

Abstract
Cancer can metastasize from early lesions without detectable tumors. Despite extensive studies on metastasis in cancer cells from patients with detectable primary tumors, mechanisms for early metastatic dissemination are poorly understood. Her2 promotes breast cancer early dissemination by inhibiting p38, but the downstream pathway in this process was unknown. Using early lesion breast cancer models, we demonstrate that the effect of p38 suppression by Her2 on early dissemination is mediated by MK2 and Hsp27. The early disseminating cells in the MMTV-Her2 breast cancer model are Her2highp-p38lowp-MK2lowp-Hsp27low, which also exist in human breast carcinoma tissues. Suppression of p38 and MK2 by Her2 reduces MK2-mediated Hsp27 phosphorylation, and unphosphorylated Hsp27 binds to β-catenin and enhances its phosphorylation by Src, leading to β-catenin activation and disseminating phenotypes in early lesion breast cancer cells. Pharmacological inhibition of MK2 promotes, while inhibition of a p38 phosphatase Wip1 suppresses, early dissemination in vivo. These findings identify Her2-mediated suppression of the p38-MK2-Hsp27 pathway as a novel mechanism for cancer early dissemination, and provide a basis for new therapies targeting early metastatic dissemination in Her2+ breast cancer.
Keywords: MK2, Hsp27, Wip1, early dissemination, Her2+ breast cancer
Introduction
Metastasis is the leading cause of breast cancer death[1] and was classically viewed as the final step of cancer progression. This concept was challenged by clinical observations. In many cases, although the tumors are very small and no metastasis is detected at the time of resection, metastasis occurs years later[2]. Also, 4–5% of breast cancer patients with metastasis had unknown primary tumors[3]. It was thus recently proposed that metastasis is an early step in multiple cancers such as breast cancer, melanoma and pancreatic cancer[4–6]. While the pathways mediating metastasis have been studied extensively in established cancer cell lines, it is unclear whether the mechanisms derived from these cells isolated from patients with detectable primary tumors are also responsible for metastatic dissemination in early lesion cancer cells. Only a few studies have been focused on the mechanisms of cancer cell early dissemination.
Her2 is an oncogene amplified in 20% of breast cancer and associated with poor prognosis[7, 8]. In the MMTV-Her2 mouse model expressing rat neu (Erbb2) under the control of the mouse mammary tumor virus (MMTV) promoter/enhancer, 50% of female mice develop mammary tumors within 205 days on average, and 72% of tumor bearing mice develop lung metastasis at over 8-month of age[9]. In this model, tumor cells disseminate systemically from early epithelial lesions (hyperplasia and intraepithelial neoplasia) when the mammary glands have normal ductal structures[4, 10]. The early disseminated cancer cells (eDCCs) are Her2+p-p38lowp-ATF2lowTwisthighE-cadlow with an active epithelial-mesenchymal transition (EMT)-like program and can spread to target organs[11]. Moreover, p38 suppresses metastatic dissemination in early breast cancer lesions, and Her2 promotes breast cancer early dissemination by inhibiting p38[11]. However, it is unclear what acts downstream of p38 in this process.
EMT, characterized by loss of cell-cell junctions and increased cell motility, is one of the important steps for cancer cells of epithelial origins to disseminate from primary tumors and colonize the target tissues[12]. In normal epithelial cells, the cell-cell junctions are maintained by the E-cadherin-β-catenin-α-catenin complex[13]. Tyr654 of β-catenin is essential for its binding to E-cadherin[14, 15]. Phosphorylation of Tyr654 by Src results in dissociation of β-catenin from membrane-bound E-cadherin, leading to disruption of E-cadherin junctions, EMT and cell motility[15]. β-catenin released from the membrane upon Src phosphorylation is translocated into nucleus where it activates expression of the target genes, including Slug and Zeb1 that suppress E-cadherin expression[16–19]. Reduced E-cadherin expression further disrupts cell-cell junctions and leads to nuclear translocation of more β-catenin, resulting in a positive feedback loop[17, 20].
p38 is a MAPK that regulates many cellular processes. The 4 isoforms (α, β, γ and δ) of p38 are activated through phosphorylation by MAPK kinases (MAP2Ks), MKK3 and MKK6[21], and have multiple substrates including Ser/Thr protein kinases such as MK2, PRAK, and MNK1 and transcription factors such as ATF2[22]. MK2 and PRAK can phosphorylate heat shock protein 27 (Hsp27)[23, 24]. Increased Hsp27 expression contributes to malignant progression of multiple cancers[23, 25, 26]. In prostate cancer cells, Hsp27 interacts with β-catenin leading to reduced binding of β-catenin to GSK3β and stabilization of β-catenin protein; Hsp27 is required for EGF-induced β-catenin phosphorylation at Tyr654, a site phosphorylated by Src, both of which contribute to EMT[27]. Nevertheless, the effect of phosphorylation status on the tumor promoting function of Hsp27 remains unclear.
The p38 pathway can be either tumor-promoting or tumor-suppressing depending on the context. Based on studies in established cancer cell lines isolated from patients with palpable primary tumors, p38 activation promotes migration and invasion[28, 29]. However, active p38 is essential for tumor-suppressing mechanisms such as oncogene-induced senescence, contact inhibition and DNA damage responses[30]. A recent study demonstrated that p38 suppresses breast cancer cell early dissemination[11]. Thus, p38 may play differential roles in metastatic dissemination of early lesion tumors and that of detectable primary tumors at later stages.
Wild type p53-induced phosphatase (Wip1, or PPM1D) is a type 2C serine/threonine protein phosphatase that inactivates p38, but not the other MAPKs, through direct dephosphorylation of Thr182, one of the dual phosphorylation sites required for activation[31, 32]. Wip1 is an oncogene overexpressed in multiple cancers, including breast cancer[33]. Transgenic Wip1 overexpression promotes late stage mammary carcinoma development by suppressing p38 in the MMTV-Her2 mouse model[34]. Besides p38, Wip1 also dephosphorylates p53, MDM2 and DNA damage response mediators (ATM, Chk1, Chk2) to impact cancer cell proliferation and drug resistance[35]. GSK2830371 is the first orally active Wip1 inhibitor[36] that inhibits tumor growth and sensitizes cancer cells to chemotherapy by reactivating p53 through restoration of p53 phosphorylation[37–39]. Neither Wip1 nor Wip1 inhibitors have been previously tested on the ability of p38 to suppress cancer cell early dissemination.
While p38 has been linked to suppression of early dissemination of breast cancer cells[11], it is unclear what p38 downstream pathway mediates this process and whether the p38 pathway can be targeted for blocking early dissemination and metastasis of breast cancer. Here, we demonstrate using cell and mouse models for early lesion breast cancer that Her2 induces disseminating phenotypes in early lesion breast cancer cells by suppressing the p38-MK2-Hsp27 pathway, and that unphosphorylated Hsp27, accumulated in Her2+ cells, binds to β-catenin and promotes its interaction with and phosphorylation by Src, leading to loss of cell-cell junctions, EMT and disseminating phenotypes. Furthermore, GSK2830371 activates the p38-MK2-Hsp27 pathway, suppresses the disseminating phenotypes in early lesion breast cancer cells, and reduces early dissemination of breast cancer in vivo. These studies have provided new insights into the mechanism of cancer early dissemination, and proof-of-principle for a novel therapeutic strategy that blocks early dissemination by targeting the p38 pathway.
Results
Her2 suppresses phosphorylation of p38 downstream effectors MK2 and Hsp27 in cell and mouse models of breast cancer early lesions and human breast cancer tissues
To investigate the p38 downstream effectors involved in Her2-mediated early dissemination, we surveyed the effect of Her2 on the activation of p38 substrates in normal MCF-10A mammary epithelial cells transduced with Her2 and mammary epithelial cells isolated from MMTV-Her2 mice. These cells were used as models of early lesion breast cancer cells since there is no early lesion breast cancer cell line available that naturally overexpresses Her2 and all the existing Her2+ breast cancer cell lines were derived from patients with detectable primary tumors and thus are unlikely to represent early lesion cancer cells. The levels of ectopic Her2 expression in these cells were comparable to or less than that in a naturally occurring Her2+ breast cancer cell line SK-BR-3 (Fig. S1A), which rule out the possibility that the effects of Her2 observed in these cells are artifacts of unnaturally high Her2 expression. Her2 expression was barely detectable in normal control breast epithelial cells and a Her2− breast cancer cell line MDA-MB-231.
