

Abstract
Cancer cachexia patients experience significant muscle wasting, which impairs the quality of life and treatment efficacy for patients. Skeletal muscle protein turnover is imparted by increased expression of ubiquitin-proteasome pathway components. Mitogen-activated protein kinases p38 and ERK have been shown to augment E3 ubiquitin ligase expression. Utilizing reverse phase protein arrays, we identified pancreatic cancer cell-conditioned media-induced activation of JNK signaling in myotubes differentiated from C2C12 myoblasts. Inhibition of JNK signaling with SP600125 reduced cancer cell-conditioned media-induced myotube atrophy, myosin heavy chain 2 protein turnover, and mRNA expression of cachexia-specific ubiquitin ligases Trim63 and Fbxo32. Furthermore, utilizing an orthotopic pancreatic cancer cachexia mouse model, we demonstrated that treatment of tumor-bearing mice with SP600125 improved longitudinal measurements forelimb grip strength. Post-necropsy measurements demonstrated that SP600125 treatment rescued body weight, carcass weight, and gastrocnemius muscle weight without impacting tumor growth. JNK inhibitor treatment also rescued myofiber degeneration and reduced the muscle expression of Trim63 and Fbxo32. These data demonstrate that JNK signaling contributes to muscle wasting in cancer cachexia and its inhibition has the potential to be utilized as an anti-cachectic therapy.
Keywords: Cancer cachexia, Muscle wasting, Ubiquitin ligases, JNK signaling
1. Introduction
Cancer remains a global burden despite advances in diagnostics and therapeutics, and it is the second leading cause of deaths worldwide [1]. Cancer lethality can be attributed to factors such as late diagnosis, drug resistance, metastasis, and recurrence. However, cancer cachexia plays an under-appreciated role in cancer patient survival. Cancer cachexia is a complex metabolic syndrome that manifests through systemic inflammation, elevated protein catabolism, energy imbalance, and uncontrollable loss of lean body mass [2]. The syndrome not only impacts the quality of life of cancer patients but also reduces the tolerance of treatments and survival [3]. Cachexia is highly prevalent in cancer patients, as about half of all cancer patients are afflicted by the disease [4]. Cancer cachexia is most prevalent in gastrointestinal malignancies [5]. Compared to other cancers, pancreatic cancer patients experience the syndrome at the highest rate of 80% [5]. Approximately a third of pancreatic cancer patients ultimately succumb to cachexia-associated complications [3]. When cachectic patients advance to a refractory stage, they no longer respond to treatments aimed at improving appetite, mitigating inflammation, or nutritional supplementation [2]. Therefore, there is a dire need to identify molecular targets that are effective in combating cancer cachexia.
Cancer cachexia is distinguished by several molecular characteristics that may be therapeutically exploited. A prominent feature of muscle wasting is an elevated state of proteolysis, which is due to increased amounts of autophagy and the induction of ubiquitin ligases that cause muscle protein degradation via the ubiquitin proteasomal pathway [6]. E3 ubiquitin ligases that are implicated in the pathogenesis of cachexia and contribute to proteolysis include Atrogin1 (Fbxo32) and MuRF1 (Trim63) [7, 8]. Previous studies have identified the role of p38 and extracellular signal-regulated kinase (ERK) mitogen-activated protein kinases (MAPKs) in contributing to elevated protein degradation in cachexia by inducing the expression of Trim63 and Fbxo32 [9, 10]. However, the potential roles of other MAPKs in cancer cachexia, such as c-Jun N-terminal Kinase (JNK), are yet to be fully elucidated and may provide novel therapeutic targets for better management of the disease.
JNK has known roles in normal muscle physiology. Activation of JNK signaling negatively regulates skeletal muscle differentiation and is downregulated during myogenesis [11]. Additionally, JNK regulates muscle remodeling by inhibiting myostatin and SMAD2 [12]. A deficiency in JNK also has a metabolic impact, and it causes cultured myotubes to increase fatty acid utilization, reduce glucose oxidation, and improves insulin sensitivity [13]. The pharmacological inhibition of toll-like receptors with IMO-8503 has been recently demonstrated to abrogate JNK-dependent muscle cell death in culture conditions and in mice implanted with Lewis lung carcinoma cells [14]. However, the role of JNK in other aspects of muscle homeostasis during cancer cachexia, such as protein catabolism, remains unknown and the utility of direct targeting of JNK in ameliorating cancer cachexia has not been established.
Here, we investigated the role of JNK in muscle catabolism in pancreatic cancer-induced cachexia. We performed reverse-phase protein array-based profiling of phospho-proteome to identify key signaling pathways in differentiated C2C12 myotube activated upon treatment with pancreatic cancer cell-conditioned media (CM). We identified an upregulation in the phosphorylation of JNK and JNK-pathway proteins in myotubes subjected to the CM. We confirmed that CM activates JNK in myotubes via immunoblotting, and identified that pharmacological inhibition of JNK mitigates myotube thinning and diminishes the expression of cachexia-related E3 ubiquitin ligases. Intraperitoneal injection (IP) of the JNK inhibitor SP600125 in an orthotopic pancreatic cancer mouse model significantly reduced cachexia as it abrogated body and muscle weight loss, improved forelimb grip strength, and rescued myofiber atrophy. JNK inhibition mitigated cancer cachexia, at least in part, by reducing the expression of proteolysis-inducing E3 ubiquitin ligases.
2. Materials and methods
2.1. Cell Culture
All cells were cultured at 37 °C in a humidified cell culture incubator with 5% CO2. S2–013 pancreatic cancer cells were obtained from Dr. Michael A. Hollingsworth [15], and were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 100 I.U./mL penicillin, and 100 μg/mL streptomycin. C2C12 myoblasts were acquired from American Type Culture Collection (ATCC) and cultured in DMEM with 20% FBS, 100 I.U./mL penicillin, and 100 μg/mL streptomycin. Myoblasts were cultured until near confluency and then differentiated with DMEM with 2% horse serum and 1 μg/mL insulin for 72 hours, as previously described [16]. For in vitro studies, the myotubes were cultured in DMEM or 100% CM, which were both supplemented with 2% horse serum and 1 μg/mL insulin [17, 18]. SP600125 was purchased from LC Laboratories (MA, USA) and dissolved in dimethyl sulfoxide (DMSO).
2.2. Cancer Cell-Conditioned Media Preparation
S2–013 pancreatic cancer cells were cultured to 80% confluency in order to produced CM, as previously described [16]. At which point, the cancer cells were rinsed twice with 1X phosphate-buffered saline (PBS) and cultured in serum-free DMEM for 24 hours. CM was then collected and centrifuged at 3,000 RPM for 10 minutes, sterile syringe-filtered, and used immediately or frozen at −80 °C for long term storage.
2.3. Phosphorylation Array
A Phospho Explorer Antibody Microarray (Full Moon BioSystems, CA, USA) was used as per the manufacturer’s instructions. In brief, cell lysates were isolated using Protein Extraction Buffer (Full Moon BioSystems). After biotin labeling, the samples were applied to pre-blocked microarray slides and washed. Protein signal was detected by using Cy3-conjugated streptavidin. Signal intensities were normalized to the average intensity of the total spots on the array, and the average median signal was used for further analysis.
