

Abstract
To mount an antipathogen response, CD4 T cells must undergo rapid cell proliferation; however, poorly controlled expansion can result in diseases such as autoimmunity. One important regulator of T-cell activity is the E3 ubiquitin ligase Itch. Itch deficient patients suffer from extensive autoinflammation. Similarly, Itch deficient mice exhibit inflammation characterized by high numbers of activated CD4 T cells. While the role of Itch in limiting CD4 T-cell cytokine production has been extensively studied, it is less clear whether and how Itch regulates proliferation of these cells. We determined that Itch deficient CD4 T cells are hyperproliferative in vitro and in vivo, due to increased S phase entry. Whole cell proteomics analysis of Itch deficient primary mouse CD4 T cells revealed increased abundance of the β-catenin coactivator WW domain-binding protein 2 (WBP2). Furthermore, Itch deficient cells demonstrate increased WBP2 protein stability, and Itch and WBP2 interact in CD4 T cells. Knockdown of WBP2 in CD4 T cells caused reduced proliferation. Together, our data support that Itch attenuates CD4 T cell proliferation by promoting WBP2 degradation. This study identifies novel roles for Itch and WBP2 in regulating CD4 T cell proliferation, providing insight into how Itch may prevent inflammation.
Keywords: Autoinflammation, Cell cycle, E3 ubiquitin ligase, Proliferation
Introduction
Autoimmune diseases are generally considered to be a family of complex heterogeneous disorders. However, there are a few examples of monogenic autoimmune diseases. One example that is only recently coming to light is due to a loss-of-function mutation in the gene encoding the E3 ubiquitin ligase Itch. Deficiency in Itch in patients was first described in a consanguineous group of 10 children, many of whom suffered from multisyndromic autoimmune disease including colitis, thyroiditis, diabetes, and autoantibodies [1]. Last year, at least two additional mutations in the ITCH locus were identified in patients with signs of immune dysregulation. One patient presented with autoimmune hepatitis [2], and the other with chronic lung disease [3]. The recent identification of these previously undiagnosed patients using whole exome sequencing suggests that Itch deficiency may be a more common cause of autoimmune disease than previously realized, highlighting the importance of understanding the role of Itch in immune regulation.
Much of our understanding of Itch function in T cells comes from studies of Itch knockout (Itch KO) mice. These mice have significant autoinflammation, including increased immune cell infiltration in the skin and gastrointestinal tract [4, 5]. This inflammation is characterized by an accumulation of activated CD4 T cells, particularly Th2 polarized cells [6]. The role of Itch in CD4 T cell differentiation has been extensively studied; the Th2 skewing of Itch deficient cells has been attributed to Itch’s role in degrading the IL-4 transcription factor JunB [7, 8]. Additionally, Itch attenuates IL-17 production and Th17 differentiation by promoting RORγt degradation [9, 10]. However, it is unknown how Itch limits the accumulation of CD4 T cell numbers in vivo.
One possibility is that Itch limits CD4 T cell proliferation. There is evidence to support a role for Itch in attenuating proliferation in other cell types. Itch deficiency leads to hyperproliferation in hematopoietic stem cells [11], keratinocytes [12], medulloblastoma cell lines [13], and breast cancer cell lines [14]. The proliferation changes in each case have been attributed to different substrates and/or biochemical mechanisms. While it has been reported that unpurified Itch deficient lymphocytes are capable of increased proliferation in vitro [6], it has not been determined which, if any, of these mechanisms apply to CD4 T cells. Furthermore, it remains unclear whether the increased numbers of T cells in Itch deficient mice is due to a direct role for Itch in CD4 T cell proliferation or an indirect effect of inflammation and altered cytokine milieu.
In this study, we sought to determine whether and how Itch regulates CD4 T-cell proliferation in vitro and in vivo. We found that Itch directly limits proliferation of activated CD4 T cells, and that it does so by restricting cell cycle progression at the stage of S phase entry. Using a whole cell proteomic approach, we identified WW domain binding protein 2 (WBP2) as a target for Itch in CD4 T cells and determined that altering the levels of WBP2 impacts T cell proliferation. These findings demonstrate a previously unappreciated role for Itch in preventing CD4 T cell hyperproliferation by limiting WBP2 protein stability, and support a novel role for WBP2 in regulating CD4 T cell proliferation.
Results
Itch restricts activated T cell accumulation and proliferation in vivo
Itch deficient mice have autoinflammation and an increased proportion of activated CD4 T cells [4, 6]. However, it is unknown whether this increase reflects an increased propensity to become activated, increased cell survival, or enhanced proliferation. To determine how Itch regulates CD4 T cell numbers, we generated mixed bone marrow chimeras by injecting Rag knockout (Rag KO) mice with a 1:1 mixture of CD45.1 wild type (WT) and CD45.2 Itch KO cells. This system allows us to examine Itch KO and WT T cells while normalizing for the level of background inflammation and cytokine milieu (Fig. 1A). After at least 8 weeks postreconstitution, we analyzed cells from the lungs, lymph nodes, and spleen using flow cytometry (Supporting information Fig. S1A).
Figure 1.
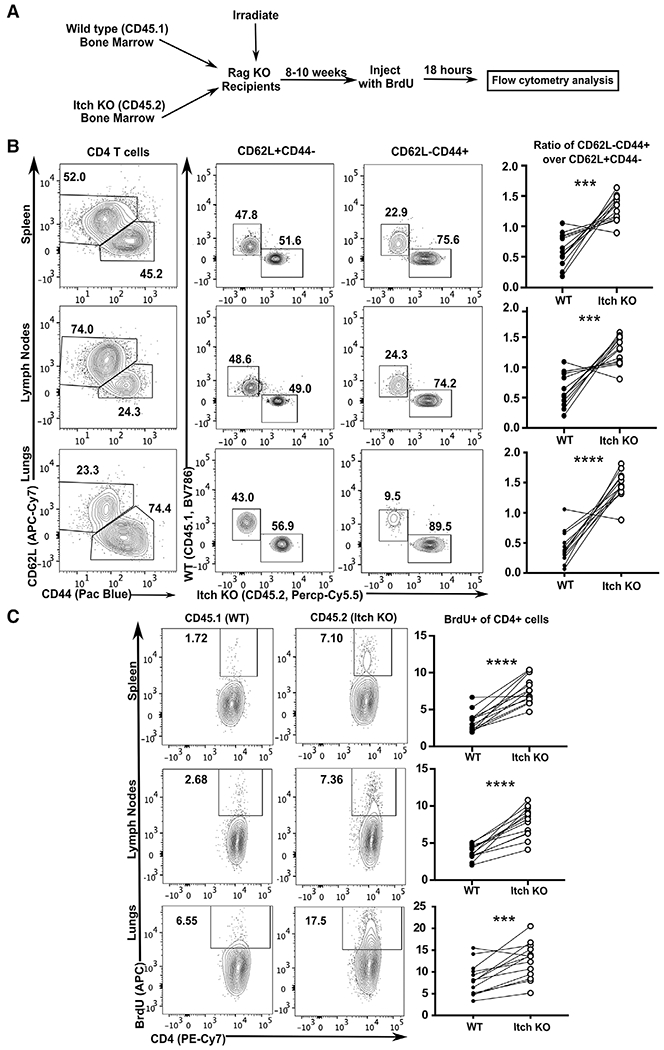
Itch limits activated CD4 T cell accumulation and proliferation in vivo. (A) Bone marrow was extracted from WT CD45.1 and Itch deficient CD45.2 mice, mixed at a 1:1 ratio of WT: Itch KO, and injected into irradiated Rag-deficient hosts. After reconstitution, mice were injected with BrdU, and 18 h later, tissues were analyzed by flow cytometry. (B) Cells from spleen, lymph nodes, and lungs were stained and gated on Live/Dead negative, singlets, lymphocytes (based on low forward and side scatter), and CD3+CD4+, as displayed in Supporting information Fig. S1A. CD4 T cells were then divided into naïve (CD62L+CD44−) or activated (CD62L−CD44+) populations, and the percent of WT (CD45.1) was compared to the percent of Itch deficient (Itch KO, CD45.2) cells. Quantification shows activated WT cells over naïve WT cells compared to activated Itch KO cells over naïve Itch KO cells. p-values were determined by paired t-test (paired by recipient mouse). p = 0.002 (spleen), p = 0.0005 (lymph nodes), p < 0.0001 (lungs). (C) Cells from spleen, lymph nodes, and lungs were stained and gated on Live/Dead negative, singlets, lymphocytes (based on low forward and side scatter), CD3+CD4+, and CD44+CD62L−, as shown in Supporting information Fig. S1A, representing activated CD4 T cells. The percent BrdU positive was analyzed separately for WT CD45.1 and Itch KO CD45.2 cells. Quantification shows percent of BrdU+ cells within the CD44+CD62L− population for each genotype. p-values were determined by paired t-test (paired by recipient mouse). p < 0.0001 (spleen and lymph nodes), p = 0.0005 (lungs). (BC) n = 13 total mixed chimeras generated from two pairs of bone marrow donors, analyzed over 4 BrdU injection experiments.
We first analyzed naïve CD4 T cells (defined as CD62L+CD44−) and found that the relative contributions of the Itch deficient cells and WT control cells were similar. In contrast, analysis of activated (CD62L−CD44+) CD4 T cells showed a strong bias toward the Itch deficient cells. This finding was reflected both in relative percentages of WT and Itch deficient cells (Fig. 1B), as well as the absolute cell numbers (Supporting information Fig. S2A). This observation suggested that, while the Itch deficient progenitors were no more likely than WT cells to differentiate into naïve CD4 T cells, the Itch deficient cells had a competitive advantage once they were activated and had undergone proliferation.
To determine if this competitive advantage was due to an increased rate of proliferation, we injected the mixed chimeras with BrdU 18 h prior to sacrifice (Fig. 1A). By gating on these activated cells and measuring the percent that incorporated BrdU (Supporting information Fig. S1A), we were able to quantify the percent that had synthesized DNA, an indication that they entered cell cycle in the prior 18 h. We observed that, in the spleen, lymph nodes, and lungs within each mouse, a higher percentage of the Itch deficient CD4+CD62L−CD44+ T cells had entered cell division compared to WT cells (Fig. 1C). While very few naïve (CD4+CD62L+CD44−) cells demonstrated BrdU incorporation, a similar pattern was observed in these cells (Supporting information Fig. S2B). It is possible that the proliferating CD4+CD62L+CD44− cells were newly activated CD4 T cells that had not yet upregulated CD44. The increased BrdU incorporation in Itch deficient cells implies that these CD4 T cells are intrinsically hyperproliferative in vivo, and that this behavior could account for the accumulation of activated CD4 T cells in Itch deficient mice. Interestingly, activated CD8 T cells analyzed from the same experiments showed increased BrdU incorporation in the Itch deficient cells compared to WT controls, particularly in the primary lymphoid organs (Supporting information Fig. S2C), suggesting these findings might also apply to CD8 T cells.
