

Abstract
In solid tumors, vascular structure and function varies from the core to the periphery. This structural heterogeneity has been proposed to influence the mechanisms by which tumor cells enter the circulation. Blood vessels exhibit regional defects in endothelial coverage, which can result in cancer cells directly exposed to flow and potentially promoting intravasation. Consistent with prior reports, we observed in human breast tumors and in a mouse model of breast cancer that approximately 6% of vessels consisted of both endothelial cells and tumor cells, so-called mosaic vessels. Due in part to the challenges associated with observing tumor-vessel interactions deep within tumors in real-time, the mechanisms by which mosaic vessels form remain incompletely understood. We developed a tissue-engineered model containing a physiologically realistic microvessel in co-culture with mammary tumor organoids. This approach allows real-time and quantitative assessment of tumor-vessel interactions under conditions that recapitulate many in vivo features. Imaging revealed that tumor organoids integrate into the endothelial cell lining, resulting in mosaic vessels with gaps in the basement membrane. While mosaic vessel formation was the most frequently observed interaction, tumor organoids also actively constricted and displaced vessels. Furthermore, intravasation of cancer cell clusters was observed following the formation of a mosaic vessel. Taken together, our data reveal that cancer cells can rapidly reshape, destroy, or integrate into existing blood vessels, thereby affecting oxygenation, perfusion, and systemic dissemination. Our novel assay also enables future studies to identify targetable mechanisms of vascular recruitment and intravasation.
Keywords: tumor microenvironment, tumor vasculature, mosaic vessels, vascular engineering, intravasation, breast cancer
INTRODUCTION
Intravasation is an early step in the metastatic cascade and is accomplished when cancer cells gain access to the circulation (1,2). Both circulating tumor cells (CTCs) and circulating tumor cell clusters (CTC clusters) have been detected in mouse models and human patients (3,4). CTC clusters have higher metastatic potential compared to single tumor cells (5–7). Intravital imaging revealed single cancer cells migrating into vessels, a process termed transendothelial migration (TEM) (8,9). However, solid tumors recruit their own vasculature as they grow (9–11). Newly formed vessels are diverse in structure and permeability (“leakiness”) and vary regionally with the tumor (11–13). Therefore, the mechanisms by which cancer cells access the circulation remain incompletely understood (14).
Our understanding of metastasis is largely derived from images of fixed tissues. Structural studies revealed tumor vessels with defective organization and loosely connected endothelial cells (15). Analysis of tumor xenografts revealed vessels with intercellular openings up to 5 μm. These gaps facilitate local extravasation of therapeutic agents and red blood cells, via the enhanced permeation and retention (EPR) effect (15,16). Tumor vasculature in colon carcinoma xenografts exhibits a hybrid structure, with both cancer cells and endothelial cells contributing to the vessel wall (termed “mosaic vessels”) (17,18). Studies in melanoma, breast, ovary, lung, prostate and glioblastoma have described the de novo formation of perfusable vasculogenic-like networks formed entirely of tumor cells, termed vascular mimicry (VM) (19,20). Across these contexts, the loss of endothelial coverage represents an opportunity for tumor cells to enter circulation without TEM (17).
Despite these intriguing observations, very little is known about tumor-vessel dynamics. We therefore developed a 3D tissue-engineered model in which we co-cultured primary tumor organoids with functional microvessels and imaged in real-time (21–24). We observed three distinct types of interaction: (1) formation of mosaic vessels, as tumor cells displace endothelial cells, disrupt the basement membrane, and facilitate intravasation of CTC clusters, (2) organoids enwrapping and constricting microvessels, limiting flow and forming “dead ends”, and (3) organoids “pulling” on microvessels. In addition, co-culture with cancer cells changed endothelial cell proliferation, permeability, and survival, which could also influence tumor-vessel interactions, and, thereby, the likelihood that CTCs or CTC clusters enter circulation.
MATERIALS AND METHODS
Tumor organoid isolation and culture and immunofluorescent staining of samples were done in close accordance with our prior publications and so the details are presented in the Supplemental Methods.
Mice and cell lines
Mice were maintained on the FVB/n background in a specific pathogen-free facility. FVB/N-Tg(MMTV-PyVT)634Mul/J (MMTV-PyMT) and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) lines were acquired from Jackson Laboratory (25). Procedures were conducted according to a protocol approved by the JHU School of Medicine Institutional Animal Care and Use Committee. For confocal time-lapse experiments, MMTV-PyMT mice were crossed with FVB/NJ, mT/mG mice (26).
VeraVec HUVEC-TURBO-GFP cells (HUVEC-GFP) (HVERA-UMB-202100; Angiocrine Bioscience, New York, NY) were seeded in the cylindrical channel of the microvessel platform (27). Endothelial cells were grown in “normal growth medium” (NGM): MCDB 131 (Caisson Labs, Carlsbad, CA) supplemented with 10% heat inactivated fetal bovine serum (F0926; Sigma), 25 mg/mL endothelial mitogen (BT-203, Biomedical Technologies), 2 U/mL heparin (H3149, Sigma), 1 μg/mL hydrocortisone (H0888, Sigma), 0.2 mM ascorbic acid 2-phosphate (49752, Sigma), and 1% penicillin-streptomycin-glutamine (10378016, ThermoFisher). Culture was at 5% CO2 and 37°C. HUVEC-GFP cells were authenticated by their manufacturer and tested negative for mycoplasma (M7006; ThermoFisher) prior to use at passages 5 to 8.
Human breast tumor samples
Primary human breast tumor specimens (T01-T03) were received from the Cooperative Human Tissue Network (CHTN), in accordance with a protocol (NA_00077976) that was acknowledged by the JHU School of Medicine IRB as exempt / not human subjects research. Specimens were deidentified by CHTN, shipped in DMEM on wet ice, and accompanied by limited prespecified clinical information. Samples were fixed in 4% paraformaldehyde overnight at 4°C, transferred to 30% sucrose and kept overnight at 4°C, embedded in Tissue Tek Optimal Cutting Temperature compound (OCT, Sakura), and then frozen at −80°C.
Fabrication of perfusable microvessel device
The microvessel platform was adapted from a previous design (21). Briefly, high concentration rat tail collagen I (354249, Corning) is diluted to 7 mg/mL and neutralized with DI water, 10x PBS, and 1 N sodium hydroxide (S2770, Sigma). After neutralization, tumor organoids are embedded into the collagen gel solution to a final concentration of 5 organoids/μL and randomly seeded surrounding a 150 μm diameter super-elastic nitinol wire (Malin Co.) within a Sylgard 184 polydimethylsiloxane (PDMS; Dow Corning) housing which is patterned using a custom-made aluminum mold. After 45 min gelation at 37°C, the rod is removed, leaving behind a cylindrical channel within the collagen gel. The channel is perfused with “organoid media” for about 1–2 days. The channel is subsequently coated with fibronectin (50 μg/mL, F2006; Sigma) to promote endothelial adhesion and spreading. Endothelial cells in NGM are introduced into the channel at a concentration of 5 × 107 cells/mL and allowed to settle and adhere to the channel walls.
After the endothelial cells have spread for 2 h, the device is placed under constant flow (~1 mL/h) maintained using fluid reservoirs separated by a height difference, as previously reported (23,28). Shear stress was calculated to be 4 to 5 dyne/cm2 using Poiseuille’s equation assuming a straight cylindrical tube (21). For time-lapse imaging, both the device and the fluid reservoirs were placed in the microscope incubator and maintained at 37°C under constant humidity and 5% CO2 (19). The devices are then tested for vessel permeability and marker expression.
