF1B, by immunocytochemistry (Supplementary Figure S12).
Figure 4.

Unique cellular characteristics and transcriptomic profile with loss of two alleles of DICER1. (A) Western blot showed decreased DICER1 protein levels in DICER1+/− and complete loss of DICER1 protein in DICER1−/− cells. Both DICER1+/− and DICER1−/− cells showed a significant decrease in (B) proliferation and (C) colony formation relative to DICER1+/+. Mean ± 95% CI, *, P<0.05, ***, P<0.001. Proliferation data using two-way ANOVA and colony formation data using two-tailed Student's t-test. Expression of cadherin 1 (CDH1 also known as E-cadherin) was high in DICER+/+ cells by qPCR (D) and immunocytochemistry (E), showing a well-differentiated phenotype. In particular, E-cadherin staining was high in the cell membrane in cells touching each other. CDH1 was decreased by qPCR (D) and immunocytochemistry (E), in DICER1−/− cells, showing a loss of well-differentiated phenotype. Mean ± SEM; ***, P < 0.001; two-tailed Student’s t-test; n=6. RQ, relative quantity of CDH1 transcript by qPCR relative to endogenous control gene GAPHD. Immunofluorescent staining of E-cadherin (green) and DAPI. Scale bars, 100 μm. (F) Principal component analysis revealed distinct clustering of human samples in DICER1+/+ (triangles), DICER1+/− (squares), and DICER1−/− (plus sign). (G) Principal component analysis for mouse showed close clustering of Pten cKO (triangles) and Pten-Dicer het (squares) but distinct clustering of Pgr+/+ (circles) and dcKO (plus sign).
Poly-A RNA sequencing (RNA-seq) was performed. Principal Component (PC) analysis showed distinct clustering. DICER1+/+, DICER1+/−, and DICER1−/− differed significantly in PC2, while DICER1+/+ and DICER1+/− were most similar in PC1 (Figure 4F). Determination of differentially expressed genes between DICER1+/+ and DICER1−/− revealed 2444 unique protein-coding genes with log2fold change >ǀ1ǀ significantly dysregulated [false discovery rate (FDR)<0.05, Supplementary Table S4], 1154 genes upregulated, and 1290 genes downregulated (Supplementary Figure S13A).
Poly-A RNA sequencing on mouse uteri was then performed. Three-week uteri were selected because: 1) PgrCre/+-mediated deletion occurs primarily in the luminal and glandular epithelium [33]; 2) the limited molecular contributions of steroid hormones in pre-adolescent female mice; 3) the ability to explore early molecular contributions; and 4) the similar ratio of cell populations (i.e., epithelium, stroma, and myometrium) across genotypes. As early as three weeks, Pten cKO and dcKO uteri showed adenocarcinoma (Supplementary Table S3) with activation of phospho-AKT limited to the epithelium (Supplementary Figure S14). PC analysis showed distinct clustering. However, Pten cKO and Pten-Dicer het mRNA profiles were most similar to each other and only varied slightly in PC2. Both dcKO and Pgr+/+ varied significantly in PC1, and dcKO varied in PC2 (Figure 4G). Because all experimental mice have a deletion of Pten, transcriptomic profiles were compared to Pten cKO. Determination of differentially expressed genes between dcKO and Pten cKO showed 1635 unique protein-coding genes with log2fold change >ǀ1ǀ significantly dysregulated (FDR<0.01, Supplementary Figure S14B), 1059 genes upregulated, and 576 genes downregulated (Supplementary Table S4).
Gene set enrichment analysis with estrogen-responsive gene sets [37] showed no significant overlap with mouse dcKO or DICER1−/− transcriptomic profile (Supplementary Table S5). The genes downregulated in mouse dcKO showed a trend (P=0.087) towards enrichment in progesterone-responsive gene sets [38], but no significant enrichment in upregulated progesterone-responsive genes. Both up- and downregulated genes in the DICER1−/− transcriptomic profile showed significant enrichment in progesterone responsive genes (Supplementary Table S5).
Web Gestalt [30] revealed that the upregulated DICER1−/− transcriptomic profile was enriched in ephrin-receptor signaling pathway genes (Supplementary Table S4). Studies have shown that high expression of EPH receptor A2 (EPHA2) in endometrial cancer was associated with high tumor grade, reduced survival, high microvessel density, and high Ki67 expression [39, 40]. QPCR of DICER1−/− cells showed that EPHA2 was 7.6-fold upregulated. Other EPH receptors, including EPH receptor A7 (EPHA7) and EPH receptor B2 (EPHB2), were 8-10-fold upregulated (Figure 5A). Further, both the non-canonical WNT signaling molecule, WNT5B, and p21 (RAC1) activated kinase 3 (PAK3) downstream effectors of EPHA2 [41, 42], were 9.6- and 9.3-fold upregulated. LIM kinase 2 (LIMK2), an essential molecule in p21 signal transduction and involved in castration-resistant prostate cancer [43], was similarly upregulated (Figure 5A). QPCR on dcKO uteri showed that EPH receptor B1 (Ephb1) was 3.5-fold upregulated, and Pak3 was 5.3-fold upregulated. Cofilin 2 (Cfl2), a potential downstream effector of LIMK2 and PAK3 [44], was 2.7-fold upregulated (Figure 5B). Pathway analysis on the upregulated dcKO uteri transcriptomic profile revealed hedgehog signaling and prostaglandin synthesis genes were upregulated (Supplementary Table S4). QPCR of dcKO uteri showed Indian hedgehog signaling molecule (Ihh) 20-fold upregulated and patched 1 (Ptch1) and GLI family zinc finger 1 and 2 (Gli1 and Gli2) were 5-fold upregulated (Figure 5C). High CFL2 expression was associated with poor survival in gastric cancer [45]. Both high CFL2 and EPHA2 have been associated with increased proliferation in cancers [39, 40, 45]. Examination of dcKO uteri showed increased expression of Ki67 compared to Pten cKO uteri (Figure 5D).
