

“Progress lies not in enhancing what is, but in advancing toward what will be.”
- Khalil Gibran
Introduction
The treatment of metastatic melanoma has undergone a dramatic transformation over the past decade with the advent of molecular targeted therapy and immunotherapy. Today, 1 in 2 patients with metastatic are alive 5 years after diagnosis when treated with combination immunotherapy (Larkin et al., 2019) and over 1 in 3 patients are alive following years of combination BRAF/MEK targeted therapy (Robert et al., 2019) or single-agent PD-1 blockade (Hamid et al., 2019b). This is in contrast to 10 years ago, when metastatic melanoma was considered uniformly fatal with an overall survival <5% (Dickson and Gershenwald, 2011). Despite these advances, additional therapeutic approaches are needed for patients resistant to available targeted and immune treatments. Here, we review the currently approved systemic and local therapies for advanced melanoma and emphasize areas of uncertainty and unmet needs.
Melanoma in 2020: The Scope of the Problem
Melanoma arises from a malignant transformation of melanocytes, the cells throughout the body that synthesize melanin, a photoprotective pigment (Lo and Fisher, 2014). Melanoma can arise from pigment producing cells in the eye, the gastrointestinal tract, genitalia, sinuses, and meninges, but most commonly arises in the skin in the setting of ultraviolet (UV) injury. Melanoma is the 5th most common form of cancer in adults (men and women) and is the deadliest form of skin cancer (NCI-SEER-Database, 2019). The incidence of melanoma has been rising in the United States and worldwide (Karimkhani et al., 2017, Schadendorf et al., 2018), with an estimated 96,480 adults (57,220 men and 39,260 women) diagnosed with melanoma in the United States in 2019, accounting for 5.5% for all new cancer cases, and resulting in 7,230 deaths (1.2% of all cancer deaths).
Melanoma is categorized by TNM staging to define patients with local disease (stage I-II), node-positive disease (stage III), and advanced/metastatic disease (stage IV). Current staging utilizes the AJCC 8th Edition (Gershenwald et al., 2017). Tumor thickness (Breslow depth), presence/absence of ulceration, mitotic rate, presence/absence of microsatellites/in-transit lesions, burden of lymph node disease, and presence/absence of distant metastasis are the key clinico-pathologic features for assigning a stage and/or assessing risk of recurrence. The majority of cutaneous melanomas are localized at the time of initial clinical presentation and are successfully treated with surgical excision with adequate margins (Joyce and Skitzki, 2020).
Roughly half of cutaneous melanomas harbor oncogenic driver mutations in BRAF (Cancer Genome Atlas, 2015, Davies et al., 2002). RAS-RAF pathway alterations are frequently encountered in cutaneous melanoma with 40–50% of melanoma harboring BRAF mutations, 20–30% with NRAS mutations, and 10–15% with mutations in NF1 (Cancer Genome Atlas, 2015) (Fig. 1). BRAF V600E/K mutations are the most common (90%) abnormality in the BRAF gene. Non-V600E/K mutations have been observed, but the responsiveness to BRAF/MEK inhibition is less clear. At this time, the presence/absence of a BRAF V600E/K mutation is the primary actionable genomic data that influences eligibility for treatment. Other genomic alterations (e.g. mutations and/or amplifications in KIT) are observed in a proportion of melanoma patients and can be used to guide treatment with KIT tyrosine kinase inhibitors (Carvajal et al., 2011). Other clinico-pathologic features associated with response to immunotherapy (e.g. PD-L1 expression, tumor mutational burden, etc. – discussed below) are not sufficiently robust to drive clinical decision-making. Thus, TNM stage and presence/absence of a BRAF V600E/K mutation (for patients with stage III-IV) are the most crucial features used in determining eligibility for FDA-approved immunotherapy or targeted therapy options.
Fig. 1 – Dysregulation of the mitogen-activated protein kinase (MAPK) signaling pathway in melanoma.
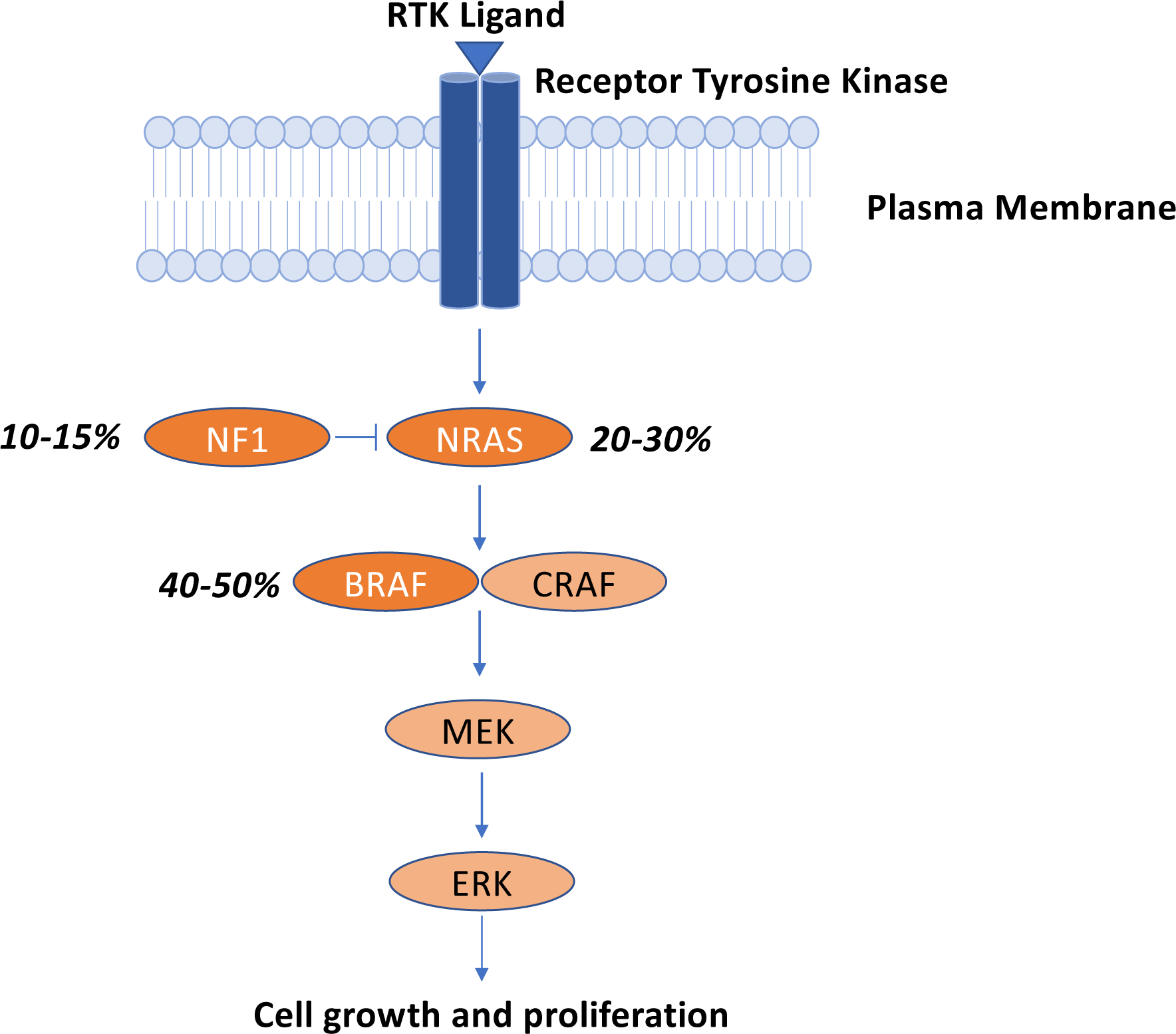
Over 80% of melanomas possess genetic abnormalities in at least one key node in the MAPK signaling pathway. Oncogenic driver mutations in BRAF (V600E or V600K) are the most common genomic abnormalities observed in cutaneous melanoma, followed by mutations in NRAS, and the RAS GTPase activating protein (GAP), neurofibromin 1 (NF1).
