

Abstract
Delayed treatment of cholinergic seizure results in benzodiazepine-refractory status epilepticus that is thought, at least in part, to result from maladaptive trafficking of N-methyl-D-aspartate (NMDA) and gamma aminobutyric acid type A (GABAA) receptors, the effects of which may be ameliorated by combination therapy with the NMDA receptor antagonist ketamine. Our objective was to establish whether ketamine and midazolam dual therapy would improve outcome over midazolam monotherapy following soman exposure when evaluated in a mouse model that, similar to humans, lacks plasma carboxylesterase, greatly reducing endogenous scavenging of soman. In the current study, continuous cortical electroencephalographic activity was evaluated in male and female plasma carboxylesterase knockout mice exposed to a seizure-inducing dose of soman and treated with midazolam or with midazolam and ketamine combination at 40 min after seizure onset. Ketamine and midazolam combination reduced soman-induced lethality, seizure severity and the number of mice that developed spontaneous recurrent seizure compared to midazolam monotherapy. In addition, ketamine-midazolam combination treatment reduced soman-induced neuronal degeneration and microgliosis. These results support that combination of anti-epileptic drug therapies aimed at correcting the maladaptive GABAA and NMDA receptor trafficking reduce the detrimental effects of soman exposure. Ketamine may be a beneficial adjunct to midazolam in reducing the epileptogenesis and neuroanatomical damage that follows nerve agent exposure and pharmacoresistant status epilepticus.
Keywords: ketamine, neuroinflammation, spontaneous recurrent seizure, organophosphorus, benzodiazepine-resistant status epilepticus, anti-epileptic
1. Introduction
Acute exposure to acetylcholinesterase inhibitors, such as chemical warfare nerve agents (CWNA), may induce self-sustaining seizure, or status epilepticus (SE), which can become pharmacoresistant when treatment is delayed [1, 2], a highly possible scenario in unprepared civilian populations or cases of mass casualties. Animal models of CWNA-induced seizures reveal that the consequences of prolonged seizure activity as a result of delayed anticonvulsant treatment include severe neuropathology, neuroinflammation, and behavioral deficits, among others [3–6]. The prompt control of seizure activity is, therefore, critical for the amelioration of the effects of CWNA exposure and adjunct therapy that replaces or complements current anticonvulsant countermeasures is needed.
Currently, the standard treatment against a CWNA exposure consists of the anticholinergic compound atropine to reduce peripheral side effects, an oxime for reactivation of acetylcholinesterase (such as pralidoxime chloride; 2-PAM), and a benzodiazepine anticonvulsant (such as diazepam or midazolam). Benzodiazepines are commonly used as a first-line therapy to treat acute seizures and SE, including CWNA-induced seizures [7], (reviewed in Reddy and Reddy [8]). Recent clinical studies have demonstrated a superior anticonvulsant activity of midazolam for the treatment of SE [9] and have encouraged the replacement of diazepam by midazolam (reviewed in Newmark [10]).
The discovery of pharmacological interventions that are effective at controlling self-sustaining seizures requires an understanding of the mechanisms associated with the self-sustaining seizure. Research suggests that seizure activity causes the internalization and transient inactivation of synaptic gamma-aminobutyric acid A (GABAA) receptors resulting in a decrease in neuronal inhibition (reviewed in Niquet et al. [11, 12], [13]). Furthermore, an increase in the trafficking of N-methyl-D-aspartate (NMDA) receptors to synapses is observed which promotes an increase in glutamatergic excitation, leading to excitotoxicity. Ketamine is a noncompetitive NMDA receptor antagonist that may benefit the control of refractory SE (reviewed in Niquet et al. [11], [13], reviewed in Dorandeu [14, 15]). In line with the paradigm of the pathophysiology of SE is the observation that the delay of first-line anticonvulsive treatment in clinics is sometimes mitigated by the administration of ketamine as several retrospective studies have found promising results (reviewed in Amengual-Gual et al. [16]). Moreover, in animal models of cholinergic-induced seizure, the addition of ketamine to delayed midazolam treatment reduces the development of spontaneous recurrent seizures (SRS), cognitive impairment and neuropathology compared to midazolam treatment [5, 17–19].
In the present study, we investigated the potential benefits of adding ketamine as adjunct to midazolam treatment that is delayed to 40 minutes after seizure onset in the carboxylesterase knockout (Es1−/−) mouse, which may be an improved model since, similar to humans, they lack plasma carboxylesterase activity and have reduced endogenous scavenging of certain organophophorus compounds, including soman (GD) [20]. We previously characterized the seizurogenic, epileptogenic, neuropathological, and neuroinflammatory responses of male Es1−/− mice exposed to various doses of GD and administered midazolam at 15 min after seizure onset [4]. Additionally, we characterized the midazolam dose-response in Es1−/− mice exposed to a seizure-inducing dose of GD with anticonvulsant administration further delayed to 40 min after seizure onset, which may more closely model first responder intervention in a mass casualty CWNA event [21]. Consistent with prior research in rats, delayed midazolam monotherapy is unable to halt progression of SE and prevent the development of SRS and the neuropathology associated with GD exposure. Results from our current study demonstrate that midazolam/ketamine combination as delayed dual therapy offers the advantages of efficiently reducing seizure severity, the incidence of SRS development, and the extent of neuronal cell loss and neuroinflammation following GD-induced seizure.
2. Materials and Methods
2.1. Animals:
Male (n=38; 24–28 g) and female (n=27; 17–22 g) Es1−/− mice were obtained at 8–9 weeks of age from the United States Army Medical Research Institute of Chemical Defense (USAMRICD) breeding colony. Animals were single-housed following telemetry implantation surgery, with food and water available ad libitum, on a 12 h:12 h light-dark cycle with lights on at 0600. The experimental protocol was approved by the Institute Animal Care and Use Committee at USAMRICD, and all procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals [22], the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and the Animal Welfare Act of 1966 (P.L. 89–544), as amended.
2.2. Surgeries:
Mice were implanted subcutaneously (SC) under 2%−5% isoflurane with ETA-F10 or F20-EET telemetry transmitters (Data Sciences International; DSI; St. Paul, MN) with wires wrapped around cortical stainless steel screws (11.5 mm right and/or left of the midline, and 1.5 mm anterior, and 3.0 mm posterior to bregma), as previously described [4]. Buprenex SR (ZooPharm) was administered (0.5 mg/kg, SC) to minimize pain. All mice were given 2 weeks of recovery from surgery before exposure.
2.3. GD exposures and treatments:
Mice were exposed SC to either saline (No GD group) or 80 μg/kg soman (GD; pinacolyl methylphosphonofluoridate; United States Army Combat Capabilities Development Command Chemical Biological Center, Aberdeen Proving Ground, MD), as previously described [4]. GD-exposed mice were treated intraperitoneally (IP) with an admix of atropine sulfate (4 mg/kg) and HI-6 (50 mg/kg) at 1 min after exposure. Saline-exposed mice received saline injections in lieu of an atropine/HI-6 treatment. Cage bedding was removed and replaced with an isopad, and food was removed minutes prior to exposure. GD-exposed animals were randomly divided into one of two treatment groups consisting of midazolam (3 mg/kg; GD + MDZ; IP) or midazolam combined with ketamine (30 mg/kg; GD + MDZ/KET; IP) at 40 min after seizure onset. Control (No GD) animals received midazolam (3 mg/kg; IP) at 50 minutes after saline administration.
2.4. Behavioral and electroencephalographic (EEG) seizure activity
Following GD exposure, behavioral seizure severity was scored using a modified Racine scale [23] of 6 stages: 0, no abnormality; 1, mastication, tongue fasciculations, oral tonus; 2, head nodding and/or tremors; 3, forelimb clonus or tonus, body tremors; 4, rearing with convulsions; and 5, rearing and falling with convulsions. Behavioral seizure activity was monitored by an observer blinded to treatment using the Noldus Pocket Observer program (Noldus Information Technology, The Netherlands). In addition, EEG signals were monitored in real-time to determine the onset of seizure activity, defined as the appearance of rhythmic high-amplitude spikes (>2 × baseline) that lasted at least 10 seconds (based on Nissinen et al. [24]). Electroencephalographic (EEG) activity and body temperature were continuously recorded using Dataquest Art Acquisition software (DSI) from 3 days before exposure up until euthanasia.
2.5. EEG scoring and power spectra analysis:
EEG activity was processed as previously described with a MATLAB-based algorithm designed to analyze large datasets of EEG [25]. Seizure activity was determined by detection thresholds previously described in detail [4, 21]. Following identification of events by the algorithm, visual inspection of candidate seizures was performed by a blinded and unbiased scorer to confirm their identity. The EEG power spectrum was divided in the following bands: delta (0.1–4.0 Hz), theta (4.1–8.0 Hz), alpha (8.1–12 Hz), beta (12.1–25 Hz), and gamma (25.1–50 Hz). The mean power was calculated for each band and integrated in 10-min bins for up to 24 hours according to previous methods.[25] Power spectrum density (μV2/Hz; 0.1–125 Hz) was determined by integrating the power spectra calculated through fast Fourier transform in 60-second epochs. In addition, full EEG power spectrum data were further reduced by extracting the median power (10 min bins) in 60-minute intervals to obtain EEG power spectral density values at baseline (24 h prior to GD or saline exposure), SE (20 min before treatment), and 1, 3, 6, and 12 h after treatment.
