

Abstract
Rosmarinic acid (RA) is a polyphenolic compound with various pharmacological properties, including, anti-inflammatory, immunomodulatory, and neuroprotective, as well as having antioxidant and anticancer activities. This study evaluated the effects and mechanisms of RA in two racially different triple-negative breast cancer (TNBC) cell lines. Results obtained show that RA significantly caused cytotoxic and antiproliferative effects in both cell lines in a dose- and time-dependent manner. Remarkably, RA induced cell cycle arrest-related apoptosis and altered the expression of many apoptosis-involved genes differently. In MDA-MB-231 cells, RA arrested the cells in the G0/G1 phase. In contrast, the data suggest that RA causes S-phase arrest in MDA-MB-468 cells, leading to a 2-fold increase in the apoptotic effect compared to MDA-MB-231 cells. Further, in MDA-MB-231 cells, RA significantly upregulated the mRNA expression of three genes: harakiri (HRK), tumor necrosis factor receptor superfamily 25 (TNFRSF25), and BCL-2 interacting protein 3 (BNIP3). In contrast, in the MDA-MB-468 cell line, the compound induced a significant transcription activation in three genes, including TNF, growth arrest and DNA damage-inducible 45 alpha (GADD45A), and BNIP3. Furthermore, RA repressed the expression of TNF receptor superfamily 11B (TNFRSF11B) in MDA-MB-231 cells in comparison to the ligand TNF superfamily member 10 (TNFSF10) and baculoviral IAP repeat-containing 5 (BIRC5) in MDA-MB-468 cells. In conclusion, the data suggest that the polyphenol RA may have a potential role in TNBC therapies, particularly in MDA-MB-468 cells.
Keywords: Rosmarinic, Triple-negative breast cancer, Apoptosis, Cell cycle, Gene expression
1. Introduction
Molecular-targeted therapies require an understanding of the mechanisms linking cancer, apoptosis, and drug resistance (Fulda, 2011; Johnstone et al., 2002). One attractive strategy for cancer therapy is to trigger cancer cell death pathways through the development of apoptotic inducer drugs in concert with other anticancer medications (Jan and Chaudhry, 2019). Mounting evidence shows that cell cycle arrests and apoptosis can sensitize cancer cells to radiotherapy or chemotherapy (Ryu et al., 2018; Zhang et al., 2018).
Apoptosis is a unique cell death mechanism that regulates development and maintains homeostasis of the cell through controlled signaling pathways. Apoptosis as a genetically programmed event, however, is frequently altered and/or impaired in cancer cells, leading to malignancy, metastasis, and chemotherapy resistance (Fulda, 2009; Plati et al., 2008). Cancer cells often resist apoptosis by upregulating anti-apoptotic proteins or attenuating the expression of proapoptotic proteins (Messeha et al., 2019). Additionally, in aggressive metastatic phenotypes, overexpression of Bcl-2 protein may increase the ability of the cancer cell to resist apoptotic death by interrupting the intrinsic apoptotic signaling pathway. Likewise, in multiple cancer cells, the upregulation of the inhibitor of apoptosis (IAP) members promotes chemoresistance (Fulda, 2008). Therapeutics that target the IAP pathway might be exploited as a molecular target for apoptosis-inducing approaches to cancer treatment (LaCasse et al., 2008).
Breast cancer comprises a diverse set of malignancies with a substantial global burden. Triple-negative breast cancer (TNBC) is a very aggressive and metastatic subtype (Dai et al., 2019). In particular, many TNBC cases have a worse outcome after chemotherapy (Anders and Carey, 2009). TNBC treatment opportunities are limited because of the lack of the three specific receptors: estrogen (ER), progesterone (PR), and human epidermal growth factor (Her2/neu)) (Sadighi et al., 2017) (Hudis and Gianni, 2011).
Rosmarinic acid (RA) is one of the potent polyphenolic compounds in rosemary (Rosmarinus officinalis) extract. In cancer research, the natural compound RA has been recognized as a potent agent in various cancer cell lines, including breast cancer (Yesil-Celiktas et al., 2010). RA also has been found to have an apoptotic effect in different types of cancer, including breast (Li et al., 2018). Furthermore, RA has been observed to augment the apoptotic effect of chemotherapeutic, adriamycin (Huang et al., 2018).
Thus, this study was undertaken to explore the anticancer mechanisms of the polyphenolic compound RA on two human TNBC cell lines, MDA-MB-231 and MDA-MB-468. We assessed the potential effects of RA on cell viability, proliferation, cell cycle arrest, and apoptosis. We hypothesized that the apoptotic effects of RA might be due to its ability to alter the expression of different apoptosis-related genes that mediate these events.
2. Materials and methods
2.1. Materials and reagents
RA (purity ≥98%) and Alamar Blue® (a solution of resazurin fluorescence dye) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Trypsin-EDTA solution, penicillin/streptomycin, and Dulbecco’s phosphate-buffered saline (DPBS) were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). An Annexin V-FITC Apoptosis Detection Kit Plus (cat. no. 68FT-Ann VP-S) was purchased from RayBiotech (Norcross, GA, USA). Propidium Iodide Flow Cytometry Kit (cat. No. ab139418) was purchased from Abcam (Cambridge, MA, USA). A DNA-free™ kit (cat. no. AM1907) was purchased from Life Technologies, Inc. (Thermo Fisher Scientific, Inc., Waltham, MA, USA). An iScript™ cDNA Synthesis kit (cat. no. 170–8890), SsoAdvanced™ Universal SYBR® Green Supermix, and the Human Apoptosis PCR array (SAB Target List) H96 were purchased from Bio-Rad Laboratories (Hercules, CA, USA). Dulbecco’s modified Eagle’s medium (DMEM) and heat-inactivated fetal bovine serum (FBS) were purchased from VWR International (Radnor, PA, USA). Cell culture flasks were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Cell culture plates were purchased from Thermo USA Scientific (Ocala, FL, USA).
2.2. Cell culture
Two TNBC cell models, MDA-MB-231 (ATCC® HTB-26™) and MDA-MB-468 (ATCC® HTB-132™) were purchased from ATCC. Both cell lines were grown as monolayers in 75-ml tissue culture (TC) flasks at 37 °C in a humidified 5% CO2 incubator and subculture as required with trypsin/EDTA (0.25%). The complete growth DMEM contained 4 mM L-glutamine and was supplemented with 10% heat-inactivated FBS (v/v) and 1% penicillin/streptomycin salt solution (100 U/ml and 0.1 mg/ml, respectively). DMEM supplemented with 2.5% heat-inactivated FBS was used as the experimental media (Sato et al., 1993).
2.3. Cell viability assay
The cytotoxicity of RA on BC cells was determined in MDA-MB-231 and MDA-MB-468 cells using Alamar Blue® (Messeha et al., 2019). Cells were plated at a density of 2 × 104 cells/well in 96-well plates and incubated overnight at 37 °C. RA was solubilized in cell culture water, and both cell lines were treated for 48 h with the compound at concentration ranges from 0 to 500 μM. Wells treated in the same manner but without cells were used as blanks. The experiments were performed in triplicates. After 48 h, 20 μl Alamar Blue® was added to each well and incubated for 4h at 37 °C. The reduced resazurin dye was measured at an excitation/emission wavelength of 530/590 nm using a Synergy HTX Multi-Mode microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
2.4. Cell proliferation assay
Alamar Blue® assay was used to determine the effect of RA on cell proliferation for MDA-MB-231 and MDA-MB-468 TNBC cells. Briefly, cells were seeded in 96-well plates (1 × 104 cells/well) and incubated overnight at 37 °C. Both MDA-MB-231 and MDA-MB-468 cell lines were treated for 72 and 96 h with RA at the same concentrations ranging from the viability assay (0–500 μM) in a final volume of 200 μl/well (Citalingam et al., 2015). Equivalent wells without cells were used as a blank. At the end of each exposure period, 20 μl of Alamar Blue® was added to each well, incubated for 4 h at 37 °C and the plates were read at an excitation/emission wavelength of 530/590 nm using a Synergy HTX Multi-Mode microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
2.5. Cell cycle analysis
The effect of RA on DNA content and cell cycle distribution was determined for both MDA-MB-231 and MDA-MB-468 cell line following previously described protocols (Cao et al., 2018; Kuo et al., 2006). Briefly, cells were seeded overnight at 1.5 × 106 cells/T25 cell culture flasks and then treated with RA at 125 and 250 μM concentration levels in a final volume of 6 mL/flask of experimental media. After 48 h, both floating and attached cells were collected, pelleted, washed with PBS, and fixed in cold 70% ethanol. The cells were again pelleted, washed in DPBS, and gently resuspended in 200 μl 1X propidium Iodide (PI) + RNase staining solution followed by incubation at 37 °C in the dark for 30 min. For each sample, 1 × 104 cells were examined for cell cycle distribution using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). Data acquisition and data analysis were performed using CellQuest software (BD Biosciences, San Jose, CA, USA).
2.6. Apoptosis assay
The apoptotic effect of RA was determined in MDA-MB-231 and MDA-MB-468 cells using the previously described protocol (Messeha et al., 2019). Briefly, in separate experiments, MDA-MB-231 and MDA-MB-468 cells were plated in 6-well plates (5 × 105 cells/well) and incubated overnight at 37 °C. In experiments designed to induce apoptosis, cells were similarly treated with RA at concentrations ranging from 0 to 400 μM in a final volume of 3 ml/well of experimental media, and control cells were exposed to only experimental media. After 48 h exposure period, treated and control cells from each well were harvested, pelleted, and washed in DPBS. Subsequently, the cell pellets were suspended in 500 μl of 1X Annexin V binding buffer and labeled with 5 μl of Annexin V-FITC and PI. The apoptotic effect was measured within 5–10 min using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). For each sample, 1 × 104 cells were examined, and CellQuest software (BD Biosciences, San Jose, CA, USA) was used for acquisition and data analysis.