Figure 5.
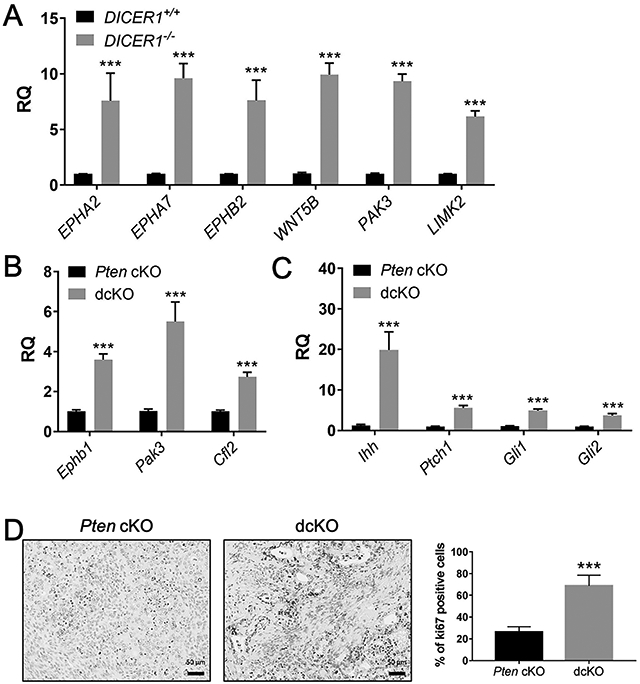
Dysregulation of ephrin receptor signaling genes with DICER1 deletion. QPCR showed upregulation of ephrin receptor signaling genes in human DICER1−/− cells (A) and dcKO mice (B). QPCR showed upregulation of hedgehog signaling genes in dcKO uteri (C). Mean± SEM; 2-tailed Student’s t-test; ***, P < 0.001; n = 6; RQ, relative quantity of each gene transcript relative to endogenous control gene GAPHD (human) and 18S (mouse) normalized to DICER1+/+ (A) or Pten cKO (B-C). (D) Elevated Ki67 expression in dcKO compared Pten cKO. (Scale bars, 50 μm.) Mean± SEM; 2-tailed Student’s t-test; ***, P < 0.001; n > 8.
To examine the effects of DICER1 deletion on miRNA expression in endometrial cancer, small RNA sequencing was performed on the same samples as poly-A RNA sequencing. Differentially expressed miRNAs (FDR<10%) in mouse dcKO and DICER1−/− cells are shown in Supplementary Table S6. DICER1−/− cells showed 193, and dcKO uteri revealed 38 mature miRNAs downregulated. There were 22 mature miRNAs downregulated in common. MiRNAs target mRNAs for repression through binding to the 3’ untranslated region of target genes [46]. Integration of upregulated miRNA-target mRNAs with downregulated miRNAs from DICER1−/− cells was performed using miRTarBase. MiRTarBase uses curated data from over 10,000 publications to make validated predictions on miRNA-target interactions (MTIs) [47, 48]. Out of 1154 upregulated genes in DICER1−/− cells, 885 were MTIs for the 193 downregulated miRNAs. Supplementary Table S6 lists the 7023 miRNA:mRNA MTIs mapped to 885 unique genes. The genes targeted by miRNAs were involved in the following pathways: microRNAs in cancer, glycosaminoglycan biosynthesis, ephrin-receptor signaling, and transforming growth factor-beta (TGFβ) signaling (Supplementary Table S6). Many TGFβ signaling genes were also dysregulated in dcKO uteri (Supplementary Table S6 and Figure S15).
Human DICER1-mutant TCGA endometrial cancers were enriched in miRNA-target genes from five miRNA families: let-7-5p, miR-17-5p, miR-16-5p, miR-29-3p, and miR-101-3p [8]. Mature miRNAs from each of these five miRNA families were significantly downregulated in the small RNA sequencing datasets from DICER1−/− cells (Supplementary Table S6). Out of 1154 upregulated genes in DICER1−/− cells, 347 unique genes were MTIs for these specific five DICER1-mutant family members. Pathways enriched included microRNAs in cancer, glycosaminoglycan biosynthesis, ephrin-receptor signaling, renal cell carcinoma, and TGFβ signaling (Supplementary Table S6). Recent work has shown that dysregulation of TGFβ signaling in the mouse uterus results in endometrial hyperproliferation and cancer [49, 50]. Members of the TGFβ signaling pathways were targeted by a number of dysregulated miRNAs. Let-7b-5p, miR-16-5p, and miR-26b-5p were mature miRNAs downregulated in both DICER1−/− cells and dcKO uteri and predicted to target TGFβ signaling genes. By QPCR, DICER1−/− cells had significant downregulation of let-7b-5p, and let-7b-5p was also 2-fold down-regulated in dcKO uteri. QPCR showed a 71-fold downregulation of miR-16-5p in DICER1−/− cells, and 2.1-fold downregulation for miR-16-5p in mouse. QPCR showed 939-fold downregulation of miR-26b-5p in human and 1.7-fold downregulation in the dcKO uteri (Figure 6A-B). These miRNAs were MTIs for the TGFβ signaling pathway genes: cyclin-dependent kinase 6 (CDK6), bone morphogenetic protein receptor 1B (BMPR1B), and transforming growth factor-beta receptor 3 (TGFBR3, Supplementary Table S6). MiR-26b-5p was also a predicted target for CDK6, transforming growth factor-beta 1 induced transcript 1 (TGFB1I1), and growth differentiation factor 10 (GDF10, Supplementary Table S6). QPCR showed higher than 6.5-fold upregulation of CDK6 in human and 2.5-fold upregulation of Cdk6 in the mouse. BMPR1B showed a greater than 14-fold upregulation in DICER1−/− cells. TGFBR3 showed 6.5-fold upregulation in human, and 4-fold upregulation in dcKO mouse. TGFB1I1 is thought to mediate progesterone resistance in endometriosis [51, 52]. TGFB1I1 was 8-fold upregulated in human and 2.9-fold upregulated in mouse. GDF10 was 8.4-fold upregulated in human and 2.3-fold upregulated in mice (Figure 6C-D). Other TGFβ signaling genes that are predicted targets of other downregulated miRNAs were upregulated (Supplementary Figure S15).