Besides ATF2, phosphorylation of MK2 at Thr222/334 (equivalent to T208/320 in mouse) and Hsp27 (at Ser15/78/82 in human and Ser15/86 in mouse), but not PRAK, was also downregulated in early lesion MMTV-Her2 cells (Fig. 1A, S1B) and MCF-10A cells transduced with Her2 (Fig. 1B), as compared to respective control cells. Her2 also reduced the levels of E-cadherin, an epithelial cell marker, and increased that of Vimentin, a mesenchymal cell marker, in MMTV-Her2 and MCF-10A cells (Fig. 1A–B), consistent with its ability to induce EMT and disseminating phenotypes.
Figure 1. Her2 suppresses phosphorylation of MK2 and Hsp27 in mammary epithelial cells and early lesions of MMTV-Her2 mouse mammary glands.

(A-B) Western blot analysis of mouse mammary epithelial cells isolated from wild type FVB mice (FVB) and MMTV-Her2 mice with early lesion (A) or MCF-10A cells transduced with vector (LXSN) or Her2 (B), detecting the status of the p38 pathway and E-cadherin, Vimentin expression.
(C) Mammary glands from 14–18-week-old MMTV-Her2 mice were co-stained for Her2 and p-MK2 (T320) (top) or p-Hsp27 (S86) (bottom) by immunofluorescence. Representative images are shown on the left with white arrows indicating Her2highp-MK2/p-Hsp27low and yellow arrows indicating Her2lowp-MK2/p-Hsp27high cells. Percentage (means±SD) of Her2high cells that were p-MK2/p-Hsp27high or p-MK2/p-Hsp27low were quantified (right), n=20 ducts from 2 mice. At least 4–25 Her2high cells were counted/duct.
(D) Mammary glands from 14–18-week-old MMTV-Her2 mice were co-stained for Her2 and p-MK2 (T320) (top) or p-Hsp27 (S86) (bottom) by immunofluorescence. Representative images with invading cells are shown with white arrows indicating Her2high-p-MK2/p-Hsp27low invading cells. Numbers on the right indicate percentage (means±SD) of Her2+p-MK2/p-Hsp27lowinvading cells in early lesions, n=2 mice. At least 50 Her2+ cells in the invading structures were counted/mice.
(E) Representative images of lung tissues from 14–18-week-old MMTV-Her2 mice co-stained for Her2 and p-MK2 (T320) (top) or p-Hsp27 (S86) (bottom) by immunofluorescence.
(F) Mammary glands from 14–18-week-old MMTV-Her2 mice were co-stained for E-cadherin and p-MK2 (T320) (top) or p-Hsp27 (S86) (bottom) by immunofluorescence. Representative images are shown on the left with white arrows indicating E-cadherinhighp-MK2/p-Hsp27high and yellow arrows indicating E-cadherinlow p-MK2/p-Hsp27low cells. Percentage (means±SD) of E-cadherinlow cells that were p-MK2/p-Hsp27low or p-MK2/p-Hsp27high were quantified (right), n=20 ducts from 2 mice. At least 4–35 E-cadherinlow cells were counted/duct.
(C, F) *** p<0.001 vs indicated controls in Mann-Whitney U-test.
We examined the levels of phosphorylated MK2 and Hsp27 in 14–18-week-old MMTV-Her2 mice. In contrast to 26-week-old mice that had developed primary tumors, those at 14–18-weeks had no detectable tumors and retained normal mammary ductal architecture, but had developed early lesions in mammary ducts (Fig. S1C) and early dissemination of Her2+ cells to the lung (Fig. S1D–E). Majority of the Her2high cells in the MMTV-Her2 mammary ducts (Fig. 1C), the Her2high invading cells in the MMTV-Her2 early lesions (Fig. 1D), and essentially all the Her2+ cells disseminated to the lung (Fig. 1E) contained very low/undetectable levels of p-MK2 and p-Hsp27. Most of the p-MK2low and p-Hsp27low (Fig. 1F) cells were also low for E-cadherin in MMTV-Her2 ducts, suggesting that these cells had an EMT and disseminating phenotype. Thus, the Her2high early disseminating cells are p-MK2lowp-Hsp27lowE-cadherinlow in the MMTV-Her2 mice. Thus, the Her2high/p-p38low/p-MK2low/p-Hsp27low cells likely contribute to early dissemination of breast cancer.
We investigated the relationship between Her2 status and the p38-MK2-Hsp27 pathway in early lesions of human breast cancer using tissue microarrays containing 16 Her2− and 12 Her2+ cases of ductal intraepithelial neoplasia (DIN) and in invasive human breast carcinoma using human breast carcinoma tissue microarrays containing 61 Her2− and 73 Her2+ cases. The Her2+ status correlated with low levels of p-p38, p-MK2 and p-Hsp27 in both early and advanced lesions (Fig. S2A–D, S2H–J). There were also positive correlations among the levels of p-p38, p-MK2 and p-Hsp27 (Fig. S2E–G, S2K–M). Thus, the Her2+p-p38lowp-MK2lowp-Hsp27low cells exist in early lesions of human breast cancer and likely contribute to the early dissemination of the cancer, and that these cells are preserved in the late stage human breast carcinoma tissues.
Her2 promotes the disseminating phenotypes in early lesion breast cancer cells through inhibition of the p38-MK2-Hsp27 pathway.
The suppression of MK2 and Hsp27 phosphorylation by Her2 in early disseminating lesions of breast cancer prompted us investigate their roles in Her2-mediated disseminating phenotypes.
Consistent with an early report[11], a p38 inhibitor SB203580, which inhibited MK2 and Hsp27 phosphorylation, reduced the levels of E-cadherin expression and junctions and increased transwell migration and percentage of organoids with outward invading cells in MCF-10A and MCF-10A-Her2 cells (Fig. S3A–D), and increased the percentage of invasive organoids in early breast cancer cells from MMTV-Her2 mice (Fig. S3E), as compared to vehicle control. p53 and its downstream targets were reported to remain wild type in MCF-10A-Her2 cells and early lesions of MMTV-Her2 mice, or even display increased activity in MMTV-Her2 mice, as compared to the controls, suggesting that Her2 accelerates cell dissemination in the presence of wild type p53[8, 40, 41]. Consistent with these findings, Her2 moderately upregulated p53 and p21 expression without altering the levels of p-p53(S15) and other target genes such as Bax and Apaf1 in MCF-10A-Her2 and HMEC-TERT-Her2 cells (Fig. S3F).
Confirming the findings using p38 inhibitor, a constitutively active mutant of a p38 upstream kinase MKK6 (MKK6E) induced p38 and Hsp27-Ser86 phosphorylation, increased E-cadherin expression and junctions in organoid cultures and reduced percentage of invasive organoids (Fig. 2A–C), while a dominant negative mutant of MKK6 (MKK6A) had the opposite effects (Fig. 2A, D–E), in early lesion MMTV-Her2 cells. Similar observations were made in MCF-10A and MCF-10A-Her2 cells, in that MKK6E increased phosphorylation of p38, MK2 and Hsp27 and E-cadherin expression and junctions in organoids and 2D culture, and reduced transwell migration in the absence or presence of a cell cycle inhibitor aphidicolin, percentage of invasive organoids and expression of an EMT signature Twist (Fig. 2F–G, 2I, 2K, S3G–I), while MKK6A had the opposite effects (Fig. 2F, 2H, 2J, 2L, S3G, S3I). As inhibition of p38 by MKK6A was sufficient to induce the disseminating phenotypes in MCF-10A cells, and constitutive activation of p38 by MKK6E reversed the stimulation of the disseminating phenotypes by Her2 in both MMTV-Her2 and MCF-10A-Her2 cells, Her2 mediates the disseminating phenotypes including migration, invasion and EMT by inhibiting p38 in early lesion breast cancer cells.
Figure 2. Her2 promotes the disseminating phenotypes by suppressing p38 in Her2+ early lesion mammary epithelial cells.