Itch restricts proliferation in response to TCR stimulation in vitro and independently of IL-4
Upon observing that Itch deficient CD4 T cells undergo more homeostatic proliferation in vivo, we next sought to determine whether we could recapitulate this finding in an in vitro activation system. We isolated naïve CD4 T cells from Itch deficient and WT mice in order to synchronize the activation of these cells. To determine whether activated Itch deficient cells have a competitive advantage in vitro, we cocultured WT CD45.1 cells with Itch deficient CD45.2 cells at a 1:1 ratio and stimulated the mixture using plate-bound anti-CD3 and anti-CD28 antibodies in IL-2 containing media. Excess IL-2 was added to the media to mitigate the potential effects of increased IL-2 production that has been reported in Itch deficient cells [6]. We then analyzed the composition of the coculture over time by flow cytometry (Supporting Information Fig. S1B) to determine the cells’ relative rate of growth. We found that Itch deficient cells out-competed WT cells by approximately 2:1 (Fig. 2A). The cells reached this ratio by day 5, and stayed stable through day 8. This finding suggests that activated Itch deficient cells have a competitive advantage over WT cells, particularly in the first 5 days after TCR stimulation.
Figure 2.
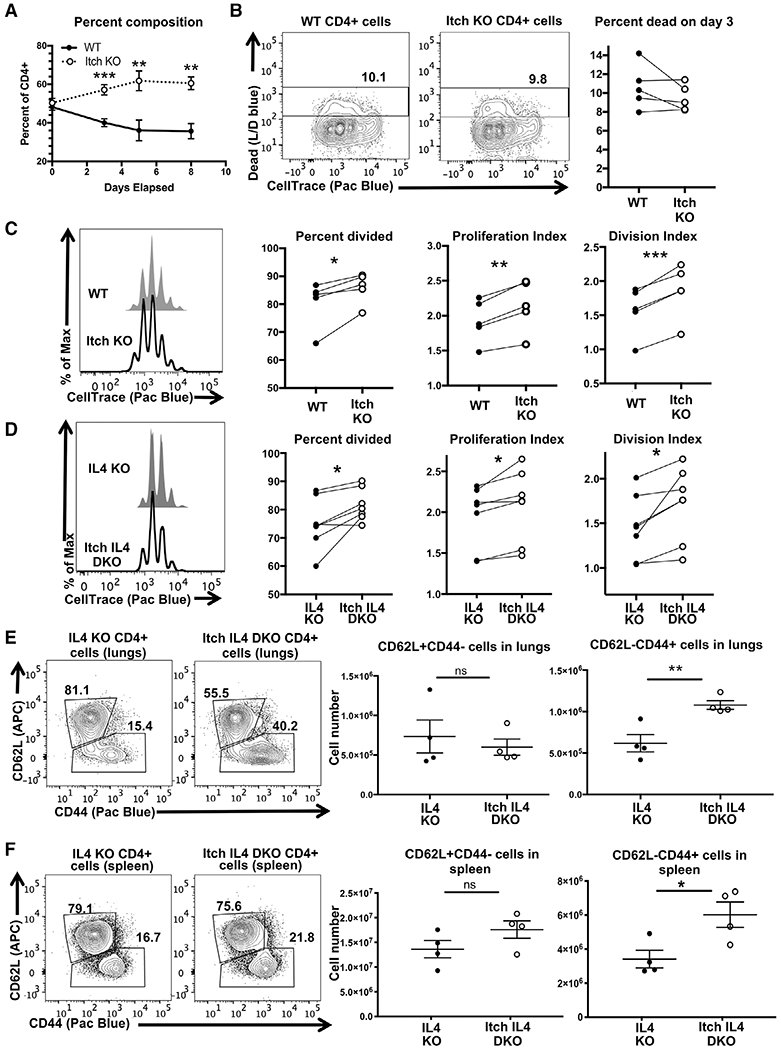
Itch limits CD4 T cell proliferation in vitro and independently of IL-4. Naïve CD4 T cells were isolated from WT CD45.1, Itch KO CD45.2, IL4 KO CD45.1, and Itch IL4 DKO CD45.2mice. Cells were mixed 1:1 of either WT and Itch KO cells (A-C) or IL4 and Itch IL4 DKO cells (D). Cocultures were labeled with CellTrace violet and cultured in IL-2-containing media on plates coated with anti-CD3 and anti-CD28 antibodies and analyzed by flow cytometry. (A) Cocultures were sampled at the indicated timepoints and stained to determine the relative composition. Cells were gated as displayed in Supporting information Fig. S1B, using the fluorophores BV786 and PerCP-Cy5.5 for CD45.1 and CD45.2, respectively. n = 5 biological replicates over four independent experiments. p = 0.0009 (day 3), p = 0.0085 (day 5), p = 0.0013 (day 8) based on multiple t-tests without correction for multiple comparisons. Error bars represent mean ± SEM. (B) Cell death and CellTrace violet dilution were analyzed on day 3 after plating. Cells were gated as described in (A) and shown in Supporting information Fig. S1, again using fluorophores BV786 and PerCP-Cy5.5 for CD45.1 and CD45.2, respectively. The Live/Dead and FSC-A versus SSC-A gates were omitted to allow for dead cell analysis of CD45.1 and CD45.2 cells separately. n = 5 biological replicates across four independent experiments. p = 0.1969 determined by paired t-test (paired by coculture). (C) The same experiments shown in (B) analyzed for proliferation. Percent divided, division index, and proliferation index were calculated using FlowJo software. n = 5 biological replicates across four independent experiments. p = 0.0245 (percent divided), p = 0.0029 (proliferation index), p = 0.0009 (division index), determined by paired t-test (paired by coculture). (D) Experiment described in (C) using IL4 KO versus Itch IL4 DKO cocultures. Cells were stained, gated, and analyzed as described in (C). n = 7 biological replicates across six independent experiments. p = 0.0232 (percent divided), p = 0.0169 (proliferation index), p = 0.0207 (division index), determined by paired t-test (paired by coculture). Cells from lungs (E) and spleens (F) of IL4 KO and Itch IL4 DKOmice were stained and gated as displayed in Supporting information Fig. S1A, replacing the fluorophore APC for CD62L. The ratio of naïve CD62L+CD44− to activated CD62L−CD44+ cells was analyzed. Cell numbers for each were quantified based on flow data and hemocytometer counts. p-values were determined by unpaired t-test. p = 0.5814 (lungs, CD62L+CD44−), p = 0.0077 (lungs, CD62L−CD44+), p = 0.17 (spleen, CD62L+CD44−), p = 0.028 (spleen, CD62L−CD44+). n = 4 mice of each genotype across three independent experiments. All error bars represent mean ± SEM.
To determine whether this competitive advantage is a result of increased survival of Itch deficient cells, we analyzed the rate of cell death by day 3 of culture. Cells were labeled with CellTrace violet, then cultured in a 1:1 mixture as described above. On day 3, cells were stained and analyzed by flow cytometry (Supporting information Fig. S1B) to measure the percent that took up Live/Dead blue. We found no difference in the percent of WT versus Itch deficient cells that were positively labeled, indicating no differences in cell death by day 3 (Fig. 2B). Furthermore, cells at all stages of division were Live/Dead blue positive in both genotypes, indicating that the WT cells were not simply dying at the start of activation and failing to divide.
To determine whether Itch deficient cells had a proliferation advantage over WT cells, we analyzed the CellTrace dilution of live cells in the same experiment. Using FlowJo software, we applied three different methods of quantification to measure proliferation: percent divided, which estimates the percent of cells that entered cell cycle, proliferation index, which measures the number of times each dividing cell has replicated, and division index, which measures the average number of times all cultured cells have divided. Across all three metrics, the Itch deficient cells proliferated more than WT cells, indicating that Itch deficient cells are more likely to enter cell cycle at the start, and those that enter cell cycle divide more frequently (Fig. 2C).
Itch deficient T cells have increased production of IL-4 in part due to accumulation of the transcription factor JunB [7, 8], and high levels of IL-4 contribute to the inflammation seen in Itch deficient mice [15]. To determine if this excess IL-4 contributed to T cell expansion, we also measured the proliferation rate of Itch IL4 double knockout (Itch IL4 DKO) CD4 T cells as compared to IL4 knockout (IL4 KO) cells, again in coculture. While the difference in proliferation was less than that observed between IL-4 sufficient cells, the Itch deficient cells still proliferated more than controls (Fig. 2D). This difference was observed at submaximal levels of TCR stimulation as well (Supporting information Fig. S3A–C). Thus, excess production of IL-4 was not sufficient to fully account for the increased proliferation. To determine whether this was also the case in vivo, we analyzed the frequencies and numbers of activated CD4 T cells in Itch IL4 DKO mice. The lungs (Fig. 2E) and spleen (Fig. 2F) of Itch IL4 DKO mice had increased percentages and total cell numbers of CD62L−CD44+ CD4 T cells when compared to IL4 KO controls, indicating that increased levels of IL-4 production and autocrine IL-4 signaling was not able to account for the accumulation of activated CD4 T cells.
Further supporting a role for Itch in directly limiting CD4 T-cell proliferation in vivo, mixed chimeras generated from IL4 KO and Itch IL4 DKO donors demonstrated phenotypic similarities to the mixed chimeras generated from WT and Itch KO donors. Itch IL4 DKO activated CD4 T cells accumulated more (Supporting information Fig. S4A) and were more likely to divide, as indicated by BrdU incorporation (Supporting information Fig. S4B), compared to Itch sufficient IL4 KO control cells. Taken together, these findings support that Itch limits CD4 T-cell proliferation in vitro and in vivo independently of its role in cytokine production.
