

Abstract
INTRODUCTION:
Abnormal gene expression patterns may contribute to the onset and progression of late-onset Alzheimer’s disease (LOAD).
METHODS:
We performed transcriptome-wide meta-analysis (N=1,440) of blood-based microarray gene expression profiles as well as neuroimaging and CSF endophenotype analysis.
RESULTS:
We identified and replicated five genes (CREB5, CD46, TMBIM6, IRAK3, and RPAIN) as significantly dysregulated in LOAD. The most significantly altered gene, CREB5, was also associated with brain atrophy and increased amyloid-β accumulation, especially in the entorhinal cortex region. cis-eQTL mapping analysis of CREB5 detected five significant associations (p<5x10−8), where rs56388170 (most significant) was also significantly associated with global cortical amyloid-β (Aβ) deposition measured by [18F]Florbetapir PET and CSF Aβ1-42.
DISCUSSION:
RNA from peripheral blood indicated a differential gene expression pattern in LOAD. Genes identified have been implicated in biological processes relevant to AD. CREB, in particular, plays a key role in nervous system development, cell survival, plasticity and learning and memory.
Keywords: Alzheimer’s disease, Microarray gene expression, Imaging genetics, CREB5, Amyloid-β, ADNI
1. Introduction
Late-onset Alzheimer’s disease (LOAD) is a progressive neurodegenerative condition characterized by brain aggregation of amyloid-β into extracellular plaques, and hyperphosphorylated tau into neurofibrillary tangles.[1, 2] While these two pathological markers in addition to impaired cognitive function are used to definitively diagnose AD post-mortem, neither marker is sufficient to cause AD.[3–5] Amyloid-β PET (positron emission tomography) imaging has revealed that some individuals with normal cognitive performance display similar amyloid deposition to other individuals with mild cognitive impairment (MCI), suggesting that the amyloid hypothesis may not be sufficient to explain AD risk or progression.[5–7] As early intervention is a goal for AD trials, there has been extensive research aimed at identifying markers of biological processes associated with AD risk or progression, especially efforts to develop more accurate ante-mortem measures of amyloid-β and tau.[8–11]
While there are studies linking AD neuropathology and cognitive performance to dysregulated gene expression in brain tissue,[12–14] there is little data regarding AD-related gene expression in peripheral blood and none based on large-scale cohorts with neuroimaging and cerebrospinal fluid (CSF) biomarkers. Given that systemic factors such as inflammation, oxidative stress, and immune function are posited to play important roles in AD risk or progression, AD diagnosis and biomarkers for AD such as brain amyloid-β load or structural atrophy may be associated with differential expression of some genes in peripheral blood.
Neuroimaging genetics has been successfully applied in the past to obtain information about which genes are involved with specific AD-related pathological processes.[15–20] Expression of genes in peripheral blood can be associated not just with AD diagnosis, but with AD-related neuroimaging biomarkers such as atrophy or amyloid-β deposition in brain. We hypothesize that a genome-wide transcriptome meta-analysis will identify dysregulated genes in AD and that expression levels of the dysregulated genes will be also associated with AD-related neuroimaging biomarkers such as amyloid-β deposition and structural atrophy. Furthermore, we hypothesize that cis-expression quantitative trait loci (cis-eQTL) analysis of the differentially expressed genes will identify associated SNPs (single nucleotide polymorphisms) and that these SNPs will also be associated with AD-related endophenotypes including neuroimaging and CSF biomarkers.
2. Methods
2.1. Study participants
All individuals used in the analysis were participants of the Alzheimer’s Disease Neuroimaging Initiative (ADNI), AddNeuroMed, or Mayo Clinic Study of Aging (MCSA) cohorts. Informed consent was obtained for all subjects, and the study was approved by the relevant institutional review board at each data acquisition site.
2.1.1. Alzheimer’s Disease Neuroimaging Initiative (ADNI):
The ADNI initial phase (ADNI-1) was launched in 2003 to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment could be combined to measure the progression of Mild Cognitive Impairment (MCI) and early AD. ADNI-1 has been extended in subsequent phases (ADNI-GO, ADNI-2, and ADNI-3) for follow-up of existing participants and additional new enrollments. Demographic information, raw scan data, APOE (apolipoprotein E) and whole-genome genotyping data, microarray gene expression data, neuropsychological test scores, and clinical information are publicly available from the ADNI data repository (http://www.loni.usc.edu/ADNI/).[21] A total of 661 ADNI participants (213 cognitively normal older adults (CN), 200 early MCI (EMCI), 145 late MCI (LMCI), and 103 AD) were available for analysis.
2.1.2. AddNeuroMed:
The AddNeuroMed study is a prospective and longitudinal multicenter collaboration for the discovery of novel biomarkers for AD. Data were collected from six medical centers across Europe. A complete description of participant recruitment, selection criteria, and characterization is available in detail elsewhere.[22–24] A total of 674 AddNeuroMed participants included 208 MCI and 223 AD as well as 243 CN.
2.1.3. Mayo Clinic Study of Aging (MCSA):
The MCSA study was launched in 2004 to investigate the prevalence, incidence and risk factors for MCI and dementia.[25] Study participants from this prospective population-based cohort, are enrolled from the community of Olmsted County, Minnesota and followed longitudinally. Initially participants aged over 70 were included in the study, more recently participants aged over 50 (since 2012) and aged over 30 (since 2014) are being enrolled. Participants are evaluated and undergo neuropsychological assessment to determine a diagnosis of clinically normal, MCI or dementia; a subset of the participants also undergo neuroimaging studies including MRI and PET. Biological samples: PAXgene blood, plasma and/or CSF, were collected for consenting participants. A total of 105 MCSA samples were selected for this study that includes 44 participants with a clinical diagnosis of AD and 61 clinically normal controls with PiB-PET amyloid imaging that scored below the threshold for PiB-PET positivity (CN-PiB-negative).
2.2. Gene expression profiling analysis
Gene expression profiling from peripheral blood samples collected using PAXgene tubes for RNA analysis was performed on the Affymetrix Human Genome U219 Array (www.affymetrix.com, Santa Clara, CA) for ADNI and on the Illumina Whole-Genome DASL assay (www.illumina.com, San Diego, CA) for AddNeuroMed and MCSA. All probe sets were mapped and annotated with reference to the human genome (hg19). Raw microarray expression values were pre-processed followed by standard quality control (QC) procedures on samples and probe sets.[26] Briefly, raw expression values were pre-processed using the robust multi-chip average normalization method.[27] We checked discrepancies between the reported sex and sex determined from sex-specific gene expression data including XIST and USP9Y.[28] We also evaluated whether SNP genotypes were matched with genotypes predicted from gene expression data.[29] After QC, the RNA expression profiles contained 21,150 probes in ADNI.
2.3. Imaging processing
As detailed in previous studies [30–33], a widely employed automated MRI analysis technique (FreeSurfer V5.1) was used to process T1-weighted structural MRI scans.[30, 31] [18F]Florbetapir PET scans were pre-processed as described previously and were intensity normalized by the whole cerebellum.[33] The normalization yielded standardized uptake value ratio (SUVR) images.