2.6. Brain tissue collection and immunohistochemistry:
Mice that survived to study endpoint (14 days after exposure) were injected with sodium pentobarbital (75 mg/kg, IP, Fatal Plus; Patterson Veterinary) and perfused with heparinized 0.9% saline in 0.1 M phosphate buffer (FD Neurotechnologies, Columbia, MD) followed by a 4% paraformaldehyde solution as previously described [4]. Brains were removed, kept in 4% paraformaldehyde for 6 h, and cryoprotected in 20% sucrose. Sectioning and staining of tissue was performed by FD Neurotechnologies using previously described methods [26]. Frozen brains were coronally cut at a thickness of 30 μm, and immunohistochemistry was performed using antibodies against the neuronal nuclear protein (NeuN; mouse anti-NeuN IgG 1:600; Millipore, Billerica, MA) and the ionized calcium-binding adaptor molecule 1 (Iba1; rabbit anti-Iba1 IgG 1:6,000; Wako Chemicals, Richmond, USA). For Iba1-stained tissue, cresyl violet was used as a counterstain for visualization of anatomic landmarks.
2.7. Cell counts:
Coverslip-mounted immunostained brain slices were scanned using an Olympus BX61IVS microscope with a Pike F-505 camera (Allied Vision, Exton, PA). Image-Pro Plus (Media Cybernetics, Inc., Rockville, MD) was used to trace regions of interest in images and obtain counts of NeuN-positive cells using the particle analysis function. Brain regions evaluated included the lateral thalamus, medial thalamus, CA1 region of the hippocampus, basolateral amygdala, and layer 3 of the piriform cortex. For each brain tissue slice, areas of interest were traced using anatomic landmarks in the region between −1.06 mm to −1.94 mm from bregma. Stereology was performed using the Stereo Investigator software (MBF Bioscience, Williston, VT) to quantify highly dense NeuN-positive neurons in the CA1 region of the hippocampus; five sections per mouse were analyzed using the optical fractionator method in the region from −1.22 to −3.88 mm from bregma [27]. Iba1 is expressed by both active and resting microglia. Therefore, analysis of the density and morphology (i.e., cell-body-to-cell-size ratio, an indication of microglial cell activation [28, 29]) of Iba1-positive cells was performed in brain regions using the ImageJ software (National Institutes of Health, Bethesda, MD); methods for quantification of cell morphology were modified, as previously described [4], from published analyses [28, 29].
2.8. Data analysis:
SPSS version 22 (IBM) was used for all statistical analyses. To determine the main effect of sex, treatment, and time on the severity of GD-induced body weight, body temperature, EEG power spectral density, and changes in EEG power frequency bands either generalized or general linear model analyses with a repeated measures paradigm were used; one-way analysis of variance (ANOVA) with Tukey’s test was performed for group comparisons at particular time points. Cox regression analysis was performed on survival data to determine the effects of sex and treatment on survival and SRS onset over time. Effects of sex and treatment on percent of survival at study endpoint were evaluated by logistic regression followed by chi square analysis of Fisher’s exact test for group comparisons. A Mann-Whitney test was used to compare the maximum mean behavioral seizure score between the GD-exposed treatment groups for each 10 min time bin after treatment. A general linear model was used to determine the effects of sex, treatment, and their interaction on duration of seizure activity, number of SRS, and NeuN and Iba1 cell density, and cell-body-size-to-cell-size ratio of Iba1-immunoreactive cells; Tukey’s test was used for group comparisons. Differences were considered statistically significant when P < 0.05.
3. Results
3.1. Midazolam/ketamine therapy increased survival and reduced weight loss following GD exposure compared to midazolam
Male and female mice were exposed to saline (No GD control) or a seizure-inducing dose of GD and administered delayed anticonvulsant therapy comprised of midazolam monotherapy or a midazolam/ketamine combination; their survival was monitored over the course of 14 days following exposure (Fig. 1). Logistic regression analysis detected a main effect of treatment without an effect of sex on survival by the end of the study. A chi square analysis with Fisher’s exact test revealed that the percentage of animals surviving at the study endpoint in the midazolam monotherapy group was significantly reduced compared to the No GD group. Percent survival in the midazolam/ketamine combination therapy group was significantly higher compared to the midazolam monotherapy group, and not significantly different from No GD group survival. Cox regression analysis revealed no effect of sex or treatment on median survival time. In addition, although both GD-exposed groups lost significant body weight from baseline compared to the No GD mice, mice treated with midazolam/ketamine therapy had lost significantly less body weight by 24 h after exposure compared to midazolam monotherapy (Suppl. Fig 1). There was no difference based on sex in the response.
Figure 1.
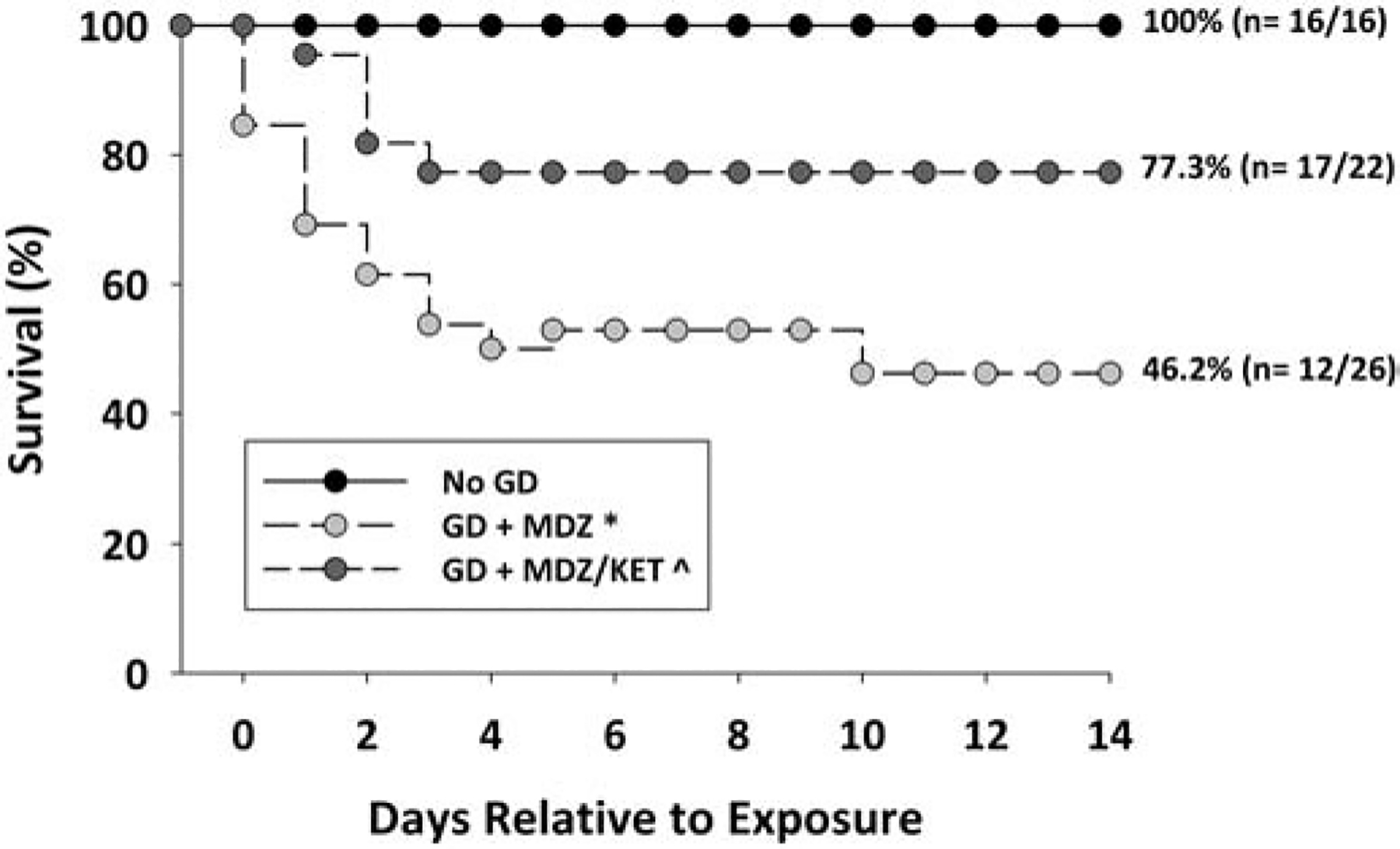
Effect of delayed midazolam treatment combined with ketamine on survival following GD-induced status epilepticus in Es1−/− mice. Mice exposed SC to 80 μg/kg of GD and treated with midazolam monotherapy (GD + MDZ) had a significantly lower percentage of survival compared to the No GD group. The animal group treated with midazolam/ketamine combination therapy (GD + MDZ/KET) had a percentage of survival that was higher than the GD + MDZ group and not significantly different from the No GD group.. *P < 0.05, compared to No GD. ^P < 0.05, compared to GD + MDZ.