Itch does not directly affect glycolytic capacity in CD4 T cells
We next investigated the cellular mechanism through which Itch regulates proliferation. One way by which CD4 T cells control their proliferation rate and effector subtype is by altering their metabolism. Upon TCR stimulation and activation, CD4 T cells switch from relying on mitochondrial respiration to glycolysis to fuel their rapid proliferation [16]. Our laboratory has previously observed that Nedd4 family-interacting protein 1 (Ndfip1) limits proliferation and glycolytic capacity of regulatory CD4 T cells [17]. Since Ndfip1 is an important activator of Itch in CD4 T cells [8], and since Itch regulates glucose metabolism in B cells [18], we tested whether Itch similarly regulates CD4 T cell proliferation by limiting glycolytic capacity. We isolated naïve CD4 T cells from IL4 KO and Itch IL4 DKO mice, in order to mitigate any effect of excess IL-4 production by Itch deficient cells. We either rested the cells in IL-2 containing media overnight or activated them using anti-CD3 and anti-CD28 bead-bound antibodies for 24 h in the IL-2 containing media. Basal glycolysis was determined by measuring the extracellular acidification rate (ECAR) of the cells after the addition of glucose. Maximal glycolytic capacity was determined by measuring the ECAR after the addition of oligomycin, which inhibits the electron transport chain, thereby forcing cells to rely exclusively on glycolysis to fuel their metabolic needs. We found that, at baseline and after 24 h of activation, Itch IL4 DKO cells had the same glycolytic capacity as IL4 KO cells (Fig. 3A and B).
Figure 3.
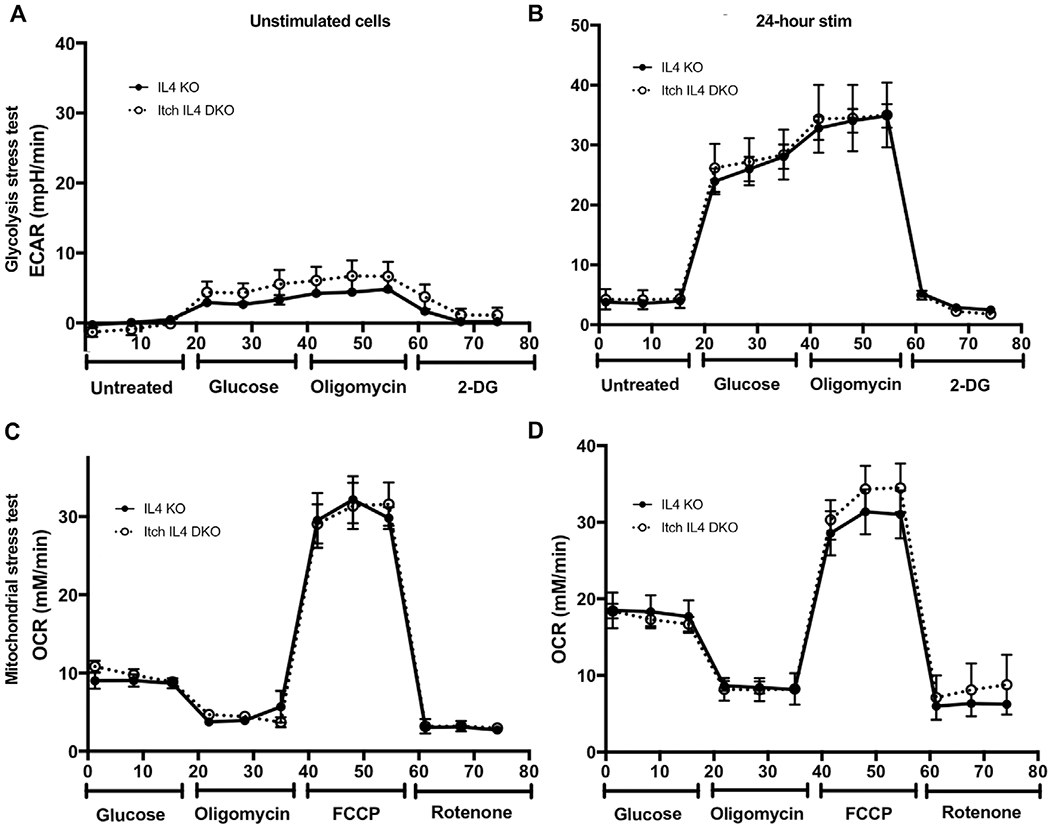
Itch does not regulate CD4 T cell metabolism during initial activation. Naïve T cells were isolated and rested in IL-2 for 24 h (A) and (C) or activated using IL-2 and anti-CD3 and anti-CD28 beads for 24 h (B) and (D). After this time, cells in (A) and (B) were tested for basal glycolysis and glycolytic activity by measuring extracellular acidification rate (ECAR) during treatment with the indicated drugs, including glucose, oligomycin, 2-deoxy-D-Glucose (2DG), and/or carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) with rotenone, using a seahorse assay. Cells in (C) and (D) were tested for basal oxidative phosphorylation and oxidative phosphorylation capacity by measuring oxygen consumption rate (OCR) during treatment with the indicated drugs, using a seahorse assay. n = 5 mice for restimulated cells (C) and (D) in three independent experiments, and n = 4 mice for unstimulated cells (A) and (B) across three independent experiments.
We also tested the effect of Itch on mitochondrial respiration under the same conditions. By measuring the oxygen consumption rate (OCR) of the cells during treatment with carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), which allows for maximal electron transport through the electron transport chain, we can approximate maximal respiratory capacity. Similarly, Itch deficient CD4 T cells had the same mitochondrial respiration capacity as WT cells (Fig. 3A and D). These findings demonstrate that Itch does not directly regulate metabolic capacity in CD4 T cells.
Itch restricts T cell cycle entry
To determine the mechanism through which Itch regulates proliferation, we next investigated its effects on cell cycle progression. To determine whether Itch regulates the progression from G1 to S phase, we stimulated Itch IL4 DKO and IL4 KO cocultured cells in IL-2-containing media and added BrdU to the media after 15 h of activation. The cells were then collected, stained for flow cytometry, and fixed at various timepoints to identify when they started incorporating BrdU, indicating that they had started DNA synthesis. We found that Itch IL4 DKO cells were more likely to be in S phase at early time points after activation, meaning that either Itch deficient cells are more likely to enter S phase, or that they enter S phase earlier, experiencing less time in G1 (Fig. 4A and B).
Figure 4.
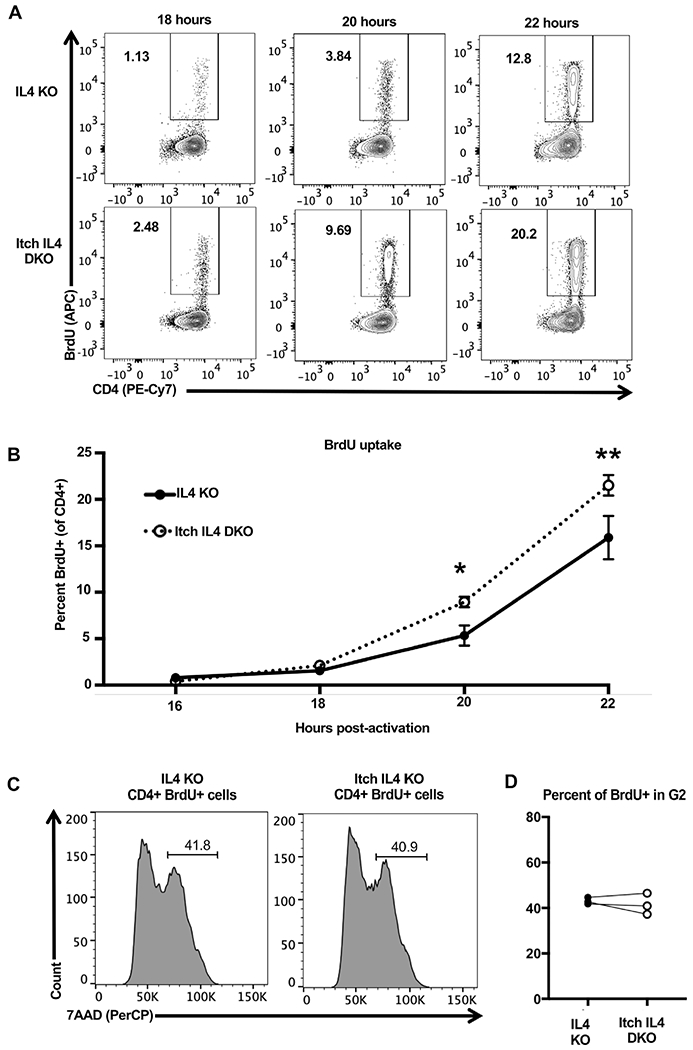
Itch limits CD4 T cell entry into S phase. Naïve CD4 T cells were isolated from CD45.1 IL4 KO and CD45.2 Itch IL4 DKO mice and activated in IL-2 containing media in coculture with plate-bound anti-CD3 and anti-CD28 antibodies for 22 h. BrdU was added at 15 h postactivation, and cells were sampled at indicated timepoints to measure BrdU uptake as an indicator of S phase entry. Cells were stained and gated on Live/Dead negative, singlets, lymphocytes (based on forward scatter and side scatter), CD4+, and CD45.1 or CD45.2, as shown in Supporting information Fig. S1. The percent BrdU+ of CD45.1 IL4 KO or CD45.2 Itch IL4 DKO cells was analyzed by flow cytometry. Representative flow plots are shown in (A) and all experiments quantified in (B). n = 5 mice of each genotype across three independent experiments. p-values were determined by multiple t-tests. p = 0.019 (20 h), p = 0.00050 (22 h). Error bars represent mean ± SEM. (C) Naïve CD4 T cells were isolated from CD45.1 IL4 KO and CD45.2 Itch IL4 DKO mice and activated in coculture with plate-bound anti-CD3 and anti-CD28 antibodies. Cells were cultured in IL-2-containing media. BrdU was added at 28 h after activation. Cells were harvested, stained, and fixed at 30 h after activation. After fixing, cells were dyed with 7AAD to measure DNA content. Cells were gated on Live/Dead negative, singlets, lymphocytes (based on forward scatter and side scatter), CD4+, CD45.1+, or CD45.2+, and BrdU+, as displayed in Supporting information Fig. S1. n = 3 mice across two independent experiments. p = 0.5588, analyzed by paired t-test (paired by coculture).
An increase in BrdU uptake in CD4 T cells at early time points could be due to increased activation sensitivity in these cells. To determine if Itch deficient cells were becoming activated earlier than their Itch sufficient counterparts, we examined the expression of the early activation markers CD69 and CD25 on the cell surface at varying doses of anti-CD3. CD69 expression occurs when cells exit quiescence but prior to S phase entry [19]. At all measured doses, we found that Itch deficient T cells exhibited the same CD69 expression kinetics as WT cells, suggesting that they do not have accelerated activation (Supporting information Fig. S5A). Likewise, Itch deficient cells exhibited similar CD25 expression kinetics as WT cells when cultured in the presence of exogenous IL-2 (Supporting information Fig. S5B). While previous studies have reported increased CD25+ cells in Itch deficient mice, this observation may be explained by the increased number of activated cells in these mice, or the increased IL-2 production by Itch deficient cells [6].