2.4. Genotyping and imputation
Participants were genotyped using several Illumina genotyping platforms. APOE genotyping was separately conducted using standard methods as described previously to yield the APOE ε4 allele defining single nucleotide polymorphisms (SNPs) (rs429358, rs7412).[21] As the cohorts (ADNI and AddNeuroMed) used different genotyping platforms, we imputed un-genotyped SNPs separately in each cohort using MACH and the 1000 Genomes Project data as a reference panel by following the ENIGMA (Enhancing NeuroImaging Genetics through Meta-Analysis) imputation protocol (http://enigma.usc.edu/wp-content/uploads/2012/07/ENIGMA2_1KGP_cookbook_v3.pdf).[34] Before the imputation, we performed standard sample and SNP quality control procedures as described previously: (1) for SNP, SNP call rate < 95%, Hardy-Weinberg p-value < 1×10−6, and minor allele frequency (MAF) < 1%; (2) for sample, sex inconsistencies, and sample call rate < 95%.[15–17, 35] Furthermore, in order to prevent spurious association due to population stratification, we selected only non-Hispanic participants of European ancestry that clustered with HapMap CEU (Utah residents with Northern and Western European ancestry from the CEPH collection) or TSI (Toscani in Italia) populations using multidimensional scaling (MDS) analysis (www.hapmap.org) in PLINK.[35, 36] Imputation and quality control procedures were performed as described previously.[15, 16] After the imputation, we imposed an r2 value equal to 0.30 as the threshold to accept the imputed genotypes.
2.5. Statistical analysis
Statistical analysis of microarray data was performed using a linear regression model to evaluate differences in gene expression between AD and CN with age, sex, batch effects, and RNA integrity number (RIN) values as covariates. From the LIMMA software, we used the function lmFit to generate the fit, and the function eBayes to generate statistical significance values (https://doi.org/10.1007/0-387-29362-0_23).[37] Meta-analysis was performed using a fixed-effect, inverse-variance-weighted model in the METAL software (https://genome.sph.umich.edu/wiki/METAL_Documentation).[38] Significant associations were determined using false discovery rate (FDR) adjustment for multiple testing.
2.6. Imaging genetics analysis
We further investigated the association of candidate gene expression levels identified from expression profiling analysis with structural and functional neuroimaging phenotypes by performing whole brain imaging genetics analyses. Multivariable analysis of cortical thickness and amyloid-β accumulation were performed to examine effects of gene expression levels on vertex-by-vertex and voxel-by-voxel bases, respectively. In MRI scans, the cortical thickness was calculated by taking the Euclidean distance between the gray and white boundary and the gray and cerebrospinal fluid (CSF) boundary at each vertex on the surface.[39] The SurfStat software package (www.math.mcgill.ca/keith/surfstat/) was used to perform a multivariable analysis of cortical thickness on a vertex-by-vertex basis using a general linear model (GLM) approach. GLMs were developed using age, sex, years of education, MRI field strength, and total intracranial volume (ICV) as covariates. The processed [18F]Florbetapir PET images were used to perform a voxel-wise statistical analysis across the whole brain using SPM8 (www.fil.ion.ucl.ac.uk/spm/). We performed a multivariate regression analysis using age and sex as covariates. Adjustment for multiple comparisons was performed using the random field theory (RFT) correction for whole brain surface-based analysis and FDR correction methods for whole brain voxel-based analysis.[40]
3. Results
Sample characteristics in each of the three datasets used in the discovery and replication analyses are presented in Supplementary Table ST1. This included 1,440 non-Hispanic older adult participants of European ancestry (517 cognitively normal controls (CN), 553 individuals with mild cognitive impairment (MCI) and 370 with AD). The MCSA sample was older and had higher mean RIN values. The ADNI and AddNeuroMed cohorts consisted of CN and AD as well as MCI, although the MCSA cohort included only CN and AD. In addition, the ADNI and AddNeuroMed samples also had structural MRI scans.
3.1. Genome-wide transcriptome analysis
In the discovery analysis using the ADNI cohort (N=661), genome-wide comparison of AD samples with CN using 21,150 probes represented on the array after standard quality controls was assessed and led to the identification of 26 significantly differentially expressed probes after controlling for multiple testing using FDR. Probes demonstrating significantly altered expression levels were shown in Supplementary Figure SF1 (a). In the Volcano plot, red open circles represented significantly differentially expressed probes in AD compared to CN. Eighteen probes were significantly up-regulated and eight probes were significantly down-regulated in AD. The 26 probes mapped to 23 corresponding genes. The top-hit up-regulated gene was MAPK14 (mitogen-activated protein kinase 14), followed by CREB5 (cAMP responsive element binding protein 5) and CD63 (CD63 molecule). The leading down-regulated gene was TMEM41A (transmembrane BAX inhibitor motif containing 6).
3.2. Replication analysis
In total, twenty-three genes in the discovery were significantly differentially expressed in AD. However, of the 23 genes, eleven genes were observed in the replication datasets and were followed-up for replication and additional meta-analysis in the independent two cohorts (AddNeuroMed and MCSA; N=571; 304 CN, 267 AD). Of 11 genes tested in the replication samples, five genes (CREB5 (p-value=1.29x10−6), CD46 (CD46 molecule; p-value=8.19x10−6), TMBIM6 (p-value=4.00x10−3), IRAK3 (interleukin 1 receptor associated kinase 3; p-value=1.81x10−4), and RPAIN (RPA interacting protein; p-value=2.37x10−3)) were replicated and significantly differentially expressed in AD after Bonferroni correction for multiple comparisons (Table 1). Of these significant genes, the most significantly altered gene was CREB5 (up-regulated), followed by CD46 and IRAK3. Supplementary Figure SF1 (b) displays the gene expression levels of CREB5 across the continuum of AD. There is a significant increase in CREB5 levels as the severity of AD increases and the expression of CREB5 was altered from the early stages of disease. Of significantly down-regulated genes, only one gene (RPAIN) was significantly replicated in the replication sample. In addition, in the combined discovery and replication sample, another gene (FLOT1 (flotillin 1)) showed an evidence for association at genome-wide significance.
Table 1.