To fix the tumor-microvessel distance, a second template rod was introduced, parallel to the first. The second rod generates a channel for the tumor organoid/collagen-I gel suspension, at a fixed distance from the microvessel channel. Once the two rods are in place, a 7mg/mL collagen-I gel solution is introduced into the device and allowed to gel for 45 min at 37°C. Tumor organoids are suspended in a 3 mg/mL collagen-I solution (354236, Corning) and introduced into the inlet port. This less dense collagen-I solution allows for better introduction of the organoid suspension into the second channel. One of the rods is then slowly removed, allowing the solution to enter the cylindrical channel. The devices are then incubated for 15–20 min at 37°C to allow gelation of the tumor organoid/collagen-I suspension. At this point, the tumor organoids are embedded in a 3mg/mL collagen-I within the channel, surrounded by a 7mg/mL collagen-I gel. Both ends of this channel are then sealed with 2% agarose. The second rod is then removed, and the steps described above for vessel formation and perfusion are followed.
Microscopy and image analysis
Whole slide tissue scans were performed on 30 μm sections mounted onto positively-charged slides. Stained slides were mounted with Fluoromount Aqueous Mounted Medium (Sigma; F4680) and covered with rectangular #1.5 High Precision 24×50mm, 170 μm-thick Microscope Cover Glasses (Thor Labs; CG15KH). Mounted slides were then scanned with AxioScan.Z1 (Zeiss) using Zen Blue 2.1 with the following configurations: Fluor 4x/0.27NA M27 objective for coarse focus mapping, Plan-Apochromat 20x/0.80NA M27 objective for fine focus mapping, Hamamatsu Orca Flash 4.0 (fluorescence) camera for image capture, and Colibri 7 VIS-LED fluorescent light source. To detect DAPI, AlexaFluor 488, mTomato, and AlexaFluor 647, the following light sources and filter sets were used, respectively: 96 HE BFP (LED module 385nm) at 50% intensity and 1.004 ms exposure, Excitation BP 390/40 and Emission BP 450/40 filter; 38 HE GFP (LED module 475nm) at 50% intensity and 31.504 ms exposure, Excitation BP 470/40 and Emission BP 525/50 filter; 43 HE DsRed (LED module 567nm) at 50% intensity and 17.204 ms exposure, Excitation BP 550/25 and Emission BP 605/70 filter; 50 Cy5 (LED module 630nm) at 50% intensity and 123.404 ms exposure, Excitation BP 640/30 and Emission BP 690/50 filter. Z-stacks were taken every 1 μm for 16 μm (15 stacks), with scaling-per-pixel at 0.163 μm x 0.163 μm x 1 μm, and stored in raw (.czi file) format with JpgXr lossless compression. Zen and Imaris software (Bitplane Scientific, Zurich, Switzerland) were used for image analysis and to adjust brightness and contrast across entire images, to maximize image clarity.
Microvessels were imaged on a Nikon TE-2000 U microscope (Nikon Instruments Inc., Melville, NY). Time-lapse fluorescence and phase-contrast images, tracer molecules for vessel permeability measurements and fluorescent proteins were excited with a Nikon Intensilight epifluorescence illuminator, detected with an ET Sedat Quad band 89000 filter set (Chroma Technology Corp, Bellows Falls, VT), using a 10x phase-contrast objective (N.A. 0.3), and captured with a Nikon DS-Qi1Mc camera. Confocal images were captured on a Zeiss 780 laser-scanning confocal microscope (Carl Zeiss Microscopy). Differential interference contrast (DIC) microscopy images were captured with an LD Plan-Neofluar 20×/0.4 Korr Ph2 objective lens and a Cell Observer system with an AxioObserver Z1 and an AxioCam MRM camera (Carl Zeiss).
Measurement of permeability and focal leaks
After maturation of endothelial vessels, permeability was simultaneously measured for three fluorescent molecular weight probes, introduced into the input port at a final concentration of 5 μg/mL 3 kDa dextran (Alexa Fluor 488, D34682, ThermoFisher), 62.5 μg/mL 10 kDa dextran (Alexa Fluor 647, D22914, ThermoFisher) and 10 μg/mL 70 kDa dextran (Texas Red, D1830, ThermoFisher), respectively. Dextran molecules were diluted in and introduced in the outer reservoirs. They reached the lumen of the microvessels in approximately 20 min. Widefield epifluorescence images of dextran arrival in microvessels and leakage into the ECM were acquired with a 4x objective over at least 1 h, at 2 min intervals. Calibration experiments were performed to optimize image acquisition such that a linear relation between concentration and fluorescence intensity was present at least 10 times above and below the perfused concentrations of tracer molecules.
The following equation was used to calculate permeability: P3D= (1/ΔIf) (dIf/dt) (d/4), where ΔIf is the initial increase in fluorescence intensity (when the vessel is being filled with the fluorescent probe), (dIf/dt) is the rate of increase in fluorescence intensity as the solute is being transported from the vessel to the ECM, and d is the diameter of the vessel (23). Permeability coefficients were calculated in cm/s. The fluorescent intensity values obtained over time were divided in 3 regions: (i) the “noise region” (prior to the vessel being filled with the fluorescent probe), (ii) when the vessel is being filled with the fluorescent probe (ΔIf), and (iii) when the fluorescent probe permeates into the ECM. An algorithm was developed to automate calculation of permeability coefficients. Each image was divided into ten regions of interest (ROI) of equal size and fluorescent intensity over time was calculated for the different probes using Fiji. The aim was to find the inflection point marking the transition when the vessel has been filled with dye and it starts diffusing into the ECM (Supplementary Fig. S1). To identify this inflection point, a polynomial equation of degree six was used to fit the data for each ROI and probe. The “noise region” was excluded to ensure a good fit of the polynomial and to properly estimate the inflection point. To properly identify the noise region the following iterative approach was used. First, it begins by using all data available and continues excluding data by applying the absolute value of the growth rate of the fluorescent intensity (using 1% increments). Second, following this iterative procedure, the data that was finally used to fit the polynomial was selected after the identified inflection points became stable after at least three iterations. If multiple inflection points were identified based on these criteria, the inflection point selected for analysis corresponds to the first one where the second derivative changes from negative to positive. If the relevant inflection point was not found, the data for that ROI were not used. Once the relevant inflection point was identified, ΔIf was determined and (dIf/dt) was calculated for all subsequent time points, using all available data and only stopping if a subsequent inflection point was found.
Focal leaks were calculated as previously described (23). Briefly, the contrast in the fluorescence images was maximized to identify any local nonuniformities in intensity along the length of the microvessel. Focal leaks (FL) were quantified over time using the following equation: FL= nleaks / (nimagesl L), where nleaks is the total number of focal leaks counted over time, nimages is the number of time points imaged, and L is the length of the microvessel imaged.
Orthotopic transplantation to mammary fat pad
Tumor organoids from MMTVPyMT;ROSAmT/mG mice were resuspended in a 50% (vol/vol) DMEM / 50% (vol/vol) Matrigel (354230; Corning) solution at a density of 25–40 organoids/μL and kept at 4°C. Orthotopic transplantations into 3- to 4-weeks-old NSG mice were performed in a laminar flow hood. In brief, mice were anesthetized with 2.5% isoflurane, immobilized and the surgical site sterilized with ethanol. The mammary gland was exposed by a 1 cm mid-sagittal cut followed by a 0.5-cm oblique cut from the initial incision to one hip. The skin was then retracted to expose the #4 mammary gland. The #5 gland and the lymph node in the #4 gland were removed. 10–20 μL of organoid suspension was injected into the #4 gland using a syringe (702RN(7636–01); Hamilton; custom 1-in needles, 26 gauge). The skin was then locally infiltrated with 5–10 μL of 0.25% bupivacaine. The procedure was repeated in the contralateral gland. Surgical wounds were closed using 9 mm autoclips and tissue glue. Triple antibiotic ointment was applied to the incision. Mice were closely monitored and autoclips were removed 10 days post-surgery. Tumors were harvested between 6- and 10-weeks post transplantation.