2.4. Immunoblotting
Protein isolation and immunoblotting were performed as described previously [19]. In brief, cells were rinsed twice with 1X PBS, shaken on ice for ten minutes with radio-immunoprecipitation assay (RIPA) lysis buffer supplemented with a cocktail of phosphatase inhibitors (10 mM sodium fluoride, 2 mM sodium orthovanadate, and 2 mM disodium beta-glycerophosphate) to lyse. Cell lysates were centrifuged at 13,000 RPM for 5 minutes and the resulting supernatant collected. Equal protein amounts for each sample were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. Primary antibodies against JNK, p-JNK, c-Jun, and p-Jun were obtained from Cell Signaling Technology (Danvers, MA). MuRF1/Trim63, Atrogin-1/Fbox32 were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). MyHC and beta-actin primary antibodies were obtained from Developmental Studies Hybridoma Bank (Iowa City, IA, USA). Beta-actin was used as a loading control. All primary antibodies were used at a dilution of 1:1000.
2.5. RNA Isolation and qRT-PCR
Total RNA was isolated from cells or tissue homogenates with TRIzol reagent (Invitrogen, Carlsbad, USA) according the manufacturer’s protocol. Complementary DNA (cDNA) was prepared with the use of a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, NY, USA). qRT-PCR was executed using SYBR Green PCR Master Mix (Applied Biosystems, NY, USA). Primers for ACTB were used for an internal control. Primer sequences are given in Supplementary Table 1. As described previously, relative gene expression analysis was completed via the ΔΔCt method [20].
2.6. Orthotopic Pancreatic Cancer Mouse Model
Six- to eight-week-old male athymic nude mice (NCr-nu/nu) were injected with 2.5 × 105 S2–013 pancreatic cancer cells into the pancreas. Age- and gender-matched healthy mice not implanted with cancer cells were used as controls. The JNK inhibitor, SP600125, was dissolved in DMSO. Ten days after orthotopic implantation, mice received IP injections of 10 mg/kg SP600125 five days a week or DMSO; all injections were 100 μL in volume. Body weight and tumor volumes estimated by caliper were recorded regularly. Twenty seven days after orthotopic implantation, the mice were euthanized and necropsied. Body weight, carcass weight, tumor weight, tumor volume, and gastrocnemius muscle weight were recorded as previously described [16]. Organs and tumor tissue were isolated, flash-frozen in liquid nitrogen or formalin-fixed. Outliers determined by a Grubb’s test (α = .05) were removed (GraphPad, CA, USA). Muscles were fixed in methacarn (60% methanol, 30% chloroform, and 10% glacial acetic acid). All animal experiments were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee (IACUC).
2.7. Measurement of Grip Strength and Rotarod test
This study utilized a grip strength meter (Columbus Instruments, OH, USA) to assess mice forelimb grip strength, as previously described [16]. Latency to fall, or rotarod performance, was determined with a rotarod apparatus (Rotamex-5, Columbus Instruments, OH, USA), as previously described [16]. Mice were acclimated to the procedure room for 15 minutes prior to rotarod testing. The average of three technical replicates was used for each mouse at each time-point.
2.8. Immunohistochemistry
Immunohistochemistry was completed by employing Novolink Polymer (Leica, Wetzlar, Germany) according to the manufacturer’s instructions. c-Jun antibody was purchased from Cell Signaling Technology (Danvers, MA, USA). The stained sections were imaged at 200X with a DMI6000 Leica microscope. Muscle fibers with c-Jun staining were counted in three random fields at 200X magnification from three biological replicates. The intensity score was generated by evaluating staining intensity of positive staining (0 = none; 1 = weak, 2 = intermediate, 3 = strong). The overall protein expression in each sample is expressed as a composite score, which is the multiplication product of the intensity score (0 – 3) by the percentage of stained fibers (100 × fibers in each intensity score divided by the total number of fibers).
2.9. Quantification and Statistical Analysis
One-way ANOVA with Tukey’s post hoc test was utilized to compare differences between multiple groups. Student’s t-test was used when appropriate. P < 0.05 was considered significant. All tests were performed using GraphPad Prism 8. For all experiments, α = 0.05 was used for significance and probability is indicated by: * p < 0.05, ** p < 0.01, and *** p < 0.001.
3. Results
3.1. In vitro model of pancreatic cancer cell-induced muscle atrophy activates JNK
To investigate the signaling pathways that contribute to muscle wasting in cancer cachexia, we utilized an in vitro cancer cell-induced muscle fiber atrophy model in which mouse C2C12-differentiated myotubes were treated with S2–013-conditioned media (CM) to induce degeneration or atrophy. A reverse phase protein array was utilized to examine phospho-proteome differences between control and CM-treated myotubes, and an elevation in JNK phosphorylation was observed in CM-treated samples (Figure 1A). Increased phosphorylation was also observed in upstream activators and downstream targets of JNK in CM-treated myotubes (Figure 1B and 1C, respectively). Consistent with the phosphorylation array data, JNK and c-Jun phosphorylation were elevated in CM-treated myotubes when examined by immunoblot analysis (Figure 1D and 1E). CM-induced c-Jun phosphorylation in myotubes was abrogated by a range of dosages of the JNK activity inhibitor SP600125 (Figure 1E) [21]. Thus, cancer cell-secreted factors in the CM induce JNK activation in myofibers in culture conditions.
Figure 1. In vitro model of pancreatic cancer cachexia activates JNK.