Meta-analysis of dysregulated genes. Five dysregulated genes were replicated in the replication sample.
| Gene | Discovery | Replication | Meta-Analysis (ALL) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ADNI | AddNeuroMed | MCSA | AddNeuroMed+MCSA | |||||||
| β (SE) | p-value | β (SE) | p-value | β (SE) | p-value | z-score | p-value | z-score | p-value | |
| CREB5 | 0.18 (0.04) | 5.03x10−6 | 0.23 (0.05) | 1.96x10−5 | 0.12 (0.05) | 2.18x10−2 | 4.84 | 1.29x10−6 | 6.61 | 3.90x10−11 |
| FLOT1 | 0.17 (0.04) | 2.50x10−5 | 0.07 (0.04) | 5.56x10−2 | 0.07 (0.06) | 1.98x10−1 | 2.28 | 2.26x10−2 | 4.35 | 1.39x10−5 |
| CD46 | 0.18 (0.04) | 2.93x10−5 | 0.20 (0.04) | 7.62x10−6 | 0.06 (0.06) | 3.31x10−1 | 4.46 | 8.19x10−6 | 6.07 | 1.26x10−9 |
| DUSP5 | −0.20 (0.05) | 3.20x10−5 | 0.03 (0.04) | 3.98x10−1 | −0.03 (0.04) | 5.07x10−1 | 0.48 | 6.32x10−1 | −2.10 | 3.59x10−2 |
| CD63 | 0.08 (0.02) | 3.68x10−5 | 0.02 (0.03) | 5.68x10−1 | 0.003 (0.061) | 9.59x10−1 | 0.54 | 5.91x10−1 | 2.89 | 3.80x10−3 |
| PELO | 0.15 (0.04) | 7.49x10−5 | 0.05 (0.03) | 7.57x10−2 | 0.04 (0.04) | 3.97x10−1 | 1.97 | 4.91x10−2 | 3.94 | 8.07x10−5 |
| TMBIM6 | 0.06 (0.02) | 9.80x10−5 | 0.07 (0.02) | 1.06x10−3 | −0.004 (0.022) | 8.53x10−1 | 2.88 | 4.00x10−3 | 4.63 | 3.58x10−6 |
| IRAK3 | 0.23 (0.06) | 9.96x10−5 | 0.17 (0.05) | 1.84x10−4 | 0.03 (0.04) | 3.93x10−1 | 3.75 | 1.81x10−4 | 5.33 | 9.96x10−8 |
| FKBP5 | 0.29 (0.07) | 1.16x10−4 | 0.13 (0.05) | 3.71x10−3 | −0.15 (0.06) | 1.17x10−2 | 1.54 | 1.24x10−1 | 3.54 | 4.06x10−4 |
| IL2R2 | 0.23 (0.06) | 1.46x10−4 | 0.08 (0.05) | 1.41x10−1 | −0.04 (0.07) | 6.08x10−1 | 1.11 | 2.68x10−1 | 3.16 | 1.60x10−3 |
| RPAIN | −0.10 (0.03) | 3.28x10−4 | −0.10 (0.03) | 1.55x10−3 | −0.01 (0.03) | 6.74x10−1 | −3.04 | 2.37x10−3 | −4.58 | 4.58x10−6 |
3.3. Follow-up for association of CREB5 gene expression levels with AD-related biomarkers
In order to investigate the effect of APOE ε4 status on levels of CREB5, the most significantly altered blood-based gene, we performed gene expression analysis after stratifying on APOE ε4 carrier status. CREB5 was significantly up-regulated in LOAD only in the APOE ε4 carrier group (β (standard error (SE)) = 0.25 (0.05), p-value = 1.37 x 10−5). We further investigated if levels of CREB5 are also associated with AD-related biomarkers. As CREB is a key component of learning and memory, we first performed an association of CREB5 gene levels with cognitive performance using composite scores for memory and executive functioning and identified significant associations (β (SE) = −0.05 (0.01), p-value = 6.73 x 10−5 and β (SE) = −0.05 (0.01), p-value = 7.74 x 10−5 for composite scores for memory and executive functioning, respectively).[41–43] Increased expression levels of CREB5 were associated with poor memory and cognitive performance.[44, 45] We then performed whole brain surface-based analysis using cortical thickness on the brain surface on vertex-by-vertex bases measured from structural MRI scans. We used the ADNI and AddNeuroMed samples as the discovery and replication samples, respectively. In the ADNI sample including MCI patients, detailed whole-brain analysis identified clusters in a widespread pattern as significantly associated with expression of CREB5 after adjusting for multiple comparisons using RFT (Fig. 1 (a)). Individuals with higher expression levels showed greater atrophy in the bilateral frontal, parietal, and temporal lobes especially including the entorhinal cortex. In the replication sample (AddNeuroMed) including MCI patients, highly significant clusters associated with CREB5 gene expression levels were found in bilateral temporal cortical regions including the entorhinal cortex, where mean cortical thickness decreased as expression levels increased, which showed consistent patterns in the same brain regions in the independent two cohorts (Fig. 1 (b)).
Figure 1.
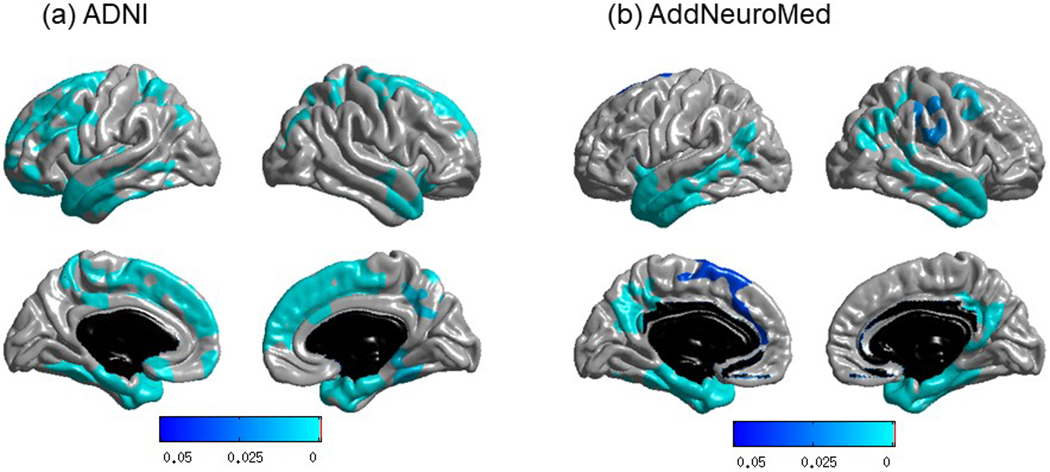
Association of CREB5 gene expression levels with brain structure atrophy using whole brain surface-based analysis in two independent cohorts: (a) ADNI (discovery sample) and (b) AddNeuroMed (replication sample). Whole-brain cortical thickness analysis demonstrated the identification and replication of brain regions, especially entorhinal cortex, significantly associated with expression levels of CREB5. Statistical maps computed using SurfStat were thresholded using random field theory (RFT) as a multiple testing correction at p-corrected < 0.05. The p-value indicates significant corrected p-values with the lightest blue color.
Next, as the amyloid-β-amyloid peptide, one of the two main pathological hallmarks of AD, mediates synapse loss through the CREB signaling pathway,[44, 45] in the ADNI cohort, we performed whole brain analysis using amyloid-β accumulation measured from [18F] Florbetapir PET scans for association of expression of CREB5 with amyloid-β load. The voxel-wise association results were similar in association direction and regional distribution to those obtained from the cortical thickness analyses (Fig. 2). Increased expression levels of CREB5 were associated with increased amyloid-β accumulation in a widespread pattern especially in the bilateral frontal, parietal, and temporal lobes after adjusting for multiple comparisons using FDR.
Figure 2.