To further elucidate the role of Itch in cell cycle progression, we also investigated the transition from S phase to G2 phase using 7AAD to stain DNA. Naïve CD4+ T cells were cocultured and activated for 30 h, with BrdU in the media for the last 2 h. Cells were then stained and analyzed by flow cytometry. We then gated on BrdU+ cells (taking in to account the difference in the percent of cells that had incorporated BrdU at this time), and determined the percentage of Itch IL4 DKO and IL4 KO cells that had completely duplicated their DNA (indicating G2 phase entry). There were no differences between Itch IL4 DKO and IL4 KO cell progression from S to G2 phase progression (Fig. 4C and D), indicating that Itch regulates cell cycle predominately at the stage of S phase entry.
Whole cell proteome reveals potential direct and indirect targets of Itch
After determining that Itch regulates S phase entry of CD4 T cells, we wanted to determine how Itch may be regulating this step. We used a proteomic approach to screen for published as well as potential unreported substrates. Given that Itch is an E3 ubiquitin ligase, known to ubiquitinate proteins in a manner that increases their rates of degradation, we predicted that Itch targets would exhibit increased protein abundance in Itch deficient T cells. To identify these targets, we performed a whole cell proteome analysis using data-independent acquisition mass spectrometry on CD4 T cells from Itch IL4 DKO and IL4 KO mice. We analyzed naïve CD4 T cells, cells that were activated for 24 h (a timepoint prior to cell division), and cells that were activated for 48 h (to assess actively dividing cells). These time points were within the 3-day range in which we observed the proliferation differences.
Consistent with the observation that Itch IL4 DKO cells had increased cell cycle progression, Itch IL4 DKO cells showed dysregulation of cell cycle proteins during active division based on Gene Ontology (GO) term analysis through the Molecular Signatures Database (Fig. 5A) [20–24]. To identify potential contributing substrates, we first focused on the expression patterns of previously reported Itch substrates that have been predicted to regulate cell cycle: WBP2 [14, 25], Rassf5 [26], MAVS [27, 28], Tab1 [29, 30], Notchl [31–33], Rassfl [34], and c-Jun [7]. Several other published substrates were investigated as well, and were either undetected in this dataset or were not predicted to play a role in cell proliferation. Of these putative direct Itch targets, WBP2 was the only protein found to be statistically significantly altered in abundance at any time point. WBP2 was higher in Itch IL4 DKO cells as compared to IL4 KO cells prior to activation (Fig. 5B and C). To obtain a comprehensive picture of WBP2 expression in CD4 T cells, taking into account the differences in cell size in activated CD4 T cells compared to naïve ones, we analyzed the WBP2 copy number per cell at each timepoint [35, 36]. According to this analysis, WBP2 abundance was greater in the Itch deficient cells at baseline, and it increased to a higher level after 24 h of stimulation when compared to control cells (Fig. 5D). These findings support the conclusion that Itch deficient cells have more WBP2 during early activation timepoints.
Figure 5.
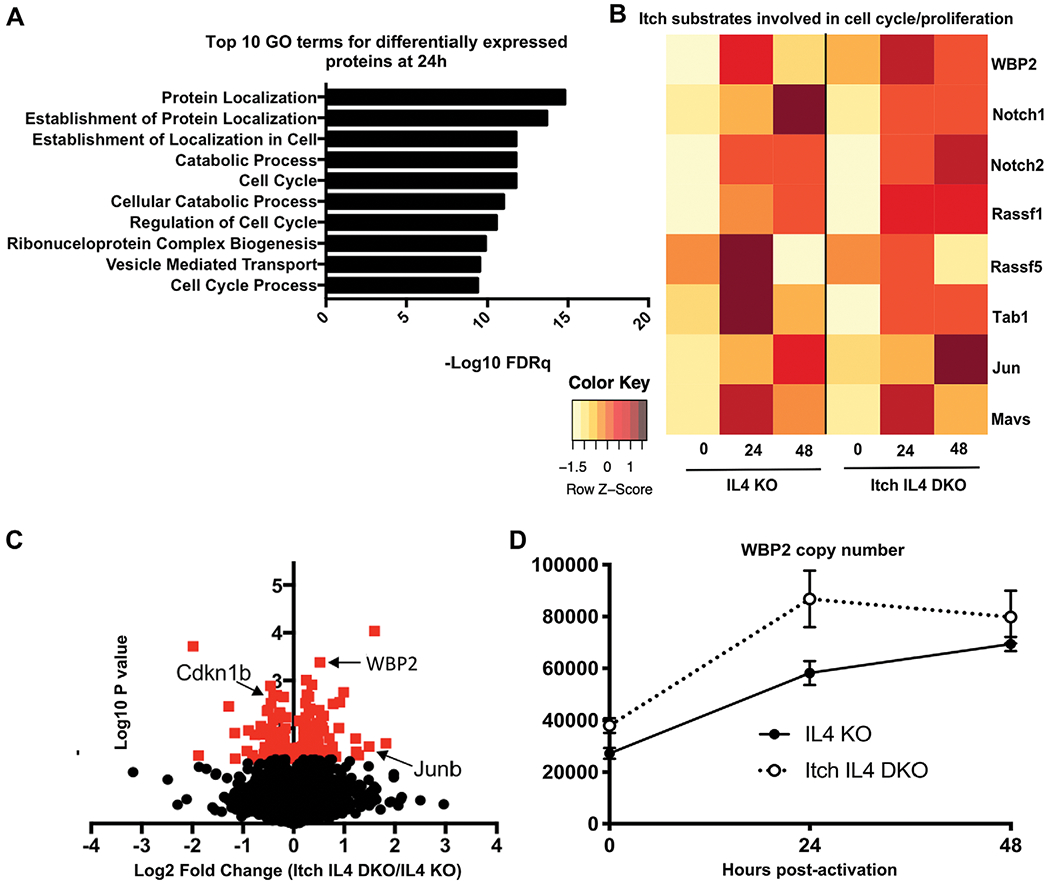
Whole cell proteome analysis reveals a role for Itch in regulating cell cycle proteins. CD4 T cells from spleen and lymph nodes of IL4 versus Itch IL4 DKO mice were activated with anti-CD3 and anti-CD28 antibody in IL-2-containing media for 0, 24, or 48 h. Whole cell proteomes were analyzed by mass spectrometry. n = 3 biological replicates (combined cells from two mice each) across three independent experiments. (A) Top 10 hits for Gene ontology (GO) term analysis on differentially expressed proteins (upregulated or downregulated) at 24 h postactivation. GO analysis was performed using the Broad Institute Molecular Signatures Databases. (B) Average abundance of select putative Itch targets at 0, 24, and 48 h postactivation in each genotype. All proteins shown here were detected in at least two of three replicates at one timepoint for both genotypes. Each undetected protein in a given replicate was assigned a value equal to the limit of detection. Average intensity values for each protein were normalized to the average intensity value for β-actin for that sample. Heat map is scaled by row. (C) Volcano plot of log2 fold change of proteins identified in naïve Itch IL4 DKO CD4 T cells over those identified in naïve IL4 CD4 T cells. Proteins normalized to the median protein in each sample. Red indicates p < 0.05 based on unpaired t-test. (D) WBP2 abundance in each sample, based on calculated protein copy number per cell. Error bars indicate mean ± SEM.
Itch attenuates WBP2 protein stability in CD4 T cells
While our proteomics dataset revealed several proteins that were increased in abundance in the Itch deficient cells compared to controls (Supporting information Fig. S6B and C), we postulated that WBP2 was the most likely to be a direct target of Itch in CD4 T cells, based on published data that Itch ubiquitinates and degrades WBP2 in breast cancer cell lines [14]. To test this, we first blotted for WBP2 in naïve CD4 T cells. As predicted by the mass spectrometry data, WBP2 abundance was higher in Itch IL4 DKO cells as compared to IL4 KO cells (Fig. 6A and B). When naïve CD4 T cells were treated for 6 h with the protein synthesis inhibitor cycloheximide or the protein degradation inhibitors MG132 and chloroquine, WBP2 abundance was only modestly decreased (in the case of cycloheximide) or increased (in the case of MG132 and chloroquine) in IL4 KO cells, suggesting a slow rate of protein turnover in unstimulated cells (Fig. 6A and B).
Figure 6.