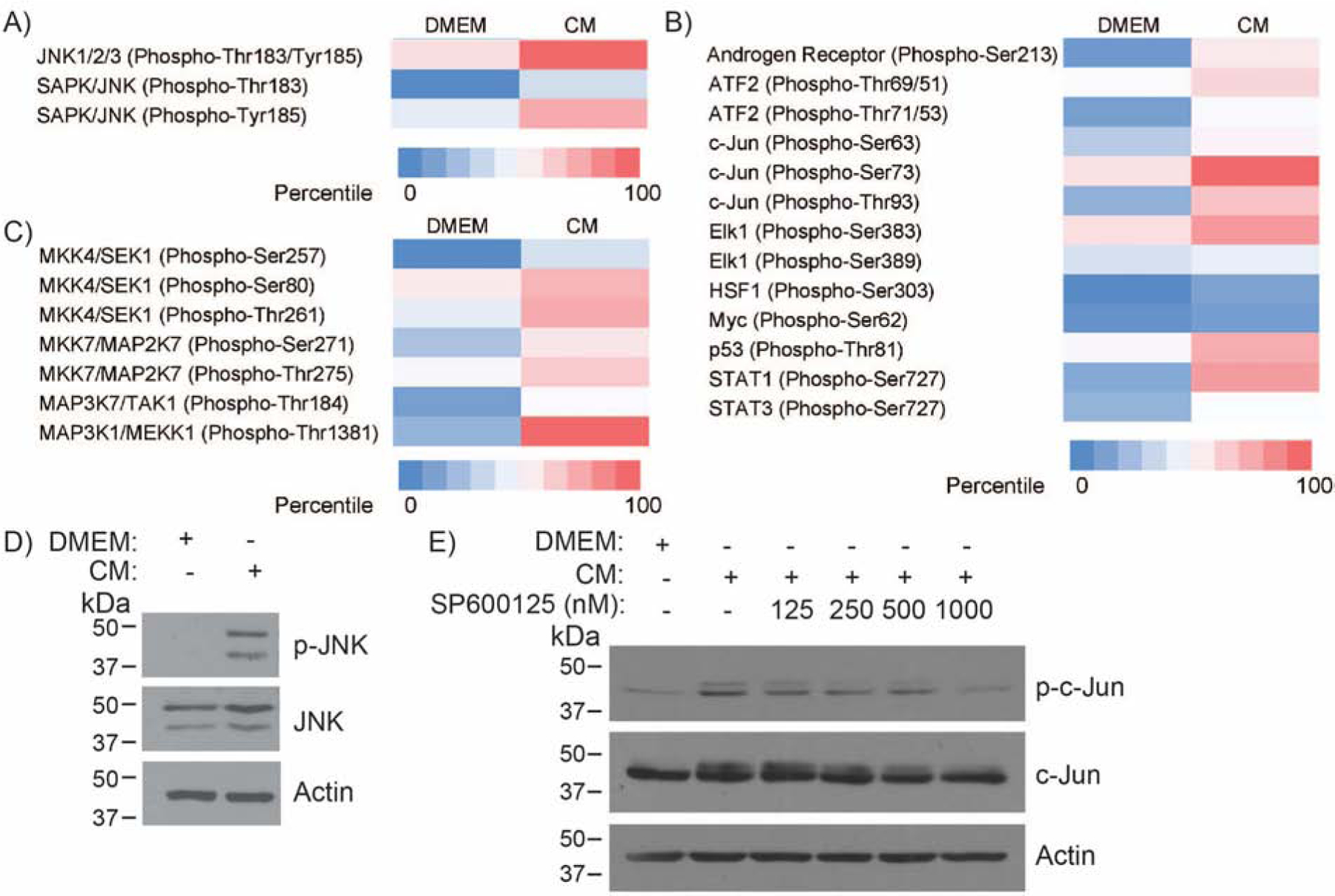
Differentiated C2C12 myoblasts were treated with control DMEM or S2–013-conditioned media (CM). The phosphorylation status of (A) SAPK/JNK proteins, (B) upstream activators of SAPK/JNK, and (C) downstream targets of SAPK/JNK was identified by a reverse phase protein array. (D) Immunoblot analysis for phosphorylated and total JNK in differentiated C2C12 myoblasts in response to treatment with control media or CM for 72 hours. (E) Immunoblot analysis of phosphorylated and total c-Jun in differentiated C2C12 myoblasts in response to treatment with control media or CM with the indicated doses of JNK inhibitor SP600125 for 72 hours.
3.2. JNK signaling contributes to myotube thinning
To investigate if JNK signaling has a functional role in myotube wasting, we examined how inhibiting JNK activity altered myotube wasting parameters in our in vitro model. JNK inhibition reduced CM-induced myotube thinning (Figure 2A and 2B). MyHC is a prominent structural protein in myotubes and a proxy for wasting status [22]. Inhibiting JNK rescued MyHC protein levels of CM-treated myotubes in a dose-dependent manner (Figure 2C). Protein degradation exhibited in cachectic muscle wasting is often partially attributed to the upregulation of cachexia-related E3 ubiquitin ligases. CM elevated the mRNA expression levels of genes coding for the E3 ubiquitin ligases, Trim63 and Fbxo32, and inhibition of JNK significantly mitigated their upregulation (Figure 2D and 2E). Therefore, inhibiting JNK activation abrogates cancer cell-induced myofiber atrophy in vitro, in part, by abrogating the expression of cachexia-associated ubiquitin ligases.
Figure 2. JNK signaling contributes to myotube thinning.
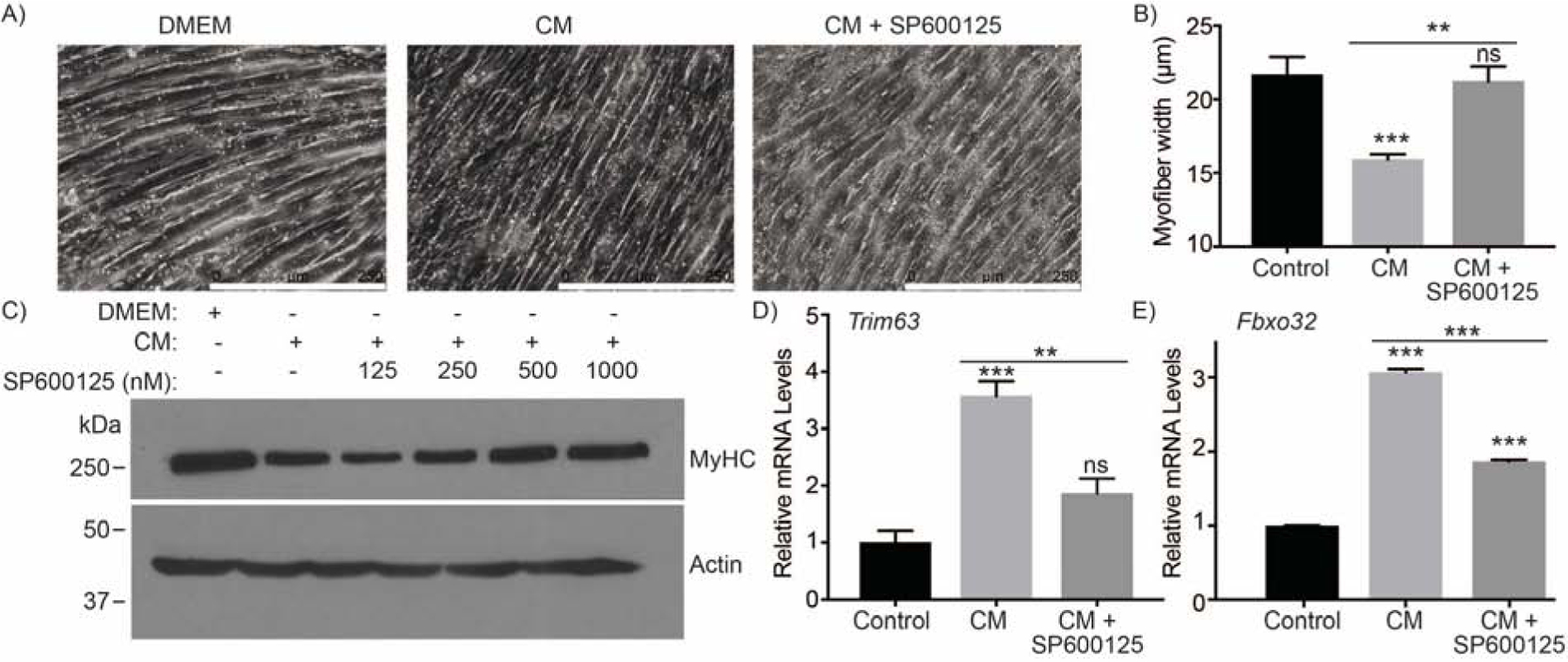
(A-B) Phase contrast micrographs (A) of differentiated C2C12 myotubes treated with DMEM, S2–013-conditioned media (CM), or CM and 500 nM SP600125 (CM + SP600125) for 72 hours (scale bar = 250 μm) and bar chart (B) representing myotube width determined by ImageJ. (C) Myotube wasting status assessed by immunoblotting for MyHC. The mRNA expression of (D) Trim63 and (E) Fbxo32 in differentiated C2C12 myotubes treated with DMEM, CM, or CM + SP600125 for 48 hours. Data are represented as mean ± SEM. Bar charts were compared by one-way ANOVA followed by Tukey’s post hoc test. **p < 0.01, and ***p <0.001.