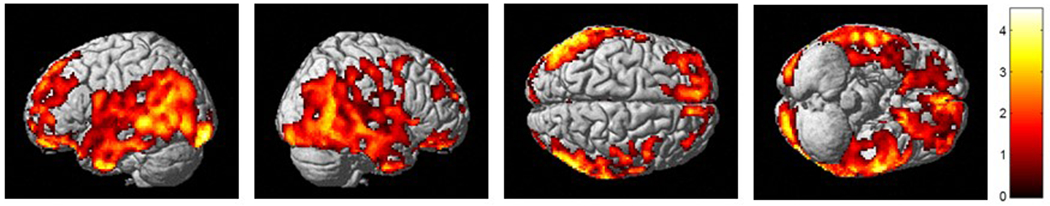
Association of CREB5 gene expression levels with amyloid-β burden measured by [18F]Florbetapir PET using whole brain analysis (FDR-corrected p-value < 0.05).
3.4. Expression quantitative trait loci (eQTL) analysis of CREB5
We performed an eQTL analysis of the most significantly altered CREB5 gene using SNPs imputed using the 1000 Genomes Project data as a reference panel in two independent cohorts (ADNI and AddNeuroMed). The analysis was limited to cis-eQTL whose SNPs were within 1 megabase (mb) distance from the transcription start or end sites of CREB5. In the ADNI sample, the eQTL mapping analysis of CREB5 detected five significant associations with genome-wide significance at p=5x10−8 (Fig. 3 (a)).
Figure 3.
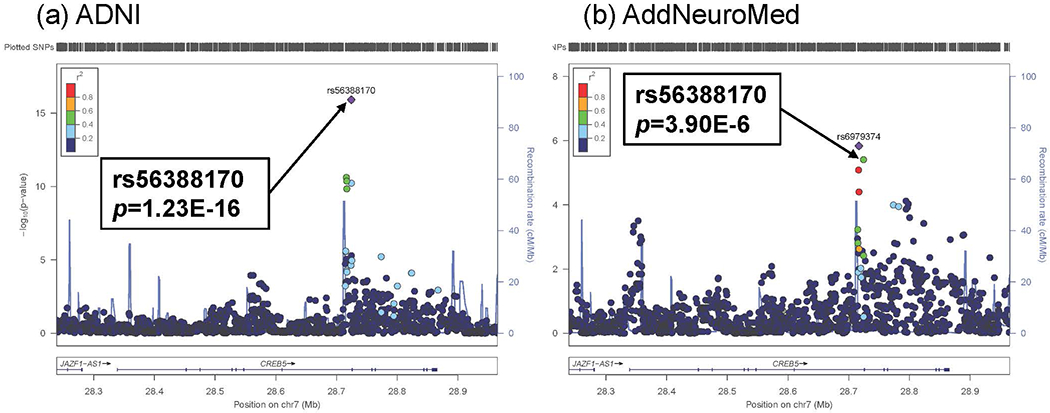
Results of cis-eQTL mapping analysis of CREB5 using two independent cohorts: (a) ADNI (discovery sample) and (b) AddNeuroMed (replication sample). cis-eQTL mapping analyses of CREB5 detected 5 significant associations with p < 5x10−8 in ADNI. The most significant cis-eQTL SNP (rs56388170) in ADNI was replicated in AddNeroMed. All SNPs are plotted based on their −log10 p-values, NCBI build 37 genomic position, and recombination rates calculated from the 1000 Genomes Project reference data.
Of these significant SNPs, rs56388170 located within a 2 kb upstream region of CREB5 was most significantly associated with expression levels of CREB5 (β (SE) = −0.15 (0.02), p=1.23 x 10−16). Individuals carrying the minor allele of rs56388170 have lower expression levels of CREB5. In addition, the most significant SNP (rs56388170) was significantly replicated in the independent AddNeuroMed sample (β (SE) = −0.22 (0.05), p=3.90 x 10−6; Fig. 3 (b)). As we showed that CREB5 expression levels are associated with amyloid-β accumulation in the brain, we determined if rs56388170 is associated with amyloid-β load in AD. The most significant eQTL SNP (rs56388170) of CREB5 was also significantly associated with global cortical amyloid-β load measured from [18F] Florbetapir PET scans and CSF amyloid-β1-42 with p=0.021 (β (SE) = −0.008 (0.001)) and p=0.035 (β (SE) = 0.013 (0.025)), respectively (Fig. 4 (a) and (b)), where the minor allele of rs56388170 conferred decreases in cortical amyloid-β levels.
Figure 4.
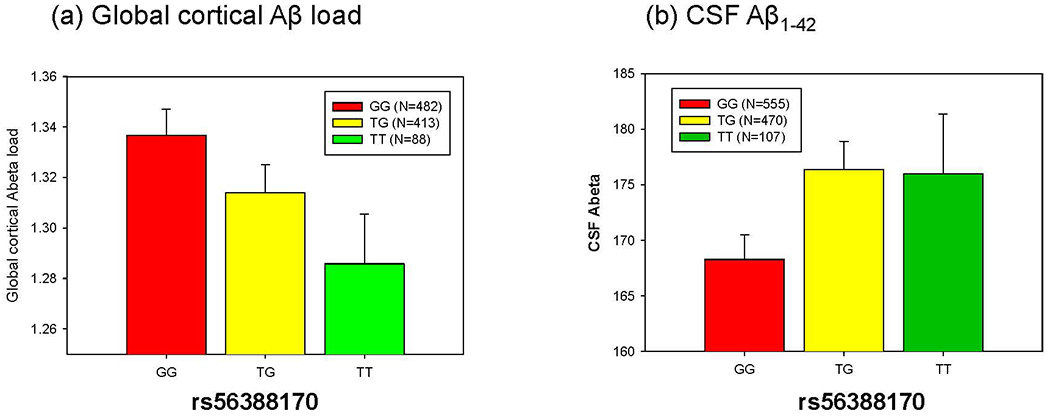
Association of the most significant cis-eQTL SNP (rs56388170) of CREB5 gene expression levels with (a) global cortical Aβ levels measured by [18F]Florbetapir PET and (b) CSF Aβ1-42 in the ADNI. rs56388170 was significantly associated with global cortical Aβ load and CSF Aβ1-42.