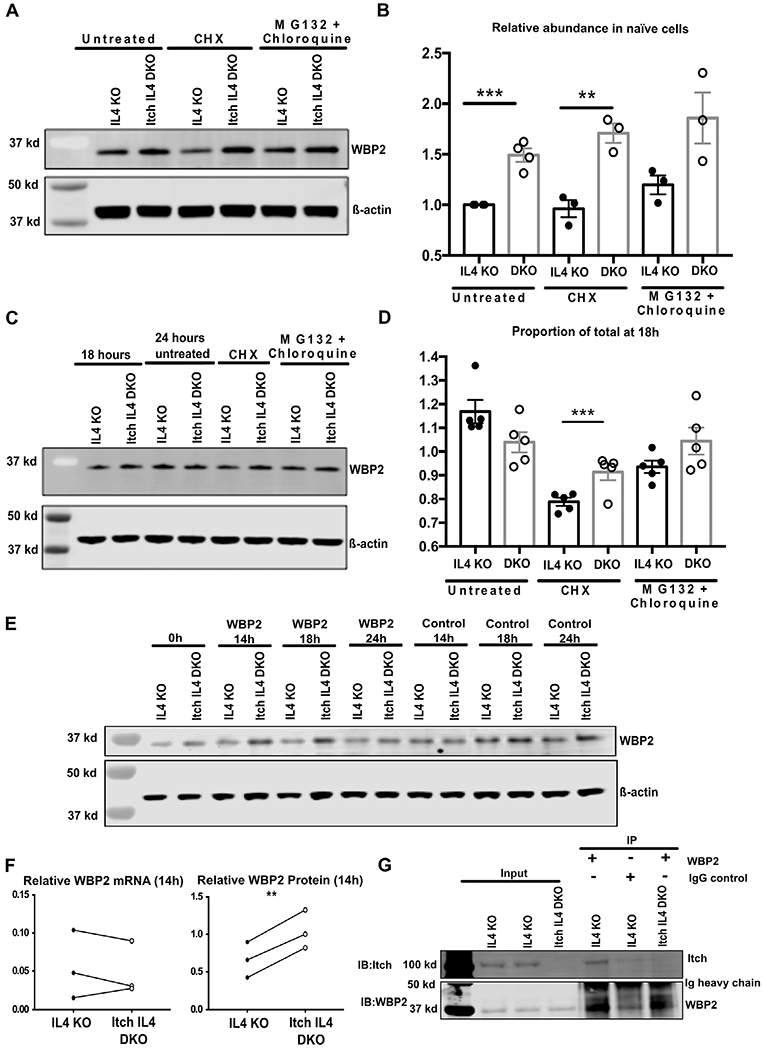
Itch attenuates WBP2 protein stability and associates with WBP2 in CD4 T cells. (A) Naïve CD4 T cells fromIL4 KO or Itch IL4 DKOmice were harvested and cultured in complete media for 6 h with no stimulation. Cells were either left untreated, or treated with the indicated drugs (CHX = cycloheximide). Western blots were performed on whole cell lysates, and membranes were probed with anti-WBP2 (rabbit polyclonal) and anti-β-actin (mouse polyclonal), and anti-rabbit 800 and anti-mouse 680 secondary antibodies were used. (B) Quantification of three biological replicates across three experiments described in (A). One additional experiment was performed on untreated cells. Each sample was normalized to β-actin, and these values were normalized to the untreated IL4 KO control sample within each experiment. p = 0.0003 (untreated), p = 0.004 (CHX-treated), p = 0.070 (MG132 and chloroquine-treated) based on unpaired t-test after the described normalizations. (C) Naïve CD4 T cells were isolated as described and activated with plate-bound anti-CD3 and anti-CD28 antibodies for 24 h in IL-2-containing media. After 18 h of activation, cells were either harvested and pelleted, left untreated, or treated with the indicated drugs for 6 h (between 18 h and 24 h). Western blots were performed as described in (A). (D) Quantification of five biological replicates in five experiments described in (C). Each sample is normalized to β-actin. Each value was normalized to its own 18 h timepoint. p-values were determined by unpaired t-test. p = 0.0009 (CHX-treated). All error bars represent mean ± SEM. (E-F) IL4 KO and Itch IL4 DKO cells were transfected with either control siRNA or WBP2 siRNA and collected at the indicated timepoints to check for protein knockdown (E) and (F) or RNA knockdown (F). n = 3 biological replicates across three independent experiments for each protein and RNA. In one of the three biological replicates, RNA and protein were analyzed in the same experiment. Data were analyzed by paired t-test, pairing IL4 KO cells with Itch IL4 DKO cells from the same knockdown experiment. WBP2 protein or RNA was normalized to β-actin protein or RNA. These values were then normalized to the relative WBP2 abundance in control-siRNA-treated cells at the same timepoint. p = 0.5641 (RNA), p = 0.0043 (protein). (G) Representative immunoprecipitation of WBP2 in CD4 T cells. After 20 h of activation, IL4 KO or Itch IL4 DKO cells were treated with MG132 and chloroquine. Cell pellets were lysed, precleared, and incubated overnight with either WBP2 antibody or Rabbit IgG control, as indicated. Pulldown was run on a gel and blotted with anti-Itch and anti-WBP2 antibody and corresponding secondary antibodies as described in (A). Blot is representative of two biological replicates done in independent experiments.
To test the rate of WBP2 turnover during T-cell activation, we activated naïve CD4 T cells for 18 h using anti-CD3 and anti-CD28 antibodies. Cells were either harvested at this time, left untreated, or treated with cycloheximide or MG132 and chloroquine for the following 6 h (corresponding to a total of 24 h of activation). Untreated cells from both IL4 KO and Itch IL4 DKO cells had a modest increase in WBP2 abundance between 18 to 24 h, indicating new protein synthesis (Fig. 6C and D). When cells were treated with cycloheximide, the abundance of WBP2 in Itch IL4 DKO cells at 24 h remained unchanged compared the abundance in these cells at 18 h, indicating no protein degradation during this time. In contrast, IL4 KO cells treated with cycloheximide showed a reduction in WBP2 abundance between 18 to 24 h, indicating WBP2 protein was degraded in these cells. No changes in abundance were observed between 18 to 24 h in MG132 and chloroquine treated cells in either genotype. Interestingly, the absolute abundance of WBP2 at 24 h postactivation was not different between Itch IL4 DKO cells as compared to IL4 KO controls, when both were normalized to β-actin (Fig. 6C). This finding seemed to contradict the proteomics analysis showing increased WBP2 abundance in Itch IL4 DKO cells at this timepoint (Fig. 5D). To determine whether the normalization method may be obscuring differences in protein abundance, we analyzed the proteomics data by normalizing WBP2 abundance to β-actin abundance at each timepoint. According to this analysis method, WBP2 was increased in Itch IL4 DKO cells in naïve cells, but this difference became negligible by 24 h after activation (Supporting information Fig. S6D), suggesting that normalizing to β-actin may not accurately capture absolute WBP2 abundance. According to any of these analyses, however, WBP2 was increased in abundance in the Itch deficient cells at early timepoints.
These observations supported that Itch promotes WBP2 protein degradation. To test this hypothesis more specifically, we treated IL4 KO and Itch IL4 DKO cells with WBP2 siRNA to suppress new protein synthesis. While both genotypes displayed similarly efficient knockdown of WBP2 RNA, the Itch IL4 DKO cells had higher WBP2 protein levels at 14 h post-transfection as compared to IL4 KO cells, indicating longer persistence of the protein product (Fig. 6E and 7F). Together, these findings suggest that Itch is required for optimal WBP2 degradation.
Figure 7.
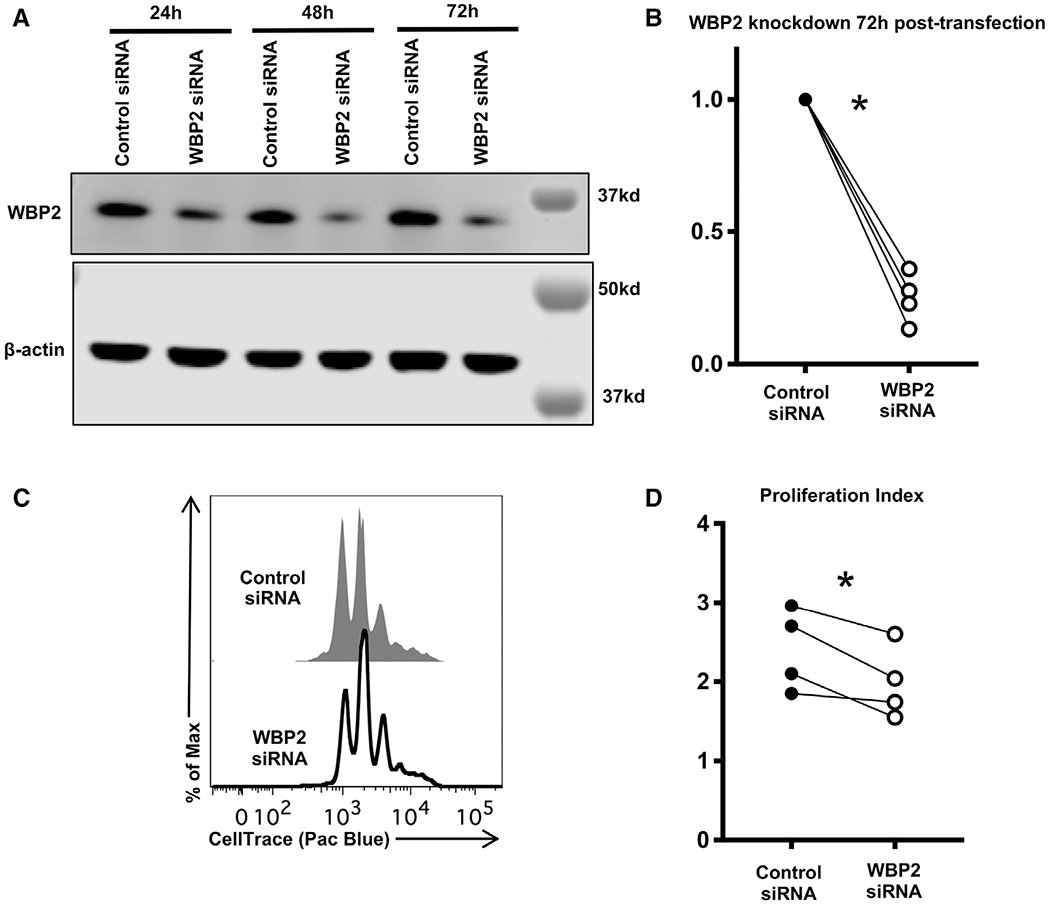
WBP2 promotes CD4 T-cell proliferation. Naïve T cells were isolated from WT mice, labeled with CellTrace violet and activated using plate-bound anti-CD3 and anti-CD28 in IL-2-containing media for 24 h, then transfected with either nontargeting control siRNA or siRNA specific for WBP2. (A) Representative Western showing WBP2 levels in transfected cells. Western blots were probed as described in Fig. 6. Timepoints refer to timing post-transfection, which was 24 h after initial activation. (B) Quantification of WBP2 protein knockdown. Each sample is normalized to β-actin. Each WBP2 siRNA-treated sample was normalized to its corresponding control siRNA-treated timepoint. p-values were calculated by paired t-test, pairing cells from the same mouse. p = 0.0006 (72 h). (C) Representative flow analysis of proliferation 72 h post-transfection. Cells were not stained to avoid further stress and cell loss; live lymphocytes were identified on the basis of forward scatter and side scatter as shown in Supporting information Fig. S1. (D) Quantification of proliferation across all experiments. Proliferation index was calculated using FlowJo software, which determines the number of cells in each peak. p = 0.0399, determined by paired t-test, pairing control versus WBP2 siRNA-treated cells from the same mouse. (A-D) n = 4 biological replicates across three independent experiments.
To determine whether Itch interacts directly with WBP2, we performed coimmunoprecipitation at the same timepoint at which degradation was observed. We stimulated IL4 KO in the presence of IL-2 for 24 h, and treated the cells with MG132 and chloroquine for the last 4 h of stimulation. When anti-WBP2 antibody was used to immunoprecipitate WBP2 from these samples, it was found to be associated with Itch (Fig. 6G). This finding implies that Itch and WBP2 interact directly, or are part of the same complex, in CD4 T cells.