3.3. Physiological impact of JNK inhibition in an orthotopic pancreatic cancer model
Athymic nude mice were orthotopically implanted with S2–013 pancreatic cancer cells (tumor-bearing; TB), and non-implanted mice served as healthy controls (HC). Mice were given DMSO or SP600125 (10 mg/kg) via IP injections daily for five days a week (Figure 3A). Forelimb grip strength was also evaluated to assess muscle physiological capacity, and JNK inhibition with SP600125 (TB + SP600125) partially abrogated the reduction of grip strength observed in tumor-bearing mice (Figure 3B). The rotarod performance in different groups showed trends similar to that of the grip strengths; however, the latency to fall in JNK inhibitor-treated tumor-bearing mice was not statistically significant, in comparison to the solvent control-treated tumor-bearing mice (Figure 3C). Longitudinal measurements of tumor size showed no significant differences between solvent control-treated and JNK inhibitor-treated tumor-bearing mice (Figure 3D). Thus, inhibition of JNK enhances body weight of tumor-bearing mice in an orthotopic pancreatic cancer mouse model and improves some physiological parameters of muscle function.
Figure 3. Physiological impact of JNK inhibition on orthotopic pancreatic cancer model.
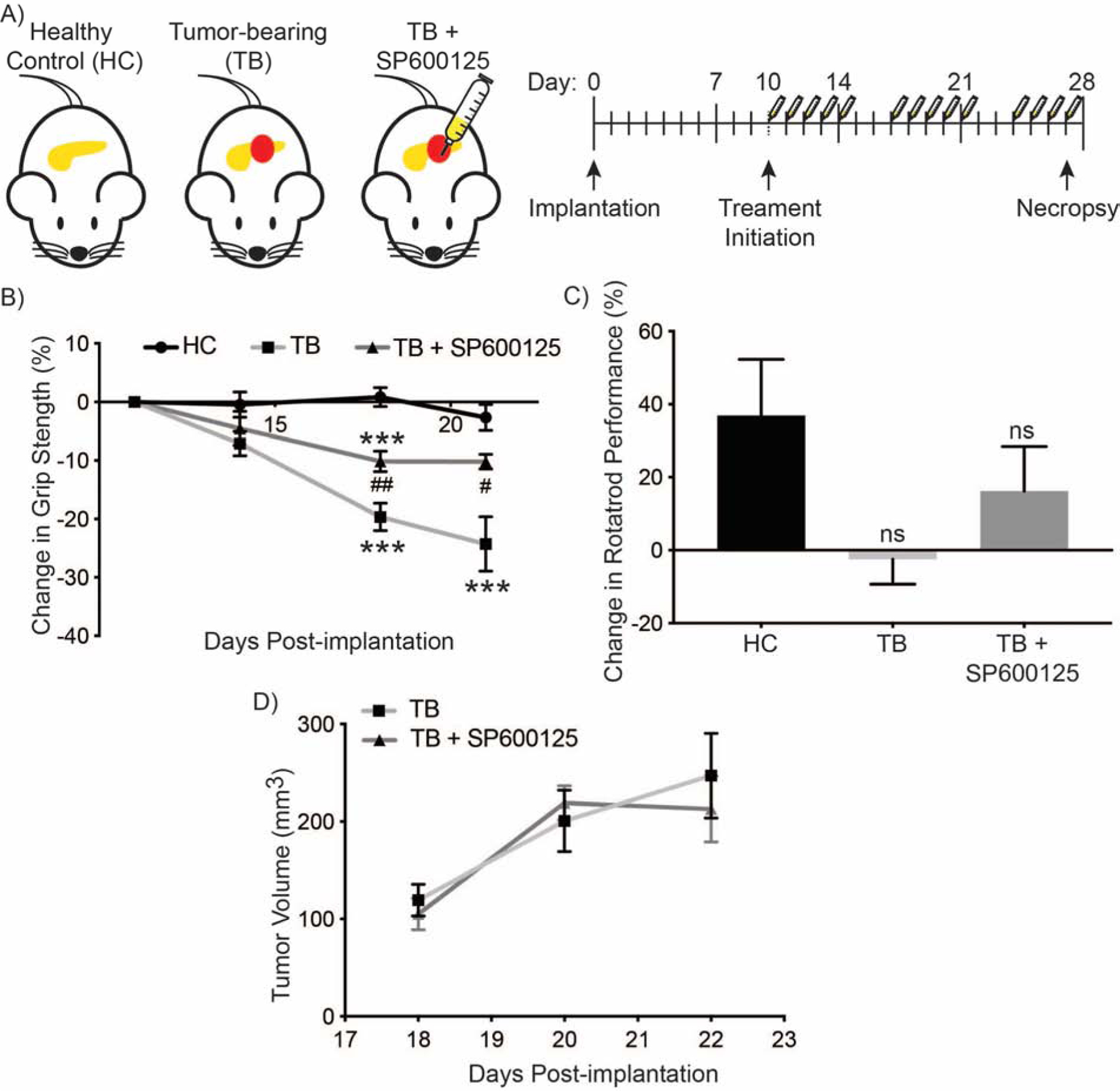
(A) Schematic illustration of experimental cohorts, experimental outline, and course of treatment. Experimental groups of female athymic nude mice included tumor-bearing (TB) mice orthotopically implanted with S2–013 pancreatic cancer cells, TB mice treated with SP600125 (TB + SP600125), or unimplanted healthy control (HC) mice. Ten days after implantation, the mice were treated once daily for five days a week with SP600125 (10 mg/kg) or solvent control and sacrificed at 27 days post-implantation. (B) Forelimb grip strength at indicated times as determined by a grip meter. (C) Percent change in rotarod performance on day 21, compared to intial measurements on day 11. (D) Longitundinal caliper measurements of tumors volume. Data in figures B, C, and D are represented as mean ± SEM. The charts in (B and C) were compared by one-way ANOVA followed by Tukey’s post hoc test. ***p <0.001 compared with healthy control mice group. #p < 0.05 and ##p < 0.01 compared with tumor-bearing mice group. Data in (D) were compared by Student’s t-test at each time point.