2.7. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) apoptosis array
Both MDA-MB-231 and MDA-MB-468 cells were treated with 350 μM of RA. The chosen concentration is close to the IC50 values in the viability study and did not show a significant necrotic effect in the apoptosis assay (Ramachandran et al., 2005) (Rahman et al., 2006) (Teoh et al., 2019). Briefly, for each cell line, two T-75 flasks of 6 × 106 cells representing the control and treated cells were incubated overnight at 37 °C, followed by a 48-h treatment period. The cells from each flask were harvested without trypsinization, pelleted, and washed twice with DPBS. The total RNA was extracted from treated cells with 1 ml of TRIzol reagent as recommended by the manufacturer. For each phase separation, 0.2 ml of chloroform was added to each sample, vortexed, incubated at room temperature (RT) for 2–3 min and centrifuged for 15 min at 10,000 × g at 2–8 °C. The aqueous phase was collected in fresh centrifuge tubes and mixed with 0.5 ml of isopropyl alcohol to pellet the RNA. The RNA pellets were then washed with 75% ethanol and reconstituted in nuclease-free water (~30–50 μl) and placed in an −80 °C freezer for later use. RNA quantity and purity were measured in each sample using a NanoDrop spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific, Inc.). Lastly, cDNA for the control and treated cells was synthesized using the iScript™ cDNA Synthesis kit; and the obtained cDNA was kept in a −80 °C freezer. Each well of the 96-well human apoptosis array was loaded with 10 μl each of the reconstituted cDNA (2.3 ng) and SsoAdvanced™ Universal SYBR® Green Supermix, and the plate placed for 5 min in a shaker and centrifuged at 1000 × g for 1 min. The fluorescent quantitative PCR run was established using a Bio-Rad CFX96 Real-Time System (Bio-Rad Laboratories) with 39 thermo-cycling of denaturation (Yamamoto et al., 2009) as follows: 30-s activation at 95 °C, 10-s denaturation at 95 °C; 20-s annealing at 60 °C; and 31-s extension at 65 °C. All RT-PCR results were confirmed by three independent experiments for each cell line. Gene expression was analyzed using the CFX 3.1 Manager software (Bio-Rad Laboratories) and verified with Student’s t-test.
2.8. Statistical analysis
Data from this study were analyzed using GraphPad Prism 6.2 software (GraphPad Software, Inc., San Diego, CA, USA). All data points present the average of at least two independent experiments and are expressed as the mean ± S.E.M. The IC50s values were calculated by nonlinear regression model of log (inhibitor) vs. normalized response-variable slope on the software with the R2 best fit and the lowest 95% confidence interval. The average of IC50 ± S.E.M. was calculated on an Excel sheet. The significance of the difference was determined using one-way or two-way analysis of variance (ANOVA) as indicated in the legends followed by Bonferroni’s multiple comparison test. Gene expression data were analyzed using CFX 3.1 Manager software (Bio-Rad Laboratories). Unpaired Student t-test was used for comparing two data sets. Generally, a difference was considered significant at *P < 0.05 (as indicated in the figures and legends).
3. Results
3.1. Rosmarinic acid decrease of the cell viability of triple-negative breast cancer cells
To evaluate the potential anticancer effect of RA in TNBC, we assessed cell viability in two TNBC cell lines, MDA-MB-231 and MDA-MB-468, at concentration ranges of 0–500 μM of the compound. The dose-response of the two cell lines to RA was similar, as indicated in Fig. 1A and B (IC50 = 321.75 ± 9.75 for MDA-MB-231 cells and 340.45 ± 7.57 μM for MDA-MB-468 cells). An apparent dose-dependent decrease in cell viability was detected in both cell lines at concentration levels 125–500 μM (P < 0.0001). In contrast to MDA-MB-231 cells, a small reduction in cell viability was found in MDA-MB-468 cells at lower concentrations of RA (15.62–62.5 μM, P < 0.05–P<0.01). These changes imply a higher sensitivity of this cell line to RA.
Fig. 1.
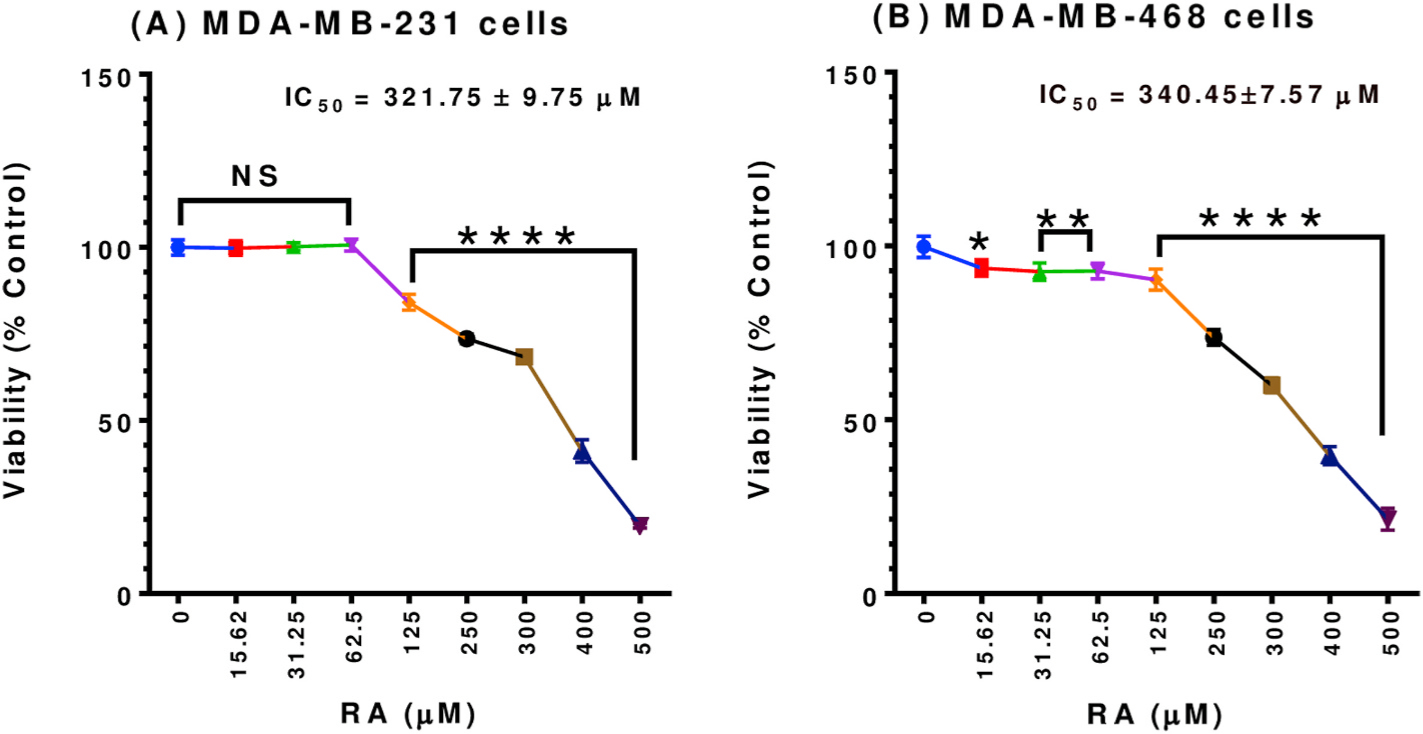
Effect of RA on the viability of (A) MDA-MB-231 and (B) MDA-MB-468 TNBC cell lines. Both cell lines were plated and treated similarly for 48 h with RA at concentration ranges of 0–500 μM. The graph shows the cell viability data expressed as percentages of cell survival compared to the control. The data points represent the mean ± S.E.M. of two independent studies, n = 5 each. One-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparisons test was used to determine the significance of the difference between the control and treated groups. The difference was considered significant at *P < 0.05, **P < 0.01, and ****P < 0.0001. NS, non-significant.
3.2. Rosmarinic acid inhibition of cell the proliferation of triple-negative breast cancer cells
Antiproliferative assays were performed to evaluate the indirect cytotoxic effect of RA on MDA-MB-231 and MDA-MB-468 TNBC cells, as indicated by the growth-inhibitory potency at more extended exposure periods. Overall, the data obtained indicated a reduction in the proliferation rate in a dose-and time-dependent pattern. In both cell lines, RA significantly inhibited cell proliferation at the 72 and 96 h treatment periods vs. control (Fig. 2A and B; P < 0.001–P<0.0001). Furthermore, the compound induced a highly significant (P < 0.0001) antiproliferative effect at 72 vs. 96h exposure period (concentration range 31.25–250 μM in MDA-MB-231 and 15.62–125 μM in MDA-MB-468 cell line) as indicated by the significant reduction in the IC50 values, in particular for MDA-MB-468 cells. The IC50 values were reduced from 134.5 to 88.0 μM in MDA-MB-231 cells (Fig. 2A) and from 128 to 64.28 μM in MDA-MB-468 cells (Fig. 2B) at the 72h vs. 96h exposure periods, respectively. On the other side, a non-significant inhibition was noticed at 15.62 as well as 300–500 μM in MDA-MB-231 cells (Figs. 2A) and 250–500 μM in its counterpart MDA-MB-468 cell line (Fig. 2B). Contrary to the viability study data, the response of the MDA-MB-468 cell line to RA antiproliferative effect was slightly higher than MDA-MB-231 cells. These different behaviors of RA against each cell line may indicate underlying different molecular mechanisms for an anticancer effect. In comparison to chemotherapy drugs, our recently published data (Messeha et al., 2019) stated the IC50 value of cells following 72 h of exposure to doxorubicin as 1.69 ± 0.11 and 0.23 ± 0.003 in MDA-MB-231 and MDA-MB-468 cells, respectively.
Fig. 2.