2D co-culture and staining
HUVEC-GFP cells were seeded in 24 well glass bottom plates (662892, Greiner Bio-one) and incubated for 24h in a humidified environment with 5% CO2 at 37°C. Organoids isolated from a MMTV-PyMT mouse were then added on top of the HUVEC-GFP cells. Control and co-culture conditions were incubated in NGM. After 24h, cells were fixed in 4% paraformaldehyde for 10 min at ambient temperature, permeabilized with 0.05% Triton X-100 for 10 min, blocked with 1% FBS / 1% BSA in D-PBS for 15 min, and then incubated with primary antibody solution for 45 min at ambient temperature in 1% BSA in D-PBS. After three washes with D-PBS for 5 min, cells were incubated with the secondary antibody solution for 45 min at ambient temperature in 1% BSA in D-PBS. VE-Cad (R&D; MAB9381; 1:100) was use as primary antibody, Goat anti-mouse AlexaFluor 647 (ThermoFisher; A32728; 1:200) and phalloidin 546 (ThermoFisher; A22283; 1:200) was used to stain F-actin.
Endothelial cell proliferation and cell death analysis
Phase-contrast and fluorescence time-lapse images were captured every 20 min for 12 h and analyzed using Fiji. Endothelial monolayers located at the top and bottom regions of the vessel were analyzed for proliferation and cell death events (29–31). Proliferating cells were distinguished by rounding of cell shape prior and a visible halo around the cell body that is evident until the two daughter cells reincorporate into the monolayer. The number of divisions over time was quantified in three different regions per vessel and divided by the total initial number of cells within the assessed region. Proliferation rate was calculated as percent per hour (%/h). Endothelial cell death was also detected by direct observation of cell detachment from the monolayer and release of cellular debris into the flow. Cell death rate was calculated as percent per hour (%/h).
Statistics
Statistical analyses were conducted using STATA or Graphpad Prism. STATA was used to create the algorithm to calculate permeability. Data was evaluated for normality using D’Agostino-Pearson omnibus test. P values were determined by one-way ANOVA or Mann-Whitney tests. P < 0.05 was considered significant. Bar graphs report s.e.m. All data represent at least three biological replicates.
RESULTS
Primary tumors are exposed to the lumen of mosaic vessels in vivo
We first sought to determine the frequency of mosaic vessels in human breast cancer tissue samples, and in a commonly used and highly metastatic mouse model of breast cancer, in which the MMTV LTR is used to drive the polyoma virus middle T oncogene (MMTV-PyMT) both in early and late stages of tumor development (25). We crossed the MMTV-PyMT cancer model with a fluorescent reporter mouse in which all cells are labeled with a membrane localized red fluorescence protein (mTomato) (26). To unambiguously identify tumor tissue in contact with the lumen of mosaic blood vessels, we performed orthotopic transplants of ROSAmTmG; MMTV-PyMT tumor organoids into non-fluorescent NSG host mice. Human tumor tissue sections were stained for actin (red). We assayed for mosaic vessels by staining for platelet-endothelial cell adhesion molecule (PECAM-1/CD31, green), and collagen IV (white). We scanned entire sections (16 μm total tissue depth) in 1 μm steps. Microvessels were considered for analysis if they intersected the tissue section with an approximately circular cross-section (i.e. orthogonal to the tissue section). We defined potential mosaic vessels by the presence of tumor cells in apparent contact with the vessel lumen in 2D, as indicated by the absence of CD31 and collagen IV staining (Fig. 1A,C and Supplementary Fig. S2A,C-F). We then confirmed the mosaic vessel structure in 3D optical reconstructions (Supplementary Fig. S2C’-F’). Based on this strategy, mosaic vessels could be readily distinguished from intact vessels (Fig. 1B,D and Supplementary Fig. S2B).
Figure 1. Identification of mosaic vessels in primary metastatic breast cancer tumors.

(A-B) 2D images of a mosaic vessel (A) and a fully lined vessel (B) in primary tumors from an orthotopic transplant of ROSAmTmG; MMTV-PyMT tumor organoids into non-fluorescent NSG host mice, stained with CD31 (green), collagen IV (white) and DAPI. Arrowheads mark gaps in basement membrane. Scale bars: 30 μm in (A) and 50 μm in (B). (C-D) 2D images of a mosaic vessel (C) and a fully lined vessel (D) in human breast tumors stained with actin (red), CD31 (green), collagen IV (white) and DAPI. Arrowheads mark gaps in basement membrane. Scale bars: 30 μm in (C) and 50 μm in (D). (E) Measurements of average distance of mosaic vessel to tumor-stroma border and the average radius of each analyzed tumor. (F) Probability density function of distance of mosaic vessels to the tumor-stroma border, normalized to tumor radius. Represents 60, 168, and 103 mosaic vessels for early murine, late murine tumors, and human primary tumors. At least 6 tissue slides were analyzed per tumor across 3 independent experiments per condition. Normalized measure ranges between 0 (at tumor-stroma border) and 100 (center of tumor). Median normalized distance of mosaic vessels to the tumor-stroma border was 17, 6, and 13 for late stage, early stage and human tumors, respectively.
In total we examined 1,627 vessels in human tumor tissue samples and identified 103 mosaic vessels (5.6%; Supplementary Table S1). In our murine primary tumor model, we also sought to identify the presence of mosaic vessels in tumors at early stages (~1cm in diameter) and late stages (2 cm in diameter) of tumor development. In the early stage tumor model, we examined a total of 1,213 vessels and identified 60 mosaic vessels (5.0%). In the late stage tumor model, we examined 2,681 vessels and identified 168 mosaic vessels (6.3%). The average mosaic luminal vessel diameter was 18 ± 8.4 μm, 23 ± 16.8 μm and 92 ± 15.6 μm for human, early and late stage tumor tissue samples, respectively (Supplementary Table S1).
Next, we sought to determine the location of mosaic vessels across tumors sections. We measured the distance of mosaic vessels to the closest tumor-stroma border (Fig. 1E). The average mosaic vessel distance to the tumor border was 976.7 ± 85.9 μm, 369.4 ± 49.8 μm, and 1,300± 74.4 μm for human, early, and late stage tumor tissue samples, respectively. To be able to compare the location of mosaic vessels across different tumor sizes, we normalized the mosaic vessel distance to the tumor-stroma border by dividing each distance to the respective tumor radius. This normalized measure ranges between 0 (on the tumor-stroma border) and 100 (center of tumor). Our findings indicate that mosaic vessels are typically located closer to the tumor-stroma border than the center of the tumor. (Fig. 1F).
A perfusable 3D tissue-engineered microvessel platform for imaging mosaic vessel formation
We next sought to understand the cellular dynamics underlying mosaic vessel formation. To accomplish this goal, we developed a tissue engineered model for real-time imaging of tumor-vessel interactions (21,23), with a functional microvessel surrounded by tumor organoids (Fig. 2A). The microvessel platform consists of a cylindrical, 150 μm diameter channel lined with a confluent monolayer of endothelial cells. The tumor organoids were first embedded in a collagen type I matrix within a PDMS housing surrounding a central metal rod which provides a template for the vessel (Fig. 2B). The rod was then removed, leaving behind a cylindrical channel that was seeded with endothelial cells, which were allowed to settle and adhere to the channel walls to form an intact vessel (Fig. 2B). The vessel was then perfused with normal endothelial growth medium (NGM) at a physiologically relevant shear stress under laminar flow. The microvessels were generated using GFP expressing human umbilical vein endothelial cells (HUVEC-GFP).