4. Discussion
This study identified and replicated five significantly differentially expressed genes as well as one marginally differentially expressed gene in AD in peripheral blood, five of which are upregulated (CREB5, FLOT1, CD46, IRAK3, and TMBIM6) and one of which is downregulated in AD (RPAIN). A literature search revealed no evidence for previous linkage of either RPAIN or TMBIM6 with AD, suggesting that further exploration of the functions of these genes may yield novel information about AD processes. RPAIN is involved in transport of replication protein A (RPA), a eukaryotic single-strand DNA binding protein with functions in DNA replication, repair, and recombination; RPAIN is an adaptor molecule that binds RPA and importin-β, and thus rehulates RPA transport into the nucleus. With this function, RPAIN, via RPA, plays an important role in cell proliferation via cell cycle regulation.[46] RPAIN is ubiquitously expressed in numerous tissues including the brain.[47] Altered RPAIN expression could potentially lead to reduced efficiency in DNA repair as well as cell cycle dysregulation, which has been previously observed in AD.[48] TMBIM6, also called Bax Inhibitor-1, is a transmembrane protein in the endoplasmic reticulum (ER). TMBIM6 is highly conserved and ubiquitously expressed in humans, and has been shown to play a role in numerous cellular pathways including ER stress, calcium imbalance, reactive oxygen species accumulation, and metabolic dysregulation. More specifically, this protein is involved in cellular calcium and pH homeostasis by mediating Ca(2+) efflux from the endoplasmic reticulum (ER), and functions to protect the cell from ER stress-induced apoptotic cell death, though the exact molecular mechanism underlying this protein function is currently unclear.[49–51] Altered TMBIM6 expression could potentially dysregulate cellular response to stress, with interesting implications for AD susceptibility and progression. Upregulation of this protein, as observed in this study, may be a compensatory response to neuronal damage in AD. There is some evidence to suggest that TMBIM6 may play a protective role in the brain, both by protecting against apoptosis as well as potentially promoting neurogenesis during development. Interestingly, TMBIM6 was reported to bind free presinilin 1; the resulting complex exhibited no proteolytic activity for amyloid beta, suggesting that this protein may play an important role in AD.[52] However, this work is based largely on animal and cellular models; more work remains to elucidate the functions of TMBIM6 in the human brain.
For three of the other genes significantly upregulated in this study (CD46, IRAK3, and CREB5), while there is evidence in the literature linking homologues or gene pathways to AD, there is a dearth of evidence directly linking CD46, IRAK3, and CREB5 to AD. CD46 has not been associated with AD; however, this ubiquitously expressed complement receptor protein has been shown to be necessary to the proper functioning of human cytotoxic CD8+ T cells in humans.[47, 53] CD46 delivers co-stimulatory signals promoting cytotoxic CD8+ T cell activity; mutations, function or regulation of this protein have been associated with viral infections as well as autoimmune diseases.[54–60] While CD46 has not yet been associated with AD, the complement system has been significantly linked to AD. CD33, another complement protein, is one of the top hits identified in the largest AD GWAS meta-analysis to date.[61] It has been postulated that the complement system works to increase inflammation, and that the AD-protective variant in CD33 truncates the protein, resulting in reduced functions including cell signaling.[62] This suggests that lower expression of complement genes may have beneficial effects on the brain, aligning with our data showing lower CD46 expression in controls compared to those with AD. IRAK3, another of the significantly upregulated genes, encodes an interleukin-1 receptor-associated kinase, which is ubiquitously expressed and functions in the Toll/IL-R immune signal transduction pathway.[47, 63] While there was no evidence from our literature search associating IRAK3 with AD, other interleukin 1 receptor associated kinases have been associated with pro-inflammatory processes in AD,[64, 65] suggesting that these molecules play important roles in AD. Finally, CREB5 binds to cAMP response element (CRE) to function as a CRE-dependent trans-activator. CREB proteins induce CRE-mediated gene transcription in response to cellular signaling.[66] cAMP signaling is involved in many cellular processes from cell growth and differentiation to gene transcription and protein expression.[67] While CREB5 has not been specifically linked to AD, CREB family proteins are known to play important roles in synaptic strengthening and memory formation.[45] Increased CREB activity was observed in the peripheral blood of patients with mild AD or those with AD and depressive symptoms,[68] and increased CREB activity and dysregulation of CREB targets were observed in AD brain tissue compared to controls.[44] These previous results from the literature support the potential roles of CD46, IRAK3, and CREB5 in AD.
In addition to these five genes, our meta-analysis of all three data sets identified FLOT1 as significantly upregulated in AD. FLOT1 is a ubiquitously expressed protein that localizes to the cavaolae, small domains on the inner cell membrane, and functions in vesicle trafficking and cell morphology pathways.[69–71] Flotillin proteins including FLOT1 have also been shown to play a role in endosomal sorting of BACE-1, a protease involved in APP processing; depletion of flotilins results in stabilized BACE-1 and increased amyloidogenic processing of amyloid precursor protein.[72] Thus, it is possible that upregulation of flotilins may be a compensatory resonse by the cell to attempt to reduce amyloidogenic processing. Future analyses should investigate the expression of this gene in additional data sets for replication, as well as exploration of the impact of this protein on BACE-1 function in conjunction with AD risk and progression.
To extend interpretation of our findings and how the identified genes might impact the brain in LOAD, we also identified several larger AD brain tissue gene expression studies, and downloaded the lists of all differentially expressed genes. While a comprehensive review of previous results was outside the scope of this study, we selected several of the larger, better powered studies to discuss. We reviewed these lists of differentially expressed genes from these studies for presence of any of the five genes we identified as differentially expressed in AD in peripheral blood. The first study, of brain tissue collected from the current Mount Sinai Medical Center Brain Bank AD cohort, included expression data from 19 brain regions for 125 individuals across the AD spectrum, excluding any brain specimens that showed non-AD related neuropathology. Gene expression was generated using the Human Genome (HG) Affymetrix U133A, U133B, or U133 Plus 2.0 microarrays. The researchers created high/normal/low groups based on AD traits: clinical dementia rating (CDR), Braak stage (tau pathology), Consortium to Establish a Registry for Alzheimer’s Disease (CERAD; amyloid pathology) diagnostic certainty, amyloid plaque density mean, sum of neuritic plaque density estimates, and sum of neurofibrillary tangle density estimates. Differences between AD trait groups in at least one brain region were observed for CREB5, TMBIM6, and RPAIN.[13] The second study, which included brain tissue gene expression from 1,647 specimens from LOAD and non-demented controls, investigated the association of gene networks with AD traits [14]. Study participants were recruited through the Harvard Brain Tissue Resource Center. Tissue specimens were from three brain regions, the dorsolateral prefrontal cortex, visual cortex, or cerebellum. Specifically, specimens were obtained from 549 brains, 376 of which were from LOAD patients, and 173 of which were from non-demented controls. In this data set, CREB5 and IRAK3 were included in networks of differentially expressed genes, but were not significantly associated with any of the AD traits analyzed. Another study, which included 87 individuals with AD and 74 controls, included lists of genes with differential expression associated with AD by brain regions.[12] The researchers analyzed differential expression in six brain regions: the entorhinal cortex, hippocampus, middle temporal gyrus, posterior cingulate cortex, superior frontal gyrus, and visual cortex; data was obtained from a study conducted by Liang et al (2008) of gene expression laser-capture microdissected non-tangle-bearing neurons in different brain regions.[73] The authors used a novel computational method to integrate gene expression information across brain regions, to identify a minimum common set of genes significantly associated with AD across brain regions. In this study, genes with differential expression associated with AD in at least one brain region included CD46, TMBIM6, IRAK3, RPAIN, and FLOT1. Finally, the last study by Ibanez et al. (2015) included two AD data sets: 1) hippocampal gene expression from nine controls and 15 individuals with AD,[74] and 2) brain tissue gene expression for varying numbers of individuals (all groups size range nine to 23) classified into each Braak stage (0–6) with 161 individuals total included.[75, 76] This meta-analysis identified differential regulation associated with AD for CREB5, IRAK3, and FLOT1, though these comparisons did not survive multiple correction. To summarize, all of the six genes of interest from our study were identified in at least one of these prior studies of AD-related differential brain tissue gene expression. These findings support potential roles for brain function of the six genes of interest from our study of blood-based gene expression.