The melanoma oncologist’s toolkit in 2020
The last ten years have witnessed a dramatic evolution in the treatment of patients with unresectable or metastatic melanoma with development if immune checkpoint blockade strategies targeting the PD-1 and CTLA-4 co-inhibitory receptors and MAP kinase (MAPK) molecular targeted therapy directed at oncogenic BRAF and MEK signaling pathways. Both approaches have proven effective in the treatment of advanced melanoma. Prior to 2010, the only primary FDA-approved treatments for advanced melanoma were high-dose interleukin-2 (hdIL2) (Atkins et al., 1999) and dacarbazine (DTIC) (Luke and Schwartz, 2013). In 2010, the results of the first phase I trial of the BRAF inhibitor PLX4032 (vemurafenib) was published (Flaherty et al., 2010). That same year, the results of the first phase 3 trial of the anti-CTLA-4 monoclonal antibody, ipilimumab, were published demonstrating improved overall survival (Hodi et al., 2010).
Since 2010, nearly a dozen new treatments/treatment regimens for melanoma (Table 1), have been approved by the FDA, including 4 systemic immunotherapy treatments/combinations (ipilimumab, nivolumab, pembrolizumab, combination ipilimumab-nivolumab), single-agent BRAF inhibitors (vemurafenib, dabrafenib), combination BRAF-MEK inhibitor regimens (dabrafenib-trametinib, vemurafenib-cobimetinib, encorafenib-binimetinib), and 1 intra-lesional immunotherapy involving a modified oncolytic herpes virus (talimogene laherparepvec, T-VEC). These treatments gained their initial indications in advanced, unresectable melanoma, and several have since gained approval in the adjuvant setting. Here we will consider the mechanism of action, efficacy, and toxicity of each class of treatments. For a more comprehensive review of the history and clinical development of these and other therapies in melanoma, we refer readers to other reviews (Luke et al., 2017, Ribas and Wolchok, 2018).
Table 1.
Targeted and Immune Therapies for Melanoma
| Agent | Mechanism | FDA-approved indications |
|---|---|---|
| Targeted Therapies | ||
| Vemurafenib | BRAF inhibitor |
|
| Cobimetinib | MEK inhibitor |
|
| Dabrafenib + trametinib | BRAF inhibitor + MEK inhibitor |
|
| Vemurafenib + cobimetinib | BRAF inhibitor + MEK inhibitor |
|
| Encorafenib + binimetinib | BRAF inhibitor + MEK inhibitor |
|
| Immunotherapies | ||
| Ipilimumab | Anti-CTLA-4 monoclonal antibody |
|
| Nivolumab | Anti-PD-1 monoclonal antibody |
|
| Pembrolizumab | Anti-PD-1 monoclonal antibody |
|
| Ipilimumab-nivolumab | anti-CTLA-4 antibody + anti-PD-1 antibody |
|
| Talimogene laherpraepvec (T-VEC) | Modified, injectable oncolytic herpes virus | Local treatment of unresectable cutaneous, subcutaneous, and nodal lesions in patients with recurrent melanoma after surgery |
Immunotherapy.
Immune checkpoint proteins (e.g. PD-1 and CTLA-4) are co-inhibitory protein receptors expressed on the cell surface of lymphocytes whose primary physiologic role is to maintain self-tolerance and limit inflammatory responses in normal tissues (Keir et al., 2008, Pardoll, 2012). The cognate ligands for PD-1 and CTLA-4 (e.g. PD-L1/PD-L2 and B7, respectively) are expressed on tumor cells or other immune cells and serve to restrain T cell function (Buchbinder and Desai, 2016). In 2010, Hodi et al. published the results of the first phase III clinical trial demonstrating an improved overall survival in patients with metastatic melanoma treated with the anti-CTLA-4 antibody (Hodi et al., 2010). Pooled analysis of patients treated with ipilimumab from 1,861 patients across 12 trials demonstrated three-year survival rates of roughly 20%, at which time the survival curve plateaued supporting the durability of responses to CTLA-4 blockade (Schadendorf et al., 2015). In 2014, the initial results of the first trials of monoclonal antibodies targeting PD-1 were published, demonstrating clinical activity in melanoma, non-small cell lung cancer, and renal cell carcinoma (Brahmer et al., 2012, Topalian et al., 2012). Longer-term follow-up in melanoma patients demonstrates overall response rates for first-line anti-PD-1 treatment are even higher (30–40% at 5 years) with ongoing durable responses in 70–80% of responding patients (Hamid et al., 2019b).