Figure 6.
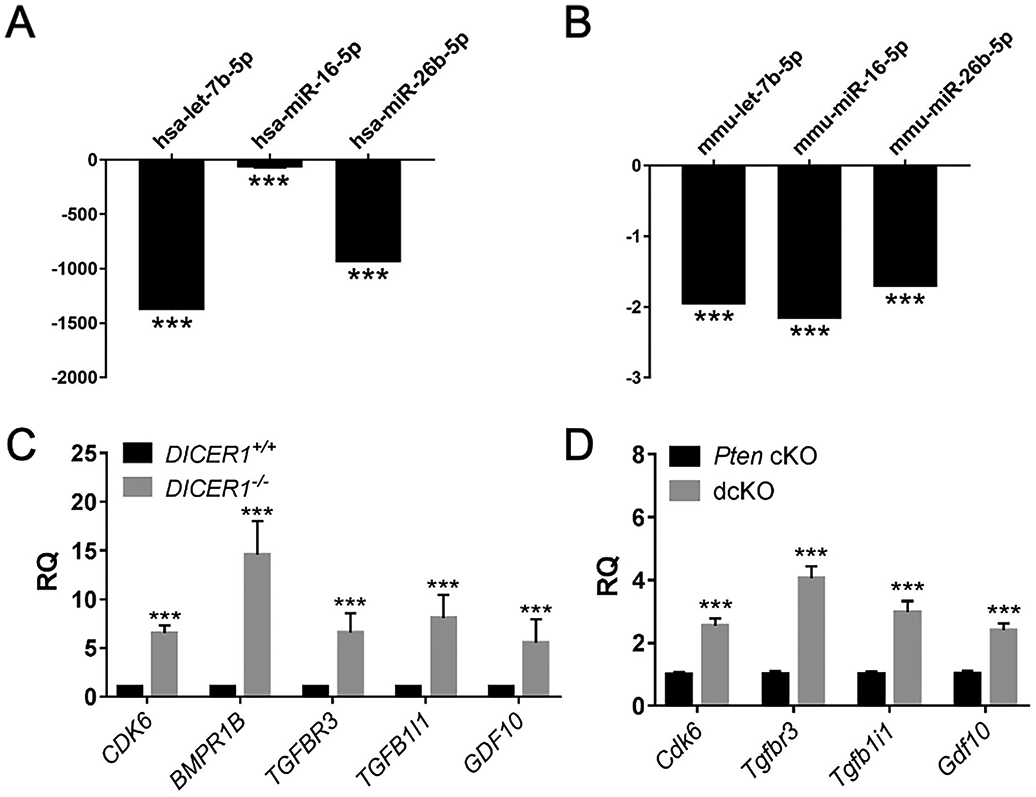
Downregulated miRNAs and upregulated TGFβ signaling genes. QPCR showed downregulation of miRNAs in human DICER1−/− cells (A) and dcKO mice (B). Mean ± SEM; 2-tailed Student’s t-test; ***, P < 0.001; n = 6; relative quantity of each miRNA by qPCR relative to endogenous control miRNA U6 snRNA (human) and snoRNA202 (mouse) normalized to DICER1+/+ (A) or Pten cKO (B). QPCR showed upregulation of TGFβ signaling genes in human (C) and mouse (D). Mean ± SEM; ***, P < 0.001; two-tailed Student’s t-test; n=6. RQ, relative quantity of each gene transcript by qPCR relative to endogenous control gene GAPHD (human) and 18S (mouse) normalized to DICER1+/+ (C) or Pten cKO (D).
Concurrent PTEN and DICER1 mutations in human disease
Four publicly available datasets of human endometrial cancer were used to determine the frequency of concurrent mutations in PTEN and DICER1 (Table 1). The TCGA PanCancer dataset (n=509) contains 75.4% endometrioid, 20.6% serous, and 4% mixed endometrioid/serous [53]. The MSK-IMPACT dataset (n=189) contains 40.6% endometrioid, 23.9% serous, 18.8% carcinosarcoma, and 6.6% clear cell [54]. Available via cBioPortal [55, 56] is an additional dataset (n=16) that contains only uterine clear-cell adenocarcinoma [57]. An additional dataset (n=30) not available via cBioPortal also contains only uterine clear cell adenocarcinoma [58]. These databases showed that mutations in PTEN and DICER1 occur concurrently in up to 11.2% of endometrial cancer samples. Concurrent mutations in PTEN and DICER1 in endometrioid endometrial cancers were frequently grade 3 and disease progression was frequent. Up to 10% of clear cell tumors contained concurrent mutations in PTEN and DICER1. These tumors were frequently grade 2 without disease progression. Many of the DICER1 mutations (26/65 samples) were known oncogenic changes, including truncating mutations, frame shift deletions, or recurrent oncogenic hotspot mutations (Supplementary Tables S7). Eight tumors were categorized as neither DNA-polymerase-ε ultra-mutated nor microsatellite instability-hypermutated tumors (Supplementary Tables S7). A majority of those contained known DICER1 recurrent oncogenic hotspot mutations.