Figure 2. Co-culturing of tumor organoids in a 3D microvessel platform.

(A) Schematic representation of workflow for isolating and imaging organoids in device. (B) Schematic side- and end-view of microvessel device during fabrication and culture. Tumor organoids embedded in collagen I are seeded around a metal rod. After gellation, the rod is removed, the channel is perfused with fibronectin-containing media, and endothelial cells are seeded. Flow is kept at 1 mL/hour, with shear stress of 4 dynes/cm2. (C) Schematic of imaging of the mid-plane of the vessel. (D) Confocal images from a 3D reconstruction of an engineered vessel stained with β-catenin (green) and DAPI. (E) Image of tumor organoids (red) embedded in collagen-I, surrounding a vessel (green). (F) Inset showing a 3D confocal reconstruction of tumor organoids near a vessel. Arrow = direction of flow. Scale bars: 50 μm (D) and 150 μm (E)
For direct visualization of tumor-vessel interactions, we imaged the mid-plane of the vessel (Fig. 2C). Immunofluorescence staining for β-catenin revealed a confluent monolayer of endothelial cells, arranged in a cylindrical geometry and with extensive intercellular adhesions (Fig. 2D). The tissue-engineered microvessels contained a single intact lumen, as demonstrated by the mid-plane and cross-section images (Fig. 2D). Tumor organoids were cultured in close proximity to the microvessel (Fig. 2E,F) and exhibited a 3-fold increase in area over three days of culture with constant vessel perfusion, consistent with survival and extensive proliferation (Supplementary Fig. S3A,B). We also found that organoids in microvessel devices without perfusion did not grow over eight days in culture (Supplementary Fig. S3C).
Co-culture requires that media conditions are acceptable to all cell types. Accordingly, we compared the growth of organoids isolated from the same mouse in three different media conditions: “organoid medium” supplemented with FGF2 (32,33), NGM, and NGM supplemented with dibutyryl-cAMP (db-cAMP; Supplementary Fig. S3D-H). Cyclic-AMP (cAMP) is an intracellular secondary messenger that has been shown to play an important role in regulating vessel function by increasing the tightness of the barrier in vivo and in vitro, as well as decreasing proliferation and apoptosis rates in endothelial cells 3D models (34–39). We used db-cAMP as a mean to verify vessel integrity and function. The growth and circularity of tumor organoids were determined by measuring the projected surface area at day 0 and day 5 in the microvessel model. We observed a significant increase in growth and circularity of tumor organoids compared to control in all media conditions (Supplementary Fig. S3I,J). In addition, organoids in NGM exhibited a more cystic morphology than in organoid media (Supplementary Fig. S3G,H).
Co-culture with tumor organoids increases vessel permeability and induces transient focal leaks
We first sought to determine the functional properties of the tissue-engineered microvessels in the presence and absence of tumor organoids. Endothelial integrity and barrier function were determined by measuring permeability (P3D, cm s−1), which describes the rate of transport of a solute from the vessel lumen across the endothelium and into the surrounding ECM (23,31). We measured vessel permeability by simultaneously perfusing three different fluorescent molecular weight probes through the vessel lumen: 3 kDa dextran (Alexa Flour-488-conjugated), 10 kDa dextran (Alexa Flour-647-conjugated), and 70 kDa dextran (Texas Red). Using three different size probes enabled assessment of the size of defects in the endothelium. From time-lapse fluorescence microscopy we identified the time prior to the arrival of the fluorescent probe (designated 0 min; Fig. 3A-D), luminal filling (typically 20 min, Fig. 3A’-D’), and the kinetics of transport of the fluorescence probes out of the vessel and into the ECM (Fig. 3A”-D”). For all three molecular weight dextrans, co-culture with tumor organoids increased vessel permeability by about 10-fold to 3 – 6 × 10−5 cm s−1 (Fig. 3E, and Supplementary Fig. S4A-J). There was no statistical difference between the permeabilities for 3 kDa, 10 kDa, and 70 kDa dextrans, suggesting that the defects in the endothelium allowed extravasation of relatively large molecules; the diameter of 70 kDa dextran is approximately 12 nm (40). Treatment with 400 μM db-cAMP decreased permeability to all three dyes, in both control and tumor co-culture microvessels (Fig. 3B-B”, D-D”, E, and Supplementary Fig. S4E-J).
Figure 3. Microvessel integrity in tumor organoid co-cultures.

(A-D”) 2D time-lapse microscopy images of control devices without tumor organoids (A-B”) and experimental devices with tumor organoids from ROSAmTmG; MMTV-PyMT mice (C-D”). Permeability was assessed by simultaneously perfusing three different labeled dextrans with different molecular weights through the vessel lumen. 70 kDa dextran is shown. Time = 0 min represents the frame prior to luminal filling, which occurs on average at 20 min. After luminal filing, the fluorescent probes perfuse out of the vessel into the surrounding ECM. A subset of both control and co-culture devices was treated with 400 μM dibutyryl-cAMP (db-cAMP) to assess vessel function (B-B” and D-D”). (E) Permeability increases in co-culture devices, relative to controls. Permeability decreases when treated with db-cAMP for 24 h. n= number of permeability values, across at least 3 biological replicates. Error bars indicate s.e.m. and p values were determined by ANOVA, with p< 0.05 considered significant. Two additional molecular probes are presented in Supplemental Fig. 4. (F-F’) 2D time-lapse images of vessel cocultured with tumor organoids showing transient focal leaks of 10 kDa dextran, with arrows indicating leaks. Transient leaks could result from disruption of cell-cell junctions. Flow as kept at 1mL/hour and shear stress at 4 dynes/cm2. Direction of flow is left to right in images. (G) Average number of focal leaks per mm of microvessel. Average is higher in co-cultures, relative to controls and lower when treated with db-cAMP. n= number of devices, across 3 biological replicates. Error bars indicate s.e.m. and p values were determined by ANOVA, with p< 0.05 considered significant. (H) Correlation between focal leaks and permeability, using the Pearson’s correlation coefficient. Corr ranges between −1 (perfect negative association) to 1 (perfect positive association).
Our dynamic imaging of vessel permeability also revealed transient focal leaks of the fluorescent probes (Fig. 3F-H and Supplementary video S1). To unambiguously identify regions of fluorescence intensity along the length of the microvessels, we used the 10 kDa dextran (Alexa Flour-647-conjugated) probe. We observed a higher number of focal leaks in co-cultures with tumor organoids and, conversely, a reduced number of focal leaks in both control and tumor co-culture microvessels following treatment with db-cAMP (Fig. 3G). In addition, we observed a positive correlation between permeability and the number of focal leaks (Fig. 3H). Consistent with our results, transient focal leaks have been detected in the tumor vasculature of mouse brains and mammary glands (8,41,42) and cAMP agonists have previously been shown to suppress focal leaks and decrease the macromolecular permeability coefficients (35,37,39). Our data suggest that focal leaks are an intrinsic property of the epithelium that is modulated by cancer cells.
Tumor organoids actively replace the endothelial lining to form mosaic vessels
Having established that our tissue-engineered microvessels were functional and that their permeability was regulated by co-culture with tumor organoids, we next sought to analyze the cellular basis of tumor-vessel interactions in real time. To accomplish this goal, we co-cultured ROSAmTmG; MMTV-PyMT organoids within the ECM of our microvessel model. We used wide-field fluorescence microscopy, focused on the mid-plane of the vessel, to follow cell behavior (Fig. 2C). We observed three different types of tumor-vessel interactions: (1) cancer cells integrating with the endothelial lining to form mosaic vessels (Fig. 4A-A’ and Supplementary video S2), (2) cancer cells wrapping around the vessel and constricting the lumen (vessel constriction; Fig. 4B-B’), or (3) cancer cells pulling on the microvessel and displacing its position (vessel pull; Fig. 4C-C’). The geometry of each type of tumor-vessel interaction was confirmed in cross-section images.