(A-E) Mammary epithelial cells isolated from 14–18-week-old MMTV-Her2 mice with early lesions were transduced with MKK6E or vector (BP) (A-C) or MKK6A or vector (BH) (A, D-E), and analyzed for the p38 pathway status and E-cadherin expression by Western blotting (A), E-cadherin junctions in organoids by immunofluorescence (B, D) and percentage of invasive organoids (C, E).
(F-L) MCF-10A cells transduced with vector (LXSN) or Her2 were transduced with MKK6E or vector (BP) (F-G, I, K) or MKK6A or vector (BH) (F, H, J, L), and analyzed for the p38 pathway status and E-cadherin expression by Western blotting (F), E-cadherin junctions in organoids by immunofluorescence (G-H), migration in transwell (I-J) and percentage of invasive organoids (K-L).
(B-E, G-L) Representative images (left) and quantification (means±SD for duplicates) of percentage of E-cadherinhigh organoids (B, D, G-H) and organoids with outward invading cells (C, E, K-L), and number of migrated cells (I-J) (right) are shown. NS, not significant; * p<0.05; ** p<0.01; *** p<0.001 vs indicated control in one-tailed unpaired t-test.
We next examined the role of MK2 in Her2-mediated disseminating phenotypes. MK2 shRNAs reduced Hsp27-Ser86 phosphorylation, E-cadherin expression and E-cadherin junctions in organoids, and increased migration and percentage of invasive organoids in MMTV-Her2 cells (Fig. 3A–D). Human MK2 shRNAs had the same effects on the disseminating phenotypes in MCF-10A and MCF-10A-Her2 cells, and in addition, increased Vimentin and Twist expression and cell migration in the presence of aphidicolin (Fig. S4A–F). In contrast, a constitutively active mutant of MK2 (MK2EE)[28, 42] enhanced Hsp27-Ser86/82 phosphorylation, increased E-cadherin junctions, and reduced percentage of invasive organoids in MMTV-Her2 and MCF-10A-Her2 cells (Fig. 3E–G, S4G–I), and reduced migration in MCF-10A and MCF-10A-Her2 cells with or without aphidicolin (Fig. S4J–K). While the MK2 shRNAs were sufficient to induce the disseminating phenotypes in MCF-10A cells (Fig. S4A–D), active MK2EE reversed suppression of Hsp27 phosphorylation by Her2 and the stimulatory effect of Her2 on the disseminating phenotypes in MMTV-Her2 and MCF-10A-Her2 cells (Fig. 3E–G, Fig. S4G–K), indicating that Her2 reduces Hsp27 phosphorylation and promotes the disseminating phenotypes by suppressing the MK2 activity. Moreover, MK2 knockdown reversed the ability of MKK6E to increase Hsp27-Ser82 phosphorylation and E-cadherin junctions and to reduce the percentage of invasive organoids in MCF-10A-Her2 cells (Fig. 3H–J), indicating that MK2 acts downstream of p38 in the pathways that suppresses the disseminating phenotypes.
Figure 3. Her2 promotes the disseminating phenotypes by suppressing p38-mediated MK2 activation in Her2+ early lesion breast cancer cells.
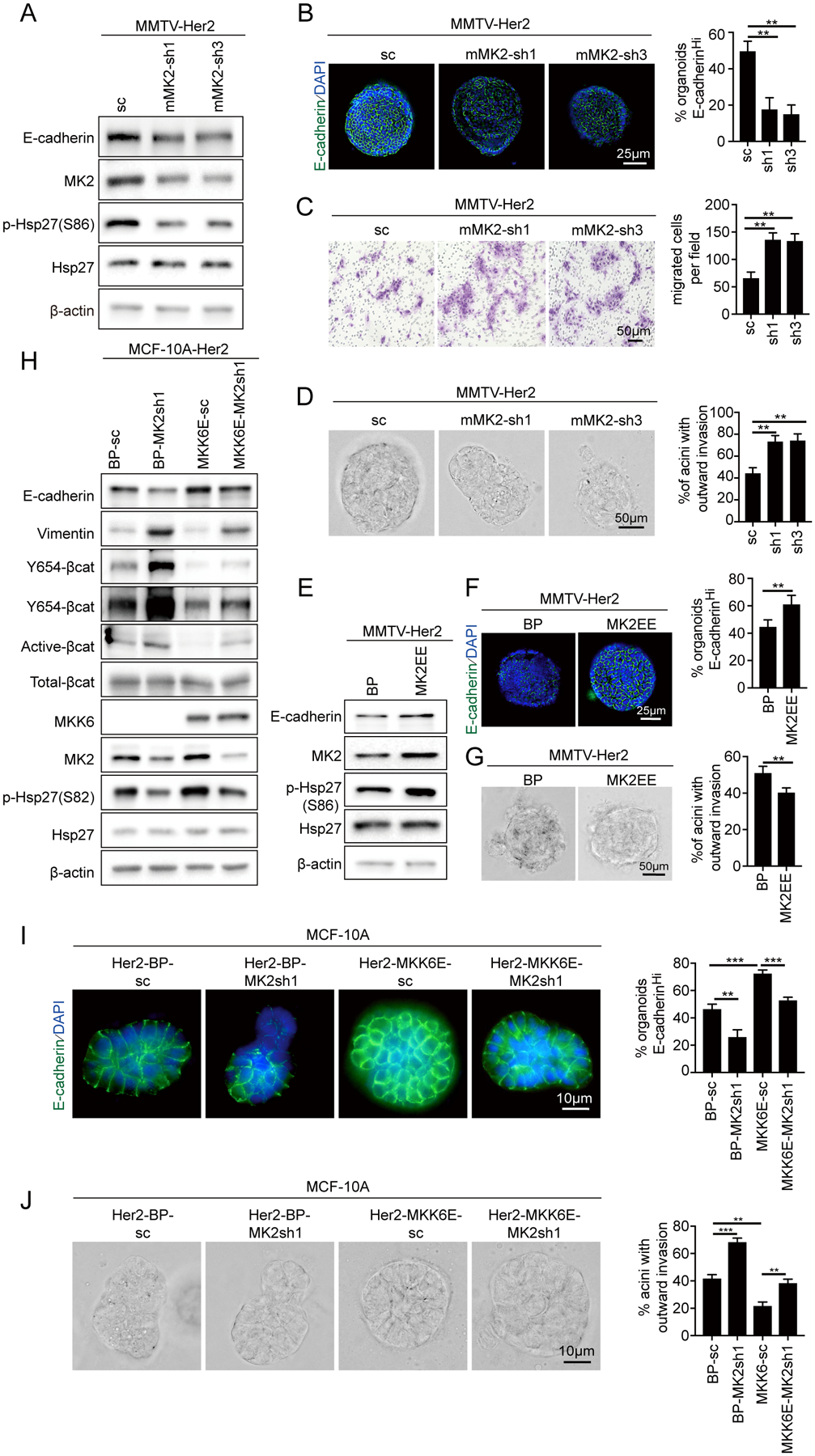
(A-D) Mammary epithelial cells isolated from 14–18-week-old MMTV-Her2 mice with early lesions were transduced with a scrambled shRNA (SC) or mouse MK2 shRNAs and analyzed for the p38 pathway status and E-cadherin expression by Western blotting (A), E-cadherin junctions in organoids by immunofluorescence (B), migration in transwell (C) and percentage of invasive organoids (D).
(E-G) Mammary epithelial cells isolated from 14–18-week-old MMTV-Her2 mice with early lesions were transduced with vector (BP) or the constitutively active MK2EE mutant and analyzed for the p38 pathway status and E-cadherin expression by Western blotting (E), E-cadherin junctions in organoids (F) and percentage of invasive organoids (G).
(H-J) MCF-10A cells transduced with Her2 were transduced with vector (BP) or constitutively active MKK6E and MK2 shRNA or a scrambled shRNA (SC), and analyzed for indicated proteins by Western blotting (H), E-cadherin junctions in organoids (I), percentage of invasive organoids (J).