Table 1:
Limited clinical characteristics of concurrent PTEN and DICER1 mutant endometrial cancers
| NCI -UCC n = 16 |
MSK -PATH n = 30 |
MSK -IMPACT n = 189 |
TCGA PanCancer n = 509 |
||
|---|---|---|---|---|---|
| Both mutations, n (%) | 0 | 3 (10%) | 5 (2.6%) | 57 (11.2%) | |
| Cancer histology, n (%) | Endometrioid | -- | -- | 4 (80%) | 54 (94.7%) |
| Uterine Carcinosarcoma | -- | -- | 1 (20%) | -- | |
| Clear cell | -- | 3 (100%) | -- | -- | |
| Mixed serous and endometrioid | -- | -- | 2 (3.5%) | ||
| Serous | -- | -- | 1 (1.8%) | ||
| Histologic grade, n (%) | Grade 1 | -- | -- | 1 (20%) | 7 (12.3%) |
| Grade 2 | -- | 3 (100%) | -- | 9 (15.8%) | |
| Grade 3 | -- | -- | 4 (80%) | 41 (71.9%) | |
| Molecular category, n (%) | MSS | -- | -- | 1 (20%) | -- |
| MSI-H | -- | -- | 2 (40%) | 17 (29.8%) | |
| POLE ultra-mutated | -- | 2 (66.7%) | 2 (40%) | 34 (59.7%) | |
| Copy-number low | -- | -- | -- | 6 (10.5%) | |
| Disease progression, n (%) | No disease progression | -- | 3 (100%) | 1 (20%) | 5 (8.8%) |
| Disease progression | -- | -- | 4 (80%) | 52 (91.2%) |
Discussion
Although nearly all common cancers have improved cancer survival since the 1970s, endometrial cancer is one of the few cancers with an increased death rate [1]. Importantly, studies have shown that the increase in incidence is mostly in high-grade or non-endometrioid histologies, including serous and clear-cell adenocarcinomas [59]. The highest risk factor linked to disease recurrence and death is a high tumor grade [5, 6]. New models are needed for high-grade tumors, as these are the tumors leading to the most substantial mortality [3, 4].
In the present study, we describe the first mouse model of poorly-differentiated endometrial adenocarcinoma. Because DICER1 mutations are frequently found in ultra-mutated tumors, DICER1 mutations in endometrial adenocarcinoma were thought to be passenger mutations. However, recent genomic profiling of tumors through TCGA PanCancer and MSK-IMPACT showed enrichment of biallelic DICER1 hotspot mutations in endometrial but not other cancers [8]. Importantly, this analysis removed POLE ultra-mutated samples [8]. DICER1 mutants play a functional role in endometrial cancers, by affecting miRNA processing, miRNA expression, and miRNA-target gene expression [8, 11-15]. These studies showed that DICER1 mutations are not passenger mutations in endometrial cancer and suggest that DICER1 may play a unique yet significant role in endometrial cancer. The frequency of concurrent mutations in PTEN and DICER1 is 2-11% (Table 1), but the number of DICER1 loss-of-function and recurrent hotspot mutations comprise nearly 50% of those concurrent mutations, advocating that our mouse model with deletion of Dicer1, has relevance to human disease. Furthermore, on the molecular level, deregulated miRNA-target genes for let-7-5p and miR-16-5p family members were significantly enriched in published DICER1-mutant TCGA datasets [8] and the present study (both dcKO mouse and DICER1−/− cells; Figure 6). Thus, the dcKO mouse model and DICER1−/− cells represent relevant models.
In humans, germline variants in DICER1 are rare and are thought to give rise to the DICER1 Syndrome. Humans with DICER1 Syndrome have an increased risk of certain cancers, but endometrial cancers are not one of those cancers [60]. However, somatic loss-of-function mutations in DICER1 have been discovered in uterine adenosarcomas and rhabdomyosarcomas [61, 62], suggesting that both the mesenchymal and the epithelial component plays a role in development of cancers. Women with Cowden Syndrome are at increased risk of endometrial cancer due to germline PTEN mutations [63], but the role of DICER1 mutations have not been published in women with Cowden Syndrome. Other mouse models suggest that deletion of Pten and Dicer1 is not sufficient for malignant transformation of all epithelial lineages. In a mouse model of prostate cancer, conditional deletion of Pten and loss of one allele of Dicer1 resulted in a more aggressive phenotype than loss of both alleles of Dicer1 [64]. Similarly, loss of two alleles of Dicer1 in an oncogenic Kras mouse model of lung cancer was protective against aggressive lung cancer [65]. Loss of Pten and one allele of Dicer1 in the uterus did not show a significantly different phenotype than Pten cKO alone, while the dcKO showed a poorly-differentiated phenotype. Thus, loss of Pten and Dicer1 leading to endometrial cancer seems to be tissue specific in our mouse model and in human datasets from TCGA and MSK-IMPACT [8]. The impact of epithelial-stromal communication and specific mutations in smaller subsets of endometrial cells deserve deeper study.