There are several plausible hypotheses regarding the mechanistic implications of the differential gene expression levels identified in blood. First, there may be genetic variants that drive expression levels of genes in both brain and blood. We have previously shown that many AD risk variants influence brain gene expression levels and are therefore likely to exert AD risk through their regulatory effects in the brain.[77–81] Some but not all eQTL have consistent effects across multiple tissue types.[82, 83] It is possible that some or all of the peripheral differential expression detected in this study are a reflection of brain eQTL for those variants that have similar eQTL effects in blood. The associations of eQTL with CREB5 levels, and the influence of the same eQTL with brain amyloid-ß levels, are consistent with this hypothesis.
The top genes identified in this study are implicated in pathways that are known to be important in AD pathophysiology such as synaptic processes (CREB5), endosomal sorting (FLOT1), immune system (CD46, IRAK3) and calcium homeostasis (TMBIM6). Hence, genetic variants that influence brain levels of these genes may also have an impact on these pathways and ultimately AD risk. Given the genetic heterogeneity of complex diseases, such as AD, eQTL and differential gene expression effects may be more readily detected than AD risk associations.[84] Therefore, expression profiling and eQTL studies provide a complementary approach to AD risk association in the identification of novel AD candidate genes and pathways.
Another plausible explanation of our results is that expression changes in blood may be reflective of cellular composition and other alterations in the brain that are downstream of disease pathology. Even though the majority of the expression levels captured by the blood measurements are likely driven by white blood cells, it is possible that these measurements also capture transcripts from other circulating cells, exosomal or cell free RNA. Single cell type, exosomal and cell free RNA based studies are necessary to fully delineate the source of these peripheral expression levels. Given this possibility, another hypothesis is that peripheral gene expression levels are biomarkers of underlying disease pathology and/or its consequences, similar to blood amyloid-ß or cytokine measurements.[85, 86]
These two hypotheses can be tested by various approaches, including multi-omics studies of large cohorts to discover eQTL, differential expression and AD risk associations implicating the same gene; longitudinal blood expression studies in cohorts that also have concurrent brain pathology biomarkers, including neuroimaging and CSF; and finally in model systems where levels of these genes are perturbed and tested for AD-related outcomes.
This study has several limitations. First, this is an observational study, where we restricted our analysis to a non-Hispanic White population. Therefore, our findings may not be generalizable to other populations. It is important for future studies to investigate our findings using large community studies that include populations with greater diversity to determine if they translate to the broader population. Second, we performed gene expression analysis based on three independent studies using two different microarray platforms; despite this, our results were replicable results across these cohorts, which indicates that our findings are unlikely to be driven by technical artefacts. In addition, we performed a meta-analysis of three datasets instead of a mega-analysis. Third, the diagnosis was based largely on clinical criteria without neuropathology confirmation, which is a common limitation for ante-mortem studies. We addressed this by using AD-related endophenotypes measured by multimodal neuroimaging and CSF biomarkers. Further, we explored the findings from brain tissue studies using neuropathologically diagnosed cases. Lastly, our study is cross-sectional and it would be important in the future to use a larger prospective cohort study to determine the potential role of the identified genes.
Overall, six genes identified in this study may have potential to provide further insight into the biological mechanisms underlying AD risk and progression. Further study is required to determine the specific roles of CREB5, CD46, TMBIM6, IRAK3, RPAIN, and FLOT1 in AD-related processes. Expression of these genes in peripheral blood should also be investigated for potential to enhance current biomarker measures of AD risk/progression.
Supplementary Material
Acknowledgements
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01AG024904) and DODADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Funding: Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). Additional support for data analysis was provided by NLM R01 LM012535, NIA R03 AG054936, NIA R01 AG19771, NIA P30 AG10133, NLM R01 LM011360, DOD W81XWH-14-2-0151, NIGMS P50GM115318, NCATS UL1 TR001108, NIA K01 AG049050, the Alzheimer’s Association, the Indiana Clinical and Translational Science Institute, and the IU Health-IU School of Medicine Strategic Neuroscience Research Initiative. This work was also supported by the National Institute on Aging RF AG051504 to N.E.T.; U01 AG046139 to N.E.T. and U01 AG006786 to R.C.P.
Abbreviations:
- AD
Alzheimer’s disease
- LOAD
late-onset Alzheimer’s disease
- MCI
mild cognitive impairment
- APOE
apolipoprotein E
- CSF
cerebrospinal fluid
- MCSA
Mayo Clinic Study of Aging
- ADNI
Alzheimer’s Disease Neuroimaging Initiative
- cis-eQTL
cis-expression quantitative trait loci
- MRI
magnetic resonance imaging
- PET
positron emission tomography
- CN
cognitively normal older adults
- RIN
RNA integrity number
- FDR
false discovery rate
- CREB5
cAMP responsive element binding protein 5
- CD46
CD46 molecule
- TMBIM6
transmembrane BAX inhibitor motif containing 6
- IRAK3
interleukin 1 receptor associated kinase 3
- RPAIN
RPA interacting protein
- SNP
single nucleotide polymorphism
- FLOT1
flotillin 1
- CD33
CD33 molecule
- APP
amyloid beta precursor protein
- BACE
beta-site amyloid precursor protein cleaving enzyme
- MDS
multidimensional scaling
- CEU
Utah residents with Northern and Western European ancestry from the CEPH collection
- TSI
Toscani in Italia
- ICV
intracranial volume
- MAPK14
mitogen-activated protein kinase 14
- CD63
CD63 molecule
- RFT
random field theory
- ENIGMA
Enhancing NeuroImaging Genetics through Meta-Analysis
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Data
Supplementary data related to this article can be found at the journal website.