Dual immune checkpoint blockade (ICB) with ipilimumab-nivolumab enhances response rates compared to single-agent ipilimumab or nivolumab in patients with metastatic melanoma (RR 58%) and both nivolumab containing arms demonstrated superior OS compared to single agent ipilimumab (Larkin et al., 2015). However, >50% of patients also experienced significant (grade 3/4) toxicity from dual ICB resulting in treatment interruption or discontinuation. Non-overlapping mechanisms of action may account for the differential clinical activity of these agents, as well as distinct toxicity profiles. Side effects from ICB therapy result from disruption of immunologic tolerance and manifest as immune-mediated reactions against healthy tissues, or so-called immune-related adverse events (irAE) (De Velasco et al., 2017). The onset of irAEs is variable and unpredictable, but the majority of grade 3/4 irAEs with ipi-nivo occur during the 12 week induction phase (Larkin et al., 2019). Importantly, PFS and OS is similar for patients who had to discontinue treatment due to irAEs compared to the overall population (Larkin et al., 2019). Five-year follow-up data confirms similar response and toxicity rates, with an impressive overall survival benefit: over half (52%) of patients in the ipi-nivo arms still alive after 5 years and impressive median treatment-free interval of 18.1 months underscoring the durability of these responses (Larkin et al., 2019). Despite these impressive data, it remains unclear which patients require dual ICB versus single-agent PD-1 blockade.
To date several putative biomarkers have shown associations with clinical response to PD-1 blockade, including pre-existing immune infiltrate (Tumeh et al., 2014), PD-L1 expression (Daud et al., 2016), and tumor mutational burden (TMB), although none of these are sufficiently robust that they drive clinical practice. Recently, integrative models incorporating clinical, genomic, and gene expression data have been developed that appear to be more robust than any of the individual features on their own (Liu et al., 2019). Given the lack of robust predictive biomarkers with a strong negative predictive value, the decision to treat with anti-PD-1 and/or anti-CTLA-4 monoclonal antibodies may be driven by patient characteristics, including age, comorbid medical conditions (including autoimmune conditions), and burden of metastatic disease, although data supporting these practices are limited. One setting in which dual ICB appears superior to single-agent anti-PD-1 treatment is in patients with asymptomatic brain metastases. Given the very robust intracranial responses observed with dual PD-1/CTLA-4 blockade in the phase II, single-arm Checkmate-204 trial (Tawbi et al., 2018), dual ICB is emerging as the preferred regimen for patients with asymptomatic brain metastases with an impressive 57% intracranial clinical benefit rate (compared to 56% extracranial benefit rate) and 26% complete response rate.
Another form of immunotherapy active in cutaneous melanoma is the injectable agent talimogene laherpraepvec (T-VEC, Imlygic). T-VEC is a modified herpes virus engineered to replicate in tumor cells to produce the growth factor, granulocyte-macrophage colony stimulating factor (GM-CSF) which facilitates immune infiltration and antigen-presentation to prime immune response (Kaufman et al., 2014). Based on the results of the phase III OPTiM trial, intralesional injection with T-VEC was associated with improved response rate compared to GM-CSF alone (26.4 vs 5.7%) (Andtbacka et al., 2015, Andtbacka et al., 2019). Injectable lesions were cutaneous and accessible lymph nodes. Importantly responses were observed in nearby and distant uninjected lesions suggesting an immune priming effect. T-VEC has been combined with ICB (Puzanov et al., 2016). A phase III trial evaluating pembrolizumab with and without T-VEC is underway (NCT02263508) and this strategy is also showing activity in soft tissue sarcoma (Kelly et al., 2020).
Targeted Therapy.
The era of molecular targeted therapy in cutaneous melanoma was ushered in following the discovery of BRAF mutations in several cancers including melanoma (Davies et al., 2002). This discovery led to the initial evaluation of BRAF inhibitors with initial trials showing 50% response rates as a single-agent in patients with metastatic melanoma (Chapman et al., 2011, Hauschild et al., 2012). Given the clinical activity of single-agent MEK inhibition (Flaherty et al., 2012), and appreciation of the importance of downstream MAPK pathway signaling, BRAF-MEK inhibitor combinations were subsequently evaluated. Dabrafenib-trametinib was the first BRAF-MEK combination approved for metastatic melanoma based of the two phase III clinical trials, COMBI-v (Robert et al., 2015) and COMBI-d (Long et al., 2015), comparing D+T to single-agent vemurafenib (V) or dabrafenib (D), respectively. D+T demonstrated response rates of 60–70% in the COMBI-v and COMBI-d trials, compared to 50% response rates with single-agent BRAF inhibitor. Furthermore, the toxicity profile of combination D+T differed from D and V, with more pyrexia with D+T, but a decreased incidence of keratoacanthoma and squamous cell carcinoma. The coBRIM trial evaluated combination vemurafenib (V) and cobimetinib (C) versus vemurafenib (and placebo) demonstrating improved ORR with V+C (70%) vs V alone (50%) (Larkin et al., 2014). The toxicity profile of V+C differs from D+T with more GI upset (diarrhea, nausea), fatigue, rash, liver enzyme abnormalities, photosensitivity (from vemurafenib), although less pyrexia compared to D+T. The randomized, phase III COLUMBUS trial evaluated a third BRAFi (encorafenib, E) and MEKi (binimetinib, B) versus vemurafenib (V) (Dummer et al., 2018) demonstrating a median PFS of 14.8 months versus 7.3 months.
The available BRAFi-MEKi combinations are comparable in terms of efficacy with response rates ranging from 60–70% and 18-month PFS rates of 30–40% (Dummer et al., 2018, Larkin et al., 2014, Long et al., 2015, Robert et al., 2015) with distinct toxicity profiles. Direct head-to-head comparison of the available regimens is unlikely to be performed, but indirect side-by-side analysis of data from V+C, D+T, E+B compared to vemurafenib monotherapy (Hamid et al., 2019a) revealed comparable PFS and OS data. Median OS was 33.6 months for patients treated with E+B compared to 25.6 months with D+T and 22.3 months with V+C, but direct comparison is not possible across trials. The availability of three approved BRAFi-MEKi regimens provides multiple treatment options for patients with stage IV BRAF-V600E/K mutant melanoma.
Combining immune therapy with targeted therapy.