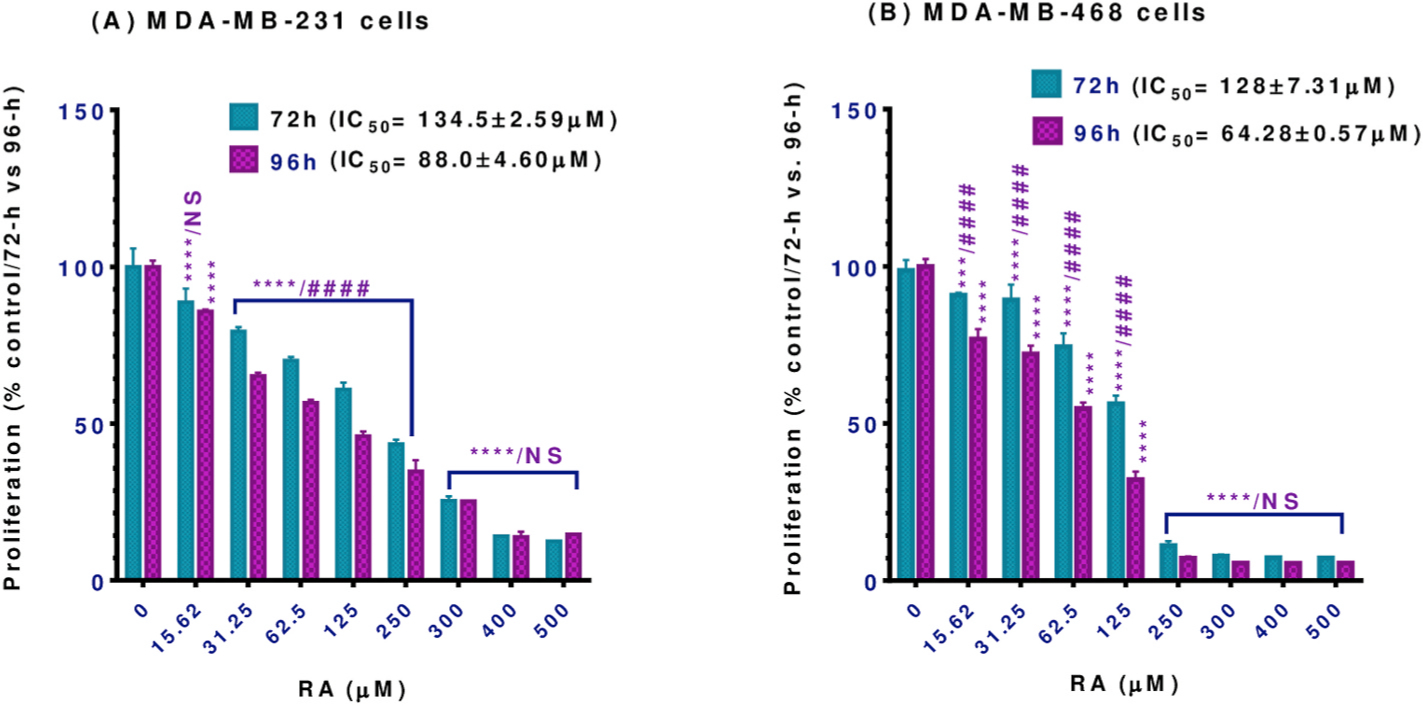
Effect of RA on proliferation in (A) MDA-MB-231 and (B) MDA-MB-468 TNBC cell lines. Both cell lines were incubated for 72 and 96 h with RA at concentration ranges of 0–500 μM. Each data point represents the mean ± S.E.M. of two independent experiments, n = 5 each. One-way ANOVA tests were used to calculate P -values for the difference between control vs. 72 or 96h exposure (*) and two-way ANOVA tests were used to calculate P-values for the difference between the different exposure periods (#). Both one-way and two-way ANOVA analyses were followed by Bonferroni’s multiple comparisons test. ***P < 0.001 and ****/####P < 0.0001 indicate a statistically significant difference between control vs. different exposure periods or between 72 vs. 96h exposure periods. NS, non-significant.
3.3. Rosmarinic acid-induced cell cycle arrest in triple-negative breast cancer cells
To gain insight into the mechanism underlying the cytotoxic and antiproliferative effects of RA, flow cytometric analysis using PI staining was performed to evaluate the cell cycle distribution in MDA-MB-231 and MDA-MB-468 cell lines after 48h exposure at 125 and 250 μM of RA. The data presented in Fig. 3A indicates that RA treatment at these exposure levels did not severely impact the cell cycle distribution in MDA-MB-231 treated cells. However, a minor but significant (˂10%, P < 0.0001) G0/G1 phase arrest was observed compared to the control, accompanied by a significant decrease in S-phase cells (P < 0.0001). There was also an increase in the number of dead MDA-MB-231 cells (Sub G1,18.56 ± 0.31), especially at the higher 250 μM RA concentration, as seen to the left of the G0/G1 peak in (Fig. 3A).
Fig. 3.
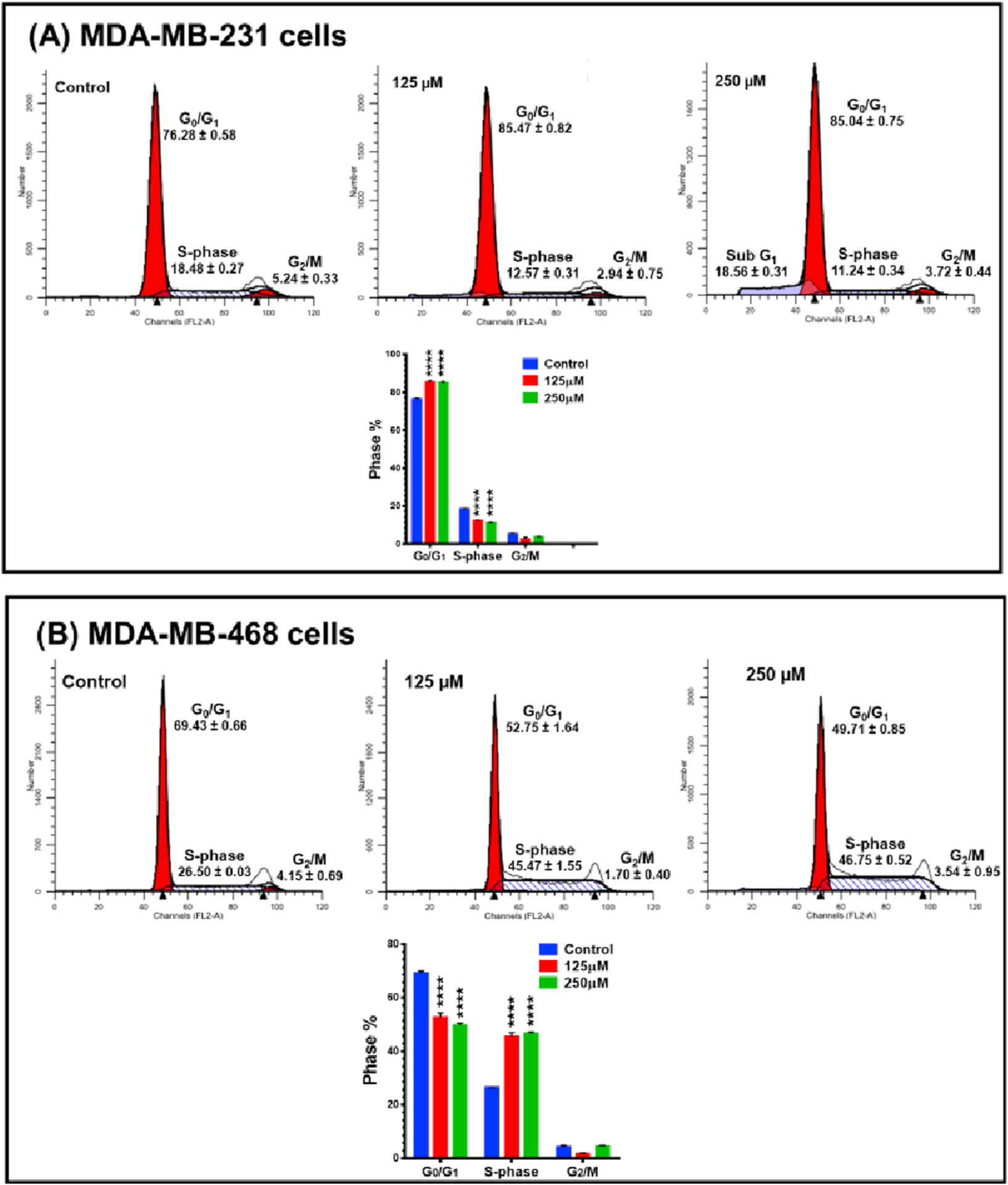
Effect of RA on cell cycle distribution in (A) MDA-MB-231 and (B) MDA-MB-468 TNBC cell lines. Both cell lines were incubated for 48 h with RA at two concentration levels, 125 and 250 μM. Flow cytometry analysis of cell distribution is shown by PI fluorescence histograms. The experiment was repeated three times. RA induced G0/G1 cell cycle arrest in MDA-MB-231 cells and S-phase arrest in MDA-MB-468 cells. One-way ANOVA, followed by Bonferroni’s multiple comparisons tests were used to determine P -values for the difference between the control and different phases of the cell cycle. The difference was considered significant at ****P < 0.0001.
The effect of RA on cell cycle progression was more evident in MDA-MB-468 cells (Fig. 3B). A highly significant 20% increase in S-phase cells (P < 0.0001) was found in RA treated cells compare to the untreated control cells (45.47 ± 1.55 or 46.75 ± 0.52 vs. 26.50 ± 0.03 in control cells) and again demonstrating a differential response to RA in MDA-MB-468 cell as compared with the MDA-MB-231 cell line.