3.2. Midazolam/ketamine therapy reduced behavioral seizure severity and reduced body temperature in GD exposed mice compared to midazolam
Exposure to GD induced behavioral seizure with mean (± SD) latency of 2.14 ± 1.32 min. Treatment with midazolam/ketamine therapy significantly reduced behavioral seizure score compared with midazolam therapy by 20 min after treatment and was significantly lower than midazolam during most of the remaining 4 h of observations (Fig.2A). Midazolam/ketamine therapy led to a greater reduction in body temperature compared to midazolam in the hours after GD exposure (Fig. 2B). Analysis showed a significant interaction between time and group; therefore each time point was evaluated separately using an ANOVA. During the initial hour after exposure (prior to treatment), GD-exposed mice had lower body temperatures compared to No GD mice. At 3 h after treatment, GD-exposed mice treated with midazolam had significantly lower body temperatures compared to the No GD group. In contrast, mice treated with midazolam/ketamine therapy had significantly lower body temperatures compared to the No GD group from 1 h to 11 h and compared to GD-exposed midazolam-treated mice.
Figure 2.
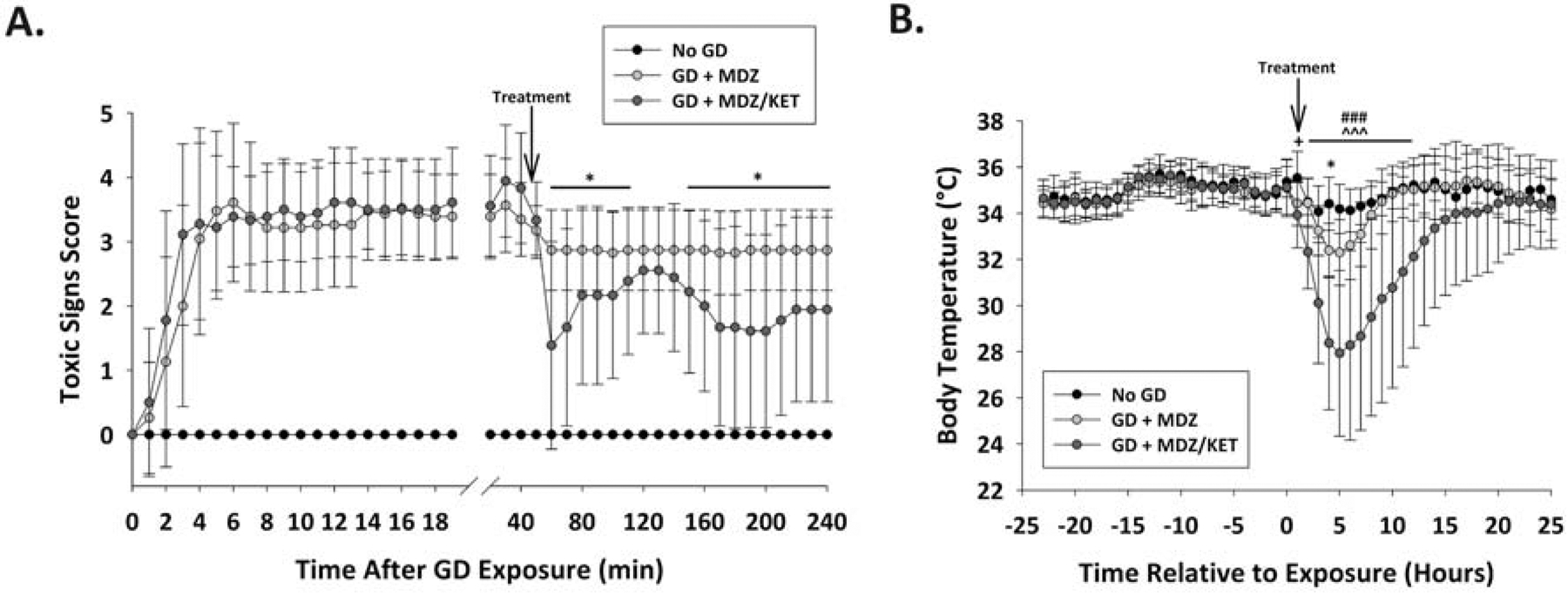
Effect of GD exposure on seizure severity and body temperature in Es1−/− mice. (A) SC exposure to 80 μg/kg of GD resulted in the appearance of behavioral seizure signs within 3 minutes of exposure. The severity of toxic signs were scored following a modified Racine scale: 0, no abnormality; 1, mastication, tongue fasciculations, oral tonus; 2, head nodding and/or tremors; 3, forelimb clonus or tonus, body tremors; 4, rearing with convulsions; and 5, rearing and falling with convulsions. Following treatment administration (arrow; 40 min after EEG seizure onset), toxic signs for the midazolam/ketamine group were reduced in severity within 20 min of treatment and for the majority of 4 hour observation period compared to midazolam monotherapy group. *P < 0.05–0.01, compared to midazolam monotherapy. (B) Prior to treatment (arrow), GD exposure reduced body temperature compared to control for both groups (+ P < 0.05). GD-exposed mice that received dual therapy of midazolam and ketamine (GD + MDZ/KET) had lower body temperature for 12 hours after treatment compared to control (No GD) and to midazolam monotherapy (GD + MDZ). Mice that received midazolam monotherapy had transient reduction in temperature at 3 h after MDZ treatment. *P < 0.05, GD + MDZ compared to control (No GD) group. ### P < 0.001 GD + MDZ/KET compared to No GD; ^^^P < 0.001, GD + MDZ compared to GD + MDZ/KET group.
3.3. Midazolam/ketamine therapy reduced GD-induced increase in EEG power density
Although midazolam/ketamine combination did not reduce initial seizure activity, both EEG power density and behavioral seizure were reduced in the hours after GD exposure, suggesting reduced seizure severity. Latency to seizure was 4.4 ± 2.1 min. Mice treated with midazolam or midazolam/ketamine combination therapy had initial seizure activity that lasted an average (± SD) of 619.6 ± 364.8 min and 528.9 ± 418.3 min, respectively, in the first 24 h following exposure with no sex or treatment differences. Representative images of EEG tracings at various time points are shown in Suppl. Fig. 2.
GD-induced SE caused an increase in the EEG power density in Es1−/− mice that was estimated to be an average (± SD) of 615 ± 181.4% in the midazolam group and 557 ± 121.1% in the midazolam/ketamine group (Fig. 3A). The midazolam monotherapy group showed increased EEG power density for up to at least 3 h compared to No GD group. In contrast, at 1, 3, and 6 h after treatment, the EEG power density in the midazolam/ketamine combination therapy was significantly lower than in the MDZ monotherapy group and was not significantly different from the No GD group. By 12 h the effect of the midazolam/ketamine therapy seemed to start wearing off, as indicated by the significantly increased power density at this time point compared to the No GD group.
Figure 3.