WBP2 promotes cell proliferation in CD4 T cells
It has been reported that WBP2 promotes tumor growth by acting as a coactivator for β-catenin downstream of Wnt signaling [14], and that it promotes cell proliferation [37]. WBP2 has been implicated specifically in S phase entry [25], consistent with the phenotype in Itch deficient CD4 T cells. However, no role for WBP2 in CD4 T cells has been described. To test the possibility that WBP2 promotes CD4 T cell proliferation, we labeled naïve WT CD4 T cells with CellTrace violet and activated them for 24 h. We subsequently treated them with WBP2 siRNA. By 72 h post-siRNA treatment (96 h of activation), WBP2 levels were reduced to about 30% abundance compared to cells treated with control siRNA for the same amount of time (Fig. 7A and B). We measured cell proliferation by CellTrace dilution at the end of 72 h post-transfection, during which time WBP2 levels were reduced (Fig. 7A). Compared to cells treated with control siRNA, cells treated with WBP2 siRNA showed reduced proliferation over this time, suggesting that WBP2 is required for optimal CD4 T cell proliferation (Fig. 7C and D). Similarly, Itch deficient cells demonstrated a trend toward decreased proliferation when WBP2 was knocked down (Supporting information Fig. 7A–D). Taken together, these findings suggest that the increased levels of WBP2 in Itch deficient cells during early T-cell activation might account, at least in part, for their increased proliferation.
Discussion
This study provides evidence that Itch is a crucial regulator of CD4 T cell proliferation. While previous studies reported that lymph node cells from Itch deficient mice proliferated more in vitro [6], this study is the first to describe a direct role for Itch in CD4 T cell proliferation following activation. Furthermore, while the prior analyses were complicated by the altered cytokine production in Itch deficient cells, here we clarified that Itch regulates CD4 cell proliferation primarily through Il-4-independent mechanisms. While the observed in vitro proliferation differences were modest, the ability of Itch deficient CD4 T cells to out-compete WT cells over time, both in vitro (Fig. 2A) and in vivo (Fig. 1B), with no differences in cell survival (Fig. 2B), suggests that these proliferation differences are biologically significant.
We found that Itch deficient cells did not show changes in glycolytic or mitochondrial respiratory capacity 24 h after activation in the absence of IL-4 (Fig. 3A–D), suggesting that Itch regulates proliferation independently of metabolism. This mechanism is different from our laboratory’s observations in Itch deficient B cells [18]. Our data suggest instead that Itch directly limits CD4 T cell proliferation by slowing cell cycle progression. Itch deficient CD4 T cells exhibited increased S phase entry at early timepoints, while progression through S phase to G2 phase was unaffected. This phenotype is unlikely to be a result of higher levels of JunB, the most well-characterized Itch substrate in CD4 T cells [7, 8], because most evidence suggests that JunB inhibits cell proliferation [38]. In fibroblasts, where JunB has been shown to promote cell cycle progression, JunB facilitates S to G2 phase progression, rather than S phase entry [39].
Our whole cell proteomics analysis and subsequent biochemical validation support that Itch regulates cell cycle progression by reducing WBP2 stability through direct association with WBP2. This is not without precedence. In breast cancer cell lines, Itch has been shown to bind WBP2 via its WW domains, and this leads to WBP2 ubiquitination and degradation [14]. Placing these observations in context with our data, we assert that WBP2 is also a substrate of Itch in CD4 T cells.
Here we show the first reported evidence that WBP2 regulates proliferation in CD4 T cells (Fig. 7C and D, Supporting information Fig. S7C and D). Although our findings demonstrate modest changes in proliferation when WBP2 levels are decreased, we may be underestimating the role of this protein due to assay limitations. The biggest differences in WBP2 abundance between WT and Itch deficient cells were observed prior to 24 h of activation (Fig. 5C and D). However, the siRNA treatment was necessarily carried out after 24 h of stimulation, and full knockdown did not occur until 24 h after siRNA treatment, 48 h after activation (Fig. 6E). Nonetheless, cells deficient in WBP2 demonstrated decreased proliferation (Fig. 7C and D, Supporting information Fig. S7C and D). These data, while correlative, suggest that the increased stability of WBP2 in Itch deficient CD4 T cells promotes proliferation. Furthermore, the increased S phase entry observed in Itch KO CD4 T cells is consistent with data that WBP2 promotes cell cycle at this stage in other cell types [25]. While further research is required to determine the mechanism through which WBP2 acts in this context, one possibility is that WBP2 serves as a coactivator for β-catenin to promote transcription of targets, such as Myc, similar to its role in breast cancer cells [14, 25, 37].
While WBP2 likely plays a prominent role in the increased CD4 T cell proliferation observed in Itch deficient cells, it is possible that other Itch substrates influence CD4 T cell proliferation. For example, we found the RNA splicing regulator Srpk2 was significantly increased in Itch IL4 DKO cells at 24 h postactivation (Supporting information Fig. S6B). This protein has been shown to promote cell proliferation [39, 40]. Furthermore, Srpk2 has a proline-rich sequence that maybe capable of binding to Itch [42], suggesting it may be a direct substrate. It is also possible that Itch regulates cell cycle through indirect mechanisms. Regulator of cell cycle (Rgcc) was upregulated in Itch deficient cells at both 24 and 48 h postactivation (Supporting information Fig. S6C and D). While Rgcc is not thought to be a substrate of Itch, it may be a downstream effector that plays a role in proliferation. Rgcc has been shown to promote S phase progression in smooth muscle cells [43], although it is thought to have antiproliferative properties in CD4 T cells [44].
In summary, here we have established that Itch is an important regulator of CD4 T-cell proliferation by reducing the levels of WBP2 and thus restricting S phase entry. Furthermore, our data identifies WBP2 as a modulator of CD4 T cell proliferation. These findings elucidate a possible mechanism by which Itch prevents autoimmune disease in mice and patients.
Materials and methods
Mice
Itch knockout (Itch KO) [4], WT, IL4 knockout (IL4 KO), Itch IL4 double knockout (Itch IL4 DKO), and Rag1 knockout (Rag KO) mice were bred in house at the Children’s Hospital of Philadelphia. IL4 KO mice were bred on a CD45.1 background, as were the WT mice used in coculture and mixed chimera experiments. Mice were used between 6-14 weeks of age, with the exception of Rag KO bone marrow chimera recipients which were injected between 6-12 weeks of age and allowed to reconstitute up to an additional 12 weeks. Male and female mice were used in most experiments and were age and sex matched within each experiment.
Mixed chimeras
Bone marrow was harvested from Itch KO CD45.2 mice and WT CD45.1 mice or Itch IL4 DKO CD45.2 and IL4 KO CD45.1 mice and frozen at −80°C until use. Upon thawing, bone marrow was T cell depleted using TCRβ PE antibody and anti-PE microbeads (Miltenyi, cat. 130-048-801). Rag KO recipients were irradiated with 400 Rad using an X-Rad Irradiator, and subsequently injected via tail vein with 2 × 106 bone marrow cells in RPMI 1640 (Hyclone, SH30027.01) with 100U/mL Penicillin-streptomycin (Gibco, cat. 15140122) at a 1:1 mixture of WT: Itch KO, or IL4: Itch IL4 DKO. The mice were maintained on antibiotics (trimethoprim-sulfamethoxazole) water for 2 weeks following irradiation and injection, and allowed to reconstitute for at an additional 6-12 weeks. They were i.p. injected with 1 mg of BrdU (BD Biosciences, cat. 552598) 18 h prior to euthanasia.
Tissue processing and staining
Spleen, lymph nodes, and lungs were harvested from mice after euthanasia by carbon dioxide and cervical dislocation. Tissues were homogenized in RPMI using 70 μm filters. Lungs were treated with collagenase I and Ia (Sigma, cat. C0130 and C9891, respectively), and 20 μg/mL DNAse I (Roche, cat. 10104159001) in 10 mL RPMI for 1 h at room temperature prior to homogenization. Spleen and lungs were treated with RBC lysis buffer and passed through an additional 70 μm filter. Single cell suspensions were stained according the guidelines for the use of flow cytometry [45]. Briefly, cells were treated with a fixable viability dye (Life Technologies, cat. L34961) and pretreated with unlabeled anti-CD16/CD32 (Fc Block, BD Pharminogen, cat. 564219). Cells were stained in FACS buffer with the following antibodies: anti-CD4 (BioLegend, clone GK1.5, PE-Cy7), anti-CD3 (Biolegend or BD biosciences, clone 17A2, AF700 or BV605), anti-CD62L (Biolegend, clone MEL-14, APC or APC-Cy7), anti-CD44 (BioLegend, clone IM7, Pac Blue), anti-CD69 (BD Biosciences, clone H1.2F3, PE), anti-CD45.1 (BioLegend, clone A20, APC-Cy7 or BV786), and anti-CD45.2 (BioLegend, clone 104, PerCP-Cy5.5 or Pac Blue). All BrdU staining was performed using the kit reagents and protocols.
Cell isolation and culture
Spleen and lymph nodes were harvested from mice as described above. Single cell suspensions were resuspended in MACS buffer consisting of 0.5% FCS (Atlanta Biologicals Premium) and 2 mM EDTA in PBS. Naïve CD4 T cells were isolated using a naïve T-cell isolation kit (Miltenyi, cat. 130–0940131) following manufacturer’s protocol. This method resulted in at least 90% CD62L+CD44−CD25−CD4+ cells. In some experiments, naïve T cells were isolated via FACs sorting using a MoFlo Astrios at the Flow Cytometry Core at Children’s Hospital of Philadelphia, gating for CD62L+CD44−CD25−CD4+ cells.
Cells were cultured in RPMI 1640 supplemented with 10% FCS, 100 U/mL Penicillin-streptomycin, 1× Glutamax (ThermoFisher, cat. 35050061), 1× nonessential amino acids (Gibco, cat. 11140-050), 2 mM HEPES (Gibco, cat. 15630-080), 1 mM sodium pyruvate (Corning, cat. 20115013), 8 μL/L 2-mercaptoethanol (Sigma), and recombinant human IL-2 at 50 U/mL (Peprotech, cat. 200–02). Cells were cultured at 37°C in 10% carbon dioxide. To activate T cells, cell culture plates were coated using 5 μg/mL anti-CD3 (BD biosciences, clone 17A2) and an equal amount of anti-CD28 (BD biosciences, clone 37.51) in PBS at 4 degrees overnight or at least 2 h at 37°C. Cells were cultured on these coated plates for up to 4 days, depending on the experiment. For proliferation experiments, cells were labeled with CellTrace violet (ThermoFisher, cat. C34571) prior to activation, and were then plated in coculture. For Western blotting and immunoprecipitation experiments, cells were treated with either cycloheximide 10 μg/mL (Sigma, C4859), or MG132 10 μM (UBPbio, F1100), and chloroquine 5 μM (Sigma, C6628) for the last 6 h (for Western) or 4 h (for immunoprecipitation) of culture.