Figure 4. Tumor organoid-vessel interactions.
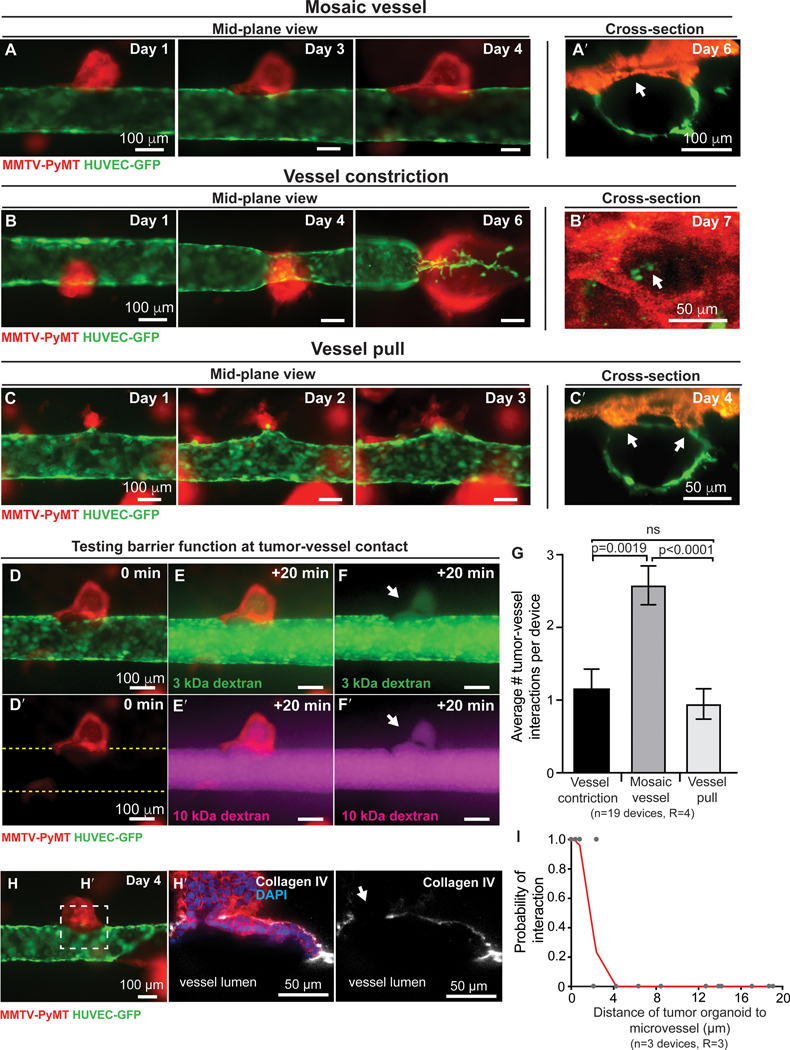
(A-C’) Time-lapse images of a ROSAmTmG; MMTV-PyMT tumor organoid growing near a microvessel (green), with flow at 1mL/hour, shear stress of 4 dynes/cm2, and images oriented with flow from left to right. 2D mid-vessel images in A-C and cross section of 3D z-stack in A’-C’. (A-A’) Organoid integrates into the vessel wall, with arrow marking gap in endothelial lining. Scale bar: 100 μm. (B-B’) Organoid wraps around and constricts the vessel (arrowhead). By day 5, the flow rate decreased to less than 0.1 mL/h, resulting in increased intraluminal pressure and upstream vessel expansion. Scale bar: 50 μm. (C-C’) Organoid pulls on and deforms the vessel wall. Scale bar: 50 μm. (D-F’) Time-lapse images showing retention of barrier function at site of tumor organoid integration into vessel wall, with 3 kDa dextran (E-F) and 10 kDa dextran (E’-F’) shown. Time= 0 min represents the frame prior to luminal filling and +20 minutes demonstrates retention of the tracer molecules. Scale bar: 100 μm. (G) Quantification of tumor-vessel interaction, revealing that mosaic vessel formation is the most frequent. n= number of vessel devices across 4 biological replicates. Error bars indicate s.e.m. and p values were determined by ANOVA, with p< 0.05 considered significant. (H) 2D image of a mosaic vessel formed when a ROSAmTmG; MMTV-PyMT tumor organoid integrated into the vessel wall. Scale bar: 100 μm. (H’) Inset showing staining for collagen IV (white) and DAPI at the site of tumor vessel-contact reveals a gap in the basement membrane. Scale bar: 50 μm. (I) Probability of tumor-vessel interaction as a function of the initial distance of the organoid to the microvessel. A non-linear probit regression analysis was conducted, which considered the dependent variable to be binary (121 observations: 101 with no interaction and 20 with interaction). Observed distances ranged from 0 to 711 μm, while the plot reports from 0 to 20 μm. The probit regression analysis Pseudo R2 was 0.95, suggesting high empirical model fit.
For quantification purposes, we identified mosaic vessels using two criteria: (i) clear retention of fluorescent dye at the site of tumor-vessel contact, and (ii) no interruption of flow following tumor-vessel interactions. In contrast, vessel constriction resulted in diminished or abolished downstream flow. We next tested the functional status of the mosaic vessels from measurements of solute permeability. We observed dye retention in the lumens of the microvessels at the sites of tumor-endothelial cell contact, revealing that the cancer cells contributed to barrier function (Fig. 4D-F, D’-F’ and Supplementary video S3). Quantification of 19 devices revealed that mosaic vessel formation was the most frequent type of tumor-vessel interaction (Fig. 4G). Moreover, we confirmed gaps in the basement membrane at the site of mosaic vessel formation by the absence of collagen IV staining, consistent with observations in vivo (Fig. 4H-H’). Furthermore, we determined that VE-cadherin is not expressed in tumor organoids in coculture with endothelial cells based on the absence of detectable immunofluorescence staining (Supplementary Fig. S5A-C).
Next, we sought to determine if the initial location of tumor organoids relative to the microvessel affected the probability of interaction. By measuring the initial distance of all tumor organoids with respect to the microvessel, we found that tumor organoids that initiate at < 4 μm away from the microvessel have an increase probability of interaction compared to tumor organoids at longer distances where the probability of interaction is negligible (Fig. 4I). Either growth or migration could bring an organoid inside the interaction distance.
Mosaic vessels as a route for intravasation of tumor cells
The presence of tumor cells in the endothelium (mosaic vessels) exposes them directly to flow and represents a potential mechanism for intravasation. Once mosaic vessels are formed, individual CTCs or CTC clusters could then be released directly into circulation. We next assayed for intravasation in mosaic regions along the microvessel. As above, we visualized tumor-vessel interactions using wide-field fluorescence microscopy focused at the mid-plane of the microvessel (Fig. 5A,B). As an example, we observed an organoid approaching a microvessel, making contact with and incorporating into the endothelium, then releasing a CTC cluster into circulation (Fig. 5A’,B’). The CTC cluster appears to roll on the luminal side of the endothelium as it moves in the direction of flow (Fig. 5A’-B’ and Supplementary video S4).
Figure 5. Intravasation of a tumor cluster following mosaic vessel formation.