(B-D, F-G, I-J) Representative images (left) and quantification (means±SD for duplicates) of percentage of E-cadherinhigh organoids (B, F, I) and organoids with outward invading cells (D, G, J), and number of migrated cells (C) (right) are shown. ** p<0.01; *** p<0.001 vs indicated control in one-tailed unpaired t-test.
ATF2 knockdown reduced E-cadherin junctions, and increased migration and percentage of invasive organoids in MCF-10A and MCF-10A-Her2 cells (Fig. S5A–D). In contrast, knockdown of another p38 downstream kinase MNK1 had no effect on E-cadherin junctions or migration (Fig. S5E–G). Thus, in addition to the MK2, ATF2 may also contribute to the disseminating phenotypes, although whether it acts downstream of Her2 and/or p38 is unclear, while MNK1 does not.
To investigate the role of the MK2 downstream substrate Hsp27 in early disseminating phenotypes, we tested the effects of wild type Hsp27 (Hsp27 WT) and Hsp27 containing phosphomimetic (Hsp27-TriD, or -S15D/S78D/S82D) or unphosphorylatable (Hsp27-TriA or -S15A/S78A/S82A) mutations at the MK2 phosphorylation sites Ser15/78/82. In MMTV-Her2 cells, ectopic expression of Hsp27-WT reduced E-cadherin expression and junctions, and increased migration and percentage of invasive organoids compared to vector control; ectopic expression of Hsp27-TriA in general led to a further increase in EMT, migration and invasion as compared to Hsp27-WT, while Hsp27-TriD suppressed these disseminating phenotypes (Fig. 4A–D). The same observations were made in MCF-10A-Her2 cells, in that Hsp27-WT promoted and Hsp27-TriA further promoted, but Hsp27-TriD suppressed, EMT (E-cadherin junctions and Twist expression), migration with or without aphidicolin and invasion (Fig. S6A–G). Thus, Hsp27-Ser15/78/82 phosphorylation by MK2 leads to inhibition of Her2-mediated disseminating phenotypes, and the unphosphorylatable Hsp27-TriA mutant and the unphosphorylated portion of the ectopically expressed wild type Hsp27 promote these phenotypes. In MCF-10A cells, both Hsp27-WT and –TriA promoted the disseminating phenotypes (Fig. S6), indicating that the unphosphorylated Hsp27 protein is sufficient to induce these phenotypes in cells without Her2. Meanwhile, the Hsp27-TriD mutant that mimics phosphorylation by MK2 disrupts Her2-induced EMT, migration and invasion in Her2+ cells. Therefore, Her2 promotes the disseminating phenotypes by inhibiting MK2-mediated phosphorylation of Hsp27.
Figure 4. Her2 promotes the disseminating phenotypes by suppressing MK2-mediated phosphorylation of Hsp27 in Her2+ early lesion breast cancer cells.

(A-D) Mammary epithelial cells isolated from 14–18-week-old MMTV-Her2 mice with early lesions were transduced with vector (MCS), wild type Hsp27 (Hsp27-WT) or Hsp27 containing phosphomimetic (Hsp27-TriD) or non-phosphorylatable (Hsp27-TriA) mutations at the MK2 phosphorylation sites, and analyzed for Hsp27 and E-cadherin expression by Western blotting (A), E-cadherin junctions in organoids by immunofluorescence (B), migration in transwell (C) and percentage of invasive organoids (D).
(E-G) MCF-10A cells transduced with Her2 were transduced with vector (BP) or MK2EE and vector (MCS) or Hsp27-TriA and analyzed for indicated proteins by Western blotting (E), E-cadherin junctions in organoids (F) and percentage of invasive organoids (G).
(B-D, F-G) Representative images (left) and quantification (means±SD for duplicates) of percentage of E-cadherinhigh organoids (B, F) and organoids with outward invading cells (D, G), and number of migrated cells (C) (right) are shown. NS, not significant; * p<0.05; ** p<0.01; *** p<0.001 vs indicated control in one-tailed unpaired t-test.
Moreover, the unphosphorylatable Hsp27-TriA mutant reversed the ability of MK2EE to increase E-cadherin expression and junctions and reduce Vimentin expression and the percentage of invasive organoids (Fig. 4E–G), indicating that phosphorylated Hsp27 acts downstream of MK2 to suppress the disseminating phenotypes. Together, our findings indicate that Her2 induces the disseminating phenotypes in early lesion breast cancer cells through inhibition of the p38-MK2-Hsp27 pathway.
The constitutively active or phoshomimetic mutants of the p38 pathway components, including MKK6E, MK2EE and Hsp27-TriD, did not have an obvious effect on some of the disseminating phenotypes, especially the loss of E-cadherin junctions and formation of invasive organoids, in MCF-10A cells without Her2 (Fig. 2G, 2K, S4H–I, S6B, S6E), although they reversed the Her2-induced disseminating phenotypes. It is likely that in the normal MCF-10A cells, the p38 pathway activity is relative high without suppression by Her2, and that the high levels of active p38 and MK2 and MK2-phosphorylated Hsp27 maintain very low levels of disseminating phenotypes in these cells, which cannot be further suppressed by additional activation of the p38 pathway.
Suppression of the p38-MK2-Hsp27 pathway by Her2 activates β-catenin through Src-mediated phosphorylation
EMT is resulted from dissociation of the E-cadherin-β-catenin-α-catenin complex and nuclear translocation of β-catenin[14]. Src induces β-catenin activation and nuclear translocation by phosphorylating its Tyr654 residue[15]. In addition, p38 suppresses early dissemination by inhibiting β-catenin activation and nuclear translocation[11]. We thus tested the effect of the p38-MK2-Hsp27 pathway on β-catenin activation by Src.
In MMTV-Her2 cells, p38 activation by MKK6E suppressed, while p38 inhibition by MKK6A increased, the levels of Src-phosphorylated (pY654) and the active forms of β-catenin[43] (Fig. 5A). In MCF-10A cells, Her2 and p38 inhibitor SB203580 alone increased, and the combination of both further increased, the levels of the Src phosphorylated and the active forms of β-catenin and nuclear translocation of β-catenin (Fig. S7A–D). Furthermore, the constitutively active MK2EE reduced, and MK2 shRNAs increased, the levels of the Src-phosphorylated and the active forms of β-catenin in MMTV-Her2 cells (Fig. 5B). In MCF-10A cells, the constitutively active MK2EE reversed Her2-mediated induction of the Src phosphorylated and the active forms of β-catenin and the nuclear translocation of β-catenin (Fig. S7E–H). Moreover, MK2 knockdown alone was sufficient to increase the levels of the Src phosphorylated and the active forms of β-catenin and nuclear translocation of β-catenin, further upregulated these forms of β-catenin to greater levels in combination with Her2 (Fig. S7I–L), and reversed MKK6E-induced reduction in the Src phosphorylated and the active form of β-catenin (Fig. 3H). Thus, Her2 activates β-catenin by inhibiting p38-mediated activation of MK2 in early lesion breast cancer cells.
Figure 5. Suppression of the p38-MK2-Hsp27 pathway by Her2 activates β-catenin through Src-mediated phosphorylation.

(A-C) Western blot analysis of Src-phosphorylated β-catenin (Y654-βcat), active β-catenin (Active-βcat) and total β-catenin in mammary epithelial cells isolated from 14–18-week-old MMTV-Her2 mice with early lesions and transduced with vector (BP) or MKK6E or vector (BH) or MKK6A (A), vector (BP) or MK2EE or a scrambled shRNA (SC) or mouse MK2 shRNAs (B), or vector (MCS), Hsp27-WT, Hsp27-TriD or Hsp27-TriA (C).
(D-E) Organoids of mammary epithelial cells isolated from 14–18-week-old MMTV-Her2 mice with early lesions and transduced with vector (MCS), Hsp27-WT, Hsp27-TriD or Hsp27-TriA were stained for active (D) or total (E) β-catenin by immunofluorescence. Percentage (means±SD for duplicates) of organoids with high active β-catenin levels (active-βcathigh) (D) or high membrane localized total β-catenin levels (βcatMEM+) were quantified (right). * p<0.05; ** p<0.01; *** p<0.001 vs indicated control in one-tailed unpaired t-test.