Clinically, miR-16-5p and let-7-5p represent important molecular targets for cancer therapy. Treatment with liposomal miR-16 is being tested for mesothelioma [66] and is frequently considered a tumor suppressor [67]. Let-7b-5p is a likely tumor suppressor in endometrial cancer [68]. Also, targets of these miRNA molecules may be very important clinically. For example, CDK6 is a predicted target for multiple robustly downregulated miRNA molecules (Supplementary Table S6). Recent studies suggest that high expression of CDK6 may be a biomarker for poor prognosis endometrial cancers [69]. Specifically, the high expression of CDK6 with high Ki67 expression was associated with shorter progression-free survival in women with endometrial cancer [69]. Intact dcKO uterine tumors showed a significantly high frequency of Ki67-staining cells (Figure 5D). The use of CDK6 inhibitors, such as palbociclib, has shown promise in vitro and in orthotopic and xenograft models, particularly when tumors are PTEN mutant [70]. Additionally, high expression of LIMK2 (Figure 5) suggests activation of novel signaling cascades that have not yet been explored in endometrial cancer. Studies indicate that the use of T56-LIMKi, a LIM kinase inhibitor, leads to decreased proliferation and migration in cancer cells [44]. Future studies will focus on testing these precision therapies in our mouse and cell models.
Poorly-differentiation endometrial tumors frequently develop within an atrophic, or postmenopausal endometrium [22]. The PgrCre/+; Dicer1flox/flox uteri represent atrophic endometrium [19]. Additional deletion of Pten in this background of Dicer1 deletion leads to poorly-differentiated adenocarcinoma. While hyper-proliferation of endometrial epithelium from endogenous or exogenous estradiol with mutation of PTEN is a biologically plausible mechanism for the development of endometrial cancer [22], little is known about the mechanism of development of endometrial cancer from atrophic endometrium. In most dcKO mice, the tumors seemed to arise from individual areas of the uterus rather than homogenously along the entire uterus. This mechanism is consistent with type II endometrial tumors primarily derived from heterogeneous endometrial polyps in women [22]. The reason for the heterogeneous nature of the tumor development along the uterine horns is unknown. We suspect that a small portion of cells, potentially a hormone-independent stem cell population, maybe playing a role as decreased expression of DICER1 in human endometrial cancers is associated with increased expression of stem cell markers [16]. These studies will be the focus of future investigations.
Further studies are needed on Dicer1 in the adnexa. High-grade serous ovarian cancer develops in the oviduct of mice with mesenchymal deletion of both Pten and Dicer1 [71] . Moreover, activation of WNT/β-catenin through deletion of adenomatous polyposis coli (Apc) with PgrCre/+ showed frequent endometrioid ovarian cancer that began in the epithelial cells of the oviduct [72]. However, we believe that Pten cKO, Pten-Dicer het, and dcKO mice have metastatic endometrial cancer to the adnexa. There are a number of reasons for this conclusion: 1) the frequency of tumors in the adnexa is of lower penetrance than the uterus; 2) the histology of the adnexal tumors is similar to the uterine tumors; 3) the location of cancer in the adnexa (i.e., ovarian hilum or adjacent to normal) suggests invasion or direct spread; 4) the large endometrial tumors with myometrial invasion; 5) multinodular location of adenocarcinoma along the oviduct with some areas of normal oviduct; and 6) no evidence of typical spread of oviductal or ovarian cancer such as peritoneal metastasis or ascites. Because PgrCre leads to recombination in the oviduct and granulosa cells of the ovary [33, 72], it is possible that either the oviduct and/or the ovary are concurrent primary tumors of lower penetrance that exhibit similar histology as the uterine adenocarcinoma. Further, metastatic disease lands in a unique “soil” and may exhibit different histology. Thus, the subtle differences between adnexal and uterine disease may develop from the unique tumor microenvironment.
The poorly-differentiated adenocarcinomas of dcKO mice also exhibit features of clear-cell adenocarcinoma. Clinically, clear-cell endometrial adenocarcinomas are challenging to distinguish from high-grade endometrioid endometrial adenocarcinomas with clear-cell changes [35]. In our mouse model, dcKO uteri show high-intensity staining for Napsin A, which is a sensitive marker for clear-cell carcinoma [34]. Additionally, dcKO tumors are HNF1B positive, another important marker of clear cells [34, 35]. However, neither Napsin A nor HNF1B is specific for endometrial clear cell adenocarcinoma [73, 74]. The use of the dcKO model as a model for clear-cell adenocarcinoma would be improved with comparison to transcriptomic studies from clear-cell endometrial adenocarcinoma from women. However, the only existing datasets of clear-cell endometrial cancer profiled five samples, used older microarray technology, and profiled a limited number of genes [75]. We are encouraged that our gene sets were enriched in let-7 and miR-16 target genes similar to derepressed gene sets from DICER1-mutant TCGA tumors [8]. Further studies are needed to transcriptomically interrogate pathologically well-characterized clear-cell endometrial adenocarcinoma from women for comparison to dcKO tumors.
In women, poorly-differentiated endometrial adenocarcinomas are associated with worse survival [3-6]. Our dcKO mice had poorly-differentiated endometrial adenocarcinomas that were associated with worse survival than normal control mice, but improved survival compared to Pten cKO mice. This discrepancy is largely based on the humane endpoint for euthanasia being palpable tumor of >1.5 cm. In women who are surgically staged, the size of the uterus is not clinically useful, but higher tumor grade and deeper myometrial invasion portend worse outcomes [5, 6]. Importantly, dcKO mice had adenocarcinomas with increased myometrial invasion, a correlate of higher stage in women, in addition to poorly-differentiated tumors. We speculate that if death were an endpoint in our mouse survival studies that dcKO mice would succumb to disease faster than Pten cKO. We speculate that DICER1 loss may result in more indolent, smaller, yet more aggressive tumors. This speculation is supported by published data from women that showed that downregulation of DICER1 expression was associated with increased myometrial invasion, higher FIGO grade tumors, and increase disease recurrence [16-18].