Given the high response rates observed with targeted therapies and the durable responses observed with immunotherapies, the combination of these effective therapeutic strategies was a logical next step for patients with BRAF-mutant melanoma. Unfortunately, the initial experience combining ICB with targeted therapy proved more of a cautionary tale than a success story. Ribas and colleagues published their experience with patients treated with ipilimumab and vemurafenib with the observation of grade 3 hepatotoxicity (Ribas et al., 2013). However, after the subsequent success of BRAF-MEK combinations and PD-1 blockade, combination approaches were revisited. Backed by pre-clinical evidence of improved efficacy (Frederick et al., 2013, Hu-Lieskovan et al., 2015), several anti-PD-(L)1/BRAFi/MEKi triplet therapy combinations have been evaluated in early-phase clinical trials with response rates greater than 70% and comparable rates of grade 3/4 toxicity (Ascierto et al., 2019, Ribas et al., 2019, Sullivan et al., 2019). Whether these combination approaches, with/without alterations in dose/schedule of BRAF/MEK targeted therapy, provide an overall survival benefit compared to either PD-1 blockade or combination BRAF/MEK inhibition in a prospective fashion remains to be seen.
Frontline Treatment of Metastatic Melanoma in 2020.
For most patients with metastatic melanoma, immunotherapy with PD-1 +/− CTLA-4 blockade is the preferred 1st line regimen given the improved overall survival, response rate, and durability of response, which may allow patients to discontinue treatment. For patient unable to tolerate systemic immunotherapy, BRAF/MEK targeted therapy is active and associated with durable responses in 1/3 of patients, especially in patients with normal LDH and fewer than 3 different organ sites involved. Additionally, responses to BRAF/MEK inhibitor therapy are usually brisk and may provide more rapid disease control for patients requiring urgent treatment. Whether efficacy and/or tolerability of BRAF/MEK targeted therapy as a second-line treatment is comparable to the data from the front-line data of published phase III trials remains to be seen, but emerging retrospective data suggests tolerability and efficacy may be diminished following PD-1 blockade (Saab et al., 2019). Immunotherapy with PD-(L)1 blockade combined with BRAF/MEK targeted has demonstrated improved response rates in early phase clinic trials, but incidence of toxicity is increased. Phase III trials are ongoing evaluating anti-PD-(L)1-BRAFi-MEKi triplets, and other trials evaluating alternate dosing schedules and lead-ins are underway. Strategies for patients who have progressed on front-line treatments include clinical trials, most of which are evaluating treatment combinations.
Adjuvant Systemic Therapy for Resected Cutaneous Melanoma
For high-risk patients with resected melanoma, adjuvant systemic therapy is offered to reduce the risk of melanoma recurrence after surgery (Eggermont et al., 2018b). Patients with high risk stage II melanoma (i.e. those that are >4 mm thick, or >2 mm thick with ulceration, i.e. stage IIB-C) and patients with node-positive disease are at increased risk of recurrence and demonstrate worse melanoma-specific survival (Gershenwald et al., 2017). Prior to 2015, high-dose interferon alpha 2b (HDI) was the only approved therapy for adjuvant treatment of resected high-risk stage II and stage III melanoma, with a consistent albeit modest benefit in recurrence-free survival (RFS) and to a lesser extent an overall survival benefit (Ives et al., 2017, Kirkwood et al., 2004, Mocellin et al., 2013). In 2015, ipilimumab (anti-CTLA-4) was approved for the adjuvant treatment of all patients with stage III melanoma based on the results of EORTC 18071 (CA184–029) which demonstrated that high-dose ipilimumab (10mg/kg) given every three weeks for a total of four doses improved RFS compared to placebo (Eggermont et al., 2016). The results of E1609 demonstrated that standard dose (3mg/kg) ipilimumab improve RFS and OS compared to HDI (Tarhini et al., 2019).
In 2017, the Checkmate-238 study demonstrated improved RFS with nivolumab (anti-PD-1) compared to ipilimumab and with a more favorable toxicity profile (Weber et al., 2017). Comparable improvement in RFS was noted in the KEYNOTE-054 study, a randomized, placebo-controlled phase III trial of pembrolizumab (Eggermont et al., 2018a). Based on the results of these trials both nivolumab and pembrolizumab are approved for adjuvant treatment of patients with resected stage III melanoma and patients with resected, oligometastatic stage IV melanoma. The efficacy of combined BRAF/MEK inhibitor therapy has also been evaluated in the phase III COMBi-AD trial which demonstrated improved recurrence-free and overall survival compared to placebo for patients with BRAF V600E/K melanoma (Long et al., 2017). The results of this trial led to the approval of dabrafenib and trametinib for the adjuvant treatment of stage III melanoma. A recent exploratory biomarker analysis revealed increased tumor mutational burden (TMB) and/or interferon gamma gene signature may identify patients more likely to respond to adjuvant D+T (Dummer et al., 2020), although further validation is needed.
The adjuvant options and surgical management of melanoma in 2020 look quite different from ten years ago (Cohen and Buchbinder, 2019, Eggermont et al., 2018b). CLND is no longer standard of care for patients with positive SLNB and PD-1 mAb ICB treatment or BRAF/MEK inhibitor therapy are the new frontline options for adjuvant therapy. Despite these advances, several unanswered questions remain. First, for patients with stage III BRAF V600E/K melanoma, it is unclear if PD-1 blockade or BRAF/MEK inhibitor therapy is preferable. BRAF/MEKi inhibitor use is supported by a clear RFS and OS benefit, whereas PD-1 blockade with either nivolumab or pembrolizumab is supported only by RFS data currently. However, the 18-month RFS rates are comparable based on the available data (D+T is 67% compared with 66–71% for PD-1 blockade). While adjuvant PD-1 blockade is generally more tolerable with ~15% patients stopping therapy due to adverse events, compared to ~25% with D+T, the risk of severe or irreversible immune-related AEs (irAEs) with PD-1 blockade in patients with modest risk of recurrence can limit enthusiasm for this approach. Of note, all adjuvant trials to date (including COMBI-AD, KEYNOTE-054, and Checkmate-238) completion lymph node dissections were required before beginning adjuvant treatment. CLND is no longer standard of care (Faries et al., 2017) and omitting CLND in favor or adjuvant therapy is reasonable, effective, and spares patients significant risk of morbidity (Eggermont et al., 2018b). Risk stratification analysis of a cohort of 1009 patients with 15 year follow-up further demonstrated low risk of recurrence (9%) for patients with a single melanoma deposit measuring <0.1mm in a single sentinel lymph node compared to patient with deposits measuring 0.1–1.0mm (16%) and >1.0mm (25%) (van der Ploeg et al., 2011). A model incorporating primary tumor ulceration status and sentinel lymph node burden has been developed with a 1.0 mm threshold for distinguishing low/intermediate- versus high-risk patients (Verver et al., 2018).