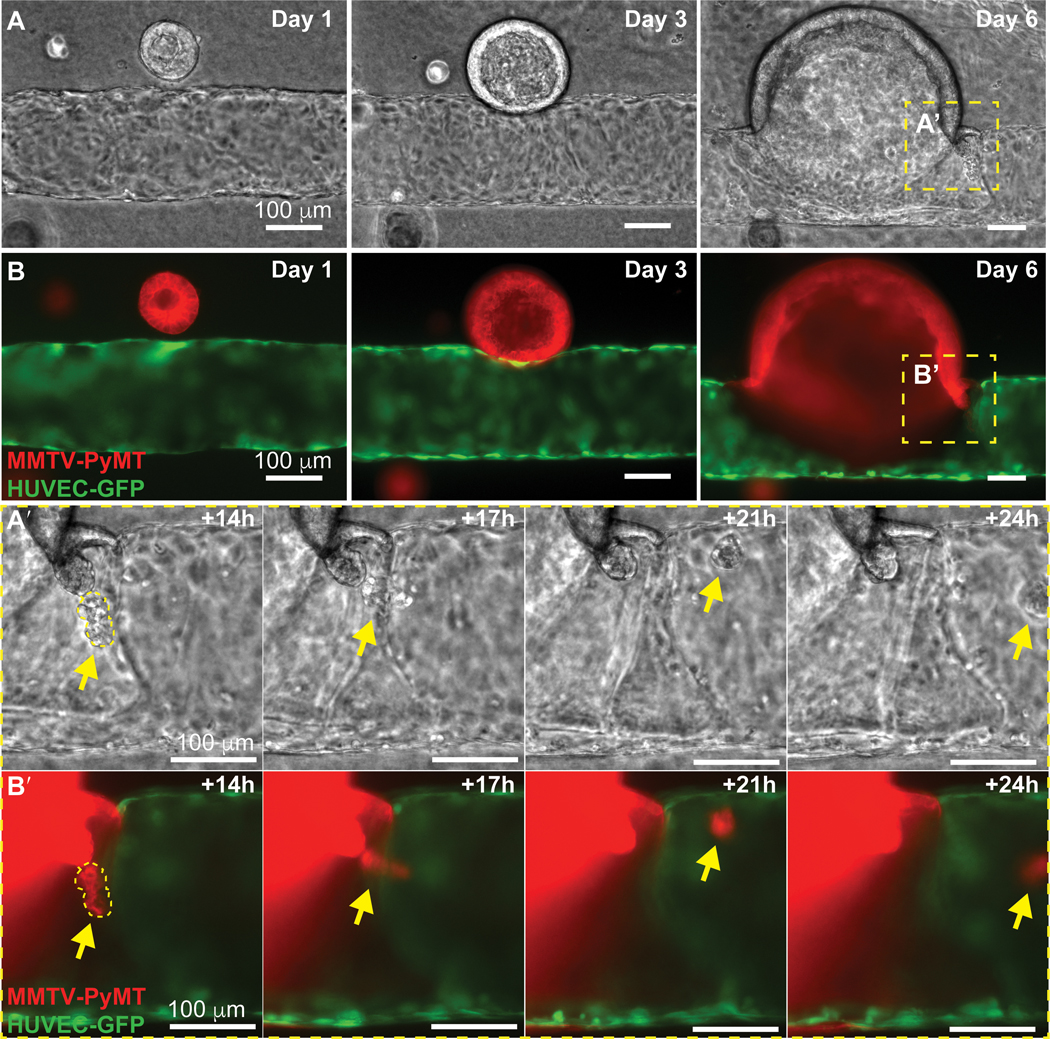
Frames from a representative (A) phase contrast and (B) fluorescence time-lapse movie of a ROSAmTmG; MMTV-PyMT tumor organoid and a HUVEC microvessel (GFP). Insets of phase (A’) and fluorescent (B’) time-lapse images showing a tumor cluster shedding into the vessel lumen under continuous flow (yellow arrows). The flow rate is kept at 1mL/hour and shear stress at 4 dynes/cm2. Direction of flow is from left to right. Scale bars: 100 μm.
An alternative geometry for modeling mosaic vessel formation
We next sought to design a microvessel model that would enable reproducible placement of tumor cells relative to the engineered microvessel. In our previous model, tumor organoids were seeded randomly throughout the collagen matrix and hence tumor-vessel interactions were dependent on the initial location of the organoids (Fig. 4I). To fix the distance between the tumor organoid and the microvessel, we introduced a modification to the microfluidic platform. A second cylindrical channel was introduced within the PDMS housing, parallel to the microvessel channel (Fig. 6A,B). Once the two template rods were placed into the PDMS housing, the collagen solution was introduced and allowed to gel. We first introduced the tumor organoids into one of the channels by slowly removing one rod leaving the other in place. At this point, the tumor organoids inside the channel are embedded in a collagen-I matrix that is surrounded by a bare collagen-I matrix. Both ends of this channel are shut with an agarose gel, thereby completely sealing this compartment. The second cylindrical rod was then removed following the steps previously described for vessel formation and perfused with NGM at a physiologically relevant shear stress under laminar flow. Using wide-field fluorescence microscopy focused at the mid-plane of the microvessel (Fig. 6C), we observed tumor invasive strands that initiate and grow in the direction of the vessel, eventually incorporating into the endothelial lining (Fig. 6D,E and Supplementary video S5).
Figure 6. Mosaic vessel formation in a 3D microvessel platform with a parallel tumor-vessel geometry.

(A) Schematic representation of workflow for isolating and imaging organoids in parallel tumor-vessel device. (B) Top- and end-view of microvessel device fabrication. To fix the tumor-vessel distance, a second cylindrical template rod is added into the device, with the collagen solution surrounding both rods. Tumor organoids at a high concentration are injected into one inlet and then the rod is slowly removed, allowing the introduction of the tumor/collagen gel suspension into the cylindrical channel. After the tumor/collagen solution gels, the second rod is removed, resulting in a bare channel which is perfused with media and coated with fibronectin. Endothelial cells are then seeded, resulting in a confluent 3D microvessel. The perfusion system is by gravity flow by differential pressure. Flow is 1 mL/h and shear stress at 4 dynes/cm2. (C) Schematic of the mid-plane of the tumor and vessel. (D) Fluorescence image of a ROSAmTmG; MMTV-PyMT tumor organoid-filled channel and a HUVEC microvessel (GFP), focusing at mid-plane. (E) Frames from a fluorescent time-lapse movie of a collective tumor strand that integrates into the vessel wall, forming a mosaic vessel. The tumor cells are exposed to flow, which is kept at 1mL/hour and shear stress at 4 dynes/cm2. Scale bars: (D) 200 μm, (E) 100 μm.
Endothelial proliferation and cell death in response to tumor organoids
To analyze the influence of tumor organoids on the endothelium, we quantified endothelial cell proliferation and cell death. We measured the frequency of cell proliferation and death within the microvessels using time-lapse phase-contrast microscopy focused at the vessel poles (i.e., top and bottom) (Supplementary Fig. S6A). Endothelial cell divisions were visualized by the increase in a cell’s optical intensity and by cell rounding followed by the integration of the two daughter cells back into the endothelium (Supplementary Fig. S6B). The proliferation rate was calculated as the percentage of endothelial cells dividing per hour (%/h). We found that tumor organoids increased endothelial cell proliferation by approximately 2-fold compared to control. In contrast, treatment with 400 μM db-cAMP decreased endothelial cell proliferation in both control and tumor co-culture microvessels (Supplementary Fig. S6C). Similarly, cell death was observed by the pronounced cell contraction and removal from the endothelium in the direction of flow (Supplementary Fig. S6D). We found that tumor organoids increased cell death by 4-fold compared to control and decreased upon treatment with db-cAMP (Supplementary Fig. S6E). Based on these results, we found that the net change in the number of endothelial cells was −0.8 %/h in the presence of tumor organoids. In particular, the negative sign in the net change demonstrates that endothelial turnover is dominated by cell death in the presence of tumor organoids.