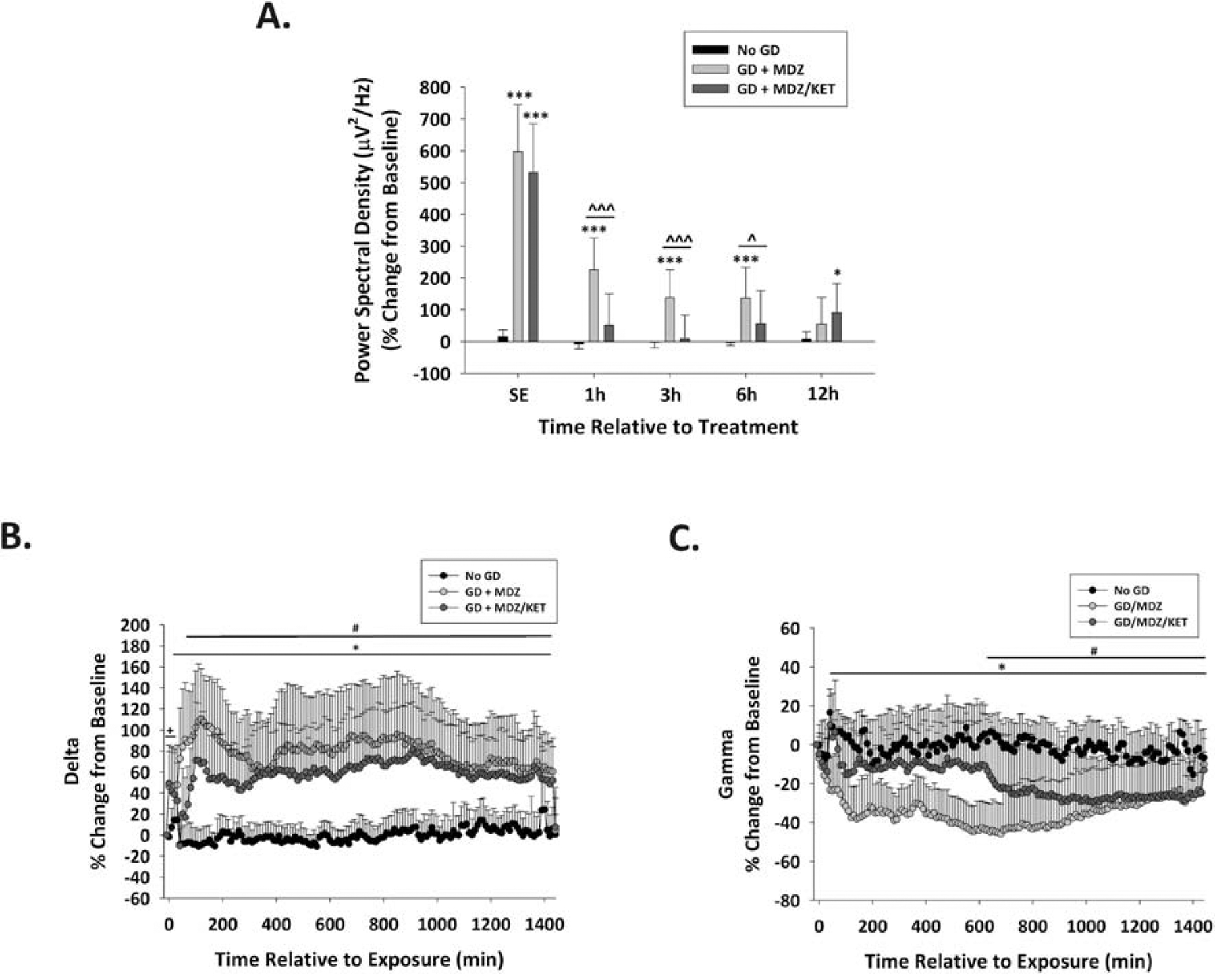
Effect of ketamine as an adjunct to delayed midazolam on EEG power spectral density and frequency bands following GD exposure. (A) GD (80 μg/kg; SC) exposure increased EEG power spectral density during SE (−20 min relative to treatment) compared to the No GD group. At 1, 3, and 6 h after treatment, the power density of the group of mice that received midazolam monotherapy after GD exposure (GD + MDZ) continued to be increased, while the midazolam/ketamine group (GD + MDZ/KET) had reduced EEG power spectral density compared to the No GD group; the effect of midazolam/ketamine treatment seems to wear off, indicated by the significant increased EEG power density at 12 h compared to the No GD group. *P < 0.05, ***P < 0.001, compared to No GD group. ^P < 0.05, ^^^P < 0.001, compared to GD + MDZ group. Tracings of average percentages of relative change in power for (B) delta (0.1–4 Hz) and (C) gamma (25.1–50 Hz) EEG frequencies are shown over a period of 1,440 min (24 h). The effect of midazolam monotherapy or midazolam/ketamine combination on frequency power spectra was compared to the No GD group; no effect of sex was found on changes in power. In Es1−/− mice, the midazolam/ketamine combination therapy was only briefly able to prevent the increase in delta power, while it transiently prevented the increases in the power of gamma caused by GD-induced status epilepticus. + P < 0.05, GD-exposed (prior to treatment) compared to No GD; *P < 0.05, GD + MDZ compared to No GD group; # P < 0.05, GD + MDZ/KET compared to No GD group.
GD-exposed mice show an increase in the power of delta (Fig. 3B) and a decrease in the power of gamma (Fig. 3C). Following administration, the midazolam/ketamine therapy caused a brief 40-min decrease in delta power such that it was not significantly different from the No GD group, whereas midazolam therapy did not reduce GD-induced increase in delta. However, neither midazolam nor midazolam/ketamine treatments were able to prevent the prolonged increase in the power of delta. The combination treatment of midazolam/ketamine also transiently maintained the power of gamma at similar levels to the No GD group for up to 600 min after treatment administration. No significant effect of treatment was detected in the power of theta, alpha, or beta in the first 24 hours after seizure onset.
3.4. Midazolam/ketamine therapy reduced the incidence of animals developing spontaneous recurrent seizure following GD exposure
In the midazolam monotherapy group, 11 out of 12 (91.7%) surviving animals developed SRS; one of these animal developed SRS prior to its death at 10 days after exposure and is included in the analysis. In the midazolam/ketamine group, 8 out of 17 (47.1%) surviving animals developed SRS. Cox regression analysis revealed a significant effect of treatment on the median onset of SRS development, without a main effect of sex (Fig. 4A). In animals that developed SRS, the midazolam monotherapy group presented with an average of 11.5 ± 9.6 SRS, while the midazolam/ketamine group had an average (± SD) of 5.8 ± 9.0 SRS (Fig. 4B). A general linear model analysis did not detect main effects of treatment or sex in the average total number of SRS events. A chi-square analysis with Fisher’s exact test detected a significant difference between treatment groups in the final incidence (percentages) of animals developing SRS.
Figure 4.

Effect of ketamine as adjunct to delayed midazolam on incidence and number of spontaneous recurrent seizures following GD-induced status epilepticus. Es1−/− mice were SC exposed to 80 μg/kg of GD and treated at 40 min after seizure onset with midazolam monotherapy (GD + MDZ) or midazolam/ketamine combination therapy (GD + MDZ/KET), and EEG activity was monitored for 14 days after exposure. (A) The onset of the first detected SRS for each surviving animal is graphed to indicate the percentage of mice in each group that developed SRS. (B) The average (± SD) number of SRS events is graphed for each group. One animal presented with SRS before dying at post-exposure day 10 and was not included in the total count of SRS during the 14 days after exposure. * P < 0.05, compared to GD + MDZ group.
3.5. Midazolam/ketamine therapy reduced neuronal loss following GD exposure compared with midazolam
Subcutaneous exposure to a seizure-inducing dose of GD resulted in neuropathology at two weeks following exposure in both male and female Es1−/− mice. A significant effect of treatment on neuronal cell density was detected, without an effect of sex. A significant decrease in neuronal (NeuN-positive) cell density was detected in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, layer 3 of the piriform cortex, and the hilus and CA1 regions of the hippocampus of GD-exposed mice treated with midazolam monotherapy compared to the No GD group (Fig. 5A). In contrast, neuronal cell density was significantly higher in the thalamic nuclei, basolateral amygdala and CA1 of the hippocampus in the midazolam/ketamine treatment group compared to the midazolam group. In addition, midazolam/ketamine resulted in significant neuroprotection in the piriform cortex and the CA1 region of the hippocampus, which was not significantly different from the No GD group. Representative images of brain slices immunohistochemically processed for NeuN are shown in Fig. 5B.
Figure 5.