For patients eligible for both adjuvant PD-1 blockade or BRAF/MEKi therapy deciding between these options is a challenge given the comparable improvement in RFS. In practice, factors influencing the choice between targeted therapy and ICB therapy incorporates stage and associated risk of recurrence, comorbid conditions, and side effect profile. For example, patients with stage IIIA melanoma (AJCC 8th Ed.) can consider BRAF/MEK inhibitor treatment if available or pursue surveillance, but rarely opt for adjuvant PD-1 blockade. Given the improved activity of ICB therapy compared to BRAF/MEKi targeted therapy in stage IV disease (discussed above), some clinicians favor PD-1 blockade in this patient population although the current data are inadequate to guide formal recommendations. Management of patients who recur after adjuvant therapy includes surgical resection of locoregional disease, alternative active agents, and clinical trials.
FUTURE DIRECTIONS AND UNANSWERED QUESTIONS
Despite the dramatic improvement in clinical outcomes over the past decade owing to immunotherapy and targeted therapy in melanoma, not all patients respond to approved systemic therapies. Extensive pre-clinical, translational, and clinical research is ongoing to better understand the mechanisms of response and resistance to current therapies, develop rational next-generation treatments (and combinations), and develop more sophisticated models of melanoma that will support further pre-clinical and translational research.
Biomarkers.
While progress has been made in the identification of features associated with response and resistance to cancer immunotherapy, more work is clearly needed to establish reliable predictors of long-term response to ICB therapy. Development of a robust biomarker or gene signature to predict response and/or resistance will likely require sophisticated, integrated multi-parameter analysis of genomic, transcriptomic, and clinical data, using matched patient samples obtained before and during ICB therapy in lesions that are responding and failing to respond to therapy.
The Role of the Tumor.
With the advent of gene editing techniques and technologies, several genome-scale or sub-genome scale CRISPR screens have been performed nominating novel genes and pathways that can render melanoma cells more sensitive immune attack (Manguso et al., 2017, Pan et al., 2018). To identify novel therapeutic targets in melanomas with impaired interferon γ (IFNγ) signaling, Vredevoogd and colleagues conducted a CRISPR screen using melanoma cell lines lacking the IFNGγ receptor (IFNGR1) (Vredevoogd et al., 2019). Such screens are valuable to sift through available targets and nominate druggable candidates for further exploration. It is important to note that the majority of these CRISPR screens to date evaluate the effect of gene deletion in tumor cells only, which cannot readily account for the effect of pharmacologic targeting of a given gene product in which tumor cells, immune cells, and stromal cells are all simultaneously engaged.
Another key feature of tumors, including melanoma, is the adaptation to treatment leading to resistance. Several mechanisms of resistance to BRAF/MEK targeted have been reported, largely involving bypass or alternative activation of the MAPK pathway (Lim et al., 2017). Recently, alterations in cell state have been associated with differential drug sensitivity to BRAF/MEK inhibition, and concurrently drug screening efforts have been directed at the cells and cell states resistant to BRAF/MEK inhibition (Eskiocak et al., 2017, Tsoi et al., 2018). Among these are epigenetic states characterized by low-MITF expression, usually associated with high expression of the EMT marker AXL. Resistance owing to altered melanoma cell states has been associated with minimal residual disease using patient-derived xenograft (PDX) models (Rambow et al., 2018). The interplay between altered cell states acquired in the short-term following BRAF+/− MEK inhibition and later accumulation of key mutations is incompletely understood, but may be related to alterations in multiple genes/pathways, associated with upregulation of error-prone polymerases and downregulation of DNA repair mechanisms (Russo et al., 2019).
The Role of the Immune System.
Successful anti-tumor immune responses following ICB presumably requires reactivation and clonal proliferation of antigen-experienced T-cells present in the tumor microenvironment (TME) (Pardoll, 2012). Recently, single-cell characterization of tumor-infiltrating immune cells has permitted comprehensive of immune cell populations and immune cell states within a given immune cell population. CD8 T cell states associated with response to PD-1 blockade include TCF7+ stem-like CD8 T cells with low expression of T cell dysfunction/exhaustion markers (Sade-Feldman et al., 2019). Orthogonal data using a murine melanoma model supported these findings and further demonstrated that ICB does not ‘reverse’ exhaustion of CD8 T cells (Miller et al., 2019). Whether terminally exhausted, dysfunctional CD8 T cells participate directly in the anti-tumor immune response remains an area of active investigation, but given the altered epigenetic state of dysfunctional CD8 T cells, these changes may be irreversible (Miller et al., 2019, Sen et al., 2016) and require recruitment of naïve immune cells to the tumor microenvironment. Interestingly, several recent papers described the presence of tumor-associated tertiary lymphoid structures (TA-TLS) and associated B cells with improve clinical outcomes in patients with melanoma (Cabrita et al., 2020, Helmink et al., 2020) and soft-tissue sarcoma (Petitprez et al., 2020). TA-TLS (also known as ectopic lymph nodes) may be a site of enhanced antigen-presentation where naïve lymphocytes can become antigen-experienced and primed for anti-tumor immune response following PD-1 blockade.
Another key question focuses on the identity of the tumor-targeted antigens. There is broad consensus that UV-induced tumor-specific neoantigens may be recognized by CD8 T cells, but the overall mutational burden is an imperfect predictor of clinical response. It is also notable that autoimmune vitiligo—with some distinctive clinical features—is commonly observed in patients experiencing major tumor regressions with ICB. While this could represent an epiphenomenon indicating overall immune activation by checkpoint inhibition, it is notable that vitiligo is virtually never seen when ICB is used to treat tumors other than melanoma. It thus remains to be seen whether melanocyte lineage antigens may also represent important functional immune targets in responding patients.
Pre-clinical cancer models.
Development of more sophisticated pre-clinical tumor models, including evaluation of patient-derived human samples capable of preserving features of the tumor immune microenvironment, and improved understanding of other extra-tumoral features that influence immune responses may facilitate and accelerate pre-clinical and translational research efforts (Friedman et al., 2015, Zitvogel et al., 2016). Patient-derived tumor models that preserve the immune contexture of the tumor microenvironment, including patient-derived organoids (Neal et al., 2018), patient-derived organotypic tumor spheroids (Jenkins et al., 2018a), as well as models in which tumor material is combined with peripheral immune cells (Dijkstra et al., 2018) have been described in recent years. Such models and assays have shown promise in liquid malignancies (Tyner et al., 2018) and more recently with solid tumors and conventional cytotoxic chemotherapy (Ooft et al., 2019, Vlachogiannis et al., 2018). Novel, function precision medicine approaches may be ideally suited to identify specific therapies, or therapeutic combinations, to optimize clinical activity and durability of clinical response for individual patients (Smyth et al., 2016, Spranger and Gajewski, 2013).