DISCUSSION
Metastasis must involve extensive interactions between cancer cells and blood vessels but little is known about their dynamics in vivo. Prior studies reported a range of vascular abnormalities in tumors (17,43), including loops, dead-ends, irregular capillaries diameters, and cancer cells integrated within the vessel wall (12,44,45). Mosaic vessels could change the mode and frequency of intravasation but it was not clear how they formed. Our model combines a perfusable microvessel with freshly isolated tumor organoids embedded in ECM (21) and recapitulates microvessel geometry, endothelium-matrix interactions, and realistic shear stress and flow (33).
We visualized cancer cell-endothelial interactions in real-time and observed mosaic vessel formation, vessel constriction, and vessel pull, with mosaic vessels being the most frequent (Fig. 7A,B). Mosaic vessels have been observed in glioblastoma, melanoma, and gastric cancer and their presence is associated with increased distant metastasis (19,46,47). Mechanistically, this increase could be explained by persistent dissemination of cancer cells that are integrated in the vessel walls. A leading concept for generation of vascular defects is vascular mimicry (VM), in which cancer cells directly form perfused vascular-like networks without endothelial cells. VM was identified in 3D cultures of human melanoma cells and correlated with matrix-rich networks in aggressive tumors (20). In VM, cancer cells co-express endothelial markers (e.g. VE-cadherin) and build tubular structures that carry blood and are surrounded by basement membrane protein (e.g. laminin) (19). Our results suggest a VM-independent path to mosaic vessel generation. In our models, tumor organoids were VE-cadherin– and collagen IV was absent at mosaic vessel sites both in vivo and in vitro. However, a longer culture time could result in more complete replacement of the endothelium or expression of vascular markers in the cancer cells. Also, we used HUVECs and they may not model all features of tumor endothelium. Indeed, cancer cell-vascular interactions could vary depending on the functional state of the endothelium or based on organ site-specific differences in blood vessels. We could only observe acute cultures with flow due to organoid overgrowth and without flow the organoids did not grow or interact with vessels. The observed interactions were therefore dependent on flow and associated nutrients. Taken together, our data suggest that cancer cells do not require vascular gene expression to generate mosaic vessels.
Figure 7. Applications of the 3D tumor-microvessel co-culture platform.

(A) Our devices allow convenient, reductive modeling of cancer cell-blood vessel interactions within a physiologically relevant 3D platform. Stromal cells can also be introduced into tumor organoid-blood vessels co-cultures. The devices can be used to study many aspects of cancer progression. (B) Schematics of three observed tumor-vessel interactions.
We observed a CTC cluster detach from a mosaic vessel and roll on the luminal side of the endothelium in the direction of flow. CTCs and CTC clusters can be shed during surgery and in response to tumor manipulation (48–51). CTC clusters have higher metastatic potential than individual CTCs (5–7,32). Our results suggest that mosaic vessel formation could precede CTC cluster intravasation and bypass the need for TEM by single tumor cells. Previous work using MDA-MB-231 cells co-cultured with a microvessel revealed single cancer cells invading the ECM/vessel interface but did observe mosaic vessel formation (21). Individual cancer cells remained at the interface and did not proliferate. Interactions may therefore be regulated by the expression of quiescence-inducing angiocrine factors by the endothelium. In our tumor organoid-microvessel model, we showed collective interactions between groups of cancer cells and the endothelium. Cancer cells could therefore employ different mechanisms for intravasation in different regions of a tumor. CTC cluster size could also be regulated by vessel diameter (52). We observed a 50 μm CTC cluster detach from a 150 μm microvessel. These values are consistent with the 100 μm average vessel diameter in vivo and with brain tumors exhibiting vascular invasion in vessels >200 μm. Notably, vascular invasion correlated with increased metastasis and poor outcomes (52).
We also observed vascular constriction and vessel pull. Previous work suggested that tumors passively create stress on vessels due to growth in a confined space (53,54). We suggest that vascular constriction is an active mechanism by which cancer cells induce vascular defects, such as dead-ends. Furthermore, vascular constriction could generate hypoxic regions and alter the balance of pro- and anti- angiogenic signals. Vascular pulling could indicate sites of co-option of existing vessels by cancer cells (21). For example, melanoma cells metastasizing to brain use vessel co-option to obtain nutrients without an angiogenic switch, suggesting limitations to anti-angiogenic therapies (55). Tumor vasculature is also typically leakier than normal vasculature, which is termed the enhanced permeation and retention (EPR) effect. This effect is exploited in systemic delivery of therapeutic agents to solid tumors. We showed that co-culture with tumor organoids increased permeability 10-fold across the 3–70 kDa range (56), suggesting paracellular transport at vascular defects (16,57).
Finally, we propose that the existing state of the endothelium influences the outcome of tumor-vessel interactions. For example, increased endothelial cell death could increase the rate of integration of cancer cells into the vessel wall. Conversely, increased endothelial proliferation could block mosaic vessel formation and cancer cells might instead constrict or pull on the vessel. Our model enables a convenient study of multiple metastatic processes that are difficult to observe in vivo. Our study provides first steps towards understanding the dynamics of tumor-vessel interactions and their implications for collective intravasation. Our model could also be adapted and customized to incorporate additional aspects of the tumor microenvironment or alternate cancer types.
Supplementary Material
Statement of Significance:
A tissue-engineered microdevice that recapitulates the tumor-vascular microenvironment enables real-time imaging of the cellular mechanisms of mosaic vessel formation and vascular defect generation.
Acknowledgments
We thank the Ewald and Searson laboratories for helpful comments. We thank Veena Padmanaban for in vivo work, Juan Carlos Ramirez for image scans, and Seyvonne Ip for image analysis. A.J.E. received support from: The Breast Cancer Research Foundation (BCRF-19-048), the Metastatic Breast Cancer Network, the Commonwealth Foundation, and the NIH/NCI (U01CA217846, U01CA221007, U54CA2101732, 3P30CA006973). P.C.S. received support for this work from DTRA (HDTRA1-15-1-0046) and NIH (R01NS106008). V.S. received support from the Isaac and Lucille Hay Graduate Fellowship. RML acknowledges a NSF Graduate Fellowship (DGE1746891).
Footnotes
Conflict of Interest Statement: VS, EH, RL, AW, PS declare no conflicts. AE is listed as an inventor on a patent application related to the use of keratin-14 as a prognostic indicator for breast cancer outcomes and a patent application related to the use of antibodies as cancer therapeutics. AE’s spouse is an employee of ImmunoCore.