(F) Interactions among endogenous Hsp27, β-catenin and Src assessed by reciprocal co-immunoprecipitation (IP). Hsp27, β-catenin or Src were immunoprecipitated from MCF-10A-LXSN or MCF-10A-Her2 cells using respective antibodies or IgG and detected by Western blotting.
(G-I) Effects of phosphomimetic and nonphosphorylatable mutations of the MK2 phosphorylation sites of Hsp27 on interactions among Hsp27, β-catenin and Src. Hsp27 (G), β-catenin (H) or Src (I) were immunoprecipitated from MCF-10A-Her2 cells transduced with vector (MCS), Hsp27-WT, Hsp27-TriD or Hsp27-TriA using respective antibodies or IgG. Presence of Hsp27, β-catenin and Src in the IPs and inputs were detected by Western blotting. Quantifications of signals for β-catenin immunoprecipitated by the anti-Hsp27 antibody (G), Src immunoprecipitated by the anti-β-catenin antibody (H) and β-catenin immunoprecipitated by the anti-Src antibody (I) are shown in the bar graphs on the right.
Consistent with their effects on EMT, migration and invasion, Hsp27-WT and to a greater extent, Hsp27-TriA increased, while Hsp27-TriD reduced, the level of Src-phosphorylated β-catenin and the level of active β-catenin in organoids (Fig. 5C–D) in MMTV-Her2 cells (Fig. 5C–D). Hsp27-WT and Hsp27-TriA overexpression also reduced, and Hsp27-TriD increased, total membrane-localized inactive β-catenin in MMTV-Her2 cells (Fig. 5E). In MCF-10A-Her2 cells, Hsp27-WT promoted and Hsp27-TriA further promoted, but Hsp27-TriD suppressed, the level of Src-phosphorylated β-catenin and active β-catenin and nuclear translocation of β-catenin (Fig. S7N–P), indicating that phosphorylation of Hsp27 by MK2 leads to inhibition of Her2-mediated activation of β-catenin, and that the unphosphorylatable Hsp27-TriA mutant and the unphosphorylated portion of the ectopically expressed wild type Hsp27 promote β-catenin activation. In MCF-10A cells, both Hsp27-WT and -TriA upregulated the Src-phosphorylated and the active β-catenin and nuclear translocation of β-catenin (Fig. S7M–P), indicating that the unphosphorylated Hsp27 is sufficient to induce β-catenin activation. Hsp27-TriD in general did not further reduce the Src-phosphorylated or the active form or nuclear translocation of β-catenin in MCF-10A cells, all of which were already at very low levels due to the absence of Her2 in these normal cells (Fig. S7M–P), consistent with its effect on the disseminating phenotypes (Fig. S6). Moreover, Hsp27-TriA reversed the ability of MK2EE to reduce the levels of the Src-phosphorylated and the active β-catenin (Fig. 4E), indicating that phosphorylated Hsp27 acts downstream of MK2 to suppress β-catenin activation. Thus, Her2 promotes β-catenin activation by inhibiting p38/MK2-mediated phosphorylation of Hsp27.
Phosphorylation by MK2 abrogates the ability of Hsp27 to bind to β-catenin and to promote interaction of β-catenin with Src
To investigate the mechanism by which Hsp27 regulates β-catenin-Tyr654 phosphorylation by Src, we examined the interaction among Hsp27, Src and β-catenin using co-immunoprecipitation (co-IP) assays. An antibody against Hsp27 immunoprecipitated both Hsp27 and β-catenin in MCF-10A and more β-catenin in MCF-10A-Her2 cells, but not Src in either cell lines; an anti-β-catenin antibody co-immunoprecipitated both Hsp27 and Src together with β-catenin in MCF-10A and more of these 2 proteins in the presence of Her2; and an anti-Src antibody brought down β-catenin in MCF-10A cells and more β-catenin in MCF-10A-Her2 cells, along with Src, but not Hsp27 (Fig. 5F), Thus, β-catenin interacts with Hsp27 and Src in separate complexes, and Her2 enhances both interactions, suggesting that the unphosphorylated form of Hsp27 in Her2 cells may preferentially bind to β-catenin, enhancing its interaction with Src. Indeed, co-IP assays in MCF-10A-Her2 cells ectopically expression Hsp27 revealed that Hsp27-TriD reduced, and Hsp27-WT and Hsp27-TriA increased, interaction of Hsp27 with β-catenin, as compared to the vector control (Fig. 5G). Like the endogenous Hsp27, overexpressed Hsp27 did not interact with Src (Fig. 5G). In these same cells, ectopic expression of Hsp27-WT and -TriA enhanced, while Hsp27-TriD reduced or failed to significantly increase, interaction between endogenous β-catenin and Src in reciprocal co-IP assays (Fig. 5H–I). Thus, Hsp27-TriA, and the unphosphorylated wild type Hsp27 in Her2+ cells where the p38-MK2 activity is low, bind preferentially, as compared to Hsp27-TriD and MK2-phosphorylated Hsp27, to β-catenin, which enhances its interaction with Src, leading to increased Tyr654 phosphorylation and activation of β-catenin. Since no Hsp27-Src interaction was detected, Hsp27 may be released from β-catenin once it has bound to and locked β-catenin in a Src-amiable conformation. Alternatively, the Hsp27-Src interaction may be below the detection limit of the assay.
Inhibition of MK2 promotes early dissemination of breast cancer in vivo.
We investigated the role of MK2 in early dissemination of breast cancer in vivo, using a specific MK2 inhibitor PF3644022[44]. In MMTV-Her2 cells, PF3644022 reduced Hsp27-Ser86 phosphorylation, increased Src-mediated phosphorylation of β-catenin-Tyr654 and active β-catenin levels, reduced E-cadherin expression and junctions in organoids and increase percentage of invasive organoids (Fig. S8A–C). PF3644022 had similar effects on Hsp27, β-catenin and disseminating phenotypes in MCF-10A and MCF-10A-Her2 cells (Fig. S8D–I).
Treatment of 14–18-week-old MMTV-Her2 mice with PF3644022 reduced Hsp27-Ser86 phosphorylation and percentage of E-cadherinhigh ducts and increased β-catenin nuclear translocation in mammary glands, compared to the control (Fig. 6A–C). PF3644022 did not alter the percentage of ducts with early lesions in mammary glands, but increased the Her2+ eDCCs in blood, bone marrow and lung (Fig. 6D–G), demonstrating that pharmacological inhibition of MK2 enhances early dissemination of breast cancer in vivo.
Figure 6. Pharmacological inhibition of MK2 promotes early dissemination in the MMTV-Her2 model.

(A) Western blot analysis of mammary epithelial cells isolated from 14–18-week-old MMTV-Her2 mice treated with vehicle or MK2i for 14 days.
(B-C) Mammary glands were isolated from 14–18-week-old MMTV-Her2 mice treated with vehicle or MK2i for 14 days and stained for E-cadherin (B) or β-catenin (C) by IHC. Percentage of E-cadherinhigh ducts (means±SD, n=4 mice/group) (B) or percentage of cells with nuclear β-catenin per duct (means±SD, n=42 ducts from 3 mice) (C) was quantified (right). All the ducts from 4 mice in each group were counted in (B).
(D) HE staining of mammary glands isolated from 14–18-week-old MMTV-Her2 mice treated with vehicle or MK2i for 14 days. Percentage of ducts with early lesions (means±SD, n=75 ducts for vehicle group, n=90 ducts for MK2i group, each group from 3 mice) were quantified (right)
(E-G) Early disseminating cancer cells (eDCCs) were detected by immunofluorescence (E-F) or IHC (G) staining for Her2 in cytospin preparations of blood (E) or bone marrow (F) or tissue sections of lungs (G) isolated from 14–18-week-old MMTV-Her2 mice treated with vehicle or MK2i for 14 days. Nuclei were stained with DAPI (E-F) or Haematoxylin (G). Arrows indicate Her2+ eDCCs (G). Her2+ eDCCs/ml of blood (means±SD, n=4 mice/group) (E), Her2+ eDCCs/bone marrow (means±SD, n=5 mice/group) (F) or percentage of Her2+ eDCCs/field (means±SD, n= 40 fields from 4 mice/group) (G) were quantified (right).