Seahorse assays
Naïve CD4 T cells were isolated and cultured in complete RMPI 1640 as described above. Cells were either stimulated with anti-CD3 anti-CD28 dynabeads at a 1:1 beads:cell ratio in the presence of IL-2, or kept “resting” in IL-2 media. After 24 h, cells were washed and resuspended in Seahorse media pH 7.4 (Agilent, cat. 102353-100), supplemented with 1× Glutamax (in the case of the glycolysis stress test) or 1× Glutamax, 1 mM sodium pyruvate, and 25 mM glucose (for the mitochondrial respiration stress test), and plated on a Seahorse plate (Agilent, 100777-004) at a density of 200K cells per well. The dynabeads were transferred to the plate with the cells, so stimulation continued for approximately 1.5 more hours during the assay. The Seahorse plates were prepared by precoating at 37°C in 10% carbon dioxide overnight with a PBS solution containing CellTak (Corning, CB40240), NaOH, and NaHCO3. Fluxplates (Agilent, cat., 103022–100) were hydrated in calibrant (Agilent, cat. 100840-000) overnight before the experiment at 37°C without carbon dioxide. Glucose, oligomycin, 2-deoxy-D-Glucose (2DG), and FCCP/rotenone were prepared from the glycolysis stress (Agilent, cat. 103020–100), or mito stress kits (Agilent, cat. 103015–100). ECAR and OCR values were measured by the Agilent Seahorse XFe96 instrument at the University of Pennsylvania Diabetes Research Center, Islet Cell Biology Core, and data were analyzed by Wave software (Agilent).
In vitro BrdU labeling
Naïve CD4 T cells were isolated using the Naïve CD4 T-cell isolation kit as described above, and activated in coculture using plate-bound anti-CD3 and anti-CD28 antibody. For S phase entry experiments, BrdU was added after 15 h of activation, and 0.5 × 106 cells were harvested, stained, and fixed every 2 h. For S to G2 phase experiments, cells were activated for 30 h and BrdU was added to the media for the last 2 h. Cells were then harvested, stained and fixed, then subsequently dyed with 7AAD (10 uL per 1 × 106 cells).
Proteomics
Naive CD4 T cells were isolated using the Naïve CD4 T-cell isolation kit as described above, and verified to have at least 95% purity. Cells were then either pelleted immediately (for the 0 h timepoint) or stimulated using plate-bound anti-CD3 and anti-CD28 antibody as described, and harvested at 24 and 48 h postactivation. Three biological replicates in three independent experiments were collected for each genotype at each time point. Each biological replicate consisted of two mice pooled.
Samples were analyzed by the Proteomics Core at the Children’s Hospital of Philadelphia on a QExactive HF mass spectrometer (Thermofisher Scientific San Jose, CA) coupled with an Ultimate 3000 nano UPLC system and and EasySpray source. Whole proteome samples were collected using data independent acquisition (DIA) and the spectral library was generated using data dependent acquisition (DDA). Tryptic digests were spiked with iRT standards (Biognosys) and separated by RP-HPLC on a nanocapillary column, 75 μm id × 50 cm 2 μm PepMap RSLC C18 column at 50°C. Mobile phase A consisted of 0.1% formic acid and mobile phase B of 0.1% formic acid/acetonitrile. Peptides were eluted into the mass spectrometer at 210 nL/min with each RP-LC run comprising a 125 min gradient from 1 to 5 % B in 15 min, 5-45% B in 110 min for DDA and 140 min for DIA. To generate the spectral library for the DIA analysis, biological replicates for each time point were pooled and data were acquired in a single shot using DDA mode. For DDA, the mass spectrometer was set to repetitively scan m/z from 300 to 1,400 (R = 240,000) followed by data-dependent MS/MS scans on the 20 most abundant ions, minimum AGC 1e4, dynamic exclusion with a repeat count of 1, repeat duration of 30 s, (R = 15,000) FTMS full-scan AGC target value was 3e6, while MSn AGC was 1 × 105, respectively. MSn injection time was 160 ms; microscans were set at one. Rejection of unassigned and 1+, 6-8 charge states was set. DIA data collection was as follows: one full-MS scan at 120,000 resolution and a scan range of 300-1,650 m/z with an AGC target of 3 × 106 and a maximum inject time of 60 ms. This was followed by 22 (DIA) isolation windows with varying sizes at 30,000 resolution, an AGC target of 3 × 106, injection times set to auto, loop count and msx count of 1. The default charge state was 4, the first mass was fixed at 200 m/z and the normalized collision energy (NCE) for each window was stepped at 25.5, 27, and 30.
Protein identification and quantification were based on intensity readings. Values were compared between IL4 KO and Itch IL4 DKO mice. Data were filtered, normalized, and statistically analyzed using R software. A protein was considered “detected” in a particular genotype and timepoint if it was identified in at least two of the three replicates. Statistical significance was determined by t-test.
Western blotting
Cell pellets were lysed using RIPA buffer (ThermoFisher, cat. 89900) supplemented with ETDA complete miniprotease inhibitor cocktail (Sigma, cat. 11836170001), and HALT phosphatase inhibitor cocktail (ThermoFisher, cat. 78426). Protein was quantified by BCA assay prior to gel loading. Samples were then boiled for 5-10 min with Laemelli 6× loading dye and 1,4-dithiothreitol, then loaded into 12 or 15-well 4-12% gradient gels (Novus biologicals). Gels were run in 1× running buffer (Novus Biologicals) at 120 volts for 2 h and transferred in 1× transfer buffer (Novus Biologicals) with 10% methanol at 45 volts for 75 min. Gels were blocked in Odyssey blocking buffer diluted 1:1 with PBS. Gels were incubated with anti-WBP2 (rabbit polyclonal; Proteintech cat. 12030-1-AP) and anti-β-actin (mouse monoclonal; Santa Cruz, cat. sc-47778) overnight in blocking buffer. Gels were washed with PBS-T, incubated with anti-mouse 680, and anti-rabbit 800 secondary antibodies, and imaged using a Li-Cor Imager. Images were quantified using ImageStudioLite.
Immunoprecipitation
Bulk CD4 T cells were isolated from the spleens and lymph nodes (about 50 × 106 cells per condition) of IL4 KO and Itch IL4 DKO mice using a CD4 isolation kit (Miltenyi, 130-104-454) and stimulated with plate-bound anti-CD3 and anti-CD28 in enriched RPMI in the presence of IL-2, as described above. Cells were treated with MG132 and chloroquine for the last 4 h of culture, as described above. These cells were pelleted and frozen until ready for use. Immunoprecipitation was carried out according to the protocol described for Tandem Ubiquitin Binding Entities (TUBE) described previously [46]. Briefly, cells were lysed using a lysis buffer supplemented with deubiquitinase inhibitors, and treated with Rabbit IgG control antibody (Cell signaling, 2729S) bound to Protein A Dynabeads (ThermoFisher, 10008D) for 2 h at 4°C. After this preclear, the lysate was diluted 1:5 with PBS supplemented transferred to either WBP2 antibody (15 mg) or additional Rabbit IgG control (15 mg) bound to protein A beads and the mixture was rotated overnight at 4°C. The beads were then washed with PBS + 0.1% Tween and bead-bound proteins were eluted using the Tandem Ubiquitin Binding Entities (TUBE) elution buffer (LifeSensors, UM411). The eluate was treated with the Pan-deubiquitinase supplied with the LifeSensors kit for 2 h at 30°C. This mixture was then boiled with Laemelli 6× loading dye and 2-mercaptoethanol, then run on a gel as described above.
siRNA knockdown
SMARTpool Accell siRNA specific to WBP2 as well as GFP-tagged nontargeting control siRNA were purchased from DharmaCon (cat. E-043579 and D-001940, respectively). Naïve CD4 T cells were purified and activated with plate-bound antibody as described above in complete RPMI with IL-2. After 24 h, the media was replaced with serum-free Accell siRNA delivery media (DharmaCon, cat. B-005000) with 600 μg/mL of either WBP2 or control siRNA resuspended in 1× resuspension buffer (DharmaCon, cat. B-002000-UB-100) diluted with nuclease-free water. After another 24 h, the transfection media was pipetted off and replaced with complete RPMI with IL-2. In each experiment, protein-level knockdown was tested by Western blot at various time points after transfection. For proliferation experiments, cells were labeled with CellTrace violet prior to plating. Proliferation of all live cells was analyzed by flow cytometry on unstained, unfixed cells at 72 h post-transfection. Live cells were gated on the basis of forward scatter and side scatter.
Quantitative PCR
CD4 T cells from IL4 and Itch IL4 DKO mice were transfected as described above. At various timepoints, cells were lysed in TRIZol (Invitrogen, cat. 15596018). RNA was extracted using chloroform and precipitated with isopropanol and Glycol blue (Invitrogen, cat. AM9515). RNA was quantified by nanodrop and converted into cDNA using a reverse transcription kit (ThermoFisher, cat. 4368814). Primers for exons 2-3 of WBP2 were purchased from IDT (cat. Mm.PT.58.42353310). Primers for β-actin were purchased from Applied Biosystems (cat. 4352341E). Quantitative PCR was performed in triplicate for each sample with each primer using TaqMan master mix (Applied Biosystems, cat. 4369016). Plates were run on an Applied Biosystems RT-PCR 7500 system at the Nucleic Acid and PCR Core at the Children’s Hospital of Philadelphia.
Statistics
Statistics were calculated using Prism7 for Mac OS X software (GraphPad). *, **, ***, or **** denotes p < 0.05, p < 0.01, p < 0.001, or p < 0.0001, respectively.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [47] partner repository with the dataset identifier PXD019272.
Supplementary Material
Acknowledgments:
We would like to thank Keisuke Sawada for help with animal husbandry, as well as lab members Binod Kumar and Varshini Gali for useful discussions and technical advice, and Christina Wright for help with R software. Thank you also to the expert technical help of the Flow Cytometry Core and Nucleic Acid and PCR Core at Children’s Hospital of Philadelphia, and the Islet Cell Biology Core at the University of Pennsylvania. This project was funded by the National Institutes of Health (NIH), Grant numbers R01AI114515 and R01AI093566.
Abbreviations:
- DDA
data dependent acquisition
- DIA
data independent acquisition
- DKO
double knockout
- WBP2
WW-domain binding protein 2
Footnotes
Conflict of interest: The authors declare no commercial or financial conflict of interest.