Our data, as presented, show that the dcKO mouse represents poorly-differentiated adenocarcinoma. Mouse models of endometrial cancer that recapitulate human disease represent translational tools for a better understanding of the aggressive disease. We anticipate that this model will be highly relevant, not only to study the molecular characteristics of the rarer forms of human endometrial cancer but also to study the preclinical development of therapeutics.
Supplementary Material
Acknowledgments
We acknowledge the Indiana Center for Musculoskeletal Health Histology Core at Indiana University School of Medicine and the Human Tissue and Acquisition and Pathology Core at the Dan L. Duncan Comprehensive Cancer Center at Baylor College of Medicine for histology services; the Center for Genomics and Bioinformatics for RNA sequencing, and the Center for Medical Genomics for RNA quality control analysis. We appreciate Dr. Ken Nephew for thoughtful review and comments. These studies were supported by the Indiana Clinical Translational Sciences Institute funded, in part, by Award Number UL1TR002529 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. These studies were supported by a Uterine SPORE Career Development Award from MD Anderson Cancer Center P50-CA098258 (to SMH) and the Intramural Research Program of the National Institute of Environmental Health Sciences supported FJD: Project Z1AES103311-01.
Footnotes
Competing Interests
The authors have no competing financial interests to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020; 70: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Creasman WT, Odicino F, Maisonneuve P, Quinn MA, Beller U, Benedet JL et al. Carcinoma of the Corpus Uteri. Int J Gynaecol Obstet 2006; 95 Suppl 1: S105–S143. [DOI] [PubMed] [Google Scholar]
- 3.Vogel TJ, Knickerbocker A, Shah CA, Schiff MA, Isacson C, Garcia RL et al. An analysis of current treatment practice in uterine papillary serous and clear cell carcinoma at two high volume cancer centers. J Gynecol Oncol 2015; 26: 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Versluis MA, de Jong RA, Plat A, Bosse T, Smit VT, Mackay H et al. Prediction model for regional or distant recurrence in endometrial cancer based on classical pathological and immunological parameters. Br J Cancer 2015; 113: 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrow CP, Bundy BN, Kurman RJ, Creasman WT, Heller P, Homesley HD et al. Relationship between surgical-pathological risk factors and outcome in clinical stage I and II carcinoma of the endometrium: a Gynecologic Oncology Group study. Gynecol Oncol 1991; 40: 55–65. [DOI] [PubMed] [Google Scholar]
- 6.Creutzberg CL, van Putten WL, Koper PC, Lybeert ML, Jobsen JJ, Warlam-Rodenhuis CC et al. Surgery and postoperative radiotherapy versus surgery alone for patients with stage-1 endometrial carcinoma: multicentre randomised trial. PORTEC Study Group. Post Operative Radiation Therapy in Endometrial Carcinoma. Lancet 2000; 355: 1404–1411. [DOI] [PubMed] [Google Scholar]
- 7.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018; 174: 1034–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vedanayagam J, Chatila WK, Aksoy BA, Majumdar S, Skanderup AJ, Demir E et al. Cancer-associated mutations in DICER1 RNase IIIa and IIIb domains exert similar effects on miRNA biogenesis. Nat Commun 2019; 10: 3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–297. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Ivan M, Hawkins SM. The role of MicroRNA molecules and MicroRNA-regulating machinery in the pathogenesis and progression of epithelial ovarian cancer. Gynecol Oncol 2017; 147: 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Wang Y, McMonechy MK, Anglesio MS, Yang W, Senz J et al. Recurrent DICER1 hotspot mutations in endometrial tumours and their impact on microRNA biogenesis. The Journal of pathology 2015; 237: 215–225. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Chen J, Yang W, Mo F, Senz J, Yap D et al. The oncogenic roles of DICER1 RNase IIIb domain mutations in ovarian Sertoli-Leydig cell tumors. Neoplasia 2015; 17: 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heravi-Moussavi A, Anglesio MS, Cheng SW, Senz J, Yang W, Prentice L et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 2012; 366: 234–242. [DOI] [PubMed] [Google Scholar]
- 14.Anglesio MS, Wang Y, Yang W, Senz J, Wan A, Heravi-Moussavi A et al. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. The Journal of pathology 2013; 229: 400–409. [DOI] [PubMed] [Google Scholar]
- 15.Gurtan AM, Lu V, Bhutkar A, Sharp PA. In vivo structure-function analysis of human Dicer reveals directional processing of precursor miRNAs. RNA 2012; 18: 1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang XJ, Jiang FZ, Tong H, Ke JQ, Li YR, Zhang HL et al. Dicer1 dysfunction promotes stemness and aggression in endometrial carcinoma. Tumour Biol 2017; 39: 1010428317695967. [DOI] [PubMed] [Google Scholar]
- 17.Torres A, Torres K, Paszkowski T, Jodlowska-Jedrych B, Radomanski T, Ksiazek A et al. Major regulators of microRNAs biogenesis Dicer and Drosha are down-regulated in endometrial cancer. Tumour Biol 2011; 32: 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zighelboim I, Reinhart AJ, Gao F, Schmidt AP, Mutch DG, Thaker PH et al. DICER1 expression and outcomes in endometrioid endometrial adenocarcinoma. Cancer 2011; 117: 1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawkins SM, Andreu-Vieyra CV, Kim TH, Jeong JW, Hodgson MC, Chen R et al. Dysregulation of uterine signaling pathways in progesterone receptor-Cre knockout of dicer. Mol Endocrinol 2012; 26: 1552–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tashiro H, Blazes MS, Wu R, Cho KR, Bose S, Wang SI et al. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res 1997; 57: 3935–3940. [PubMed] [Google Scholar]