(B-G) NS, not significant; * p<0.05; ** p<0.01; *** p<0.001 vs indicated control in Mann-Whitney U-test.
A Wip1 inhibitor GSK2830371 actives the p38 pathway and reduces early dissemination of breast cancer in vivo.
Wip1 is a p38 phosphatase that inactivates p38 by dephosphorylating Thr182, one of the dual phosphorylation sites[31, 32]. We thus tested whether Wip1 inhibitor abrogates early dissemination and metastasis of breast cancer in the MMTV-Her2 model. GSK2830371 (GSK) upregulated phosphorylation of p38, MK2 and Hsp27-Ser86 and downregulated the levels of the Src phosphorylated and the active forms of β-catenin, increased E-cadherin junctions in organoids, and reduced percentage of invasive organoids in MMTV-Her2 cells (Fig. S9A–C). GSK also induced p38 and Hsp27 phosphorylation and downregulated Src phosphorylated β-catenin in MCF-10A cells with or without Her2, and reversed Her2-induced disseminating phenotypes and activation and nuclear translocation of β-catenin (Fig. S9D–J). These findings demonstrate the efficacy of GSK in activating the p38 pathway and suppressing the Her2-mediated disseminating phenotypes in vitro. GSK had no significant effect on the disseminating phenotypes in MCF-10A cells (Fig. S9E–G), again likely due to the high p38 pathway activity and already weak disseminating phenotypes in these cells without Her2.
Treatment of 14–18-week-old MMTV-Her2 mice with GSK led to significant upregulation in phosphorylation of p38, MK2 and Hsp27-Ser86 in mammary tissues as compared to vehicle (Fig. 7A). IHC staining showed that E-cadherin was upregulated in mammary glands of mice treated with GSK (Fig. 7B). Moreover, although the percentage of ducts with early lesions did not change in mammary glands (Fig. 7C), the Her2+ eDCCs were decreased by GSK as compared to vehicle control in bone marrow and lung (Fig. 7D–E). Therefore, pharmacological inhibition of Wip1 activates the p38 pathway and reduces Her2-mediated early dissemination of breast cancer in vivo.
Figure 7. Pharmacological inhibition of Wip1 inhibits early dissemination in the MMTV-Her2 model.
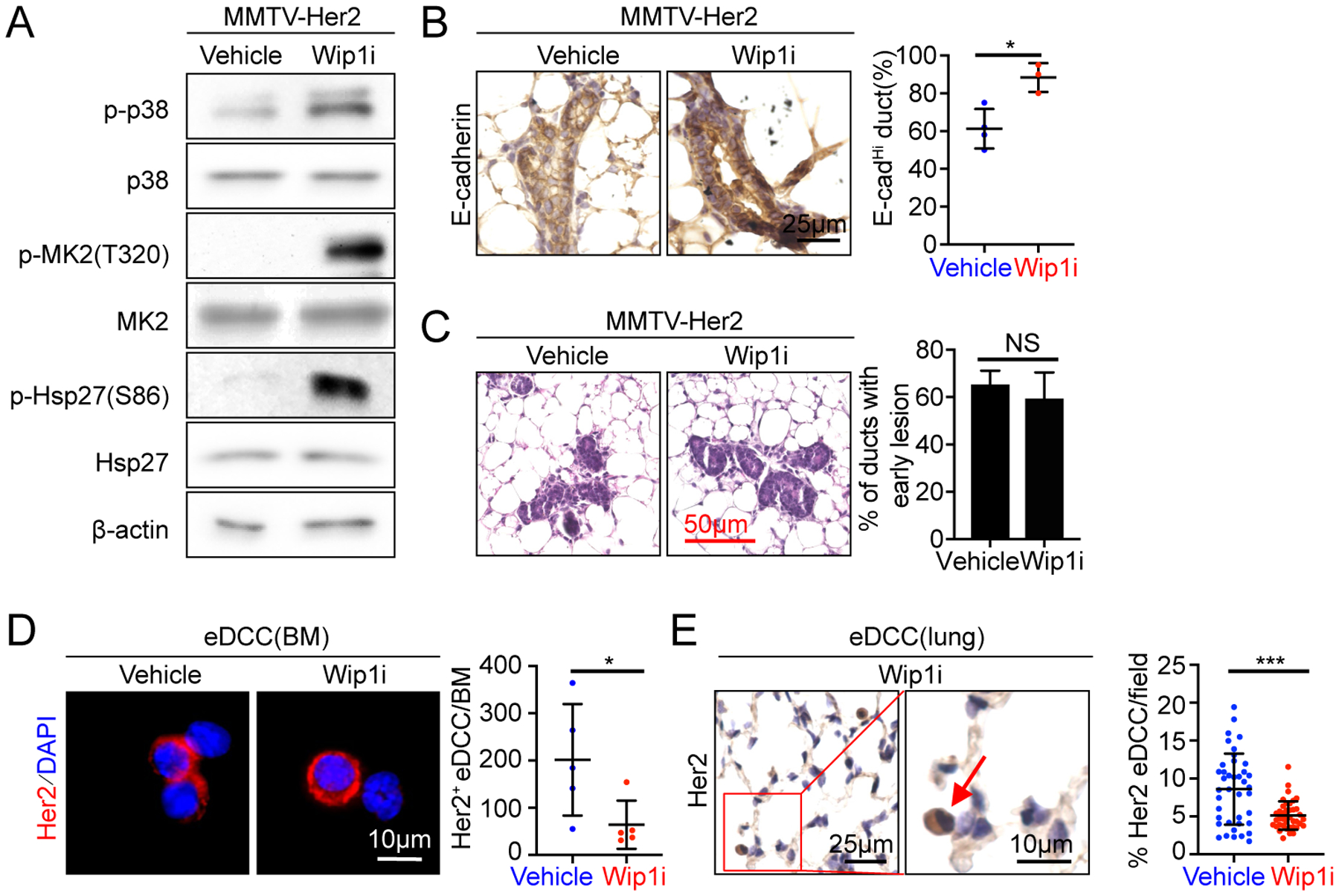
(A) Western blot analysis of mammary epithelial cells isolated from 14–18-week-old MMTV-Her2 mice treated orally with vehicle or GSK2830371 for 21 days.
(B) Mammary glands isolated from 14–18-week-old MMTV-Her2 mice treated with vehicle or Wip1i for 21 days were stained for E-cadherin by IHC. Percentage of E-cadherinhigh ducts (means±SD, n=4 mice for vehicle, 3 mice for Wip1i) was quantified (right). All the ducts in all mice in each group were counted.
(C) HE staining of mammary glands isolated from 14–18-week-old MMTV-Her2 mice treated with vehicle or Wip1i for 21 days. Percentage of ducts with early lesions (means±SD, n=60 ducts for vehicle group, n=75 ducts for Wip1i group, each group from 3 mice) were quantified (right)
(D-E) Early disseminating cancer cells (eDCCs) were detected by immunofluorescence (D) or IHC (E) staining for Her2 in cytospin preparations of bone marrow (D) or tissue sections of lungs (E) isolated from 14–18-week-old MMTV-Her2 mice treated with vehicle or Wip1i for 21 days. Nuclei were stained with DAPI (D) or Heamotoxylin (E). Arrows indicate Her2+ eDCCs (E). Her2+ eDCCs/bone marrow (means±SD, n=5 mice/group) (D) or percentage of Her2+ eDCCs/field (means±SD, n= 40 fields from 4 mice/group) (E) were quantified (right).
(B-E) NS, not significant; * p<0.05; *** p<0.001 vs indicated control in Mann-Whitney U-test (B-E).
Discussion
Using cell and mouse models for early lesion breast cancer, we identified MK2 and Hsp27 as novel mediators of the disseminating phenotypes in early lesion breast cancer cells. Supporting the key role of this pathway in early dissemination in vivo, in MMTV-Her2 mice, the early disseminating cells present in the early lesions in mammary glands and lung are Her2highp-p38lowp-MK2lowp-Hsp27low, and pharmacological inhibition of MK2 increases early dissemination. Existence of Her2highp-p38lowp-MK2lowp-Hsp27low cells in early breast cancer lesions from patients suggests that these cells likely contribute to early dissemination in the human disease. Interestingly, this group of cells were detected in human breast carcinoma, suggesting that the Her2-p38-MK2-Hsp2 network is preserved in late-stage breast cancer, although the metastasis-suppressing function of this pathway may not be preserved at the late stage, given previous reports that p38 promotes migration and invasion in established cancer cell lines[45, 46].