3.4. JNK inhibition improves mouse body and skeletal muscle weights measured upon necropsy
Healthy control mice and mice orthotopically implanted with pancreatic cancer cells with or without JNK inhibitor treatment were sacrificed and evaluated. We observed that JNK inhibition rescued the body weight loss in live tumor-bearing mice when compared to treatment with solvent control at day 21 (Figure 4A). Measurement of carcass weight at day 27 also demonstrated that JNK inhibition abrogates weight loss in tumor-bearing mice (Figure 4B). JNK inhibitor treatment also rescued gastrocnemius muscle weight in tumor-bearing mice (Figure 4C). We observed no significant differences in tumor volume or weight between JNK inhibitor- and solvent control-treated mice (Figures 4D and 4E). These data suggest that JNK inhibition ameliorates the cachectic phenotype in tumor-bearing mice.
Figure 4. JNK inhibition improves mouse body weight and muscle weight.
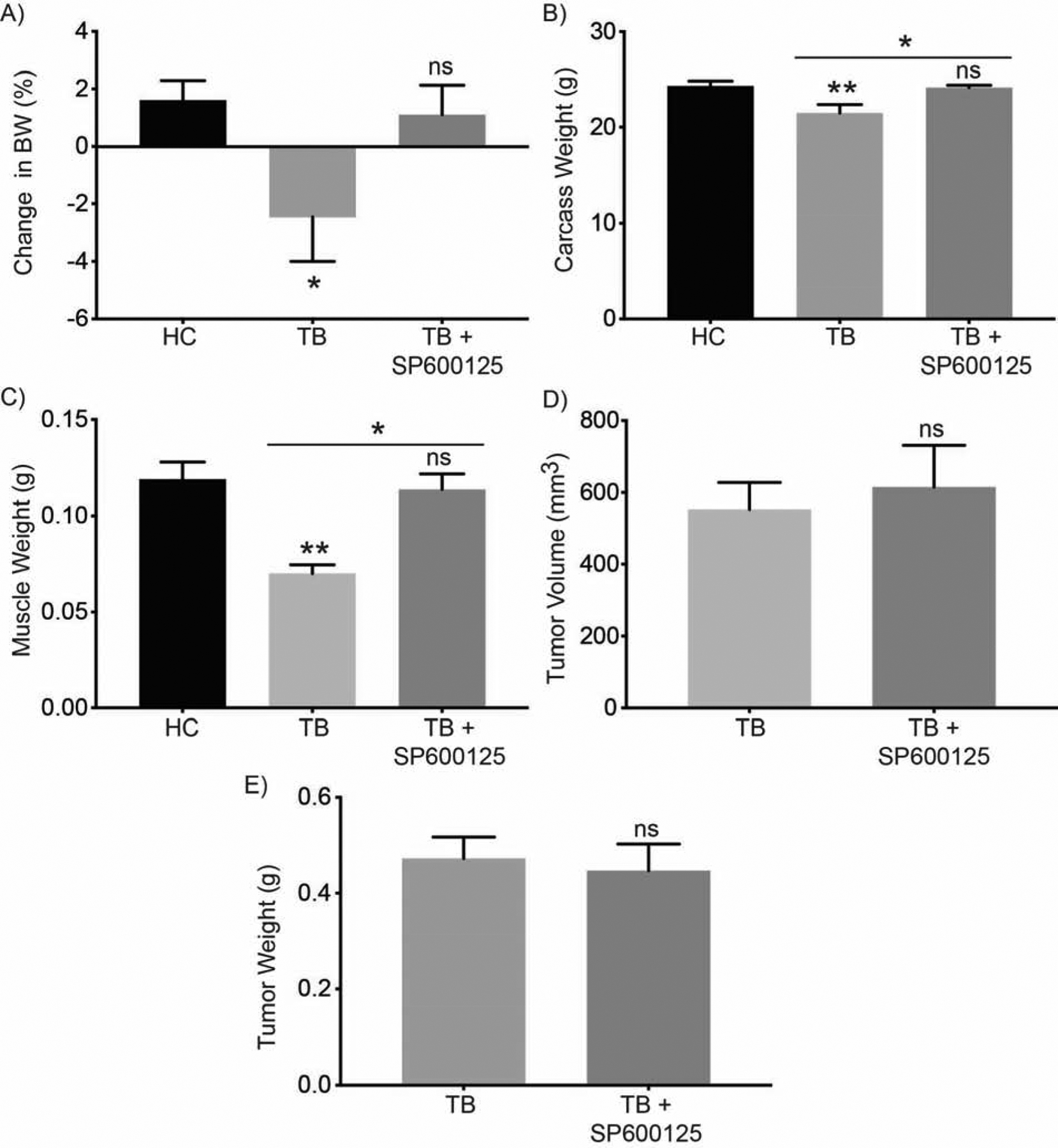
Mouse necropsy measurements for female athymic nude mice included tumor-bearing (TB) mice orthotopically implanted with S2–013 pancreatic cancer cells, TB mice treated with 10 mg/kg SP600125 (TB + SP600125), or unimplanted healthy control (HC) mice. (A) Body weight (BW) at day 21 in comparison to the intial measurement on day 4. Bar charts represent (B) carcass weight, (C) muscle weight, (D) tumor volume, and (E) tumor weight. Data are represented as mean ± SEM. The bar charts in (A, B, C) were compared by one-way ANOVA followed by Tukey’s post hoc test. Data in (D and E) were compared by Student’s t-test. *p < 0.05 and **p < 0.01.
3.5. JNK inhibition ameliorates muscle wasting in an orthotopic pancreatic tumor model
To examine if the elevated JNK activity was consistent in our orthotopic pancreatic tumor model, we examined JNK activity via probing for c-Jun levels, a marker for JNK activity [23], in the mouse muscle. Total c-Jun levels were upregulated in the gastrocnemius muscle from tumor-bearing mice, but the upregulation was abrogated in the JNK inhibitor-treated tumor-bearing group (Figure 5A and 5B). To examine the functional impact of this altered JNK activity, we measured the cross-sectional area of gastrocnemius myofibers. The muscle fiber cross-sectional area was significantly reduced in tumor-bearing mice as compared to healthy controls, and treatment of tumor-bearing mice with SP600125 significantly rescued the cross-sectional area (Figures 5A and 5C). To evaluate the molecular contributions that could explain the differences in the muscle fiber cross-sectional area, we examined the expression of cachexia-related E3 ubiquitin ligases. The gastrocnemius muscle from tumor-bearing mice exhibited elevated mRNA expression levels of the E3 ubiquitin ligases Trim63 and Fbxo32, and inhibition of JNK abrogated their upregulation (Figure 5D and 5E). Therefore, these data suggest that the inhibition of JNK ameliorates muscle wasting in tumor-bearing mice by abolishing the tumor-induced upregulation of cachexia-associated ubiquitin ligases.
Figure 5. Inhibition of JNK ameliorates muscle wasting in tumor-bearing mouse model.