Next-generation therapies and combination approaches.
Clinical trials are already underway evaluating novel immune modulatory agents in combination with anti-PD-1/PD-L1 therapies in an effort to overcome innate resistance (Jenkins et al., 2018b, O’Donnell et al., 2017, Sharma et al., 2017). Despite increasing reports of ‘rational’ combination strategies, these therapies remain “one size fits all”, due to the lack of robust biomarkers to guide clinical decision-making. Recently, the results of two phase III trials comparing novel, promising combination strategies were reported. The combination of the IDO inhibitor epacadostat with pembrolizumab showed no survival benefit compared with single-agent PD-1 blockade in the ECHO-301/KEYNOTE-252 phase III, placebo-controlled, randomized clinical trial (Long et al., 2019). The results of the phase III IMPSIRE 170 trial comparing combination treatment with cobimetinib (MEKi) and atezolizumab (anti-PD-L1 mAb) versus pembrolizumab (anti-PD-1) are not yet published, but presentation at the ESMO 2019 meeting indicated the atezolizumab-cobimetinib combination failed to meet its primary endpoint in patients with BRAF-wild-type melanoma (A.M. Arance, 2019). With these recent high profile negative trials and the expanding number of combination trials (Tang et al., 2018), there is renewed focus on the pre-clinical and early-phase clinical development of combination strategies.
CONCLUSION
Advances in molecular targeted therapy and immune checkpoint inhibition (ICI) have led to unprecedented improvement in overall survival for patients with advanced melanoma. Single-agent PD-1 blockade and combination BRAF/MEK inhibitor therapy have both demonstrated long-term, 5-year overall survival benefit of 30–40%. Superior response rates have been demonstrated with combined PD-1/CTLA-4 blockade, with a numerically higher although not statistically significant OS benefit compared to single-agent PD-1 blockade. BRAF/MEK therapy and PD-1 blockade have supplanted HDI and ipilimumab as the preferred adjuvant treatment options for patients with stage III melanoma. Intense investigation is ongoing to identify effective treatment strategies for patients for whom ICB therapy and/or BRAF/MEK targeted therapy are ineffective. Given the durability of responses observed in patients successfully treated with ICB therapy, the vast majority of current melanoma clinical trials include an ICB ‘backbone’ in an effort to match the high response rates seen with some targeted therapy responses with the durability of responses evidence with cancer immunotherapy. Given the recent failures of several initially high-profile phase III combination immunotherapy trials coupled with the ever-increasing number of novel therapies and combination trials, there is an unmet need for novel approaches, tools, techniques, and methods for pre-clinical evaluation to better understand mechanisms of response and resistance to immune checkpoint inhibitors and next-generation anti-tumor immune modulatory drugs (O’Donnell et al., 2017, Sharma et al., 2017). Just as the advances of 2010 (BRAF targeted therapy, CTLA-4 blockade) represented new treatment paradigms rather than incremental improvement over the standard of care, progress over the next decade will likely require more than simply “enhancing what is” (e.g. anti-PD-1 therapy), and focusing on “advancing toward what will be” with an eye on overcoming resistance, novel biomarker strategies, and advances in precision functional medicine.
Fig. 2 –
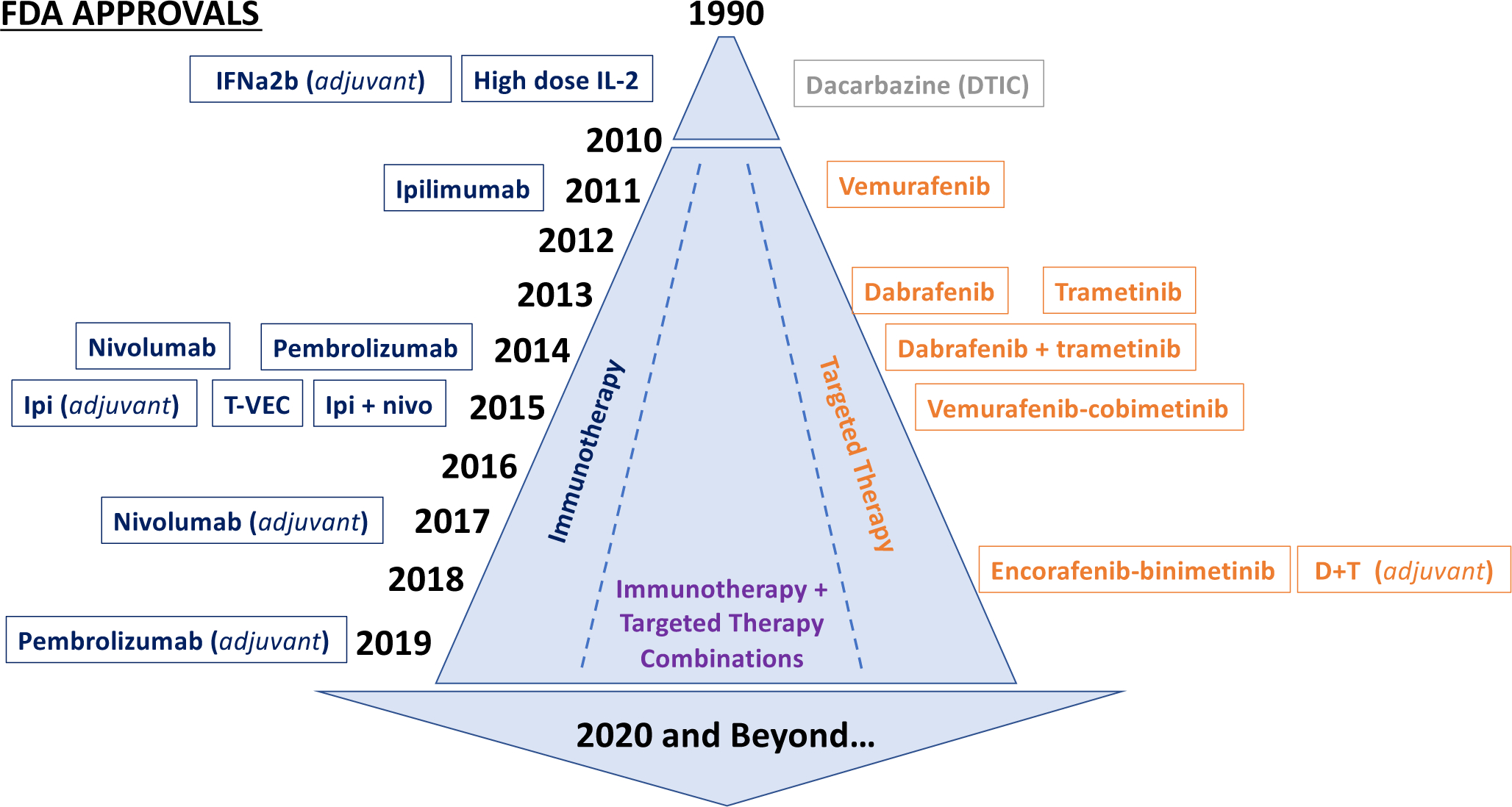
Timeline of FDA-approved therapies for melanoma.