Among the p38 substrates[47], MK2 suppression is necessary for Her2-induced disseminating phenotypes and sufficient to trigger them by itself, but PRAK and MNK1 are not involved. In the initial report[11], ATF2 phosphorylation was reduced by Her2, but its function in early dissemination was unknown. We confirmed suppression of ATF2 phosphorylation by Her2 and showed that ATF2 silencing induces the disseminating phenotypes. It remains to be determined whether ATF2 acts downstream of p38 to inhibit the disseminating phenotypes and whether ATF2 suppression contributes to Her2-induced early dissemination. Further studies are also needed to investigate the roles of the other p38 substrates in cancer early dissemination. Despite the possible involvement of other downstream effectors, data from epistatic analyses indicate that p38 suppresses the disseminating phenotypes at least partly through MK2 and Hsp27. The exact roles of different p38 isoforms also need to be examined, despite the enhancement of early dissemination in vitro and in vivo by SB203580, an inhibitor mainly for p38α/β[11].
Src-mediated phosphorylation of β-catenin-Tyr654 results in its dissociation from membrane-bound E-cadherin, leading to disruption of E-cadherin junctions, its nuclear translocation, and EMT[14, 15]. We showed that Her2 suppresses p38/MK2 activity and reduces MK2-mediated Hsp27 phosphorylation, leading to accumulation of unphosphorylated Hsp27 that binds to β-catenin and enhances Src-β-catenin interaction and Src-mediated phosphorylation and nuclear translocation of β-catenin. These findings provide a mechanism for the essential role of Hsp27 in β-catenin activation. Hsp27 is a molecular chaperon[48] that promotes cell migration and invasion in cancer cells[49, 50]. Contrary to a report that Hsp27 binds and stabilizes β-catenin protein in prostate cancer cells[27], neither Hsp27-WT nor Hsp27-TriD/A altered total β-catenin levels in early lesion breast cancer cells (Fig. 5C, 5G–I, S7M), possibly due to tissue specificity. Hsp27 can be tumor-promoting[51] or tumor-suppressing[52]. Little is known about the role of Hsp27 phosphorylation in cancer. We found that MK2-mediated Hsp27 phosphorylation abrogates its function in promoting Her2+ breast cancer early dissemination. Hsp27-TriD failed to bind β-catenin and reduced disseminating phenotypes, suggesting that TriD is dominant negative over the endogenous Hsp27. Possibly, TriD sequesters endogenous unphosphorylation Hsp27 or other proteins required for its function, preventing its interaction with β-catenin.
While targeted therapies have been developed for breast cancer[33, 53], we propose a novel therapeutic strategy to block dissemination and metastasis at early stages of cancer or in asymptomatic high-risk individuals by restoring the p38 pathway, based on our observation that pharmacological inhibition of Wip1, a p38 phosphatase[32], restores the p38-MK2-Hsp27 activity and suppresses early dissemination in both cell and mouse models of early lesion Her2+ breast cancer. As a oncogene with amplification/overexpression in cancers[35], Wip1 has other substrates besides p38, including p53[32]. Wip1 inhibitors are being pursued as anti-cancer drugs based on their ability to reactivate p53 in patients with wild type p53[36–38]. Since suppression of early dissemination by the p38-MK2-Hsp27 pathway is independent of p53, Wip1 inhibitors may be effective in patients with either wild type or mutant p53. Despite improved specificity and toxicity compared to other Wip1 inhibitors[54, 55], GSK2830371 is unstable. Better Wip1 inhibitors need to be developed. While our study provides a proof-of-principle for a potential therapy for breast cancer metastasis, it needs to be validated and further investigated in additional preclinical studies. Reactivation of p38 by Wip1 inhibitors, although inhibits early dissemination, may not eventually lead to attenuation of metastasis due to selection of compensatory mechanisms such as group migration[56] or activation of other metastasis-promoting pathways. Thus, Wip1 inhibitors may need to be combined with other drugs targeting those compensatory mechanisms or cancer cell proliferation or survival.
Materials and Methods
Reagent
MCF-10A cells were treated with 5μM SB203580 (Sigma), 0.5μM GSK2830371 (Selleckchem), 5μM PF3644022 (R&D System) or 10μM aphidicolin (Sigma) for the inhibition of p38, Wip1, MK2 or DNA synthesis, respectively[36, 57, 58]
For treatment in mice, GSK2830371 was dissolved in 2% DMSO, 30% PEG300 and 5% Tween80[36]; MK2 PF3644022 was dissolved in 20% polyethylene glycol-400,10% ethanol in 70% normal saline[44, 57]. Additional materials and methods are provided in Supplementary Information
Supplementary Material
Acknowledgements
This study was supported by NIH/NCI grants CA131231, CA172115 (PS) and Bilateral Inter-Governmental S&T Cooperation Project grants from Ministry of Science and Technology of China (81972882 and 2018YFE0114300, RX), and by Cell Engineering and Tumor Tissue and Pathology Shared Resources of WFBCCC, supported by NCI’s Cancer Center Support Grant (P30CA012197). PS is supported by the Anderson Oncology Research Professorship.
Footnotes
Conflict of Interests
Authors declare no competing interests in relation to the work described.
Reference
- 1.Koniali L, Hadjisavvas A, Constantinidou A, Christodoulou K, Christou Y, Demetriou C et al. Risk factors for breast cancer brain metastases: a systematic review. Oncotarget 2020; 11: 650–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer 2008; 8: 329–340. [DOI] [PubMed] [Google Scholar]
- 3.Greco FA, Hainsworth JD. Introduction: unknown primary cancer. Semin Oncol 2009; 36: 6–7. [DOI] [PubMed] [Google Scholar]
- 4.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008; 13: 58–68. [DOI] [PubMed] [Google Scholar]
- 5.Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 2010; 120: 2030–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012; 148: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987; 235: 177–182. [DOI] [PubMed] [Google Scholar]
- 8.Taneja P, Maglic D, Kai F, Sugiyama T, Kendig RD, Frazier DP et al. Critical roles of DMP1 in human epidermal growth factor receptor 2/neu-Arf-p53 signaling and breast cancer development. Cancer Res 2010; 70: 9084–9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A 1992; 89: 10578–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardiff RD. Validity of mouse mammary tumour models for human breast cancer: comparative pathology. Microsc Res Tech 2001; 52: 224–230. [DOI] [PubMed] [Google Scholar]
- 11.Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature 2016; 540: 588–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 13.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest 2002; 109: 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brembeck FH, Rosario M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev 2006; 16: 51–59. [DOI] [PubMed] [Google Scholar]
- 15.Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem 1999; 274: 36734–36740. [DOI] [PubMed] [Google Scholar]
- 16.Polakis P Wnt signaling and cancer. Genes Dev 2000; 14: 1837–1851. [PubMed] [Google Scholar]
- 17.Gavert N, Ben-Ze’ev A. beta-Catenin signaling in biological control and cancer. J Cell Biochem 2007; 102: 820–828. [DOI] [PubMed] [Google Scholar]
- 18.Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res 2002; 62: 1613–1618. [PubMed] [Google Scholar]
- 19.Sanchez-Tillo E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci U S A 2011; 108: 19204–19209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci 1999; 112 ( Pt 8): 1237–1245. [DOI] [PubMed] [Google Scholar]
- 21.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001; 410: 37–40. [DOI] [PubMed] [Google Scholar]
- 22.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005; 15: 11–18. [DOI] [PubMed] [Google Scholar]