3.4. Rosmarinic acid-induced apoptotic effect in triple-negative breast cancer cells
Next, flow cytometer apoptosis analysis, using Annexin V-FITC/PI double staining, was performed to further comprehend the mechanism of growth inhibition in RA-treated TNBC cell lines. After 48h exposure period to RA at concentration ranges 0–400 μM, a significant (P < 0.05–P<0.0001) dose-dependent apoptotic effect was observed in both cell lines (Fig. 4A and B). The apoptotic effect was slower in MDA-MB-231 cells compared with its counterpart MDA-MB-468 cell line, which was twice as sensitive to RA. When treated with 400 μM of RA, 77.61% of the MDA-MB-468 cells analyzed were in an apoptotic phase, whereas 37.74% of MDA-MB-231 cells exhibited apoptotic effects. A minor component of the necrotic cell (7.44% in MDA-MB-231 cells and 0.32% in MDA-MB-468 cells) were also detected. While RA has similar cytotoxic effects in both cell lines, the apoptosis analysis again demonstrated that the MDA-MB-468 cell line is more sensitive to this agent than are the MDA-MB-231 cells. RA has a more significant antiproliferative effect on these cells. Flow cytometry apoptosis analysis suggests that RA causes MDA-MB-231 cells to be arrested in the G0/G1 phase, while the MDA-MB-468 becomes arrested in the S-phase and then undergo apoptosis rather than proceed to G2/M.
Fig. 4.
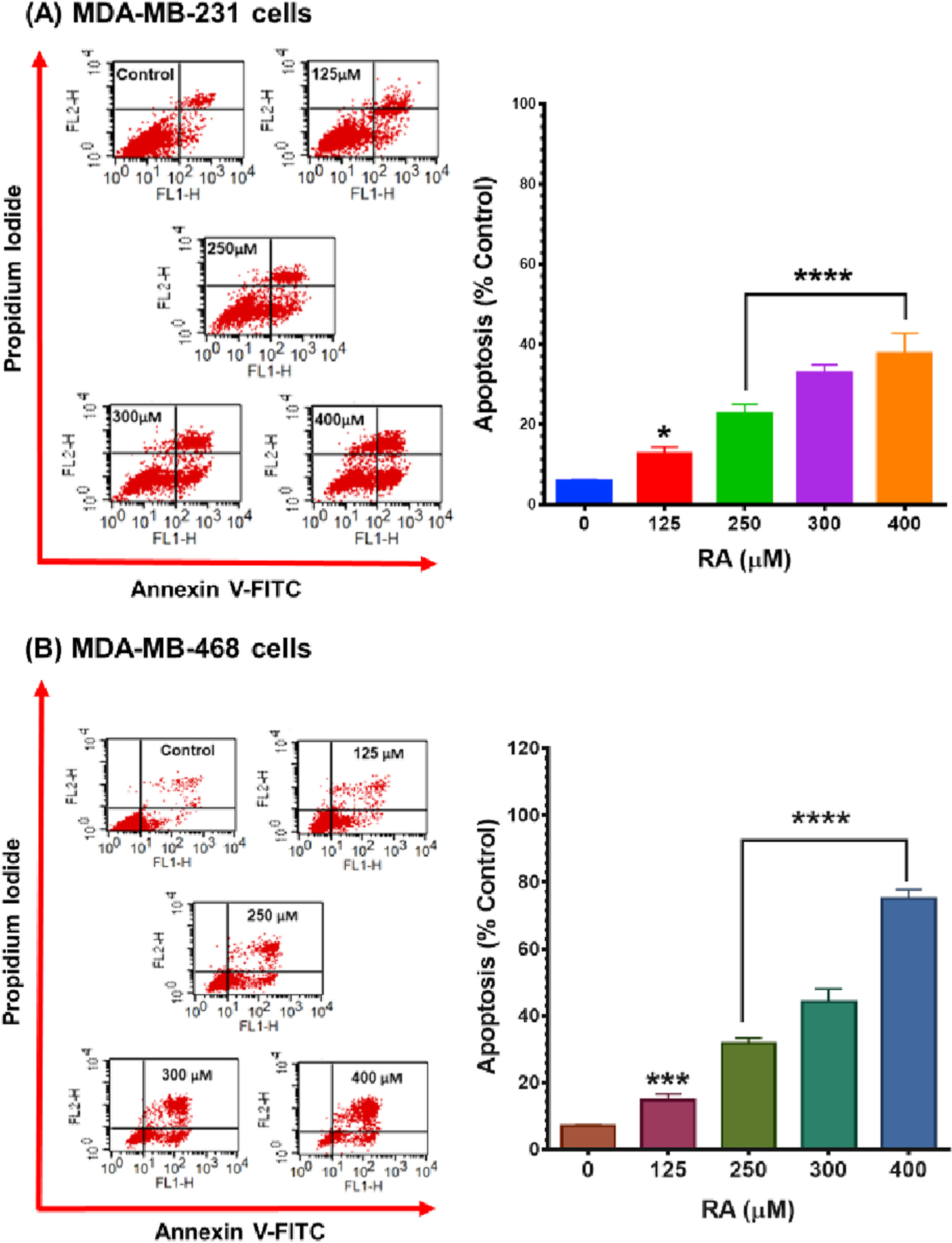
Apoptotic effect of RA in (A) MDA-MB-231 and (B) MDA-MB-468 TNBC cell lines. Both cell lines were exposed to RA for 48 h at concentrations ranging from 0 to 400 μM. Control cells were exposed only to experimental media. An Annexin V-FITC apoptosis kit was used to label treated/control cells, and a FACSCalibur flow cytometer was used to analyze the percentage of apoptotic cells in RA-treated samples as well as the control. Each data point in the bar graphs represents the mean ± S.E.M. of two independent studies, n = 3 each. The significance of the difference between control and each treatment was calculated using one-way ANOVA followed by Bonferroni’s multiple comparisons test. *P < 0.05, ***P < 0.001, and ****P < 0.0001 indicated the statistically significant difference.
3.5. Rosmarinic acid altered the expression of apoptosis-related genes in triple-negative breast cancer cells
In further analysis, quantitative reverse transcription-PCR (qRT-PCR) for an array of apoptosis genes was performed to determine the molecular effects of RA on MDA-MB-231 and MDA-MB-468 cell lines. The profiled normalized mRNA expression revealed the influence of RA on several apoptosis-related genes. Those significantly upregulated/downregulated genes with >2-fold change in transcription are presented in Figs. 5–7. In both cell lines, the fold changes in expression and P values are given in Table 1. The data suggest the ability of RA to alter the expression of five genes in MDA-MB-468 cells significantly, and four genes in MDA-MB-231 cells.
Fig. 5.
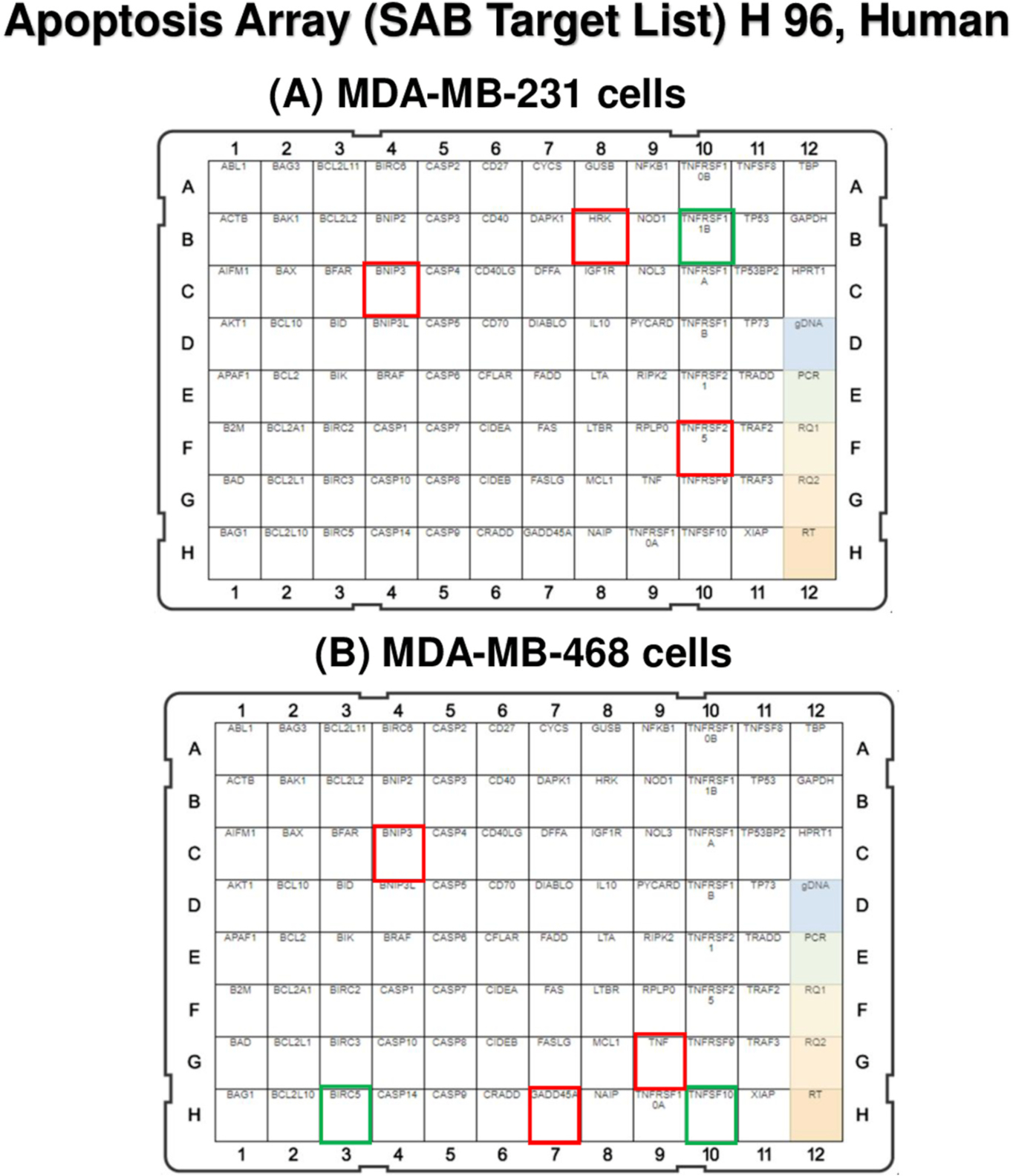
Effect of RA on mRNA gene expression in (A) MDA-MB-231 and (B) MDA-MB-468 TNBC cell lines using qRT-PCR. Human apoptosis array was used to measure the transcription activation in various apoptosis-involved genes. In MDA-MB-231 cells (A), the upregulated genes, HRK, TNFRSF25, and BNIP3 are framed with red color, while the downregulated gene, TNFRSF11B, is presented with a green square. Similarly, in MDA-MB-468 cells (B), the upregulated TNF, BNIP3, and GADD45A are bordered by red, and the repressed TNFSF10 and BIRC5 are shown with green-colored frames. HRK, harakiri; TNFRSF25, tumor necrosis factor receptor superfamily 25; BNIP3, BCL2 interacting protein 3; TNFRSF11B, tumor necrosis factor receptor superfamily 11B; TNF, tumor necrosis factor; BIRC5, baculoviral IAP repeat-containing 5; GADD45A, the growth arrest and DNA damage-inducible 45 alpha; TNFSF10, ligand tumor necrosis factor superfamily member 10.