Acknowledgements:
Dr. Jenkins is supported by the National Cancer Institute (1K08CA226391), the Melanoma Research Alliance, the Karin Grunebaum Cancer Research Foundation, the V Foundation, and the Henri and Belinda Termeer Fund for Early Career Investigators in Systems Pharmacology.
Dr. Fisher is supported by grants from NIH: 2P01 CA163222-06; 5R01 AR043369-23; 5R01CA222871-03; 5R01AR072304-03, and a grant from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
Abbreviations:
- HDI
high dose interferon α2b therapy
- PD-1
programmed cell death-1 (PDCD1
- PD-L1
programmed cell death ligand-1 (CD274)
- CTLA-4
cytotoxic T lymphocyte antigen-4 (CTLA4)
- T-VEC
talimogene laherpraepvec
- RR
response rate
- PFS
progression-free survival
- OS
overall survival
Footnotes
Conflict of Interest Statement:
RWJ - Advisory Board: XSphera Biosciences. Research support: Monopteros Therapeutics
DEF- Board of Directors and Consultant: Soltego Inc
DEF has a financial interest in Soltego, Inc., a company developing SIK inhibitors for topical skin darkening treatments that might be used for a broad set of human applications. Dr. Fisher’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies.
RWJ has a financial interest in XSphera Biosciences Inc., a company focused on using ex vivo profiling technology to deliver functional, precision immune-oncology solutions for patients, providers, and drug development companies. RWJ’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies.
References
- Arance HG AM, Dreno B, Flaherty KT, Demidov L, Stroyakovskiy D, Eroglu Z, Ferrucci PF, Pigozzo J, Rutkowski P, Mackiewicz J, Rooney I, Voulgari A, Troutman S, Pitcher B, Yan Y, Larkin JMG. COMBINATION TREATMENT WITH COBIMETINIB (C) AND ATEZOLIZUMAB (A) VS PEMBROLIZUMAB (P) IN PREVIOUSLY UNTREATED PATIENTS (PTS) WITH BRAFV600 WILD TYPE (WT) ADVANCED MELANOMA: PRIMARY ANALYSIS FROM THE PHASE 3 IMSPIRE170 TRIAL, https://oncologypro.esmo.org/meeting-resources/esmo-2019-congress/Combination-treatment-with-cobimetinib-C-and-atezolizumab-A-vs-pembrolizumab-P-in-previously-untreated-patients-pts-with-BRAFV600-wild-type-wt-advanced-melanoma-primary-analysis-from-the-phase-3-IMspire170-trial; 2019. [accessed 30 (suppl_5): v851–v934. 10.1093/annonc/mdz394]. [DOI] [Google Scholar]
- Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 2015;33(25):2780–8. [DOI] [PubMed] [Google Scholar]
- Andtbacka RHI, Collichio F, Harrington KJ, Middleton MR, Downey G, hrling K, et al. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J Immunother Cancer 2019;7(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med 2019;25(6):941–6. [DOI] [PubMed] [Google Scholar]
- Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999;17(7):2105–16. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366(26):2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol 2016;39(1):98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020;577(7791):561–5. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N Genomic Classification of Cutaneous Melanoma. Cell 2015;161(7):1681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011;305(22):2327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364(26):2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JV, Buchbinder EI. The Evolution of Adjuvant Therapy for Melanoma. Curr Oncol Rep 2019;21(12):106. [DOI] [PubMed] [Google Scholar]
- Daud AI, Wolchok JD, Robert C, Hwu WJ, Weber JS, Ribas A, et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J Clin Oncol 2016;34(34):4102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417(6892):949–54. [DOI] [PubMed] [Google Scholar]
- De Velasco G, Je Y, Bosse D, Awad MM, Ott PA, Moreira RB, et al. Comprehensive Meta-analysis of Key Immune-Related Adverse Events from CTLA-4 and PD-1/PD-L1 Inhibitors in Cancer Patients. Cancer Immunol Res 2017;5(4):312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson PV, Gershenwald JE. Staging and prognosis of cutaneous melanoma. Surg Oncol Clin N Am 2011;20(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018;174(6):1586–98 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2018;19(5):603–15. [DOI] [PubMed] [Google Scholar]
- Dummer R, Brase JC, Garrett J, Campbell CD, Gasal E, Squires M, et al. Adjuvant dabrafenib plus trametinib versus placebo in patients with resected, BRAF(V600)-mutant, stage III melanoma (COMBI-AD): exploratory biomarker analyses from a randomised, phase 3 trial. Lancet Oncol 2020. [DOI] [PubMed] [Google Scholar]
- Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, et al. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N Engl J Med 2016;375(19):1845–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N Engl J Med 2018a;378(19):1789–801. [DOI] [PubMed] [Google Scholar]
- Eggermont AMM, Robert C, Ribas A. The new era of adjuvant therapies for melanoma. Nat Rev Clin Oncol 2018b;15(9):535–6. [DOI] [PubMed] [Google Scholar]
- Eskiocak B, McMillan EA, Mendiratta S, Kollipara RK, Zhang H, Humphries CG, et al. Biomarker Accessible and Chemically Addressable Mechanistic Subtypes of BRAF Melanoma. Cancer Discov 2017;7(8):832–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faries MB, Thompson JF, Cochran AJ, Andtbacka RH, Mozzillo N, Zager JS, et al. Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N Engl J Med 2017;376(23):2211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363(9):809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367(2):107–14. [DOI] [PubMed] [Google Scholar]
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19(5):1225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer 2015;15(12):747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long GV, Ross MI, et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin 2017;67(6):472–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Cowey CL, Offner M, Faries M, Carvajal RD. Efficacy, Safety, and Tolerability of Approved Combination BRAF and MEK Inhibitor Regimens for BRAF-Mutant Melanoma. Cancers (Basel) 2019a;11(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol 2019b;30(4):582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380(9839):358–65. [DOI] [PubMed] [Google Scholar]
- Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020;577(7791):549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363(8):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med 2015;7(279):279ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ives NJ, Suciu S, Eggermont AMM, Kirkwood J, Lorigan P, Markovic SN, et al. Adjuvant interferon-alpha for the treatment of high-risk melanoma: An individual patient data meta-analysis. Eur J Cancer 2017;82:171–83. [DOI] [PubMed] [Google Scholar]
- Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 2018a;8(2):196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 2018b;118(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce D, Skitzki JJ. Surgical Management of Primary Cutaneous Melanoma. Surg Clin North Am 2020;100(1):61–70. [DOI] [PubMed] [Google Scholar]