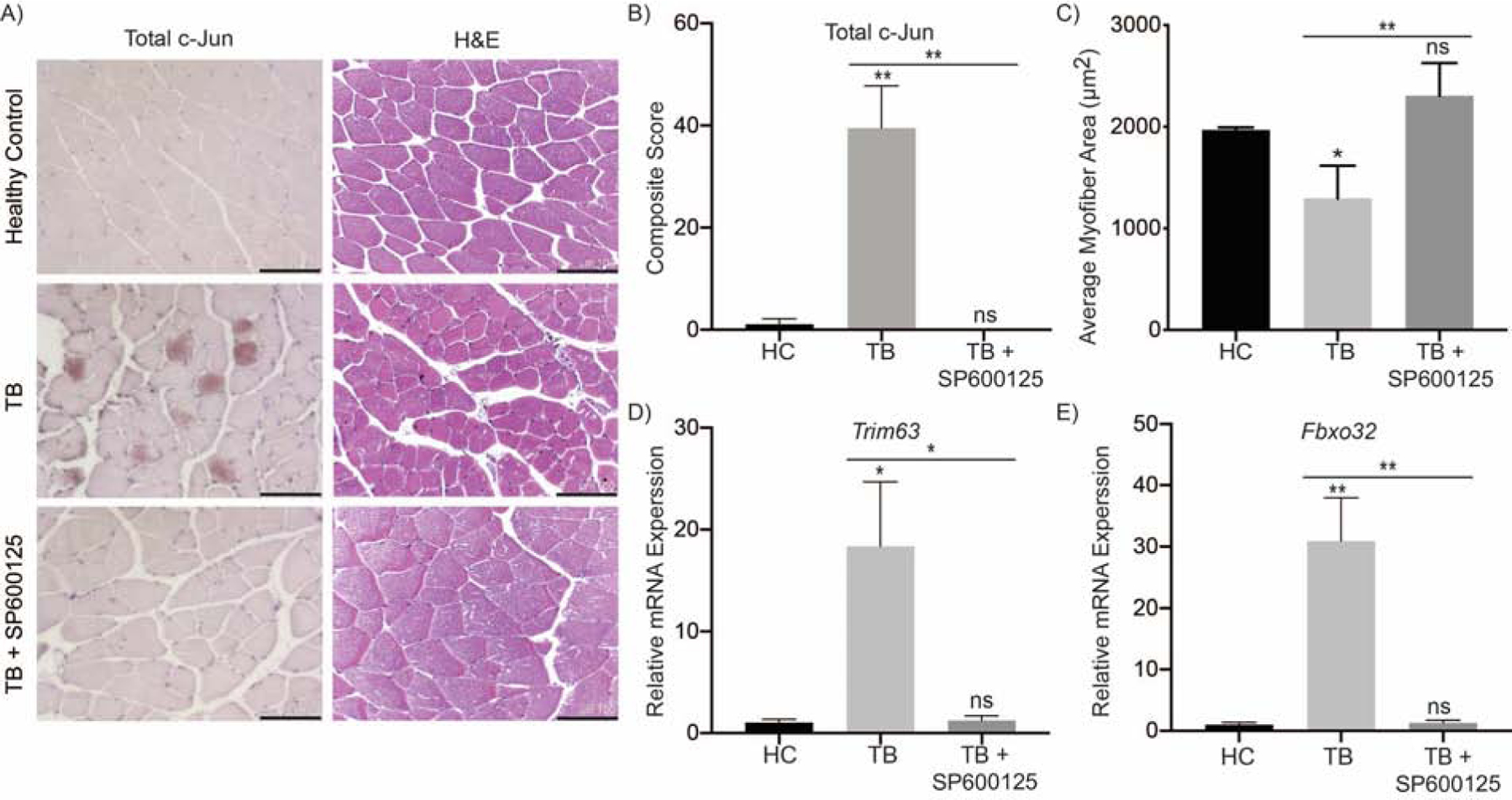
(A) IHC staining for c-Jun and H&E in methacarn-fixed muscle sections from female athymic nude mice; experiemental groups included tumor-bearing (TB) mice orthotopically implanted with S2–013 pancreatic cancer cells, TB mice treated with 10 mg/kg SP600125 (TB + SP600125), or unimplanted healthy control (HC) mice (scale bar = 100 μm). (B) Quantification of c-Jun staining and (C) muscle fiber cross-sectional area. The mRNA expression of (D) Trim63 and (E) Fbxo32 from flash frozen mouse gastrocnemius muscle. Data are represented as mean ± SEM. Data were compared by one-way ANOVA followed by Tukey’s post hoc test. *p < 0.05 and **p < 0.01.
4. Discussion
Cachexia frequently occurs in cancer and particularly in many gastrointestinal malignancies, such as pancreatic cancer [24]. The occurrence of cancer cachexia not only lessens the quality of life for cancer patients but also impairs tolerance of treatments and overall survival [25]. The multifaceted syndrome is characterized by uncontrollable weight loss, especially due to muscle wasting [5]. Muscle homeostasis is a balance of anabolic and catabolic processes, and cancer cachexia tilts the equilibrium towards catabolism. Muscle wasting is in part due to excessive protein turnover, which can be the result of autophagy and proteasome-dependent protein degradation [6, 26]. Previous studies have identified that the MAPKs p38 and ERK are known to induce the expression of E3 ubiquitin ligases Fbxo32 and Trim63, but the role of the MAPK JNK in cachexia is not fully elucidated [27, 28].
This study demonstrates that JNK signaling directly contributes to skeletal muscle protein turnover in pancreatic cancer cachexia and substantiates JNK as a possible therapeutic target. Our studies suggest that JNK, as well as its upstream activators and downstream targets, are activated in both in vitro and in vivo models of cancer cachexia. JNK signaling is significant for the pathogenesis of cancer cachexia as inhibiting JNK mitigated CM-induced thinning of differentiated C2C12 myotubes. Recently, it has been identified that a TLR7/8/9 antagonist resulted in reduced levels of phosphorylated JNK and also lessened cancer cachexia in multiple models through inhibiting microRNA-induced JNK-dependent cell death [14]. However, this study supports the notion that the improved cancer cachexia phenotype seen from inhibiting JNK signaling is in part due to the reduction of protein turnover. We assert this because the inhibition of JNK abrogated the loss of the structural protein MyHC, and the treatment caused a drastic reduction in the expression of cachexia-related E3 ubiquitin ligases Trim63 and Fbxo32. This in vitro data supported the validity of JNK as a therapeutic target in cancer cachexia, and provided rationale for further examination in a mouse model.
To further validate the significance of JNK signaling in muscle wasting and cancer cachexia, we also utilized an orthotopic mouse model of pancreatic cancer. JNK inhibitor treatment improved the body weight of tumor-bearing mice. Differences in body weight can be in part attributed to the fact that JNK inhibitor treatment reduced the muscle wasting as evident by improved muscle mass and myofiber cross-sectional area in tumor-bearing mice. Importantly, the improvement in muscle mass had functional significance as it improved the forelimb grip strength of tumor-bearing mice. Surprisingly, we did not observe any reduction in tumor volume with JNK inhibitor treatment as previously reported, but this may be due to differences in the mouse models and the higher dosages previously used [27]. Overall, our data suggest that JNK signaling contributes to elevated protein turnover in skeletal muscle in cancer cachexia, but JNK may have other roles in skeletal muscle or other organs.