Fig. 7.
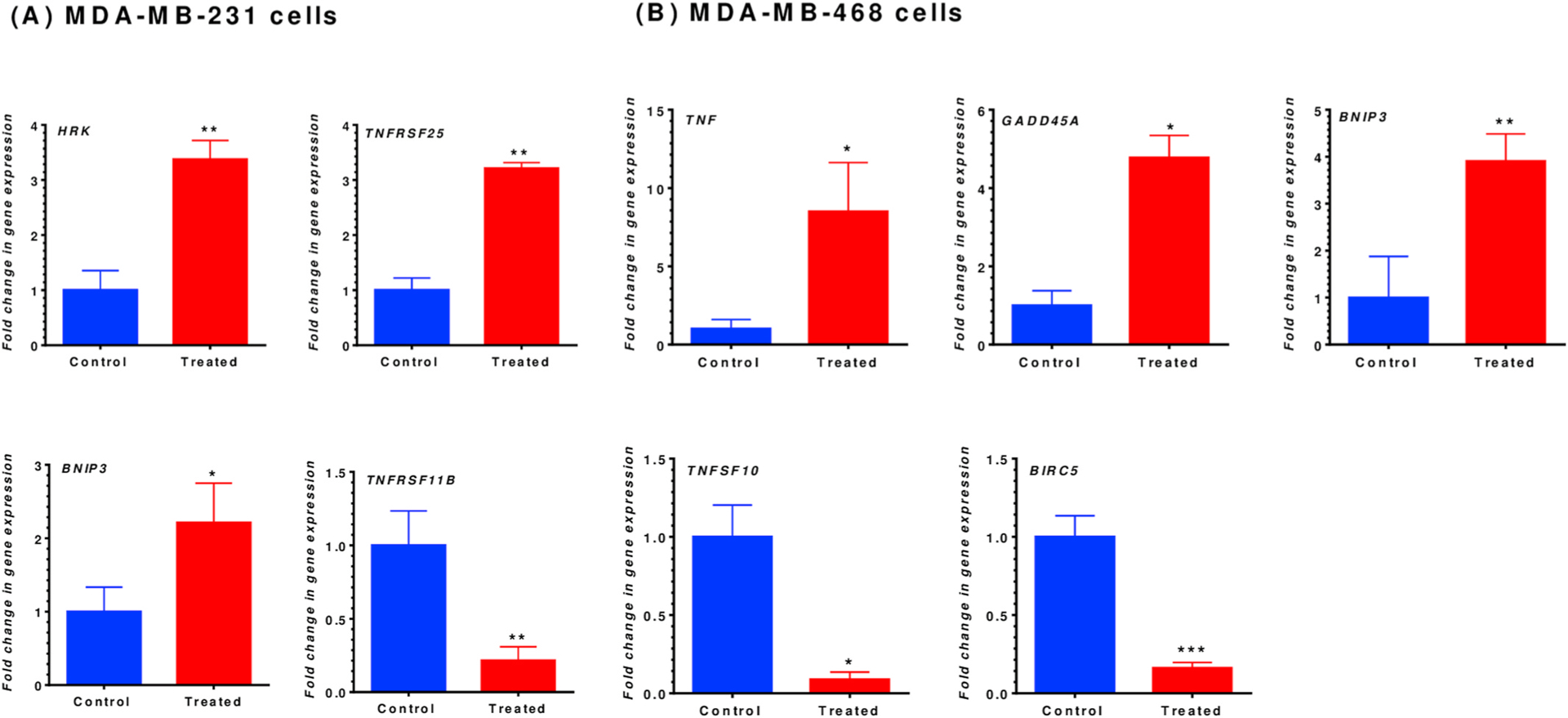
Gene expression quantification for MDA-MB-231 (A) and MDA-MB-468 (B) TNBC cells. Both cell lines were exposed to RA for 48-h at a concentration of 350 μM. Normalized mRNA expression data for the control vs. treated cells showed a significant impact on gene expression in both cell lines. In MDA-MB-231 cells, RA upregulated three genes (HRK, TNFRSF25, and BNIP3), while TNFRSF11B was significantly inhibited. Similarly, in MDA-MB-468 cells, three genes were significantly upregulated, including TNF, GADD45A, and BNIP3, while TNFSF10 and BIRC5 were repressed. The data points represented the mean ± S.E.M. of at least four independent experiments. The significance of the difference was determined using an unpaired t-test between the control vs. treated cells. The difference was considered significant at *P < 0.05, **P < 0.01, and ***P < 0.001. RA, rosmarinic acid; MDA-MB-231, MDA-MB-231; HRK, harakiri; TNFRSF25, tumor necrosis factor receptor superfamily 25; BNIP3, BCL2 interacting protein 3; TNFRSF11B, tumor necrosis factor receptor superfamily 11B; TNF, tumor necrosis factor; BIRC5, baculoviral IAP repeat-containing 5; GADD45A, the growth arrest, and DNA damage-inducible 45 alpha; TNFSF10, ligand tumor necrosis factor superfamily member 10.
Table 1.
mRNA gene expression fold changes in MM-231 and MM-468 TNBC cells after exposure to RA for 48 h. In MM-231 cells (left panel), HRK, TNFRSF25, and BNIP3 were upregulated, while TNFRSF11B was repressed. Comparably, MM-468 cells (right panel) TNF, GADD45A and BNIP3, were upregulated, while TNFSF10 and BIRC5 were inhibited.
| Control vs. treated MM-231 cells | Control vs. treated MM-468 cells | ||||
|---|---|---|---|---|---|
| Target gene | Fold changes | p-value | Target gene | Fold changes | p-value |
| HRK | +3.38 | 0.0011 | TNF | +8.50 | 0.0496 |
| TNFRSF25 | +3.21 | 0.0060 | GADD45A | +4.78 | 0.0496 |
| BNIP3 | +2.21 | 0.0295 | BNIP3 | +3.91 | 0.0087 |
| TNFRSF11B | − 4.65 | 0.0057 | TNFSF10 | − 11.25 | 0.0496 |
| BIRC5 | − 6.19 | 0.0005 | |||
The expression of BCL-2 interacting protein 3 (BNIP3) was significantly upregulated in both cell lines (Fig. 7A and B), but the BNIP3 mRNA was upregulated in MDA-MB-468 cells ~2-fold higher than it was for the MDA-MB-231 cells (3.91 (P < 0.0087) vs. 2.21 (P < 0.0295)-fold increase; Table 1). An increase in apoptosis-related gene expression was also found in MDA-MB-231 cells for two more genes (Fig. 7A). Harakiri (HRK) was upregulated by 3.38-fold, and TNF receptor superfamily 25 (TNFRSF25) had a 3.21-fold increase in its mRNA. Meanwhile, more than 4-fold repression in TNF receptor superfamily 11B (TNFRSF11B) gene expression was detected in MDA-MB-231 cells (Table 1).
Considerably more apoptosis-related genes changes were found in RA-treated MDA-MB-468 cells, as compared to MDA-MB-231. Two members of the TNF family of genes were greatly and significantly impacted (P < 0.05) (Fig. 7B and Table 1). TNF expression was upregulated by 8.50-fold, while the ligand TNF superfamily member 10 (TNFSF10) was repressed by higher than 11-fold, as shown in Table 1. Also, a member of the inhibitor of apoptotic proteins (IAPs) family, baculoviral IAP repeat-containing 5 (BIRC5) inhibition was highly significant (6.19-fold; P < 0.0005). Moreover, growth arrest and DNA damage-inducible 45 alpha (GADD45A) expression were (~5-fold) increased. RA did not show significant alterations in the expression of the different caspases (data not shown).
4. Discussion
Targeting the cell cycle-mediate apoptosis pathway is a rational approach for enhancing tumor sensitivity in coordination with anticancer agents (Katsman et al., 2009). The present study provides an analysis of apoptotic proteins participating in the anticancer effect of the natural polyphenol compound RA. The results suggest two markedly different mechanisms in altering apoptosis-related gene expressions between two racially different TNBC cell lines: MDA-MB-231 and MDA-MB-468.
The study indicated that in the two cell lines, RA differently induced cell cycle arrest and apoptosis. Interestingly, RA arrested the cell cycle of MDA-MB-468 cells early in mitosis and had a more significant apoptotic effect than it did in MDA-MB-231 cells. Further, RA significantly upregulated the mRNA expression of three apoptosis-related genes in the MDA-MB-468 cells, two of which were not affected in MDA-MB-231 cells. Instead, and to a lesser extent, two apoptosis-related genes in MDA-MB-231 cells were upregulated. Also, RA repressed the expression for two genes in MDA-MB-468 cells, while only one apoptosis-related gene was inhibited in the MDA-MB-231 cell line. Initially, the current data indicate that RA shows two distinct molecular mechanisms to inhibit TNBC cell proliferation.
RA showed cytotoxic and antiproliferative effects in both TNBC cell lines, parallel to other reports (Yesil-Celiktas et al., 2010). Similarly, several in-vitro studies have shown the ability of RA to inhibit cell proliferation and induce cell cycle arrest and caspase-independent apoptosis in various cancer cells (Jang et al., 2018; Li et al., 2018). Consistently, we found that RA arrested MDA-MB-468 cell cycle progression at the S-phase, induced apoptosis, and decreased proliferation. In contrast, in MDA-MB-231 cells, the compound principally induced G0/G1 cell cycle arrest, having a weaker apoptotic effect as compared with MDA-MB-468 cells. Therefore, we presume that the RA antiproliferative effect in MDA-MB-231 cells does not rely solely on apoptosis.