- 21.Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP et al. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res 2008; 68: 5619–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandita P, Wang X, Jones DE, Collins K, Hawkins SM. Unique Molecular Features in High-Risk Histology Endometrial Cancers. Cancers (Basel) 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ullman-Cullere MH, Foltz CJ. Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab Anim Sci 1999; 49: 319–323. [PubMed] [Google Scholar]
- 24.Wang X, Khatri S, Broaddus R, Wang Z, Hawkins SM. Deletion of Arid1a in Reproductive Tract Mesenchymal Cells Reduces Fertility in Female Mice. Biol Reprod 2016; 94: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soslow RA, Tornos C, Park KJ, Malpica A, Matias-Guiu X, Oliva E et al. Endometrial Carcinoma Diagnosis: Use of FIGO Grading and Genomic Subcategories in Clinical Practice: Recommendations of the International Society of Gynecological Pathologists. Int J Gynecol Pathol 2019; 38 Suppl 1: S64–S74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hawkins SM, Creighton CJ, Han DY, Zariff A, Anderson ML, Gunaratne PH et al. Functional microRNA involved in endometriosis. Mol Endocrinol 2011; 25: 821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 2017; 45: W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res 2011; 39: D163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurman RJ, Norris HJ. Evaluation of criteria for distinguishing atypical endometrial hyperplasia from well-differentiated carcinoma. Cancer 1982; 49: 2547–2559. [DOI] [PubMed] [Google Scholar]
- 33.Soyal SM, Mukherjee A, Lee KY, Li J, Li H, DeMayo FJ et al. Cre-mediated recombination in cell lineages that express the progesterone receptor. Genesis 2005; 41: 58–66. [DOI] [PubMed] [Google Scholar]
- 34.Lim D, Ip PP, Cheung AN, Kiyokawa T, Oliva E. Immunohistochemical Comparison of Ovarian and Uterine Endometrioid Carcinoma, Endometrioid Carcinoma With Clear Cell Change, and Clear Cell Carcinoma. Am J Surg Pathol 2015; 39: 1061–1069. [DOI] [PubMed] [Google Scholar]
- 35.Murali R, Davidson B, Fadare O, Carlson JA, Crum CP, Gilks CB et al. High-grade Endometrial Carcinomas: Morphologic and Immunohistochemical Features, Diagnostic Challenges and Recommendations. Int J Gynecol Pathol 2019; 38 Suppl 1: S40–S63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Yang D, Cogdell D, Hu L, Xue F, Broaddus R et al. Genomic characterization of gene copy-number aberrations in endometrial carcinoma cell lines derived from endometrioid-type endometrial adenocarcinoma. Technol Cancer Res Treat 2010; 9: 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Creighton CJ, Cordero KE, Larios JM, Miller RS, Johnson MD, Chinnaiyan AM et al. Genes regulated by estrogen in breast tumor cells in vitro are similarly regulated in vivo in tumor xenografts and human breast tumors. Genome biology 2006; 7: R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong JW, Lee KY, Kwak I, White LD, Hilsenbeck SG, Lydon JP et al. Identification of murine uterine genes regulated in a ligand-dependent manner by the progesterone receptor. Endocrinology 2005; 146: 3490–3505. [DOI] [PubMed] [Google Scholar]
- 39.Kamat AA, Coffey D, Merritt WM, Nugent E, Urbauer D, Lin YG et al. EphA2 overexpression is associated with lack of hormone receptor expression and poor outcome in endometrial cancer. Cancer 2009; 115: 2684–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merritt WM, Kamat AA, Hwang JY, Bottsford-Miller J, Lu C, Lin YG et al. Clinical and biological impact of EphA2 overexpression and angiogenesis in endometrial cancer. Cancer Biol Ther 2010; 10: 1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ji H, Goode RJ, Vaillant F, Mathivanan S, Kapp EA, Mathias RA et al. Proteomic profiling of secretome and adherent plasma membranes from distinct mammary epithelial cell subpopulations. Proteomics 2011; 11: 4029–4039. [DOI] [PubMed] [Google Scholar]
- 42.Hussain NK, Thomas GM, Luo J, Huganir RL. Regulation of AMPA receptor subunit GluA1 surface expression by PAK3 phosphorylation. Proc Natl Acad Sci U S A 2015; 112: E5883–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nikhil K, Chang L, Viccaro K, Jacobsen M, McGuire C, Satapathy SR et al. Identification of LIMK2 as a therapeutic target in castration resistant prostate cancer. Cancer Lett 2019; 448: 182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mashiach-Farkash E, Rak R, Elad-Sfadia G, Haklai R, Carmeli S, Kloog Y et al. Computer-based identification of a novel LIMK1/2 inhibitor that synergizes with salirasib to destabilize the actin cytoskeleton. Oncotarget 2012; 3: 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bian Y, Guo J, Qiao L, Sun X. miR-3189–3p Mimics Enhance the Effects of S100A4 siRNA on the Inhibition of Proliferation and Migration of Gastric Cancer Cells by Targeting CFL2. Int J Mol Sci 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009; 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res 2018; 46: D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang HY, Lin YC, Li J, Huang KY, Shrestha S, Hong HC et al. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res 2020; 48: D148–D154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kriseman M, Monsivais D, Agno J, Masand RP, Creighton CJ, Matzuk MM. Uterine double-conditional inactivation of Smad2 and Smad3 in mice causes endometrial dysregulation, infertility, and uterine cancer. Proc Natl Acad Sci U S A 2019; 116: 3873–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monsivais D, Peng J, Kang Y, Matzuk MM. Activin-like kinase 5 (ALK5) inactivation in the mouse uterus results in metastatic endometrial carcinoma. Proc Natl Acad Sci U S A 2019; 116: 3883–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gennarino VA, Sardiello M, Avellino R, Meola N, Maselli V, Anand S et al. MicroRNA target prediction by expression analysis of host genes. Genome Res 2009; 19: 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aghajanova L, Velarde MC, Giudice LC. The progesterone receptor coactivator Hic-5 is involved in the pathophysiology of endometriosis. Endocrinology 2009; 150: 3863–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018; 173: 291–304 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017; 23: 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le Gallo M, Rudd ML, Urick ME, Hansen NF, Zhang S, Program NCS et al. Somatic mutation profiles of clear cell endometrial tumors revealed by whole exome and targeted gene sequencing. Cancer 2017; 123: 3261–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeLair DF, Burke KA, Selenica P, Lim RS, Scott SN, Middha S et al. The genetic landscape of endometrial clear cell carcinomas. The Journal of pathology 2017; 243: 230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clarke MA, Devesa SS, Harvey SV, Wentzensen N. Hysterectomy-Corrected Uterine Corpus Cancer Incidence Trends and Differences in Relative Survival Reveal Racial Disparities and Rising Rates of Nonendometrioid Cancers. J Clin Oncol 2019; 37: 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schultz KAP, Stewart DR, Kamihara J, Bauer AJ, Merideth MA, Stratton P et al. DICER1 Tumor Predisposition In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K et al (eds). GeneReviews((R)): Seattle (WA), 1993. [Google Scholar]
- 61.Bean GR, Anderson J, Sangoi AR, Krings G, Garg K. DICER1 mutations are frequent in mullerian adenosarcomas and are independent of rhabdomyosarcomatous differentiation. Mod Pathol 2019; 32: 280–289. [DOI] [PubMed] [Google Scholar]
- 62.de Kock L, Yoon JY, Apellaniz-Ruiz M, Pelletier D, McCluggage WG, Stewart CJR et al. Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod Pathol 2020; 33: 1207–1219. [DOI] [PubMed] [Google Scholar]
- 63.Schultz KAP, Rednam SP, Kamihara J, Doros L, Achatz MI, Wasserman JD et al. PTEN, DICER1, FH, and Their Associated Tumor Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res 2017; 23: e76–e82. [DOI] [PubMed] [Google Scholar]
- 64.Zhang B, Chen H, Zhang L, Dakhova O, Zhang Y, Lewis MT et al. A dosage-dependent pleiotropic role of Dicer in prostate cancer growth and metastasis. Oncogene 2014; 33: 3099–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev 2009; 23: 2700–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Munson PB, Hall EM, Farina NH, Pass HI, Shukla A. Exosomal miR-16–5p as a target for malignant mesothelioma. Sci Rep 2019; 9: 11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang E, Liu R, Chu Y. miRNA-15a/16: as tumor suppressors and more. Future Oncol 2015; 11: 2351–2363. [DOI] [PubMed] [Google Scholar]
- 68.Ihira K, Dong P, Xiong Y, Watari H, Konno Y, Hanley SJ et al. EZH2 inhibition suppresses endometrial cancer progression via miR-361/Twist axis. Oncotarget 2017; 8: 13509–13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ikeda Y, Oda K, Ishihara H, Wada-Hiraike O, Miyasaka A, Kashiyama T et al. Prognostic importance of CDK4/6-specific activity as a predictive marker for recurrence in patients with endometrial cancer, with or without adjuvant chemotherapy. Br J Cancer 2015; 113: 1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dosil MA, Mirantes C, Eritja N, Felip I, Navaridas R, Gatius S et al. Palbociclib has antitumour effects on Pten-deficient endometrial neoplasias. The Journal of pathology 2017; 242: 152–164. [DOI] [PubMed] [Google Scholar]
- 71.Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A 2012; 109: 3921–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van der Horst PH, van der Zee M, Heijmans-Antonissen C, Jia Y, DeMayo FJ, Lydon JP et al. A mouse model for endometrioid ovarian cancer arising from the distal oviduct. Int J Cancer 2014; 135: 1028–1037. [DOI] [PubMed] [Google Scholar]
- 73.Al-Maghrabi JA, Butt NS, Anfinan N, Sait K, Sait H, Marzouki A et al. Infrequent Immunohistochemical Expression of Napsin A in Endometrial Carcinomas. Appl Immunohistochem Mol Morphol 2017; 25: 632–638. [DOI] [PubMed] [Google Scholar]
- 74.Han G, Sidhu D, Duggan MA, Arseneau J, Cesari M, Clement PB et al. Reproducibility of histological cell type in high-grade endometrial carcinoma. Mod Pathol 2013; 26: 1594–1604. [DOI] [PubMed] [Google Scholar]
- 75.Zorn KK, Bonome T, Gangi L, Chandramouli GV, Awtrey CS, Gardner GJ et al. Gene expression profiles of serous, endometrioid, and clear cell subtypes of ovarian and endometrial cancer. Clin Cancer Res 2005; 11: 6422–6430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.