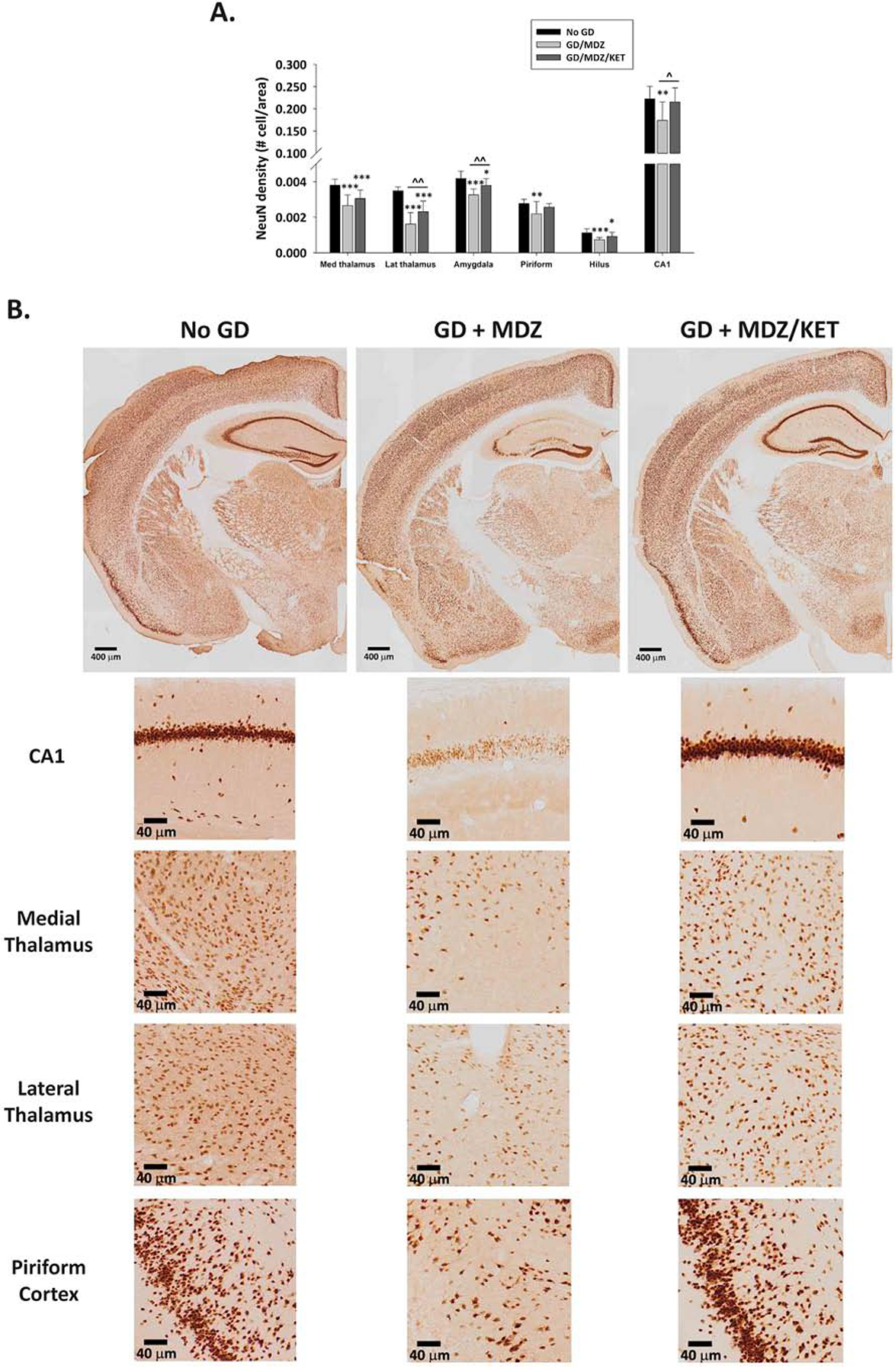
Effect of GD-induced status epilepticus and delayed midazolam monotherapy or midazolam/ketamine combination treatment on mature neuronal cell population of Es1−/− mice. At 2 weeks following SC exposure to GD and delayed midazolam (GD + MDZ) or midazolam/ketamine (GD + MDZ/KET) treatment, Es1−/− mice were perfused and brains collected for immunohistochemistry processing with an antibody against a neuronal nuclear protein (NeuN) to visualize mature neurons. (A) NeuN-positive (NeuN+) cells were counted in the bregma range of −1.28 to −1.64 mm, and cell densities estimated in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, and layer 3 of the piriform cortex. NeuN+ cell density in the CA1 region of the hippocampus was quantified following stereology methods in the region of −1.22 to −3.88 mm from bregma. (B) Representative images are shown. *P < 0.05, **P < 0.01, ***P < 0.001, compared to control (No GD) group. ^P < 0.05, ^^P < 0.01, GD + MDZ compared to GD + MDZ/KET group.
3.6. Midazolam/ketamine therapy reduced reactive microgliosis following GD exposure compared with midazolam
A robust neuroinflammatory response, indicated by the presence of reactive microglia (Iba1-positive cells), was observed at two weeks following GD-induced seizures and delayed anticonvulsant therapy. A significant effect of treatment on Iba1-positive cell density was detected, without an effect of sex. GD-exposed mice receiving midazolam monotherapy showed an increase in Iba1-positive cell density in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, layer 3 of the piriform cortex, and the CA1 region of the hippocampus compared with the No GD group (Fig. 6A). The midazolam/ketamine therapy resulted in significantly reduced Iba1-positive cell density in the piriform cortex and the CA1 region of the hippocampus compared to midazolam monotherapy; these densities were not significantly different from the No GD group. Furthermore, Iba1-positive cell densities in the basolateral amygdala and dorsomedial thalamus of GD-exposed mice treated with midazolam/ketamine combination were not significantly different from those of the No GD group.
Figure 6.
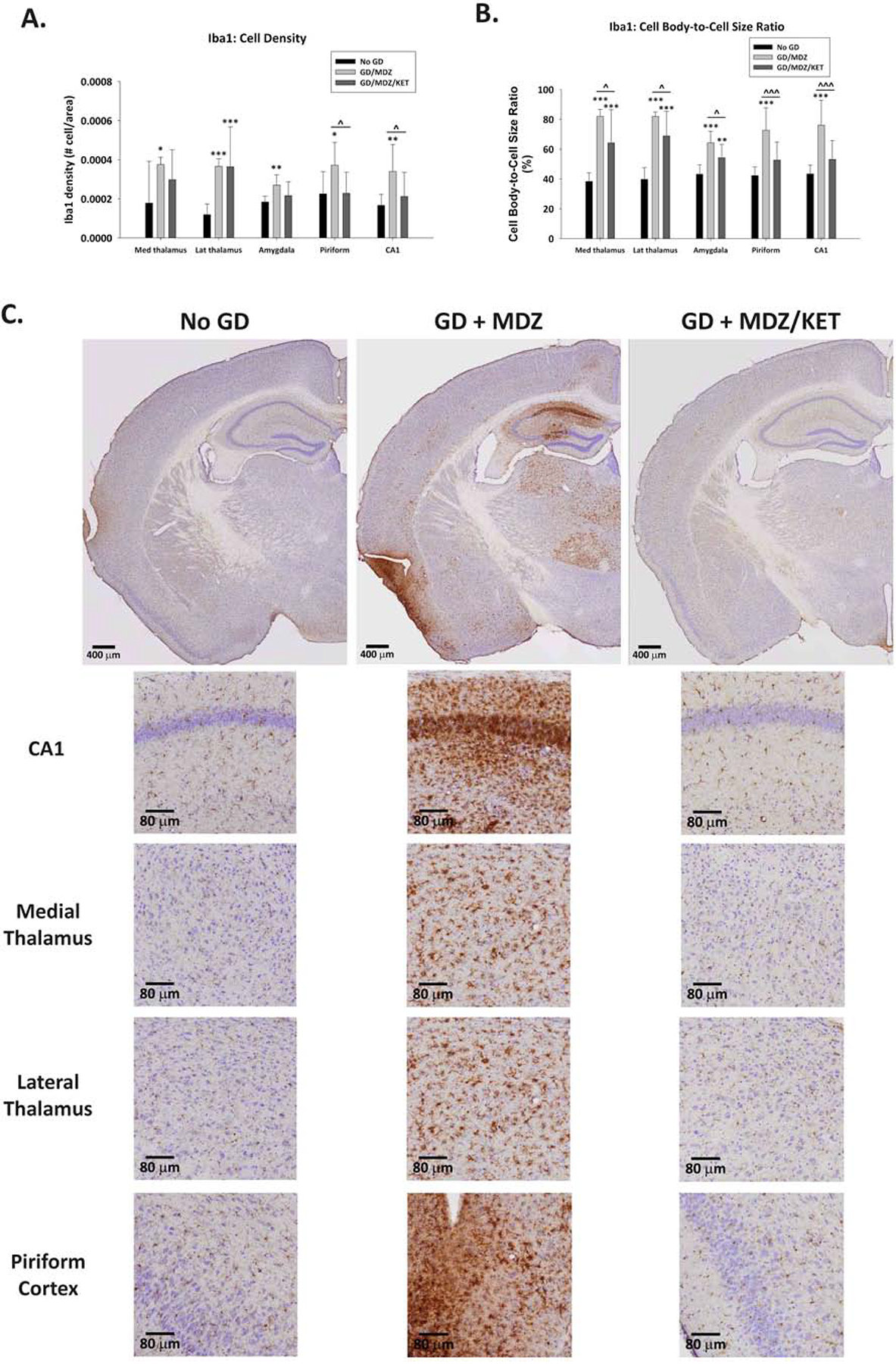
Effect of GD-induced status epilepticus and delayed midazolam monotherapy or midazolam/ketamine combination treatment on microgliosis and microglia activation in Es1−/− mice. At 2 weeks following SC exposure to GD and delayed midazolam (GD + MDZ) or midazolam/ketamine (GD + MDZ/KET) treatment, ES1−/− mice were perfused and brains collected for immunohistochemistry processing with an antibody against ionized calcium binding adaptor molecule 1 (Iba1) to visualize microglia. Cresyl violet was used as counterstain for visualization of anatomic landmarks. Measures of (A) cell density and (B) cell-body-to-cell-size ratio and cell density of Iba1-positive (Iba1+) cells were estimated in the bregma range of −1.28 to −1.64 mm in the CA1 region of the hippocampus, dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, and layer 3 of the piriform cortex. (C) Representative images are shown. *P < 0.05, **P < 0.01, ***P < 0.001, compared to control (No GD) group. ^P < 0.05, ^^^P < 0.001, GD + MDZ compared to GD + MDZ/KET group.
Since Iba1 is a protein that is expressed in both resting and reactive microglia, a quantitative analysis of the average cell body-to-cell size ratio was performed to assess the state of microglia present in the brain regions of interest. At two weeks following exposure to GD, a significant increase in the average cell-body-to-cell-size ratio was detected in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, layer 3 of the piriform cortex, and the CA1 region of the hippocampus of GD-exposed mice treated with midazolam only (Fig. 6B). The midazolam/ketamine therapy significantly reduced the average cell-body-to-cell-size ratio in all brain regions of interest when compared to midazolam monotherapy group. The average cell body-to-cell size ratio in Iba1-positive cells in the piriform cortex and CA1 was not significantly different from that in the No GD group. Representative images of brain slices immunohistochemically processed for Iba1 are shown in Fig. 6C.