In this study, RA induced apoptosis by altering the expression of several genes that regulating the extrinsic and intrinsic apoptotic pathways. In the TNBC cells, RA impacted the mRNA expression of TNF-α and TNFSF10 in MDA-MB-468, while it altered TNFRSF25 and TNFRSF11B genes in MDA-MB-231 cells. One reported molecular mechanisms of RA-induced apoptosis involves the mitochondrial apoptotic pathway (Hur et al., 2004).
Thus, the current data adds insight into the potential role of TNF/TNF receptor superfamily in RA-mediated apoptotic effect in human cancer, in agreement with a previous in vivo study, (Silva et al., 2014). In MDA-MB-231 cells, RA induced a significant increase in transcription activation of TNFRSF25 (3.21-fold), while inhibited TNFRSF11B by 4.65-fold. These genes were not significantly affected in its counterpart cell line, MDA-MB-468. The gene TNFRSF25/DR3 is involved in several programmed cell death signaling, including the nuclear factor kB (NF-қB), p38 mitogen-activated protein kinase (MAPK), and caspase activation. Compared to healthy tissues, reduced TNFRSF25 expression is present in the MDA-MB-231 cell line, and this deficit correlates with a poorer prognosis and a significantly shorter survival rate (Ge et al., 2013). On the other hand, the TNFRSF11B expression inhibits the extrinsic apoptotic pathway and enhancing cancer cell survival (Silva et al., 2014). Thus, inhibiting the gene expression in RA-treated MDA-MB-231 cells may be a specific target in TNBC therapy.
In contrast, 8.50-fold upregulation of the TNF/TNF-α gene expression was noticed in RA-treated MDA-MB-468 cells. The multifunctional TNF-α cytokine is the most potent inducer of apoptosis in the TNF superfamily (Rath and Aggarwal, 1999). This finding may explain the higher response of this cell line to the apoptotic effect induced by RA compared with the MDA-MB-231 cell line. Indeed, TNF-α has been found to increase cell survival and proliferation in some cancers, including breast cancer (Szlosarek and Balkwill, 2003) through NF-қB activation (Luo et al., 2004). Nevertheless, the protein typically maintains tissue homeostasis by activating caspase, inhibiting the cell cycle, and inducing apoptosis (Robbs et al., 2013).
In MDA-MB-231 cells, RA upregulated the expression of two members of the proapoptotic BCL-2 family: HRK and BNIP3 genes. Comparably, BNIP3 was upregulated in MDA-MB-468 cells; however, the transcription activation of the gene was greater in MDA-MB-468 cells compared with its counterpart MDA-MB-231 cell line. The previously reported association between upregulated TNF and BNIP3 upregulation (Ghavami et al., 2009) may explain the current findings.
Bcl-2 Interacting Protein (BNIP3) and Harakiri, a Bcl-2 Interacting Protein (HRK), have a C-terminal transmembrane and a motif resembling BH3 domains that are crucial for apoptosis induction (Ma et al., 2017). BNIP3 expression is downregulated in the late stage of different types of cancer, (Erkan et al., 2005b), (Murai et al., 2005a, 2005b), and was found to correlate with chemoresistance and worse prognosis (Akada et al., 2005; Erkan et al., 2005). In BC, upregulated BNIP3 induces apoptosis through (FAS) inhibition and inhibits cell proliferation (Bandyopadhyay et al., 2006; Khan et al., 2014) by arresting cells at the S-phase (Zhou et al., 2003). Although BNIP3 can activate Bcl-2 and Bcl-xL, its affinity is too low to trigger cell death (Bellot et al., 2009). Additionally, the upregulation of expressed HRK selectively blocks the function of these anti-apoptotic proteins and induces intrinsic apoptosis by altering mitochondrial membrane permeability (Imazu et al., 1999; Inohara et al., 1997). Overexpression of HRK induces both intrinsic and extrinsic pathways in the prostate, ovarian, and breast cancer cells (Kaya-Aksoy et al., 2019; Lin et al., 2001).
Interestingly, in MDA-MB-468 cells, RA inhibited the expression of two proteins with different behaviors toward apoptosis: TNFSF10 and BIRC5 mRNAs. Based on previous reports, these inhibitions may have inconsistent effects on MDA-MB-468 cells. The smallest member of the IAP family and the highly specific tumor gene BIRC5 (Hingorani et al., 2013) (Jha et al., 2012) suppresses apoptosis. In BC, overexpression of BIRC5 is considered a marker in the early diagnosis of the disease, and it is correlated with the resistance to chemo and radiotherapy and poor clinical outcomes (Gunaldi et al., 2018; Jha et al., 2012). The multifunctional BIRC5 controls mitosis (Altieri, 2013) and prevents apoptosis by inhibiting the expression of different caspases (Shin et al., 2001), leading to metastasis and decreased survival rate. Indeed, having this gene only inhibited in MDA-MB-468, compared to MDA-MB-231 cells, strongly suggests the involvement of BIRC5 attenuation in the higher caspase-independent apoptotic effects of RA in the MDA-MB-468 cell line.
The data indicated the dramatic suppression of TNFRSF10 that might weaken the RA-apoptotic effect in MDA-MB-468. The gene TNFSF10 has the potential to induce apoptotic and autophagy, the surrogates of programmed cell death. It can activate the extrinsic apoptosis pathway once binding to its death receptors, TNFRSF10A, and TNFRSF10B (He et al., 2012; Kelley and Ashkenazi, 2004). To enhance proliferation, some cancers, however, utilize specific mechanisms such as NF-қB activation to control TNFSF10-induced apoptosis, leading to upregulated expression of anti-apoptotic proteins (Lin et al., 2000; Wajant, 2004). Nonetheless, RA was able to have alternative mechanisms to enhance apoptosis, mediated by BIRC5, independent of TNFSF10.
In MDA-MB-468 cells, the proapoptotic gene GADD45A was significantly upregulated by ~5-fold in RA-treated cells. The overexpressed GADD45A induces apoptosis by activating various signaling pathways, including NF-қB, p38 MAPK, and c-Jun amino-terminal kinase (JNK) signaling pathways (De Smaele et al., 2001; McCarthy et al., 1998; Takekawa and Saito, 1998; Zhang et al., 2010).
GADD45A is a member of the stress sensor family GADD45, which involved in many cellular functions, including DNA repair, apoptosis, cell cycle regulation, genomic stability, and immune response (Zhan, 2005). GADD45A can induce G1 cell cycle arrest (Liebermann and Hoffman, 2007) and G2/M arrest (Wang et al., 1999). The hold on cell cycle advancement promotes apoptosis via p38 and JNK pathways (Liebermann et al., 2011). GADD45 genes are epigenetically inactivated in different types of cancers (Al-Romaih et al., 2008). In TNBC, the low expression of the gene is strongly related to the absence of the three receptors, ER, PR, and Her2/neu (Tront et al., 2013). Consistent with previous findings (Saha et al., 2010), the upregulated expression of GADD45A in MDA-MB-468 could mediate the observed cell cycle arrest at the S-phase and consequential apoptosis.
In conclusion, the data presented in this study demonstrated the anticancer mechanism of the natural polyphenol compound RA in two different TNBC cell lines; MDA-MB-231 and MDA-MB-468, in which this agent altered the expression of various apoptosis-involved genes. This study found that RA was more potent against MDA-MB-468 cells in inducing mitotic arrest and apoptosis. Indeed, the data underline the importance of the compound in initiating apoptosis through extrinsic and intrinsic apoptosis-related genes. RA upregulates the proapoptotic gene BNIP3 in both MDA-MB-231 and MDA-MB-468 cells. The RA mechanism of action on the MDA-MB-468 genotype involves extrinsic and intrinsic caspase-independent apoptosis pathways mainly. RA apoptotic effect is primarily implemented through the suppression of surviving protein BIRC5 and the upregulation of the proapoptotic genes; TNF, GADD45A, and BNIP3, that could arrest the cell at its S-phase. Noticeably, the RA apoptotic mechanism in the MDA-MB-231 genotype is weaker and mainly promoted by the upregulation of HRK, TNFRSF25, and BNIP3 as well as the downregulation of the anti-apoptotic gene of TNFRSF11B. Considering the 500 mg of RA per day as a safe dose in human (Noguchi-Shinohara et al., 2015), the data obtained suggest that the polyphenol RA may have potential in TNBC therapies, particularly in MDA-MB-468 cells.
Fig. 6.