While our evidence supports that JNK signaling contributes to skeletal muscle expression of ubiquitin ligases in cancer cachexia, chemical inhibition of JNK in cachectic tumor-bearing mice may have other systemic roles. Hypothalamic signaling has a pleiotropic impact as it regulates appetite, energy expenditure, body weight, and glucose homeostasis [28]. JNK inhibitor treatment in the hypothalamus has been shown to reduce nutritional uptake in rats [29]. JNK is also activated in the brain of obese mice, and JNK1-deficiency in the mouse central nervous system has also been shown to reduce body mass and epididymal fat pad weight [30, 31]. Conversely, our in vivo model does not experience over-nutrition seen in obesity. Instead, the cancer cachexia models are nutritionally challenged because of cachexia-induced anorexia, and this difference may contribute to weight gain in the JNK inhibitor-treated tumor-bearing mice.
Overall, our study identifies that pancreatic cancer induces JNK signaling in the muscle that contributes to muscle wasting. We also demonstrate that pharmacological inhibition of JNK mitigates muscle wasting, at least in part, by reducing protein turnover and the expression of E3 ubiquitin ligases. We propose that the pharmacological inhibition of JNK, alone or in combination with other MAPKs, warrants further investigation as a targeted strategy component of a multimodal treatment for cancer cachexia.
Supplementary Material
Supplementray Table 1. Primer Sequences.
Highlights:
JNK is activated in pancreatic cancer-associated cachexia mouse model
JNK inhibitor rescues body and muscle weight and improves grip strength in tumor-bearing mice
Inhibiting JNK ameliorates cachexia-induced myotube and myofiber thinning
Inhibition of JNK reduces protein turnover and E3 ubiquitin ligase expression
Significance:
This study demonstrates that pancreatic cancer induces c-Jun N-terminal Kinase (JNK) signaling in muscle and contributes to muscle wasting. JNK inhibitor treatment in tumor-bearing mice rescues body weight, carcass weight, muscle weight, and forelimb grip strength. Inhibition of JNK abrogates myotube and myofiber thinning by reducing protein turnover and decreasing the expression of the E3 ubiquitin ligases Atrogin1 (Fbxo32) and MuRF1 (Trim63).
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award numbers R01CA210439 and P30CA036727. This project is also supported by an Eppley Institute Cancer Biology Training Grant from the National Cancer Institute (T32CA009476 to S.E.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- MAPK
mitogen-activated protein kinases
- JNK
c-Jun N-terminal Kinase
- CM
cancer cell-conditioned media
- MyHC
myosin heavy chain 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest Statement:
The authors declare that no conflicts exist
References
- [1].C. Global Burden of Disease Cancer, Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, Dandona L, Fleming T, Forouzanfar MH, Hancock J, Hay RJ, Hunter-Merrill R, Huynh C, Hosgood HD, Johnson CO, Jonas JB, Khubchandani J, Kumar GA, Kutz M, Lan Q, Larson HJ, Liang X, Lim SS, Lopez AD, MacIntyre MF, Marczak L, Marquez N, Mokdad AH, Pinho C, Pourmalek F, Salomon JA, Sanabria JR, Sandar L, Sartorius B, Schwartz SM, Shackelford KA, Shibuya K, Stanaway J, Steiner C, Sun J, Takahashi K, Vollset SE, Vos T, Wagner JA, Wang H, Westerman R, Zeeb H, Zoeckler L, Abd-Allah F, Ahmed MB, Alabed S, Alam NK, Aldhahri SF, Alem G, Alemayohu MA, Ali R, Al-Raddadi R, Amare A, Amoako Y, Artaman A, Asayesh H, Atnafu N, Awasthi A, Saleem HB, Barac A, Bedi N, Bensenor I, Berhane A, Bernabe E, Betsu B, Binagwaho A, Boneya D, Campos-Nonato I, Castaneda-Orjuela C, Catala-Lopez F, Chiang P, Chibueze C, Chitheer A, Choi JY, Cowie B, Damtew S, das Neves J, Dey S, Dharmaratne S, Dhillon P, Ding E, Driscoll T, Ekwueme D, Endries AY, Farvid M, Farzadfar F, Fernandes J, Fischer F, TT GH, Gebru A, Gopalani S, Hailu A, Horino M, Horita N, Husseini A, Huybrechts I, Inoue M, Islami F, Jakovljevic M, James S, Javanbakht M, Jee SH, Kasaeian A, Kedir MS, Khader YS, Khang YH, Kim D, Leigh J, Linn S, Lunevicius R, El Razek HMA, Malekzadeh R, Malta DC, Marcenes W, Markos D, Melaku YA, Meles KG, Mendoza W, Mengiste DT, Meretoja TJ, Miller TR, Mohammad KA, Mohammadi A, Mohammed S, Moradi-Lakeh M, Nagel G, Nand D, Le Nguyen Q, Nolte S, Ogbo FA, Oladimeji KE, Oren E, Pa M, Park EK, Pereira DM, Plass D, Qorbani M, Radfar A, Rafay A, Rahman M, Rana SM, Soreide K, Satpathy M, Sawhney M, Sepanlou SG, Shaikh MA, She J, Shiue I, Shore HR, Shrime MG, So S, Soneji S, Stathopoulou V, Stroumpoulis K, Sufiyan MB, Sykes BL, Tabares-Seisdedos R, Tadese F, Tedla BA, Tessema GA, Thakur JS, Tran BX, Ukwaja KN, Uzochukwu BSC, Vlassov VV, Weiderpass E, Wubshet Terefe M, Yebyo HG, Yimam HH, Yonemoto N, Younis MZ, Yu C, Zaidi Z, Zaki MES, Zenebe ZM, Murray CJL, Naghavi M, Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study, JAMA Oncol, 3 (2017) 524–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fearon KC, Glass DJ, Guttridge DC, Cancer cachexia: mediators, signaling, and metabolic pathways, Cell Metab, 16 (2012) 153–166. [DOI] [PubMed] [Google Scholar]
- [3].Bachmann J, Ketterer K, Marsch C, Fechtner K, Krakowski-Roosen H, Buchler MW, Friess H, Martignoni ME, Pancreatic cancer related cachexia: influence on metabolism and correlation to weight loss and pulmonary function, BMC Cancer, 9 (2009) 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO, Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, Oken MM, Perlia C, Rosenbaum C, Silverstein MN, Skeel RT, Sponzo RW, Tormey DC, Prognostic effect of weight loss prior tochemotherapy in cancer patients, The American Journal of Medicine, 69 (1980) 491–497. [DOI] [PubMed] [Google Scholar]
- [5].Fearon KC, Voss AC, Hustead DS, Cancer Cachexia Study G, Definition of cancer cachexia: effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis, Am J Clin Nutr, 83 (2006) 1345–1350. [DOI] [PubMed] [Google Scholar]
- [6].Glass DJ, Signalling pathways that mediate skeletal muscle hypertrophy and atrophy, Nat Cell Biol, 5 (2003) 87–90. [DOI] [PubMed] [Google Scholar]
- [7].Gomes-Marcondes MC, Tisdale MJ, Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress, Cancer Lett, 180 (2002) 69–74. [DOI] [PubMed] [Google Scholar]