4. Discussion
In the current study, we report on the beneficial effects of ketamine as an adjunct to delayed midazolam in ameliorating the toxic effects of GD-induced SE in an Es1−/− mouse model. Our findings in Es1−/− mice agree with previous findings in rats of limited effectiveness of delayed treatment with midazolam after GD-induced SE and that the addition of the NMDA receptor antagonist ketamine improves outcome ([5], reviewed in Niquet et al. [11], [13]). The therapeutic limitations of benzodiazepines highlight the need for improved medical interventions against CWNA-induced SE. Although delayed benzodiazepine treatment dose-dependently increases survival to CWNA agent exposure, it does not prevent the development of epileptogenesis, performance deficits and brain pathology in rodent models [3–5, 21, 30–32]. Anti-epileptic drugs that can be administered as an adjunct to benzodiazepines may increase survival and reduce the neuropathological effects that follow exposure to toxic organophosphorus compounds. In agreement with our observations in the rat model of GD-induced SE, the addition of ketamine to midazolam treatment offers the benefits of an increase in survival and a reduction in seizure severity [5, 13]. Similarly, a synergism between diazepam and ketamine occurs in the rapid and sustained control (for at least 5 hours) of seizure activity in the rat model of pilocarpine-induced SE [33]. In the present study the control of seizure severity was for approximately 12 hours after midazolam/ketamine therapy, as indicated by the transient changes in EEG power density. Midazolam/ketamine therapy also reduced behavioral seizure activity within 20 min of treatment whereas midazolam was less effective.
Pathological changes in EEG power in the delta and gamma frequency bands were found following seizure onset in GD-exposed mice, even after administration of delayed anticonvulsant treatments. Increased delta band during the period after exposure correlates with neuropathological damage in GD-exposed rats [34, 35]. In the present study, the prolonged increase in delta could relate to the observations of incomplete neuroprotection in the midazolam/ketamine therapy group. A decrease in gamma power, characteristic of GD-induced SE in male Es1−/− mice [4], was observed in animals receiving midazolam monotherapy; this decrease was prevented by midazolam/ketamine therapy, but its effect starts wearing off by approximately 10 hours after its administration. Ketamine has been shown to dose-dependently increase gamma oscillations in the hippocampus [36]; these effects would explain the ability of ketamine to prevent the alterations in the power of gamma observed in the present study. Therefore, our observations suggest that, although the effect of dual therapy is rapid, the duration of the effect on seizure severity is limited and targeting another mechanism by a third antiepileptic drug may sustain for longer the control of seizures (reviewed in Niquet et al. [11]). For example, valproic acid in combination with midazolam and ketamine improves outcome in both pilocarpine- and GD-induced SE in rats [13]. In addition, valnoctamide, isomer of the valproic acid amide, and secbutylpropylacetamide, a derivative of valproic acid, have improved anticonvulsive activity over diazepam against cholinergic-induced seizure when treatment was delayed [37] and could potentially provide additional beneficial effects if evaluated in combination with a benzodiazepine and ketamine.
Our results of neuroprotective effects in brain regions associated with seizure initiation and propagation in Es1−/− mice that received midazolam/ketamine combination following GD exposure are consistent with findings in rats that ketamine and diazepam treatment given at 40 minutes after GD-induced seizure is neuroprotective in the piriform cortex and thalamic nuclei (reviewed in Ballough et al. [38]). In GD-exposed Es1−/− mice, midazolam/ketamine had less neuronal loss in the lateral thalamus and amygdala compared to mice treated with midazolam and was not different from control in the piriform cortex and CA1 region. Similar neuroprotective effects occur in the hippocampus following sarin-induced SE in rats treated with midazolam/ketamine therapy at 50 minutes after exposure [17].
Persistent neuroinflammation may play an important role in epileptogenesis and secondary neuronal degeneration (reviewed in Clossen and Reddy [39]). The secretion of proinflammatory cytokines by an activated neuroinflammatory response following seizure is well documented (reviewed in Hiragi et al. [40]) and occurs after CWNA exposure [41, 42]. Although microglia have many roles in the central nervous system, they are partly responsible for the release of some cytokines that are associated with the epileptic brain (reviewed in Hiragi et al. [40] and Devinsky et al. [43]). We have previously reported on the robust neuroinflammatory response resulting from GD-induced seizures and delayed midazolam treatment in the Es1−/− mouse [4, 21]. When midazolam was given as a monotherapy, a high dose (9 mg/kg) was needed to reduce microgliosis and microglia activation [21]. In the present study, a lower dose of midazolam (3 mg/kg) combined with ketamine reduced microglial cell density and activation after GD-induced seizure. In all brain regions the midazolam/ketamine therapy reduced neuroinflammation, indicated by the reduction in morphological changes linked to the active state of microglia, compared to midazolam monotherapy. Interestingly, the CA1 and piriform cortex, where the least neuroinflammation was observed, are also the locations of the highest level of neuroprotection, indicated by NeuN+ cell density levels that were not significantly different from those in the No GD group. The anti-inflammatory properties observed in this study are in line with those of Dhote et al. where ketamine in combination with atropine administered at a delayed time point was shown to reduce the neuroinflammatory response, indicated by a decrease in glial activation, in a mouse model of acute GD poisoning [44].
The neuroprotection of the midazolam/ketamine therapy in GD-exposed Es1−/− mice could be the result of a lower incidence of SRS development, previously shown to exacerbate neuropathology [45], or a prolonged decrease in body temperature during the acute phase of seizure activity. Ketamine results in hypothermia in mice and rats at doses higher than the 30 mg/kg used in the present study [46]. Additionally, midazolam reduces body temperature in a dose-dependent manner [21]. Our current observations suggest an additive effect of midazolam and ketamine on reducing body temperature in the hours following GD exposure. Hypothermia has been shown to be neuroprotective in animal models of epileptic, ischemic and traumatic brain injury (reviewed in Motamedi et al. [47]). Various processes are altered by hypothermia including, but not limited to, a reduction in cerebral metabolic rate, slowing of release of excitatory neurotransmitters, reduction in the breakdown of the blood-brain barrier, all of which may reduce seizure severity and the neuropathological damage resulting from it [48]. Thus, the reduction in body temperature by midazolam/ketamine combination may have been one means of providing neuroprotection. However, neuroprotective effect of midazolam/ketamine against GD exposure was incomplete, suggesting additional anti-epileptic drugs may be needed.
In regards to pre-hospital usefulness, operational constraints of nerve agent casualties may lead to delays in evacuation and diagnosis that may require medical management by first responders or medics. An effective treatment of nerve agent-induced status epilepticus should possess some key properties, including efficacy against lethality, seizure and brain damage when administered beyond 30 min of seizure duration, as well as be safe in poisoned victims (reviewed in Dorandeu et al [49]). Ketamine has been used to treat refractory status epilepticus and is considered relatively safe, with results of clinical trials pending (reviewed in Yan et al. [50]). Analysis of drug interactions in pilocarpine-exposed rats using isobolograms indicates that sub-anesthetic doses of ketamine used in combination with the benzodiazepine diazepam and the anti-epileptic drug valproate have synergistic therapeutic effects but that the toxic effects on motor function and consciousness are additive; thus, the therapeutic index is improved by using this combination [51]. The combination of drugs aimed at counteracting the effect of seizure-induced changes in GABAA and glutamate receptors is better than the sum of its parts, yet is less toxic than higher-dose monotherapy with either component. Other combinations (valproate with midazolam or ketamine) evaluated against pilocarpine did not show this synergism, although a therapeutic index was not calculated [18].
Sub-anesthetic doses of ketamine are used by the US military in prehospital settings for analgesia and are considered safe and without cardiorespiratory depressant effects commonly seen with opioid analgesics (reviewed in Butler et al. [52] and Shackelford et al. [53]). Ketamine can be administered through multiple routes, but there is also recommendation to develop an auto-injector (IM) for prehospital trauma care (reviewed in Butler et al. [52] and Shackelford et al. [53]). Such an auto-injector that administers a sub-anesthetic dose of ketamine might also be useful in the case of nerve agent-induced status epilepticus, in particular if used in combination with midazolam.
4.1. Conclusions
In summary, ketamine as adjunct to delayed midazolam treatment offers the benefits of increased survival, reduction of seizure severity, reduction in the incidence of development of spontaneous recurrent seizures, and reduction in neuropathology as a result of GD-induced status epilepticus. The present study not only validates further the use of the Es1−/− mouse as an appropriate model to screen novel medical countermeasures against CWNA-induced seizure, but also provides solid evidence of the benefits of targeting the glutamatergic system to reduce the epileptogenic and neuropathological changes associated with the toxic insult. Future studies will focus on identifying other pharmacological candidates that, in combination with those tested in the present study, offer complete neuroprotection.
Supplementary Material
Supplemental Figure 1. Representative EEG tracings of 24-h compressed signal and 10-sec recordings at baseline (24 h prior to GD exposure), status epilepticus, 20 min after treatment, and 24 h after treatment.