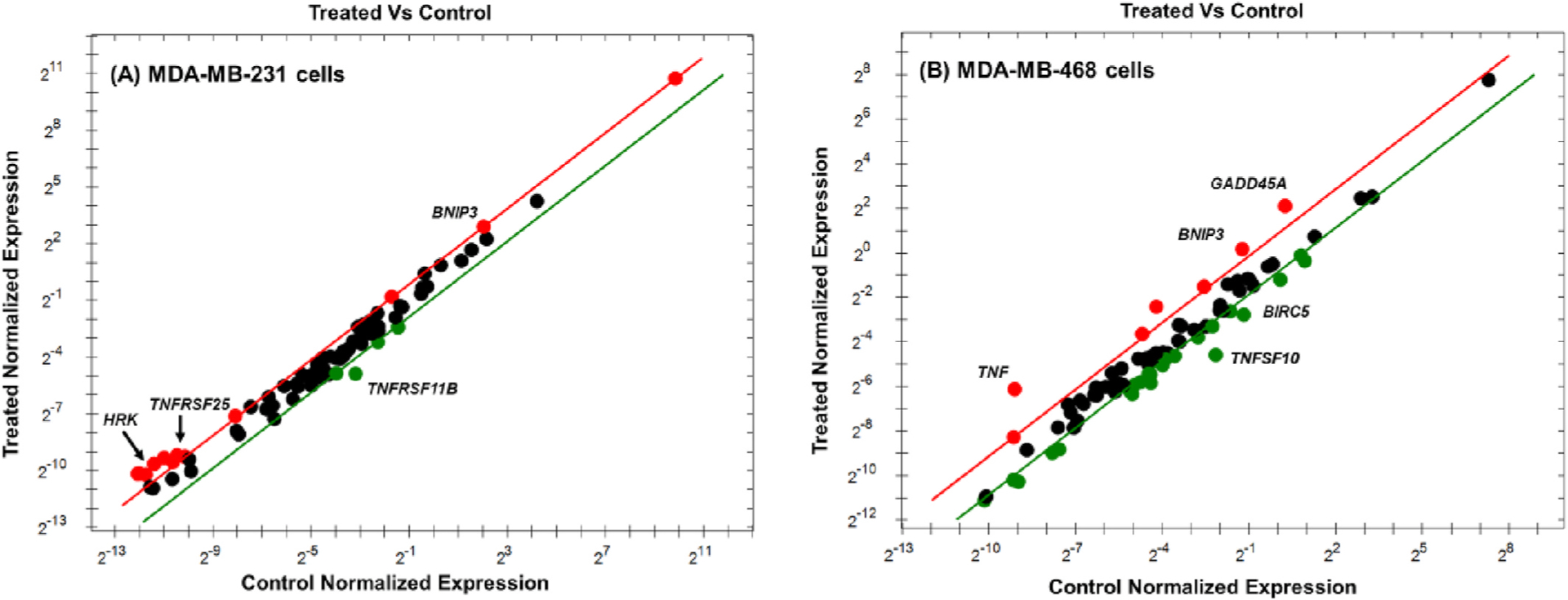
Scatter plot for MDA-MB-231 (A) and MDA-MB-468 (B) TNBC cells. Both cell lines were exposed to RA for 48 h at a concentration of 350 μM. The mRNA expression of target genes for the control vs. treated cells was normalized. Based on the threshold set, the plot images show the changes in various apoptosis-related gene expression as follows: upregulation, red dots, downregulation, green dots, and no changes, black dots.
Acknowledgments
The authors sincerely appreciate the assistance of Dr. Ramesh Badisa and the valuable editing and comments of Dr. Charles Lewis. The authors also acknowledge the biology students Mahmoud Aldawalibi and Seham Elsulami from the Dept. of Biology, Florida A&M University, for their assistance.
Funding
The present study was supported by the United States’ National Institute of Minority Health and Health Disparity through grants U54 MD007582 and P20 MD006738.
Footnotes
Declaration of competing interest
The authors state that they have no competing interest.
Authors agreement
All authors declare that there is no conflict of interest regarding the work performed in this manuscript. We further declare that the work described has not been published previously (except in the form of an abstract), that it is not under consideration for publication elsewhere, that its publication is approved by all authors and tacitly or explicitly by the responsible authorities where the work was carried out, and that, if accepted, it will not be published elsewhere in the same form, in English or in any other language, including electronically without the written consent of the copyright-holder.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
References
- Akada M, Crnogorac-Jurcevic T, Lattimore S, Mahon P, Lopes R, Sunamura M, Matsuno S, Lemoine NR, 2005. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin. Canc. Res 11, 3094–3101. [DOI] [PubMed] [Google Scholar]
- Al-Romaih K, Sadikovic B, Yoshimoto M, Wang Y, Zielenska M, Squire JA, 2008. Decitabine-induced demethylation of 5’ CpG island in GADD45A leads to apoptosis in osteosarcoma cells. Neoplasia 10, 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri DC, 2013. Targeting survivin in cancer. Canc. Lett 332, 225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders CK, Carey LA, 2009. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Canc 9 (Suppl. 2), S73–S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Zhan R, Wang Y, Pai SK, Hirota S, Hosobe S, Takano Y, Saito K, Furuta E, Iiizumi M, Mohinta S, Watabe M, Chalfant C, Watabe K, 2006. Mechanism of apoptosis induced by the inhibition of fatty acid synthase in breast cancer cells. Canc. Res 66, 5934–5940. [DOI] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM, 2009. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol 29, 2570–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao YY, Yu J, Liu TT, Yang KX, Yang LY, Chen Q, Shi F, Hao JJ, Cai Y, Wang MR, Lu WH, Zhang Y, 2018. Plumbagin inhibits the proliferation and survival of esophageal cancer cells by blocking STAT3-PLK1-AKT signaling. Cell Death Dis. 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citalingam K, Abas F, Lajis NH, Othman I, Naidu R, 2015. Antiproliferative effect and induction of apoptosis in androgen-independent human prostate cancer cells by 1,5-bis(2-hydroxyphenyl)-1,4-pentadiene-3-one. Molecules 20, 3406–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Ma R, Zhao X, Zhou F, 2019. Epigenetic profiles capturing breast cancer stemness for triple-negative breast cancer control. Epigenomics 11 (16), 1811–1825. [DOI] [PubMed] [Google Scholar]
- De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G, 2001. Induction of gadd45beta by NF-kappaB downregulates proapoptotic JNK signaling. Nature 414, 308–313. [DOI] [PubMed] [Google Scholar]
- Erkan M, Kleeff J, Esposito I, Giese T, Ketterer K, Buchler MW, Giese NA, Friess H, 2005. Loss of BNIP3 expression is a late event in pancreatic cancer, contributing to chemoresistance and worsened prognosis. Oncogene 24, 4421–4432. [DOI] [PubMed] [Google Scholar]
- Fulda S, 2008. Targeting inhibitor of apoptosis proteins (IAPs) for cancer therapy. Anti Canc. Agents Med. Chem 8, 533–539. [DOI] [PubMed] [Google Scholar]
- Fulda S, 2009. Tumor resistance to apoptosis. Int. J. Canc 124, 511–515. [DOI] [PubMed] [Google Scholar]
- Fulda S, 2011. Targeting apoptosis signaling pathways for anticancer therapy. Frontiers in oncology 1, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Z, Sanders AJ, Ye L, Mansel RE, Jiang WG, 2013. Expression of death receptor-3 in human breast cancer and its functional effects on breast cancer cells in vitro. Oncol. Rep 29, 1356–1364. [DOI] [PubMed] [Google Scholar]
- Ghavami S, Eshraghi M, Kadkhoda K, Mutawe MM, Maddika S, Bay GH, Wesselborg S, Halayko AJ, Klonisch T, Los M, 2009. Role of BNIP3 in TNF-induced cell death–TNF upregulates BNIP3 expression. Biochim. Biophys. Acta 1793, 546–560. [DOI] [PubMed] [Google Scholar]
- Gunaldi M, Isiksacan N, Kocoglu H, Okuturlar Y, Gunaldi O, Topcu TO, Karabulut M, 2018. The value of serum survivin level in early diagnosis of cancer. J. Canc. Res. Therapeut 14, 570–573. [DOI] [PubMed] [Google Scholar]
- He W, Wang Q, Xu J, Xu X, Padilla MT, Ren G, Gou X, Lin Y, 2012. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy 8, 1811–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani P, Dickman P, Garcia-Filion P, White-Collins A, Kolb EA, Azorsa DO, 2013. BIRC5 expression is a poor prognostic marker in Ewing sarcoma. Pediatr. Blood Canc 60, 35–40. [DOI] [PubMed] [Google Scholar]
- Huang Y, Cai Y, Huang R, Zheng X, 2018. Rosmarinic acid combined with adriamycin induces apoptosis by triggering mitochondria-mediated signaling pathway in HepG2 and bel-7402 cells. Med. Sci. Mon. Int. Med. J. Exp. Clin. Res.: international medical Journal of experimental and clinical research 24, 7898–7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudis CA, Gianni L, 2011. Triple-negative breast cancer: an unmet medical need. Oncol. 16 (Suppl. 1), 1–11. [DOI] [PubMed] [Google Scholar]
- Hur YG, Yun Y, Won J, 2004. Rosmarinic acid induces p56lck-dependent apoptosis in Jurkat and peripheral T cells via the mitochondrial pathway independent from Fas/Fas ligand interaction J. Immunol 172, 79–87 (Baltimore, Md. : 1950). [DOI] [PubMed] [Google Scholar]
- Imazu T, Shimizu S, Tagami S, Matsushima M, Nakamura Y, Miki T, Okuyama A, Tsujimoto Y, 1999. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with BCL-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene 18, 4523–4529. [DOI] [PubMed] [Google Scholar]
- Inohara N, Ding L, Chen S, Nunez G, 1997. harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-X(L). EMBO J. 16, 1686–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan R, Chaudhry GE, 2019. Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv. Pharmaceut. Bull 9, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YG, Hwang KA, Choi KC, 2018. Rosmarinic acid, a component of rosemary tea, induced the cell cycle arrest and apoptosis through modulation of HDAC2 expression in prostate cancer cell lines. Nutrients 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha K, Shukla M, Pandey M, 2012. Survivin expression and targeting in breast cancer. Surg Oncol 21, 125–131. [DOI] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW, 2002. Apoptosis: a link between cancer genetics and chemotherapy. Cell 108, 153–164. [DOI] [PubMed] [Google Scholar]
- Katsman A, Umezawa K, Bonavida B, 2009. Chemosensitization and immunosensitization of resistant cancer cells to apoptosis and inhibition of metastasis by the specific NF-kappaB inhibitor DHMEQ. Curr. Pharmaceut. Des 15, 792–808. [DOI] [PubMed] [Google Scholar]
- Kaya-Aksoy E, Cingoz A, Senbabaoglu F, Seker F, Sur-Erdem I, Kayabolen A, Lokumcu T, Sahin GN, Karahuseyinoglu S, Bagci-Onder T, 2019. The proapoptotic Bcl-2 family member Harakiri (HRK) induces cell death in glioblastoma multiforme. Cell death discovery 5, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley SK, Ashkenazi A, 2004. Targeting death receptors in cancer with Apo2L/TRAIL. Curr. Opin. Pharmacol 4, 333–339. [DOI] [PubMed] [Google Scholar]
- Khan A, Aljarbou AN, Aldebasi YH, Faisal SM, Khan MA, 2014. Resveratrol suppresses the proliferation of breast cancer cells by inhibiting fatty acid synthase signaling pathway. Cancer Epidemiology 38, 765–772. [DOI] [PubMed] [Google Scholar]