References
- 1.Lohr NJ,Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, Rider NL et al. , Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am. J. Hum. Genet 2010. 86: 447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleine-Eggebrecht N, Staufner C, Kathemann S, Elgizouli M, Kopajtich R, Prokisch H and Lainka E, Mutation in ITCH gene can cause syndromic multisystem autoimmune disease with acute liver failure. Pediatrics 2019. 143: 20181554. [DOI] [PubMed] [Google Scholar]
- 3.Brittain HK, Feary J, Rosenthal M, Spoudeas H, Deciphering Developmental Disorders (DDD) Study and Wilson LC, Biallelic human ITCH variants causing a multisystem disease with dysmorphic features: a second report. Am. J. Med. Genet. Part A 2019. 179: 1346–1350. [DOI] [PubMed] [Google Scholar]
- 4.Perry WL, Hustad CM, Swing DA, O’Sullivan TN, Jenkins NA and Copeland NG, The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat. Genet 1998. 18: 143–146. [DOI] [PubMed] [Google Scholar]
- 5.Hustad CM, Perry WL, Siracusa LD, Rasberry C, Cobb L, Cattanach BM, Kovatch R et al. , Molecular genetic characterization of six recessive viable alleles of the mouse agouti locus. Genetics. 1995. 140: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang D, Elly C, Gao B, Fang N, Altman Y, Joazeiro C, Hunter T et al. , Dysregulation of T lymphocyte function in itchy mice: a role for itch in TH2 differentiation. Nat. Immunol 2002. 3: 281–287. [DOI] [PubMed] [Google Scholar]
- 7.Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC and Karin M, Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 2004. 306: 271–275. [DOI] [PubMed] [Google Scholar]
- 8.Oliver PM, Cao X, Worthen GS, Shi P, Briones N, MacLeod M, White J et al. , Ndfip1 protein promotes the function of itch ubiquitin ligase to prevent T cell activation and T helper 2 cell-mediated inflammation. Immunity. 2006. 25: 929–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kathania M, Khare P, Zeng M, Cantarel B, Zhang H, Ueno H and Venuprasad K, Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-gammat ubiquitination. Nat. Immunol 2016. 17: 997–1004. [DOI] [PubMed] [Google Scholar]
- 10.Akosua A, Layman K, Sprout SL, Phillips D and Oliver PM, Ndfip1 restricts Th17 cell potency by limiting lineage stability and proinflammatory cytokine production. Sci. Rep 2016. 7 10.1038/srep39649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rathinam C, Matesic LE and Flavell RA, The E3 ligase Itch is a negative regulator of the homeostasis and function of hematopoietic stem cells. Nat. Immunol 2011. 12: 399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giamboi-Miraglia A, Cianfarani F, Cattani C, Lena AM, Serra V, Campione E, Terrinoni A et al. , The E3 ligase Itch knockout mice show hyperproliferation and wound healing alteration. FEBS J. 2015. 282: 4435–4449. [DOI] [PubMed] [Google Scholar]
- 13.Di Marcotullio L, Greco A, Mazzà D, Canettieri G, Pietrosanti L, Infante P, Coni S et al. , Numb activates the E3 ligase Itch to control Gli1 function through a novel degradation signal. Oncogene. 2011. 30: 65–76. [DOI] [PubMed] [Google Scholar]
- 14.Lim SK, Lu SY, Kang S, Tan HJ, Li Z, Ning Z, Wee A et al. , Wnt signaling promotes breast cancer by blocking Itch-mediated degradation of YAP/TAZ transcriptional coactivator WBP2. Mol. Cell Pathobiol 2016. 76: 6278–6290. [DOI] [PubMed] [Google Scholar]
- 15.Moser EK, Field NS and Oliver PM, Aberrant Th2 inflammation drives dysfunction of alveolar macrophages and susceptibility to bacterial pneumonia. Cell Mol. Immunol 2018. 15: 480–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hume DA, Radik JL, Ferber E and Weidemann MJ, Aerobic glycolysis and lymphocyte transformation. Biochem. J 1978. 174: 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Layman AAK, Deng G, O’Leary CE, Tadros S, Thomas RM, Dybas JM, Moser EK et al. , Ndfip1 restricts mTORC1 signalling and glycolysis in regulatory T cells to prevent autoinflammatory disease. Nat. Commun 2017. 8: 15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moser EK, Roof J, Dybas JM, Spruce LA, Seeholzer SH, Cancro MP and Oliver PM, The E3 ubiquitin ligase Itch restricts antigen-driven B cell responses. J. Exp. Med 2019. 216: 2170–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ziegler SF, Ramsdell F and Alderson MR, The activation antigen CD69. Stem Cells. 1994. 12: 456–465. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005. 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP and Tamayo P, The molecular signatures database hallmark gene set collection. Cell Syst. 2015. 1: 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carbon S, Douglass E, Dunn N, Good B, Harris NL, Lewis SE, Mungall CJ et al. , The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019. 47: D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP et al. , Gene ontology: tool for the unification of biology. Nat. Genet 2000. 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P. and Mesirov JP, Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011. 27: 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren Y, Wang H, Zhang Y and Liu Y, WBP2 modulates G1/S transition in ER+ breast cancer cells and is a direct target of miR-206. Cancer Chemother. Pharmacol 2017. 79: 1003–1011. [DOI] [PubMed] [Google Scholar]
- 26.Suryaraja R, Anitha M, Anbarasu K, Kumari G and Mahalingam S, The E3 ubiquitin ligase Itch regulates tumor suppressor protein RASSF5/NORE1 stability in an acetylation-dependent manner. Cit. Cell Death Dis 2013. 4: e5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z and Jiang Z, PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol 2009. 10: 1300–1308. [DOI] [PubMed] [Google Scholar]
- 28.Choi YB, Shembade N, Parvatiyar K, Balachandran S and Harhaj EW, TAX1BP1 restrains virus-induced apoptosis by facilitating itch-mediated degradation of the mitochondrial adaptor MAVS. Mol. Cell. Biol 2017. 37: 422–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Theivanthiran B, Kathania M, Zeng M, Anguiano E, Basrur V, Vandergriff T, Pascual V et al. , The E3 ubiquitin ligase Itch inhibits p38a signaling and skin inflammation through the ubiquitylation of Tab1. Sci. Signal 2015. 8: ra22. [DOI] [PubMed] [Google Scholar]
- 30.Tan TH, Chen DY, Chuang HC, Huang CY, Yang CY, Li JP, Chiu LL et al. , Signaling by inhibiting TAB1 activation (DUSP14/MKP6) negatively regulates TCR dual-specificity phosphatase 14. J. Immunol 2014. 192: 1547–1557. [DOI] [PubMed] [Google Scholar]
- 31.Qiu L, Joazeiro C, Fang N,Wang HY, Elly C, Altman Y, Fang D et al. , Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J. Biol. Chem 2000. 275: 35734–35737. [DOI] [PubMed] [Google Scholar]
- 32.Matesic LE, Haines DC, Copeland NG and Jenkins NA, Itch genetically interacts with Notch1 in a mouse autoimmune disease model. Hum. Mol. Genet 2006. 15: 3485–3497. [DOI] [PubMed] [Google Scholar]
- 33.Chastagner P, Israel A and Brou C, AIP4/Itch regulates Notch receptor degradation in the absence of ligand. PLoS One. 2008. 3: e2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pefani DE, Pankova D, Abraham AG, Grawenda AM, Vlahov N, Scrace S, O’ E. et al. , TGF-b targets the hippo pathway scaffold RASSF1A to facilitate YAP/SMAD2 nuclear translocation. Mol. Cell 2016. 63: 156–166. [DOI] [PubMed] [Google Scholar]
- 35.Wiśniewski JR, Hein MY, Cox J. and Mann M, A ‘proteomic ruler’ for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteomics 2014. 13: 3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howden AJM, Hukelmann JL, Brenes A, Spinelli L, Sinclair LV, Lamond AI and Cantrell DA, Quantitative analysis of T cell proteomes and environmental sensors during T cell differentiation. Nat. Immunol 2019. 20: 1542–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song H, Wu T, Xie D, Li D, Hua K, Hu J and Fang L, WBP2 downregulation inhibits proliferation by blocking YAP transcription and the EGFR/PI3K/Akt signaling pathway in triple negative breast cancer. Cell. Physiol. Biochem 2018. 48: 1968–1982. [DOI] [PubMed] [Google Scholar]
- 38.Passegue E and Wagner EF, JunB suppresses cell proliferation by transcriptional activation of p16INK4a expression. EMBO J. 2000. 19: 2969–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andrecht S, Kolbus A, Hartenstein B, Angel P and Schorpp-Kistner M, Cell cycle promoting activity of JunB through cyclin A activation. J. Biol. Chem 2002. 277: 35961–35968. [DOI] [PubMed] [Google Scholar]
- 40.Jang SW, Yang SJ, Ehlén Å, Dong S, Khoury H, Chen J, Persson JL et al. , SRPK2 promotes leukemia cell proliferation by phosphorylating acinus and regulating cyclin A1. Cancer Res. 2008. 68: 4559–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jang SW, Liu X, Fu H, Rees H, Yepes M, Levey A and Ye K, Interaction of Akt-phosphorylated SRPK2 with 14-3-3 mediates cell cycle and cell death in neurons. J. Biol. Chem 2009. 284: 24512–24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC and Fu XD, SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J. Cell Biol 1998. 140: 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Badea T, Niculescu F, Soane L, Fosbrink M, Sorana H, Rus V, Shin ML et al. , RGC-32 increases p34CDC2 kinase activity and entry of aortic smooth muscle cells into S-phase. J. Biol. Chem 2002. 277: 502–508. [DOI] [PubMed] [Google Scholar]
- 44.Tegla CA, Cudrici CD, Nguyen V, Danoff J, Kruszewski AM, Boodhoo D, Mekala AP et al. , RGC-32 is a novel regulator of the T-lymphocyte cell cycle. Exp. Mol. Pathol 2015. 98: 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cossarizza A, Chang H, Radbruch A, Acs A, Adam D, Adam-Klages S, Agace WW et al. , Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol 2019. 49: 1457–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Field NS, O’Leary CE, Dybas JM,Ding H andOliver PM, An integrated strategy for identifying targets of ubiquitin-mediated degradation in CD4+ T cells. . Method Mol. Biol 2020. 2111: 239–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A et al. , The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019. 47: D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [47] partner repository with the dataset identifier PXD019272.