REFERENCES
- 1.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002;2:563–72 [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006;12:895–904 [DOI] [PubMed] [Google Scholar]
- 3.Liotta LA, Saidel MG, Kleinerman J. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res 1976;36:889–94 [PubMed] [Google Scholar]
- 4.Moore GE, Sandberg AA, Watne AL. The comparative size and structure of tumor cells and clumps in the blood, bone marrow, and tumor imprints. Cancer 1960;13:111–7 [DOI] [PubMed] [Google Scholar]
- 5.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014;158:1110–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci U S A 2016;113:E854–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maddipati R, Stanger BZ. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov 2015;5:1086–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov 2015;5:932–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med 2000;6:389–95 [DOI] [PubMed] [Google Scholar]
- 10.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989;339:58–61 [DOI] [PubMed] [Google Scholar]
- 11.Ziyad S, Iruela-Arispe ML. Molecular mechanisms of tumor angiogenesis. Genes Cancer 2011;2:1085–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katt ME, Wong AD, Searson PC. Dissemination from a Solid Tumor: Examining the Multiple Parallel Pathways. Trends Cancer 2018;4:20–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev 2011;37:63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nature reviews Cancer 2011;11:512–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol 2000;156:1363–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong AD, Ye M, Ulmschneider MB, Searson PC. Quantitative Analysis of the Enhanced Permeation and Retention (EPR) Effect. PLoS One 2015;10:e0123461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proceedings of the National Academy of Sciences of the United States of America 2000;97:14608–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willis RA. Tumor seminar. Tex State J Med 1950;46:611–38 [PubMed] [Google Scholar]
- 19.Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nature reviews Cancer 2003;3:411–21 [DOI] [PubMed] [Google Scholar]
- 20.Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe’er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol 1999;155:739–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong AD, Searson PC. Live-cell imaging of invasion and intravasation in an artificial microvessel platform. Cancer Res 2014;74:4937–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchanan CF, Verbridge SS, Vlachos PP, Rylander MN. Flow shear stress regulates endothelial barrier function and expression of angiogenic factors in a 3D microfluidic tumor vascular model. Cell Adh Migr 2014;8:517–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chrobak KM, Potter DR, Tien J. Formation of perfused, functional microvascular tubes in vitro. Microvasc Res 2006;71:185–96 [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Lee H, Chung M, Jeon NL. Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip 2013;13:1489–500 [DOI] [PubMed] [Google Scholar]
- 25.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 1992;12:954–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 2007;45:593–605 [DOI] [PubMed] [Google Scholar]
- 27.Seandel M, Butler JM, Kobayashi H, Hooper AT, White IA, Zhang F, et al. Generation of a functional and durable vascular niche by the adenoviral E4ORF1 gene. Proceedings of the National Academy of Sciences of the United States of America 2008;105:19288–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linville RM, DeStefano JG, Sklar MB, Xu Z, Farrell AM, Bogorad MI, et al. Human iPSC-derived blood-brain barrier microvessels: validation of barrier function and endothelial cell behavior. Biomaterials 2019;190–191:24–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bogorad MI, DeStefano J, Wong AD, Searson PC. Tissue-engineered 3D microvessel and capillary network models for the study of vascular phenomena. Microcirculation 2017;24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeStefano JG, Williams A, Wnorowski A, Yimam N, Searson PC, Wong AD. Real-time quantification of endothelial response to shear stress and vascular modulators. Integr Biol (Camb) 2017;9:362–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huxley VH, Curry FE, Adamson RH. Quantitative fluorescence microscopy on single capillaries: alpha-lactalbumin transport. Am J Physiol 1987;252:H188–97 [DOI] [PubMed] [Google Scholar]
- 32.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013;155:1639–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen-Ngoc KV, Shamir ER, Huebner RJ, Beck JN, Cheung KJ, Ewald AJ. 3D culture assays of murine mammary branching morphogenesis and epithelial invasion. Methods in molecular biology (Clifton, NJ) 2015;1189:135–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong KH, Truslow JG, Tien J. The role of cyclic AMP in normalizing the function of engineered human blood microvessels in microfluidic collagen gels. Biomaterials 2010;31:4706–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adamson RH, Liu B, Fry GN, Rubin LL, Curry FE. Microvascular permeability and number of tight junctions are modulated by cAMP. Am J Physiol 1998;274:H1885–94 [DOI] [PubMed] [Google Scholar]
- 36.Casnocha SA, Eskin SG, Hall ER, McIntire LV. Permeability of human endothelial monolayers: effect of vasoactive agonists and cAMP. J Appl Physiol (1985) 1989;67:1997–2005 [DOI] [PubMed] [Google Scholar]
- 37.He P, Zeng M, Curry FE. Dominant role of cAMP in regulation of microvessel permeability. Am J Physiol Heart Circ Physiol 2000;278:H1124–33 [DOI] [PubMed] [Google Scholar]
- 38.Schaeffer RC Jr., Bitrick MS Effects of human alpha-thrombin and 8bromo-cAMP on large and microvessel endothelial monolayer equivalent “pore” radii. Microvasc Res 1995;49:364–71 [DOI] [PubMed] [Google Scholar]
- 39.Price GM, Chrobak KM, Tien J. Effect of cyclic AMP on barrier function of human lymphatic microvascular tubes. Microvasc Res 2008;76:46–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armstrong JK, Wenby RB, Meiselman HJ, Fisher TC. The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophys J 2004;87:4259–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst 2006;98:335–44 [DOI] [PubMed] [Google Scholar]
- 42.Price GM, Wong KH, Truslow JG, Leung AD, Acharya C, Tien J. Effect of mechanical factors on the function of engineered human blood microvessels in microfluidic collagen gels. Biomaterials 2010;31:6182–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain RK. Molecular regulation of vessel maturation. Nat Med 2003;9:685–93 [DOI] [PubMed] [Google Scholar]
- 44.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res 2004;64:3731–6 [DOI] [PubMed] [Google Scholar]
- 45.Senchukova MA, Nikitenko NV, Tomchuk ON, Zaitsev NV, Stadnikov AA. Different types of tumor vessels in breast cancer: morphology and clinical value. Springerplus 2015;4:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El Hallani S, Boisselier B, Peglion F, Rousseau A, Colin C, Idbaih A, et al. A new alternative mechanism in glioblastoma vascularization: tubular vasculogenic mimicry. Brain 2010;133:973–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang JP, Liao YD, Mai DM, Xie P, Qiang YY, Zheng LS, et al. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: a meta-analysis. Angiogenesis 2016;19:191–200 [DOI] [PubMed] [Google Scholar]
- 48.Atkin G, Chopada A, Mitchell I. Colorectal cancer metastasis: in the surgeon’s hands? Int Semin Surg Oncol 2005;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park SY, Choi GS, Park JS, Kim HJ, Ryuk JP, Choi WH. Influence of surgical manipulation and surgical modality on the molecular detection of circulating tumor cells from colorectal cancer. J Korean Surg Soc 2012;82:356–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015;163:506–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan ZC, Yan J, Liu GD, Tan XY, Weng XF, Wu WZ, et al. Real-time monitoring of rare circulating hepatocellular carcinoma cells in an orthotopic model by in vivo flow cytometry assesses resection on metastasis. Cancer Res 2012;72:2683–91 [DOI] [PubMed] [Google Scholar]
- 52.Nagakawa Y, Aoki T, Kasuya K, Tsuchida A, Koyanagi Y. Histologic features of venous invasion, expression of vascular endothelial growth factor and matrix metalloproteinase-2 and matrix metalloproteinase-9, and the relation with liver metastasis in pancreatic cancer. Pancreas 2002;24:169–78 [DOI] [PubMed] [Google Scholar]
- 53.Helmlinger G, Netti PA, Lichtenbeld HC, Melder RJ, Jain RK. Solid stress inhibits the growth of multicellular tumor spheroids. Nat Biotechnol 1997;15:778–83 [DOI] [PubMed] [Google Scholar]
- 54.Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature 2004;427:695. [DOI] [PubMed] [Google Scholar]
- 55.Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 2010;16:116–22 [DOI] [PubMed] [Google Scholar]
- 56.Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 2011;63:136–51 [DOI] [PubMed] [Google Scholar]
- 57.Wong AD, Russell LM, Katt ME, Searson PC. Chemotherapeutic Drug Delivery and Quantitative Analysis of Proliferation, Apoptosis, and Migration in a Tissue-Engineered Three-Dimensional Microvessel Model of the Tumor Microenvironment. ACS Biomaterials Science & Engineering 2019;5:633–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.