- Kuo PL, Hsu YL, Cho CY, 2006. Plumbagin induces G2-M arrest and autophagy by inhibiting the AKT/mammalian target of rapamycin pathway in breast cancer cells. Mol. Canc. Therapeut 5, 3209–3221. [DOI] [PubMed] [Google Scholar]
- LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG, 2008. IAP-targeted therapies for cancer. Oncogene 27, 6252–6275. [DOI] [PubMed] [Google Scholar]
- Li H, Zhuang HL, Lin JJ, Zhang YF, Huang H, Luo T, Yu WT, Ni F, 2018. Effect of rosmarinic acid from Sarcandra glabra in inhibiting proliferation and migration and inducing apoptosis of MDA-MB-231 cells via regulation of expressions of Bcl-2 and Bax]. Zhongguo Zhong Yao za Zhi = Zhongguo Zhong Yao za Zhi = China Journal of Chinese materia medica 43, 3335–3340. [DOI] [PubMed] [Google Scholar]
- Liebermann DA, Hoffman B, 2007. Gadd45 in the response of hematopoietic cells to genotoxic stress. Blood Cell Mol. Dis 39, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebermann DA, Tront JS, Sha X, Mukherjee K, Mohamed-Hadley A, Hoffman B, 2011. Gadd45 stress sensors in malignancy and leukemia. Crit. Rev. Oncog 16, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Page C, Jin X, Sethi AO, Patel R, Nunez G, 2001. Suppression activity of proapoptotic gene products in cancer cells, a potential application for cancer gene therapy. Anticancer Res. 21, 831–839. [PubMed] [Google Scholar]
- Lin Y, Devin A, Cook A, Keane MM, Kelliher M, Lipkowitz S, Liu ZG, 2000. The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun N-terminal kinase. Mol. Cell Biol 20, 6638–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo JL, Maeda S, Hsu LC, Yagita H, Karin M, 2004. Inhibition of NF-kappaB in cancer cells converts inflammation-induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Canc. Cell 6, 297–305. [DOI] [PubMed] [Google Scholar]
- Ma Z, Chen C, Tang P, Zhang H, Yue J, Yu Z, 2017. BNIP3 induces apoptosis and protective autophagy under hypoxia in esophageal squamous cell carcinoma cell lines: BNIP3 regulates cell death. Dis. Esophagus: official Journal of the International Society for Diseases of the Esophagus 30, 1–8. [DOI] [PubMed] [Google Scholar]
- McCarthy JV, Ni J, Dixit VM, 1998. RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J. Biol. Chem 273, 16968–16975. [DOI] [PubMed] [Google Scholar]
- Messeha SS, Zarmouh NO, Mendonca P, Alwagdani H, Cotton C, Soliman KFA, 2019. Effects of gossypol on apoptosis-related gene expression in racially distinct triple-negative breast cancer cells. Oncol. Rep 42, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai M, Toyota M, Satoh A, Suzuki H, Akino K, Mita H, Sasaki Y, Ishida T, Shen L, Garcia-Manero G, Issa JP, Hinoda Y, Tokino T, Imai K, 2005a. Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br. J. Canc 92, 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai M, Toyota M, Suzuki H, Satoh A, Sasaki Y, Akino K, Ueno M, Takahashi F, Kusano M, Mita H, Yanagihara K, Endo T, Hinoda Y, Tokino T, Imai K, 2005b. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin. Canc. Res 11, 1021–1027. [PubMed] [Google Scholar]
- Noguchi-Shinohara M, Ono K, Hamaguchi T, Iwasa K, Nagai T, Kobayashi S, Nakamura H, Yamada M, 2015. Pharmacokinetics, safety, and tolerability of melissa officinalis extract which contained rosmarinic acid in healthy individuals: a randomized controlled trial. PloS One 10 e0126422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plati J, Bucur O, Khosravi-Far R, 2008. Dysregulation of apoptotic signaling in cancer: molecular mechanisms and therapeutic opportunities. J. Cell. Biochem 104, 1124–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman KW, Li Y, Wang Z, Sarkar SH, Sarkar FH, 2006. Gene expression profiling revealed survivin as a target of 3,3’-diindolylmethane-induced cell growth inhibition and apoptosis in breast cancer cells. Canc. Res 66, 4952–4960. [DOI] [PubMed] [Google Scholar]
- Ramachandran C, Rodriguez S, Ramachandran R, Raveendran Nair PK, Fonseca H, Khatib Z, Escalon E, Melnick SJ, 2005. Expression profiles of apoptotic genes induced by curcumin in human breast cancer and mammary epithelial cell lines. Anticancer Res. 25, 3293–3302. [PubMed] [Google Scholar]
- Rath PC, Aggarwal BB, 1999. TNF-induced signaling in apoptosis. J. Clin. Immunol 19, 350–364. [DOI] [PubMed] [Google Scholar]
- Robbs BK, Lucena PI, Viola JP, 2013. The transcription factor NFAT1 induces apoptosis through cooperation with Ras/Raf/MEK/ERK pathway and upregulation of TNF-alpha expression. Biochim. Biophys. Acta 1833, 2016–2028. [DOI] [PubMed] [Google Scholar]
- Ryu H, Nam KY, Kim JS, Hwang SG, Song JY, Ahn J, 2018. The small molecule AU14022 promotes colorectal cancer cell death via p53-mediated G2/M-phase arrest and mitochondria-mediated apoptosis. J. Cell. Physiol 233, 4666–4676. [DOI] [PubMed] [Google Scholar]
- Sadighi S, Zokaasadi M, Kasaeian A, Maghsudi S, Jahanzad I, Kamranzadeh Fumani H, 2017. The effect of immunohistochemically detected p53 accumulation in the prognosis of breast cancer; A retrospective survey of the outcome. PloS One 12, e0182444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Kuzuhara T, Echigo N, Fujii A, Suganuma M, Fujiki H, 2010. Apoptosis of human lung cancer cells by curcumin mediated through up-regulation of “growth arrest and DNA damage-inducible genes 45 and 153. Biol. Pharm. Bull 33, 1291–1299. [DOI] [PubMed] [Google Scholar]
- Sato N, Beitz JG, Kato J, Yamamoto M, Clark JW, Calabresi P, Raymond A, Frackelton AR Jr., 1993. Platelet-derived growth factor indirectly stimulates angiogenesis in vitro. Am. J. Pathol 142, 1119–1130. [PMC free article] [PubMed] [Google Scholar]
- Shin S, Sung BJ, Cho YS, Kim HJ, Ha NC, Hwang JI, Chung CW, Jung YK, Oh BH, 2001. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and −7. Biochemistry 40, 1117–1123. [DOI] [PubMed] [Google Scholar]
- Silva JC, Ferreira-Strixino J, Fontana LC, Paula LM, Raniero L, Martin AA, Canevari RA, 2014. Apoptosis-associated genes related to photodynamic therapy in breast carcinomas. Laser Med. Sci 29, 1429–1436. [DOI] [PubMed] [Google Scholar]
- Szlosarek PW, Balkwill FR, 2003. Tumour necrosis factor alpha: a potential target for the therapy of solid tumours. Lancet Oncol. 4, 565–573. [DOI] [PubMed] [Google Scholar]
- Takekawa M, Saito H, 1998. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell 95, 521–530. [DOI] [PubMed] [Google Scholar]
- Teoh PL, Liau M, Cheong BE, 2019. Phyla nodiflora L. Extracts induce apoptosis and cell cycle arrest in human breast cancer cell line, MCF-7. Nutr. Canc 71, 668–675. [DOI] [PubMed] [Google Scholar]
- Tront JS, Willis A, Huang Y, Hoffman B, Liebermann DA, 2013. Gadd45a levels in human breast cancer are hormone receptor-dependent. J. Transl. Med 11, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajant H, 2004. TRAIL and NFkappaB signaling–a complex relationship. Vitam. Horm 67, 101–132. [DOI] [PubMed] [Google Scholar]
- Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, Hollander MC, O’Connor PM, Fornace AJ Jr., Harris CC, 1999. GADD45 induction of a G2/M cell cycle checkpoint. Proc. Natl. Acad. Sci. U. S. A 96, 3706–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Omelchenko I, Shi X, Nuttall AL, 2009. The influence of NF-kappaB signal-transduction pathways on the murine inner ear by acoustic overstimulation. J. Neurosci. Res 87, 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesil-Celiktas O, Sevimli C, Bedir E, Vardar-Sukan F, 2010. Inhibitory effects of rosemary extracts, carnosic acid, and rosmarinic acid on the growth of various human cancer cell lines. Plant Foods Hum. Nutr. (Dordr.) 65, 158–163. [DOI] [PubMed] [Google Scholar]
- Zhan Q, 2005. Gadd45a, a p53- and BRCA1-regulated stress protein, in the cellular response to DNA damage. Mutat. Res 569, 133–143. [DOI] [PubMed] [Google Scholar]
- Zhang D, Lin J, Han J, 2010. Receptor-interacting protein (RIP) kinase family. Cell. Mol. Immunol 7, 243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HW, Hu JJ, Fu RQ, Liu X, Zhang YH, Li J, Liu L, Li YN, Deng Q, Luo QS, Ouyang Q, Gao N, 2018. Flavonoids inhibit cell proliferation and induce apoptosis and autophagy through downregulation of PI3Kgamma mediated PI3K/AKT/mTOR/p70S6K/ULK signaling pathway in human breast cancer cells. Sci. Rep 8, 11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Simpson PJ, McFadden JM, Townsend CA, Medghalchi SM, Vadlamudi A, Pinn ML, Ronnett GV, Kuhajda FP, 2003. Fatty acid synthase inhibition triggers apoptosis during the S phase in human cancer cells. Canc. Res 63, 7330–7337. [PubMed] [Google Scholar]