References
- [1].Wang J, Gu BJ, Masters CL, Wang YJ. A systemic view of Alzheimer disease - insights from amyloid-beta metabolism beyond the brain. Nat Rev Neurol. 2017;13:703. [DOI] [PubMed] [Google Scholar]
- [2].Kumar A, Singh A, Ekavali. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep 2015;67:195–203. [DOI] [PubMed] [Google Scholar]
- [3].Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1:15056. [DOI] [PubMed] [Google Scholar]
- [5].Adlard PA, Tran BA, Finkelstein DI, Desmond PM, Johnston LA, Bush AI, et al. A review of beta-amyloid neuroimaging in Alzheimer’s disease. Front Neurosci. 2014;8:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66:1469–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Herholz K, Ebmeier K. Clinical amyloid imaging in Alzheimer’s disease. Lancet Neurol. 2011;10:667–70. [DOI] [PubMed] [Google Scholar]
- [8].Huynh RA, Mohan C. Alzheimer’s Disease: Biomarkers in the Genome, Blood, and Cerebrospinal Fluid. Front Neurol 2017;8:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Humpel C, Hochstrasser T. Cerebrospinal fluid and blood biomarkers in Alzheimer’s disease. World J Psychiatry. 2011;1:8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Identifying Humpel C. and validating biomarkers for Alzheimer’s disease. Trends Biotechnol. 2011;29:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].O’Bryant SE, Mielke MM, Rissman RA, Lista S, Vanderstichele H, Zetterberg H, et al. Blood-based biomarkers in Alzheimer disease: Current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimers Dement. 2017;13:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Puthiyedth N, Riveros C, Berretta R, Moscato P. Identification of Differentially Expressed Genes through Integrated Study of Alzheimer’s Disease Affected Brain Regions. PLoS One. 2016;11:e0152342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang M, Roussos P, McKenzie A, Zhou X, Kajiwara Y, Brennand KJ, et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 2016;8:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nho K, Corneveaux JJ, Kim S, Lin H, Risacher SL, Shen L, et al. Identification of functional variants from whole-exome sequencing, combined with neuroimaging genetics. Molecular psychiatry. 2013;18:739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nho K, Corneveaux JJ, Kim S, Lin H, Risacher SL, Shen L, et al. Whole-exome sequencing and imaging genetics identify functional variants for rate of change in hippocampal volume in mild cognitive impairment. Molecular psychiatry. 2013;18:781–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nho K, Kim S, Risacher SL, Shen L, Corneveaux JJ, Swaminathan S, et al. Protective variant for hippocampal atrophy identified by whole exome sequencing. Ann Neurol. 2015;77:547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ramanan VK, Nho K, Shen L, Risacher SL, Kim S, McDonald BC, et al. FASTKD2 is associated with memory and hippocampal structure in older adults. Mol Psychiatry. 2015;20:1197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ramanan VK, Risacher SL, Nho K, Kim S, Shen L, McDonald BC, et al. GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer’s disease implicates microglial activation gene IL1RAP. Brain. 2015;138:3076–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ramanan VK, Risacher SL, Nho K, Kim S, Swaminathan S, Shen L, et al. APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study. Molecular psychiatry. 2014;19:351–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Saykin AJ, Shen L, Yao X, Kim S, Nho K, Risacher SL, et al. Genetic studies of quantitative MCI and AD phenotypes in ADNI: Progress, opportunities, and plans. Alzheimers Dement 2015;11:792–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Westman E, Simmons A, Muehlboeck JS, Mecocci P, Vellas B, Tsolaki M, et al. AddNeuroMed and ADNI: similar patterns of Alzheimer’s atrophy and automated MRI classification accuracy in Europe and North America. Neuroimage. 2011;58:818–28. [DOI] [PubMed] [Google Scholar]
- [23].Simmons A, Westman E, Muehlboeck S, Mecocci P, Vellas B, Tsolaki M, et al. MRI measures of Alzheimer’s disease and the AddNeuroMed study. Ann N Y Acad Sci. 2009;1180:47–55. [DOI] [PubMed] [Google Scholar]
- [24].Lovestone S, Francis P, Kloszewska I, Mecocci P, Simmons A, Soininen H, et al. AddNeuroMed--the European collaboration for the discovery of novel biomarkers for Alzheimer’s disease. Ann N Y Acad Sci. 2009;1180:36–46. [DOI] [PubMed] [Google Scholar]
- [25].Roberts RO, Geda YE, Knopman DS, Cha RH, Pankratz VS, Boeve BF, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008;30:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lunnon K, Sattlecker M, Furney SJ, Coppola G, Simmons A, Proitsi P, et al. A blood gene expression marker of early Alzheimer’s disease. J Alzheimers Dis. 2013;33:737–53. [DOI] [PubMed] [Google Scholar]
- [27].Choe SE, Boutros M, Michelson AM, Church GM, Halfon MS. Preferred analysis methods for Affymetrix GeneChips revealed by a wholly defined control dataset. Genome Biol. 2005;6:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vawter MP, Evans S, Choudary P, Tomita H, Meador-Woodruff J, Molnar M, et al. Gender-specific gene expression in post-mortem human brain: localization to sex chromosomes. Neuropsychopharmacology. 2004;29:373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schadt EE, Woo S, Hao K. Bayesian method to predict individual SNP genotypes from gene expression data. Nat Genet. 2012;44:603–8. [DOI] [PubMed] [Google Scholar]
- [30].Risacher SL, Shen L, West JD, Kim S, McDonald BC, Beckett LA, et al. Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol Aging. 2010;31:1401–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Risacher SL, Saykin AJ, West JD, Shen L, Firpi HA, McDonald BC, et al. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr Alzheimer Res. 2009;6:347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jack CR Jr., , Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. Journal of magnetic resonance imaging : JMRI 2008;27:685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Risacher SL, Kim S, Nho K, Foroud T, Shen L, Petersen RC, et al. APOE effect on Alzheimer’s disease biomarkers in older adults with significant memory concern. Alzheimers Dement. 2015;11:1417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Thompson PM, Stein JL, Medland SE, Hibar DP, Vasquez AA, Renteria ME, et al. The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav. 2014;8:153–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. [DOI] [PubMed] [Google Scholar]
- [37].Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chung MK, Worsley KJ, Nacewicz BM, Dalton KM, Davidson RJ. General multivariate linear modeling of surface shapes using SurfStat. Neuroimage. 2010;53:491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Worsley KJ, Andermann M, Koulis T, MacDonald D, Evans AC. Detecting changes in nonisotropic images. Hum Brain Mapp. 1999;8:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Crane PK, Carle A, Gibbons LE, Insel P, Mackin RS, Gross A, et al. Development and assessment of a composite score for memory in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav 2012;6:502–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gibbons LE, Carle AC, Mackin RS, Harvey D, Mukherjee S, Insel P, et al. A composite score for executive functioning, validated in Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav. 2012;6:517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nho K, Risacher SL, Crane PK, DeCarli C, Glymour MM, Habeck C, et al. Voxel and surface-based topography of memory and executive deficits in mild cognitive impairment and Alzheimer’s disease. Brain Imaging Behav. 2012;6:551–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Satoh J, Tabunoki H, Arima K. Molecular network analysis suggests aberrant CREB-mediated gene regulation in the Alzheimer disease hippocampus. Dis Markers. 2009;27:239–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Teich AF, Nicholls RE, Puzzo D, Fiorito J, Purgatorio R, Fa M, et al. Synaptic therapy in Alzheimer’s disease: a CREB-centric approach. Neurotherapeutics. 2015;12:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Namkoong S, Lee EJ, Jang IS, Park J. Elevated level of human RPA interacting protein alpha (hRIPalpha) in cervical tumor cells is involved in cell proliferation through regulating RPA transport. FEBS Lett. 2012;586:3753–60. [DOI] [PubMed] [Google Scholar]