- Karimkhani C, Green AC, Nijsten T, Weinstock MA, Dellavalle RP, Naghavi M, et al. The global burden of melanoma: results from the Global Burden of Disease Study 2015. Br J Dermatol 2017;177(1):134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman HL, Ruby CE, Hughes T, Slingluff CL, Jr. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J Immunother Cancer 2014;2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CM, Antonescu CR, Bowler T, Munhoz R, Chi P, Dickson MA, et al. Objective Response Rate Among Patients With Locally Advanced or Metastatic Sarcoma Treated With Talimogene Laherparepvec in Combination With Pembrolizumab: A Phase 2 Clinical Trial. JAMA Oncol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood JM, Manola J, Ibrahim J, Sondak V, Ernstoff MS, Rao U, et al. A pooled analysis of eastern cooperative oncology group and intergroup trials of adjuvant high-dose interferon for melanoma. Clin Cancer Res 2004;10(5):1670–7. [DOI] [PubMed] [Google Scholar]
- Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371(20):1867–76. [DOI] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2019;381(16):1535–46. [DOI] [PubMed] [Google Scholar]
- Lim SY, Menzies AM, Rizos H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017;123(S11):2118–29. [DOI] [PubMed] [Google Scholar]
- Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Amon L, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 2019;25(12):1916–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo JA, Fisher DE. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science 2014;346(6212):945–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol 2019;20(8):1083–97. [DOI] [PubMed] [Google Scholar]
- Long GV, Hauschild A, Santinami M, Atkinson V, Mandala M, Chiarion-Sileni V, et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N Engl J Med 2017;377(19):1813–23. [DOI] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015;386(9992):444–51. [DOI] [PubMed] [Google Scholar]
- Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol 2017;14(8):463–82. [DOI] [PubMed] [Google Scholar]
- Luke JJ, Schwartz GK. Chemotherapy in the management of advanced cutaneous malignant melanoma. Clin Dermatol 2013;31(3):290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017;547(7664):413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 2019;20(3):326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocellin S, Lens MB, Pasquali S, Pilati P, Chiarion Sileni V. Interferon alpha for the adjuvant treatment of cutaneous melanoma. Cochrane Database Syst Rev 2013(6):CD008955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCI-SEER-Database. Cancer Stat Facts: Melanoma of the Skin. 2019.
- Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018;175(7):1972–88 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev 2017;52:71–81. [DOI] [PubMed] [Google Scholar]
- Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, van Werkhoven E, et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci Transl Med 2019;11(513). [DOI] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018;359(6377):770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12(4):252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020;577(7791):556–60. [DOI] [PubMed] [Google Scholar]
- Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J Clin Oncol 2016;34(22):2619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018;174(4):843–55 e19. [DOI] [PubMed] [Google Scholar]
- Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 2013;368(14):1365–6. [DOI] [PubMed] [Google Scholar]
- Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr., Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 2019;25(6):936–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359(6382):1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med 2019;381(7):626–36. [DOI] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372(1):30–9. [DOI] [PubMed] [Google Scholar]
- Russo M, Crisafulli G, Sogari A, Reilly NM, Arena S, Lamba S, et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019;366(6472):1473–80. [DOI] [PubMed] [Google Scholar]
- Saab KR, Mooradian MJ, Wang DY, Chon J, Xia CY, Bialczak A, et al. Tolerance and efficacy of BRAF plus MEK inhibition in patients with melanoma who previously have received programmed cell death protein 1-based therapy. Cancer 2019;125(6):884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2019;176(1–2):404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol 2015;33(17):1889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, et al. Melanoma. Lancet 2018;392(10151):971–84. [DOI] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science 2016;354(6316):1165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168(4):707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol 2016;13(3):143–58. [DOI] [PubMed] [Google Scholar]
- Spranger S, Gajewski T. Rational combinations of immunotherapeutics that target discrete pathways. J Immunother Cancer 2013;1:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat Med 2019;25(6):929–35. [DOI] [PubMed] [Google Scholar]
- Tang J, Shalabi A, Hubbard-Lucey VM. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol 2018;29(1):84–91. [DOI] [PubMed] [Google Scholar]
- Tarhini AA, Lee SJ, Hodi FS, Rao UNM, Cohen GI, Hamid O, et al. Phase III Study of Adjuvant Ipilimumab (3 or 10 mg/kg) Versus High-Dose Interferon Alfa-2b for Resected High-Risk Melanoma: North American Intergroup E1609. J Clin Oncol 2019:JCO1901381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N Engl J Med 2018;379(8):722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366(26):2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018;33(5):890–904 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018;562(7728):526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ploeg AP, van Akkooi AC, Rutkowski P, Nowecki ZI, Michej W, Mitra A, et al. Prognosis in patients with sentinel node-positive melanoma is accurately defined by the combined Rotterdam tumor load and Dewar topography criteria. J Clin Oncol 2011;29(16):2206–14. [DOI] [PubMed] [Google Scholar]
- Verver D, van Klaveren D, van Akkooi ACJ, Rutkowski P, Powell B, Robert C, et al. Risk stratification of sentinel node-positive melanoma patients defines surgical management and adjuvant therapy treatment considerations. Eur J Cancer 2018;96:25–33. [DOI] [PubMed] [Google Scholar]
- Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018;359(6378):920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 2019;178(3):585–99 e15. [DOI] [PubMed] [Google Scholar]
- Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N Engl J Med 2017;377(19):1824–35. [DOI] [PubMed] [Google Scholar]
- Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer 2016;16(12):759–73. [DOI] [PubMed] [Google Scholar]