{kind=link}
Supplemental Figure 2. Effect of GD-induced status epilepticus and delayed midazolam monotherapy or midazolam/ketamine combination on changes in body weight following GD exposure and delayed treatment in Es1?/? mice. Mice exposed to GD lost body weight during the first day after exposure, with both midazolam monotherapy (GD + MDZ) and midazolam/ketamine combination therapy showing significantly decreased body weights compared to No GD group. Mice in the GD + MDZ/KET group showed a reduced loss of body weight compared to GD + MDZ; all GD-exposed mice recovered the lost weight within the first week after exposure. ***P 003C 0.001, GD + MDZ compared to No GD group; ### P 003C 0.001, GD + MDZ/KET compared to No GD group; ^^P 003C 0.01 GD + MDZ compared to GD + MDZ/KET group.
{kind=link}
Highlights.
Benzodiazepine pharmacoresistance develops following cholinergic-induced seizure.
Limited efficacy of delayed midazolam in soman exposed carboxylesterase knockout mice.
Midazolam/ketamine therapy increases survival following soman exposure.
Midazolam/ketamine therapy reduces seizure severity and epileptogenesis after soman.
Midazolam/ketamine therapy reduces neuronal loss and neuroinflammatory response.
Acknowledgements
This research was supported by a grant from the National Institute of Neurological Disorders and Stroke [R21 NS103820-02] to Dr. Lucille A. Lange and the Geneva Foundation. The authors acknowledge Dr. Linn Cadieux, Ms. Sandra DeBus, Mr. Erik Matson, and Mr. Timothy Barry, II, for management of the USAMRICD Es1−/− mouse colony and Ms. Cindy Kronman for editorial review. Ms. Erica Kundrick and Ms. Katie Walker were supported in part by an appointment to the Research Participation Program for the U.S. Army Medical Research and Development Command administered by the Oak Ridge Institute for Science and Education through an agreement between the U.S. Department of Energy and U.S. Army Medical Research and Development Command. Regarding author contribution EK, KW, DN, CS, MS, MAF contributed to data collection and analysis; BM and LL contributed to manuscript writing, experimental design, data collection and analysis and data integrity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Conflict of Interest
The authors have no conflicts of interest. Dr. Marcio de Araujo Furtado conducted EEG analysis under a contract with BioSEad but was blinded to the treatment groups.
Supplemental Data
Access to data can be requested by contacting the corresponding author.
The views expressed in this manuscript are those of the authors and do not reflect the official policy of the Department of Army, Department of Defense, or the U.S. Government
References
- [1].Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17(19):7532–40. 10.1523/jneurosci.17-19-07532.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mazarati AM, Baldwin RA, Sankar R, Wasterlain CG. Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Res. 1998;814(1–2):179–85. 10.1016/s0006-8993(98)01080-4 [DOI] [PubMed] [Google Scholar]
- [3].Schultz MK, Wright LK, de Araujo Furtado M, Stone MF, Moffett MC, Kelley NR, et al. Caramiphen edisylate as adjunct to standard therapy attenuates soman-induced seizures and cognitive deficits in rats. Neurotoxicol Teratol. 2014;44:89–104. 10.1016/j.ntt.2014.06.002 [DOI] [PubMed] [Google Scholar]
- [4].Marrero-Rosado B, de Araujo Furtado M, Schultz CR, Stone M, Kundrick E, Walker K, et al. Soman-induced status epilepticus, epileptogenesis, and neuropathology in carboxylesterase knockout mice treated with midazolam. Epilepsia. 2018;59(12):2206–18. 10.1111/epi.14582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lumley LA, Rossetti F, de Araujo Furtado M, Marrero-Rosado B, Schultz CR, Schultz MK, et al. Dataset of EEG power integral, spontaneous recurrent seizure and behavioral responses following combination drug therapy in soman-exposed rats. Data Brief. 2019;27:104629 10.1016/j.dib.2019.104629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lumley L, Miller D, Muse WT, Marrero-Rosado B, de Araujo Furtado M, Stone M, et al. Neurosteroid and benzodiazepine combination therapy reduces status epilepticus and long-term effects of whole-body sarin exposure in rats. Epilepsia Open. 2019;4(3):382–96. 10.1002/epi4.12344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shih T, McDonough JH Jr., Koplovitz I Anticonvulsants for soman-induced seizure activity. J Biomed Sci. 1999;6(2):86–96. 10.1007/bf02256439 [DOI] [PubMed] [Google Scholar]
- [8].Reddy SD, Reddy DS. Midazolam as an anticonvulsant antidote for organophosphate intoxication--A pharmacotherapeutic appraisal. Epilepsia. 2015;56(6):813–21. 10.1111/epi.12989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Silbergleit R, Durkalski V, Lowenstein D, Conwit R, Pancioli A, Palesch Y, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med. 2012;366(7):591–600. 10.1056/NEJMoa1107494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Newmark J Therapy for acute nerve agent poisoning: An update. Neurol Clin Pract. 2019;9(4):337–42. 10.1212/CPJ.0000000000000641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Niquet J, Lumley L, Baldwin R, Rossetti F, Schultz M, de Araujo Furtado M, et al. Early polytherapy for benzodiazepine-refractory status epilepticus. Epilepsy Behav. 2019;101(Pt B):106367 10.1016/j.yebeh.2019.06.011 [DOI] [PubMed] [Google Scholar]
- [12].Niquet J, Baldwin R, Suchomelova L, Lumley L, Naylor D, Eavey R, et al. Benzodiazepine-refractory status epilepticus: pathophysiology and principles of treatment. Ann N Y Acad Sci. 2016;1378(1):166–73. 10.1111/nyas.13147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Niquet J, Lumley L, Baldwin R, Rossetti F, Suchomelova L, Naylor D, et al. Rational polytherapy in the treatment of cholinergic seizures. Neurobiol Dis. 2020;133:104537 10.1016/j.nbd.2019.104537 [DOI] [PubMed] [Google Scholar]
- [14].Dorandeu F Ketamine for the treatment of (super) refractory status epilepticus? Not quite yet. Expert Rev Neurother. 2017;17(5):419–21. 10.1080/14737175.2017.1288099 [DOI] [PubMed] [Google Scholar]
- [15].Dorandeu F, Barbier L, Dhote F, Testylier G, Carpentier P. Ketamine combinations for the field treatment of soman-induced self-sustaining status epilepticus. Review of current data and perspectives. Chem Biol Interact. 2013;203(1):154–9. 10.1016/j.cbi.2012.09.013 [DOI] [PubMed] [Google Scholar]
- [16].Amengual-Gual M, Sanchez Fernandez I, Wainwright MS. Novel drugs and early polypharmacotherapy in status epilepticus. Seizure. 2019;68:79–88. 10.1016/j.seizure.2018.08.004 [DOI] [PubMed] [Google Scholar]
- [17].Lewine JD, Weber W, Gigliotti A, McDonald JD, Doyle-Eisele M, Bangera N, et al. Addition of ketamine to standard-of-care countermeasures for acute organophosphate poisoning improves neurobiological outcomes. Neurotoxicology. 2018;69:37–46. 10.1016/j.neuro.2018.08.011 [DOI] [PubMed] [Google Scholar]
- [18].Niquet J, Baldwin R, Norman K, Suchomelova L, Lumley L, Wasterlain CG. Midazolam-ketamine dual therapy stops cholinergic status epilepticus and reduces Morris water maze deficits. Epilepsia. 2016;57(9):1406–15. 10.1111/epi.13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Niquet J, Baldwin R, Norman K, Suchomelova L, Lumley L, Wasterlain CG. Simultaneous triple therapy for the treatment of status epilepticus. Neurobiol Dis. 2017;104:41–9. 10.1016/j.nbd.2017.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Duysen EG, Koentgen F, Williams GR, Timperley CM, Schopfer LM, Cerasoli DM, et al. Production of ES1 plasma carboxylesterase knockout mice for toxicity studies. Chem Res Toxicol. 2011;24(11):1891–8. 10.1021/tx200237a [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kundrick E, Marrero-Rosado B, Stone M, Schultz C, Walker K, Lee-Stubbs RB, et al. Delayed midazolam dose effects against soman in male and female plasma carboxylesterase knockout mice. Ann N Y Acad Sci. 2020. 10.1111/nyas.14311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Council NR. Guide for the care and use of laboratory animals: National Academies Press; 2010. 10.17226/12910 [DOI]
- [23].Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32(3):281–94. 10.1016/0013-4694(72)90177-0 [DOI] [PubMed] [Google Scholar]