- [8].Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ, Identification of ubiquitin ligases required for skeletal muscle atrophy, Science, 294 (2001) 1704–1708. [DOI] [PubMed] [Google Scholar]
- [9].Quan-Jun Y, Yan H, Yong-Long H, Li-Li W, Jie L, Jin-Lu H, Jin L, Peng-Guo C, Run G, Cheng G, Selumetinib Attenuates Skeletal Muscle Wasting in Murine Cachexia Model through ERK Inhibition and AKT Activation, Mol Cancer Ther, 16 (2017) 334–343. [DOI] [PubMed] [Google Scholar]
- [10].Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB, TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle, FASEB J, 19 (2005) 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xie SJ, Li JH, Chen HF, Tan YY, Liu SR, Zhang Y, Xu H, Yang JH, Liu S, Zheng LL, Huang MB, Guo YH, Zhang Q, Zhou H, Qu LH, Inhibition of the JNK/MAPK signaling pathway by myogenesis-associated miRNAs is required for skeletal muscle development, Cell Death Differ, 25 (2018) 1581–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lessard SJ, MacDonald TL, Pathak P, Han MS, Coffey VG, Edge J, Rivas DA, Hirshman MF, Davis RJ, Goodyear LJ, JNK regulates muscle remodeling via myostatin/SMAD inhibition, Nat Commun, 9 (2018) 3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vijayvargia R, Mann K, Weiss HR, Pownall HJ, Ruan H, JNK deficiency enhances fatty acid utilization and diverts glucose from oxidation to glycogen storage in cultured myotubes, Obesity (Silver Spring), 18 (2010) 1701–1709. [DOI] [PubMed] [Google Scholar]
- [14].Calore F, Londhe P, Fadda P, Nigita G, Casadei L, Marceca GP, Fassan M, Lovat F, Gasparini P, Rizzotto L, Zanesi N, Jackson D, Mehta S, Nana-Sinkam P, Sampath D, Pollock RE, Guttridge DC, Croce CM, The TLR7/8/9 Antagonist IMO-8503 Inhibits Cancer-Induced Cachexia, Cancer Res, 78 (2018) 6680–6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, Zhang B, Mehla K, Brown RB, Caffrey T, Yu F, Johnson KR, Powers R, Hollingsworth MA, Singh PK, MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer, Proc Natl Acad Sci U S A, 109 (2012) 13787–13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shukla SK, Dasgupta A, Mulder SE, Singh PK, Molecular and Physiological Evaluation of Pancreatic Cancer-Induced Cachexia, Methods Mol Biol, 1882 (2019) 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dasgupta A, Shukla SK, Vernucci E, King RJ, Abrego J, Mulder SE, Mullen NJ, Graves G, Buettner K, Thakur R, Murthy D, Attri KS, Wang D, Chaika NV, Pacheco CG, Rai I, Engle DD, Grandgenett PM, Punsoni M, Reames BN, Teoh-Fitzgerald M, Oberley-Deegan R, Yu F, Klute KA, Hollingsworth MA, Zimmerman MC, Mehla K, Sadoshima J, Tuveson DA, Singh PK, SIRT1-NOX4 signaling axis regulates cancer cachexia, J Exp Med, 217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shukla SK, Markov SD, Attri KS, Vernucci E, King RJ, Dasgupta A, Grandgenett PM, Hollingsworth MA, Singh PK, Yu F, Mehla K, Macrophages potentiate STAT3 signaling in skeletal muscles and regulate pancreatic cancer cachexia, Cancer Lett, 484 (2020) 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shukla SK, Dasgupta A, Mehla K, Gunda V, Vernucci E, Souchek J, Goode G, King R, Mishra A, Rai I, Nagarajan S, Chaika NV, Yu F, Singh PK, Silibinin-mediated metabolic reprogramming attenuates pancreatic cancer-induced cachexia and tumor growth, Oncotarget, 6 (2015) 41146–41161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, King RJ, Abrego J, Goode GD, Dasgupta A, Illies AL, Gebregiworgis T, Dai B, Augustine JJ, Murthy D, Attri KS, Mashadova O, Grandgenett PM, Powers R, Ly QP, Lazenby AJ, Grem JL, Yu F, Mates JM, Asara JM, Kim JW, Hankins JH, Weekes C, Hollingsworth MA, Serkova NJ, Sasson AR, Fleming JB, Oliveto JM, Lyssiotis CA, Cantley LC, Berim L, Singh PK, MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer, Cancer Cell, 32 (2017) 71–87 e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW, SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase, Proc Natl Acad Sci U S A, 98 (2001) 13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, Guttridge DC, Cancer cachexia is regulated by selective targeting of skeletal muscle gene products, J Clin Invest, 114 (2004) 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kayahara M, Wang X, Tournier C, Selective regulation of c-jun gene expression by mitogen-activated protein kinases via the 12-o-tetradecanoylphorbol-13-acetate-responsive element and myocyte enhancer factor 2 binding sites, Mol Cell Biol, 25 (2005) 3784–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Laviano A, Meguid MM, Nutritional issues in cancer management, Nutrition, 12 (1996) 358–371. [DOI] [PubMed] [Google Scholar]
- [25].Aapro M, Arends J, Bozzetti F, Fearon K, Grunberg SM, Herrstedt J, Hopkinson J, Jacquelin-Ravel N, Jatoi A, Kaasa S, Strasser F, Esmo, Early recognition of malnutrition and cachexia in the cancer patient: a position paper of a European School of Oncology Task Force, Ann Oncol, 25 (2014) 1492–1499. [DOI] [PubMed] [Google Scholar]
- [26].Tisdale MJ, Cancer cachexia, Curr Opin Gastroenterol, 26 (2010) 146–151. [DOI] [PubMed] [Google Scholar]
- [27].Takahashi R, Hirata Y, Sakitani K, Nakata W, Kinoshita H, Hayakawa Y, Nakagawa H, Sakamoto K, Hikiba Y, Ijichi H, Moses HL, Maeda S, Koike K, Therapeutic effect of c-Jun N-terminal kinase inhibition on pancreatic cancer, Cancer Sci, 104 (2013) 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vallerie SN, Hotamisligil GS, The role of JNK proteins in metabolism, Sci Transl Med, 2 (2010) 60rv65. [DOI] [PubMed] [Google Scholar]
- [29].De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA, Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus, Endocrinology, 146 (2005) 4192–4199. [DOI] [PubMed] [Google Scholar]
- [30].Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, Bronneke HS, Brodesser S, Hampel B, Schauss AC, Bruning JC, Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism, Proc Natl Acad Sci U S A, 107 (2010) 6028–6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sabio G, Cavanagh-Kyros J, Barrett T, Jung DY, Ko HJ, Ong H, Morel C, Mora A, Reilly J, Kim JK, Davis RJ, Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1, Genes Dev, 24 (2010) 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementray Table 1. Primer Sequences.