- [47].Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Varvel NH, Bhaskar K, Patil AR, Pimplikar SW, Herrup K, Lamb BT. Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease. J Neurosci. 2008;28:10786–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kiviluoto S, Luyten T, Schneider L, Lisak D, Rojas-Rivera D, Welkenhuyzen K, et al. Bax Inhibitor-1-mediated Ca2+ leak is decreased by cytosolic acidosis. Cell Calcium. 2013;54:186–92. [DOI] [PubMed] [Google Scholar]
- [50].Bultynck G, Kiviluoto S, Methner A. Bax inhibitor-1 is likely a pH-sensitive calcium leak channel, not a H+/Ca2+ exchanger. Sci Signal. 2014;7:pe22. [DOI] [PubMed] [Google Scholar]
- [51].Lebeaupin C, Blanc M, Vallee D, Keller H, Bailly-Maitre B. Bax Inhibitor-1: between stress and survival. FEBS J 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wu S, Song W, Wong CCL, Shi Y. Bax inhibitor 1 is a gamma-secretase-independent presenilin-binding protein. Proceedings of the National Academy of Sciences of the United States of America. 2019;116:141–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Arbore G, West EE, Rahman J, Le Friec G, Niyonzima N, Pirooznia M, et al. Complement receptor CD46 co-stimulates optimal human CD8(+) T cell effector function via fatty acid metabolism. Nat Commun 2018;9:4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tsai YG, Wen YS, Wang JY, Yang KD, Sun HL, Liou JH, et al. Complement regulatory protein CD46 induces autophagy against oxidative stress-mediated apoptosis in normal and asthmatic airway epithelium. Sci Rep 2018;8:12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ellinghaus U, Cortini A, Pinder CL, Le Friec G, Kemper C, Vyse TJ. Dysregulated CD46 shedding interferes with Th1-contraction in systemic lupus erythematosus. Eur J Immunol. 2017;47:1200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Qiao P, Dang E, Cao T, Fang H, Zhang J, Qiao H, et al. Dysregulation of mCD46 and sCD46 contribute to the pathogenesis of bullous pemphigoid. Sci Rep 2017;7:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Johansson L, Rytkonen A, Wan H, Bergman P, Plant L, Agerberth B, et al. Human-like immune responses in CD46 transgenic mice. J Immunol. 2005;175:433–40. [DOI] [PubMed] [Google Scholar]
- [58].Stein KR, Gardner TJ, Hernandez RE, Kraus TA, Duty JA, Ubarretxena-Belandia I, et al. CD46 facilitates entry and dissemination of human cytomegalovirus. Nat Commun. 2019;10:2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu J, Boehme P, Zhang W, Fu J, Yumul R, Mese K, et al. Human adenovirus type 17 from species D transduces endothelial cells and human CD46 is involved in cell entry. Sci Rep. 2018;8:13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liszewski MK, Atkinson JP. Complement regulator CD46: genetic variants and disease associations. Hum Genomics. 2015;9:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Siddiqui SS, Springer SA, Verhagen A, Sundaramurthy V, Alisson-Silva F, Jiang W, et al. The Alzheimer’s disease-protective CD33 splice variant mediates adaptive loss of function via diversion to an intracellular pool. J Biol Chem. 2017;292:15312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Freihat LA, Wheeler JI, Wong A, Turek I, Manallack DT, Irving HR. IRAK3 modulates downstream innate immune signalling through its guanylate cyclase activity. Sci Rep. 2019;9:15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cameron B, Tse W, Lamb R, Li X, Lamb BT, Landreth GE. Loss of interleukin receptor-associated kinase 4 signaling suppresses amyloid pathology and alters microglial phenotype in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32:15112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cui JG, Li YY, Zhao Y, Bhattacharjee S, Lukiw WJ. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-kappaB in stressed human astroglial cells and in Alzheimer disease. J Biol Chem. 2010;285:38951–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Koyanagi S, Hamdan AM, Horiguchi M, Kusunose N, Okamoto A, Matsunaga N, et al. cAMP-response element (CRE)-mediated transcription by activating transcription factor-4 (ATF4) is essential for circadian expression of the Period2 gene. J Biol Chem. 2011;286:32416–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yan K, Gao LN, Cui YL, Zhang Y, Zhou X. The cyclic AMP signaling pathway: Exploring targets for successful drug discovery (Review). Mol Med Rep. 2016;13:3715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Platenik J, Fisar Z, Buchal R, Jirak R, Kitzlerova E, Zverova M, et al. GSK3beta, CREB, and BDNF in peripheral blood of patients with Alzheimer’s disease and depression. Prog Neuropsychopharmacol Biol Psychiatry. 2014;50:83–93. [DOI] [PubMed] [Google Scholar]
- [69].Langhorst MF, Reuter A, Stuermer CA. Scaffolding microdomains and beyond: the function of reggie/flotillin proteins. Cell Mol Life Sci. 2005;62:2228–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Babuke T, Tikkanen R. Dissecting the molecular function of reggie/flotillin proteins. Eur J Cell Biol. 2007;86:525–32. [DOI] [PubMed] [Google Scholar]
- [71].Xiong Q, Lin M, Huang W, Rikihisa Y. Infection by Anaplasma phagocytophilum Requires Recruitment of Low-Density Lipoprotein Cholesterol by Flotillins. mBio. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].John BA, Meister M, Banning A, Tikkanen R. Flotillins bind to the dileucine sorting motif of beta-site amyloid precursor protein-cleaving enzyme 1 and influence its endosomal sorting. FEBS J 2014;281:2074–87. [DOI] [PubMed] [Google Scholar]
- [73].Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4441–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ibanez K, Boullosa C, Tabares-Seisdedos R, Baudot A, Valencia A. Molecular evidence for the inverse comorbidity between central nervous system disorders and cancers detected by transcriptomic meta-analyses. PLoS Genet. 2014;10:e1004173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiol Genomics. 2008;33:240–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zou F, Chai HS, Younkin CS, Allen M, Crook J, Pankratz VS, et al. Brain expression genome-wide association study (eGWAS) identifies human disease-associated variants. PLoS Genet 2012;8:e1002707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Allen M, Zou F, Chai HS, Younkin CS, Crook J, Pankratz VS, et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology. 2012;79:221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Allen M, Kachadoorian M, Quicksall Z, Zou F, Chai HS, Younkin C, et al. Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimers Res Ther. 2014;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Allen M, Kachadoorian M, Carrasquillo MM, Karhade A, Manly L, Burgess JD, et al. Late-onset Alzheimer disease risk variants mark brain regulatory loci. Neurol Genet. 2015;1:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Carrasquillo MM, Allen M, Burgess JD, Wang X, Strickland SL, Aryal S, et al. A candidate regulatory variant at the TREM gene cluster associates with decreased Alzheimer’s disease risk and increased TREML1 and TREM2 brain gene expression. Alzheimers Dement. 2017;13:663–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KC, et al. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–7. [DOI] [PubMed] [Google Scholar]
- [83].Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–8. [DOI] [PubMed] [Google Scholar]
- [84].Ertekin-Taner N, De Jager PL, Yu L, Bennett DA. Alternative Approaches in Gene Discovery and Characterization in Alzheimer’s Disease. Curr Genet Med Rep. 2013;1:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Snyder HM, Carrillo MC, Grodstein F, Henriksen K, Jeromin A, Lovestone S, et al. Developing novel blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement. 2014;10:109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Blennow K, Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. J Intern Med. 2018;284:643–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.