- 23.Katsogiannou M, Andrieu C, Rocchi P. Heat shock protein 27 phosphorylation state is associated with cancer progression. Front Genet 2014; 5: 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett 1992; 313: 307–313. [DOI] [PubMed] [Google Scholar]
- 25.Tian L, Zhao ZF, Xie L, Zhu JP. Taurine up-regulated 1 accelerates tumorigenesis of colon cancer by regulating miR-26a-5p/MMP14/p38 MAPK/Hsp27 axis in vitro and in vivo. Life Sci 2019; 239: 117035. [DOI] [PubMed] [Google Scholar]
- 26.Shiota M, Bishop JL, Nip KM, Zardan A, Takeuchi A, Cordonnier T et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res 2013; 73: 3109–3119. [DOI] [PubMed] [Google Scholar]
- 27.Cordonnier T, Bishop JL, Shiota M, Nip KM, Thaper D, Vahid S et al. Hsp27 regulates EGF/beta-catenin mediated epithelial to mesenchymal transition in prostate cancer. Int J Cancer 2015; 136: E496–507. [DOI] [PubMed] [Google Scholar]
- 28.Xu L, Chen S, Bergan RC. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene 2006; 25: 2987–2998. [DOI] [PubMed] [Google Scholar]
- 29.Han Y, Zhang L, Wang W, Li J, Song M. Livin promotes the progression and metastasis of breast cancer through the regulation of epithelialmesenchymal transition via the p38/GSK3beta pathway. Oncol Rep 2017; 38: 3574–3582. [DOI] [PubMed] [Google Scholar]
- 30.Han J, Sun P. The pathways to tumor suppression via route p38. Trends Biochem Sci 2007; 32: 364–371. [DOI] [PubMed] [Google Scholar]
- 31.Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H et al. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J 2000; 19: 6517–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goloudina AR, Kochetkova EY, Pospelova TV, Demidov ON. Wip1 phosphatase: between p53 and MAPK kinases pathways. Oncotarget 2016; 7: 31563–31571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Xu J, Choi HH, Han C, Fang Y, Li Y et al. Targeting 17q23 amplicon to overcome the resistance to anti-HER2 therapy in HER2+ breast cancer. Nat Commun 2018; 9: 4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Demidov ON, Kek C, Shreeram S, Timofeev O, Fornace AJ, Appella E et al. The role of the MKK6/p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene 2007; 26: 2502–2506. [DOI] [PubMed] [Google Scholar]
- 35.Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev 2008; 27: 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilmartin AG, Faitg TH, Richter M, Groy A, Seefeld MA, Darcy MG et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat Chem Biol 2014; 10: 181–187. [DOI] [PubMed] [Google Scholar]
- 37.Esfandiari A, Hawthorne TA, Nakjang S, Lunec J. Chemical Inhibition of Wild-Type p53-Induced Phosphatase 1 (WIP1/PPM1D) by GSK2830371 Potentiates the Sensitivity to MDM2 Inhibitors in a p53-Dependent Manner. Mol Cancer Ther 2016; 15: 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pechackova S, Burdova K, Benada J, Kleiblova P, Jenikova G, Macurek L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 2016; 7: 14458–14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, Wang L, Yao D, Yang T, Cao WM, Dou J et al. Wip1 inhibitor GSK2830371 inhibits neuroblastoma growth by inducing Chk2/p53-mediated apoptosis. Sci Rep 2016; 6: 38011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang SE, Narasanna A, Whitell CW, Wu FY, Friedman DB, Arteaga CL. Convergence of p53 and transforming growth factor beta (TGFbeta) signaling on activating expression of the tumor suppressor gene maspin in mammary epithelial cells. J Biol Chem 2007; 282: 5661–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fry EA, Taneja P, Inoue K. Oncogenic and tumor-suppressive mouse models for breast cancer engaging HER2/neu. Int J Cancer 2017; 140: 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engel K, Schultz H, Martin F, Kotlyarov A, Plath K, Hahn M et al. Constitutive activation of mitogen-activated protein kinase-activated protein kinase 2 by mutation of phosphorylation sites and an A-helix motif. J Biol Chem 1995; 270: 27213–27221. [DOI] [PubMed] [Google Scholar]
- 43.Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016; 165: 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mourey RJ, Burnette BL, Brustkern SJ, Daniels JS, Hirsch JL, Hood WF et al. A benzothiophene inhibitor of mitogen-activated protein kinase-activated protein kinase 2 inhibits tumor necrosis factor alpha production and has oral anti-inflammatory efficacy in acute and chronic models of inflammation. J Pharmacol Exp Ther 2010; 333: 797–807. [DOI] [PubMed] [Google Scholar]
- 45.Montero L, Nagamine Y. Regulation by p38 mitogen-activated protein kinase of adenylate- and uridylate-rich element-mediated urokinase-type plasminogen activator (uPA) messenger RNA stability and uPA-dependent in vitro cell invasion. Cancer Res 1999; 59: 5286–5293. [PubMed] [Google Scholar]
- 46.Huang S, New L, Pan Z, Han J, Nemerow GR. Urokinase plasminogen activator/urokinase-specific surface receptor expression and matrix invasion by breast cancer cells requires constitutive p38alpha mitogen-activated protein kinase activity. J Biol Chem 2000; 275: 12266–12272. [DOI] [PubMed] [Google Scholar]
- 47.Shi Y, Gaestel M. In the cellular garden of forking paths: how p38 MAPKs signal for downstream assistance. Biol Chem 2002; 383: 1519–1536. [DOI] [PubMed] [Google Scholar]
- 48.Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, Arrigo AP. Hsp27 as a negative regulator of cytochrome C release. Mol Cell Biol 2002; 22: 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JW, Kwak HJ, Lee JJ, Kim YN, Lee JW, Park MJ et al. HSP27 regulates cell adhesion and invasion via modulation of focal adhesion kinase and MMP-2 expression. Eur J Cell Biol 2008; 87: 377–387. [DOI] [PubMed] [Google Scholar]
- 50.Di K, Wong YC, Wang X. Id-1 promotes TGF-beta1-induced cell motility through HSP27 activation and disassembly of adherens junction in prostate epithelial cells. Exp Cell Res 2007; 313: 3983–3999. [DOI] [PubMed] [Google Scholar]
- 51.Rocchi P, Jugpal P, So A, Sinneman S, Ettinger S, Fazli L et al. Small interference RNA targeting heat-shock protein 27 inhibits the growth of prostatic cell lines and induces apoptosis via caspase-3 activation in vitro. BJU Int 2006; 98: 1082–1089. [DOI] [PubMed] [Google Scholar]
- 52.Matsushima-Nishiwaki R, Takai S, Adachi S, Minamitani C, Yasuda E, Noda T et al. Phosphorylated heat shock protein 27 represses growth of hepatocellular carcinoma via inhibition of extracellular signal-regulated kinase. J Biol Chem 2008; 283: 18852–18860. [DOI] [PubMed] [Google Scholar]
- 53.Neviere Z, De La Motte Rouge T, Floquet A, Johnson A, Berthet P, Joly F. How and when to refer patients for oncogenetic counseling in the era of PARP inhibitors. Ther Adv Med Oncol 2020; 12: 1758835919897530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamaguchi H, Durell SR, Feng H, Bai Y, Anderson CW, Appella E. Development of a substrate-based cyclic phosphopeptide inhibitor of protein phosphatase 2Cdelta, Wip1. Biochemistry 2006; 45: 13193–13202. [DOI] [PubMed] [Google Scholar]
- 55.Lee JS, Park JR, Kwon OS, Kim H, Fornace AJ Jr., Cha HJ. Off-target response of a Wip1 chemical inhibitor in skin keratinocytes. J Dermatol Sci 2014; 73: 125–134. [DOI] [PubMed] [Google Scholar]
- 56.Haeger A, Wolf K, Zegers MM, Friedl P. Collective cell migration: guidance principles and hierarchies. Trends Cell Biol 2015; 25: 556–566. [DOI] [PubMed] [Google Scholar]
- 57.Dietlein F, Kalb B, Jokic M, Noll EM, Strong A, Tharun L et al. A Synergistic Interaction between Chk1- and MK2 Inhibitors in KRAS-Mutant Cancer. Cell 2015; 162: 146–159. [DOI] [PubMed] [Google Scholar]
- 58.Collins NL, Reginato MJ, Paulus JK, Sgroi DC, Labaer J, Brugge JS. G1/S cell cycle arrest provides anoikis resistance through Erk-mediated Bim suppression. Mol Cell Biol 2005; 25: 5282–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.