- [24].Nissinen J, Lukasiuk K, Pitkanen A. Is mossy fiber sprouting present at the time of the first spontaneous seizures in rat experimental temporal lobe epilepsy? Hippocampus. 2001;11(3):299–310. 10.1002/hipo.1044 [DOI] [PubMed] [Google Scholar]
- [25].de Araujo Furtado M, Zheng A, Sedigh-Sarvestani M, Lumley L, Lichtenstein S, Yourick D. Analyzing large data sets acquired through telemetry from rats exposed to organophosphorous compounds: an EEG study. J Neurosci Methods. 2009;184(1):176–83. 10.1016/j.jneumeth.2009.07.020 [DOI] [PubMed] [Google Scholar]
- [26].Hsu S-M, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. Journal of Histochemistry & Cytochemistry. 1981;29(4):577–80. 10.1177/29.4.6166661 [DOI] [PubMed] [Google Scholar]
- [27].Bonthius DJ, McKim R, Koele L, Harb H, Karacay B, Mahoney J, et al. Use of frozen sections to determine neuronal number in the murine hippocampus and neocortex using the optical disector and optical fractionator. Brain Res Brain Res Protoc. 2004;14(1):45–57. 10.1016/j.brainresprot.2004.09.003 [DOI] [PubMed] [Google Scholar]
- [28].Hovens IB, Nyakas C, Schoemaker RG. A novel method for evaluating microglial activation using ionized calcium-binding adaptor protein-1 staining: cell body to cell size ratio. Neuroimmunol Neuroinflammation. 2014;1(2):82–8. 10.4103/2347-8659.139719 [DOI] [Google Scholar]
- [29].Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, et al. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun. 2010;24(7):1058–68. 10.1016/j.bbi.2010.02.001 [DOI] [PubMed] [Google Scholar]
- [30].Schultz MK, Wright LK, Stone MF, Schwartz JE, Kelley NR, Moffett MC, et al. The anticholinergic and antiglutamatergic drug caramiphen reduces seizure duration in soman-exposed rats: synergism with the benzodiazepine diazepam. Toxicol Appl Pharmacol. 2012;259(3):376–86. 10.1016/j.taap.2012.01.017 [DOI] [PubMed] [Google Scholar]
- [31].Langston JL, Wright LK, Connis N, Lumley LA. Characterizing the behavioral effects of nerve agent-induced seizure activity in rats: increased startle reactivity and perseverative behavior. Pharmacol Biochem Behav. 2012;100(3):382–91. 10.1016/j.pbb.2011.09.011 [DOI] [PubMed] [Google Scholar]
- [32].Moffett MC, Schultz MK, Schwartz JE, Stone MF, Lumley LA. Impaired auditory and contextual fear conditioning in soman-exposed rats. Pharmacol Biochem Behav. 2011;98(1):120–9. 10.1016/j.pbb.2010.11.022 [DOI] [PubMed] [Google Scholar]
- [33].Martin BS, Kapur J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia. 2008;49(2):248–55. 10.1111/j.1528-1167.2007.01384.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McDonough JH Jr., Clark TR, Slone TW Jr., Zoeffel D, Brown K, Kim S, et al. Neural lesions in the rat and their relationship to EEG delta activity following seizures induced by the nerve agent soman. Neurotoxicology. 1998;19(3):381–91. https://www.ncbi.nlm.nih.gov/pubmed/9621344 [PubMed] [Google Scholar]
- [35].Philippens IH, Melchers BP, de Groot DM, Wolthuis OL. Behavioral performance, brain histology, and EEG sequela after immediate combined atropine/diazepam treatment of soman-intoxicated rats. Pharmacol Biochem Behav. 1992;42(4):711–9. 10.1016/0091-3057(92)90019-c [DOI] [PubMed] [Google Scholar]
- [36].Lazarewicz MT, Ehrlichman RS, Maxwell CR, Gandal MJ, Finkel LH, Siegel SJ. Ketamine modulates theta and gamma oscillations. J Cogn Neurosci. 2010;22(7):1452–64. 10.1162/jocn.2009.21305 [DOI] [PubMed] [Google Scholar]
- [37].Shekh-Ahmad T, Hen N, McDonough JH, Yagen B, Bialer M. Valnoctamide and sec-butyl-propylacetamide (SPD) for acute seizures and status epilepticus. Epilepsia. 2013;54 Suppl 6:99–102. 10.1111/epi.12290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ballough GP, Newmark J, Levine ES, Filbert MG. Neuroprotection as a treatment. Medical aspects of chemical warfare. 2008:221. [Google Scholar]
- [39].Clossen BL, Reddy DS. Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim Biophys Acta Mol Basis Dis. 2017;1863(6):1519–38. 10.1016/j.bbadis.2017.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hiragi T, Ikegaya Y, Koyama R. Microglia after Seizures and in Epilepsy. Cells. 2018;7(4). 10.3390/cells7040026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dhote F, Peinnequin A, Carpentier P, Baille V, Delacour C, Foquin A, et al. Prolonged inflammatory gene response following soman-induced seizures in mice. Toxicology. 2007;238(2–3):166–76. 10.1016/j.tox.2007.05.032 [DOI] [PubMed] [Google Scholar]
- [42].Spradling KD, Lumley LA, Robison CL, Meyerhoff JL, Dillman JF 3rd., Transcriptional responses of the nerve agent-sensitive brain regions amygdala, hippocampus, piriform cortex, septum, and thalamus following exposure to the organophosphonate anticholinesterase sarin. J Neuroinflammation. 2011;8:84 10.1186/1742-2094-8-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36(3):174–84. 10.1016/j.tins.2012.11.008 [DOI] [PubMed] [Google Scholar]
- [44].Dhote F, Carpentier P, Barbier L, Peinnequin A, Baille V, Pernot F, et al. Combinations of ketamine and atropine are neuroprotective and reduce neuroinflammation after a toxic status epilepticus in mice. Toxicol Appl Pharmacol. 2012;259(2):195–209. 10.1016/j.taap.2011.12.024 [DOI] [PubMed] [Google Scholar]
- [45].de Araujo Furtado M, Lumley LA, Robison C, Tong LC, Lichtenstein S, Yourick DL. Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague-Dawley rats. Epilepsia. 2010;51(8):1503–10. 10.1111/j.1528-1167.2009.02478.x [DOI] [PubMed] [Google Scholar]
- [46].Raith H, Schuelert N, Duveau V, Roucard C, Plano A, Dorner-Ciossek C, et al. Differential effects of traxoprodil and S-ketamine on quantitative EEG and auditory event-related potentials as translational biomarkers in preclinical trials in rats and mice. Neuropharmacology. 2020:108072 10.1016/j.neuropharm.2020.108072 [DOI] [PubMed] [Google Scholar]
- [47].Motamedi GK, Lesser RP, Vicini S. Therapeutic brain hypothermia, its mechanisms of action, and its prospects as a treatment for epilepsy. Epilepsia. 2013;54(6):959–70. 10.1111/epi.12144 [DOI] [PubMed] [Google Scholar]
- [48].Niquet J, Baldwin R, Gezalian M, Wasterlain CG. Deep hypothermia for the treatment of refractory status epilepticus. Epilepsy Behav. 2015;49:313–7. 10.1016/j.yebeh.2015.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dorandeu F, Dhote F, Barbier L, Baccus B, Testylier G. Treatment of status epilepticus with ketamine, are we there yet? CNS Neurosci Ther. 2013;19(6):411–27. 10.1111/cns.12096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yan Y, Peng X, Jing W, Wang X. How close is ketamine to routine use in refractory status epilepticus? Expert Rev Neurother. 2020;20(5):421–3. 10.1080/14737175.2020.1757433 [DOI] [PubMed] [Google Scholar]
- [51].Niquet J, Baldwin R, Suchomelova L, Lumley L, Eavey R, Wasterlain CG. Treatment of experimental status epilepticus with synergistic drug combinations. Epilepsia. 2017;58(4):e49–e53. 10.1111/epi.13695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Butler FK, Kotwal RS, Buckenmaier C 3rd, Edgar EP, O’Connor KC, Montgomery HR, et al. A triple-option analgesia plan for tactical combat casualty care: TCCC guidelines change 13–04. J Spec Oper Med. 2014;14(1):13–25. [DOI] [PubMed] [Google Scholar]
- [53].Shackelford SA, Fowler M, Schultz K, Summers A, Galvagno SM, Gross KR, et al. Prehospital pain medication use by US Forces in Afghanistan. Military medicine. 2015;180(3):304–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Representative EEG tracings of 24-h compressed signal and 10-sec recordings at baseline (24 h prior to GD exposure), status epilepticus, 20 min after treatment, and 24 h after treatment.
Supplemental Figure 2. Effect of GD-induced status epilepticus and delayed midazolam monotherapy or midazolam/ketamine combination on changes in body weight following GD exposure and delayed treatment in Es1?/? mice. Mice exposed to GD lost body weight during the first day after exposure, with both midazolam monotherapy (GD + MDZ) and midazolam/ketamine combination therapy showing significantly decreased body weights compared to No GD group. Mice in the GD + MDZ/KET group showed a reduced loss of body weight compared to GD + MDZ; all GD-exposed mice recovered the lost weight within the first week after exposure. ***P 003C 0.001, GD + MDZ compared to No GD group; ### P 003C 0.001, GD + MDZ/KET compared to No GD group; ^^P 003C 0.01 GD + MDZ compared to GD + MDZ/KET group.