

Abstract
CRISPR/Cas9 technology has revolutionized rapid and reliable gene editing in cells. Although many cell types have been subjected to CRISPR/Cas9 mediated gene editing, there is no evidence of success in genetic alteration of antigen experienced memory CD8 T cells. In this study, we show that CRISPR/Cas9 mediated gene editing in memory CD8 T cells precludes their proliferation after antigen re-encounter in vivo. This defect is mediated by the pro-apoptotic transcription factor, p53 a sensor of DNA damage. Temporarily inhibiting p53 function offers a window of opportunity for the memory CD8 T cells to repair the DNA damage, facilitating robust recall responses on antigen re-encounter. We demonstrate this by functionally altering memory CD8 T cells using CRISPR/Cas9 mediated targeted gene disruption under the aegis of p53siRNA in the mouse model. Our approach thus adapts the CRISPR/Cas9 technology for memory CD8 T cells to undertake gene-editing in vivo, for the first time.
Introduction
The CRISPR/Cas9 system is being increasingly used to edit mammalian germline sequences in cell lines and primary cells, to study gene functions as well as to sustainably alter these cells genetically and functionally(1). Although CRISPR/Cas9 has been used rather efficiently in various primary human and mouse cells to undertake targeted gene disruption, its competency has been particularly limited in immune cells, including T cells. The inability to deliver plasmids encoding Cas9 and single guide RNA (sgRNAs) to primary T cells as reliably and efficiently as in other cell types may have contributed to this. Nevertheless, the increasing use of Cas9-sgRNA ribonucleoprotein complex transfections, viral vectors and better transfection methods in general have helped make significant progress in ablating target genes in primary human and mouse T cells(2–5).
The potential to efficiently induce genetic modifications in primary T cells and return these cells to the host for analysis is a powerful experimental approach, with additional implications for immunotherapy(6–8). Although CRISPR/Cas9 mediated targeted gene disruption in T cells currently requires ex-vivo activation and prolonged maintenance in culture, this approach has helped modify CD8 T cell function to aid in immunotherapy, as well as to investigate the contributions of the targeted genes in CD8 T cell function. As a testament to these advancements, multiple scientific and clinical trials are underway to manipulate T-cell genomes(7, 9) and a recent study provided result from a phase 1 trial using CRISPR/Cas9 to both delete genes and add back specific TCR sequences to T cells that were then expanded in vitro and infused into patients(10). However, few studies have moved beyond demonstration of gene editing to actually study the biology of the gene edited T cells after their transfusion into a recipient animal or human subject, despite the tremendous research and translational significance of this method (7, 11). The physiological perturbations associated with in vitro culture and activation of CD8 T cells, that are currently indispensable to facilitate CRISPR/Cas9 mediated ablation of genes are also a caveat in learning the biology of these cells, even after their subsequent adoptive transfer into the host (2–4). While recent advances have helped overcome some of the above shortcomings in the mouse model, all of these attempts were also directed at modifying naïve CD8 T cells, to the best of our knowledge (12). Given that already expanded populations of antigen experienced memory CD8 T cells could be the primary target of personalized immunotherapy, and that our understanding of the transcriptional basis of T cell memory establishment, maintenance and recall with repeated antigen exposure is still elementary, we attempted to undertake CRISPR/ Cas9 mediated targeted gene disruption in primary, memory CD8 T cells. If targeted gene disruption can be achieved in memory CD8 T cells, without ex vivo culture or activation of the CD8 T cells, it will help investigate the contributions of individual genes to the development and maintenance of function of subsequent memory populations in vivo, as well as in mounting recall CD8 T cell responses.
To address these issues, we undertook CRISPR/Cas9 mediated targeted gene disruption in memory CD8 T cells in mice. Unexpectedly, we found that CRISPR/Cas9 gene editing in memory CD8 T cells precluded their proliferation in vivo; apparently due to p53-mediated CD8 T cell apoptosis in response to the DNA strand breaks. Temporarily subduing p53 function to offer a window of opportunity for DNA repair helped achieve CRISPR/Cas9 mediated target gene disruption in memory CD8 T cells in vivo, after adoptive transfer and restimulation in recipient mice. We here provide a roadmap for functional alteration of primary, antigen experienced memory CD8 T cells by CRISPR/Cas9 mediated targeted gene disruption.
Materials & Methods
Mice and pathogens
The C57BL/6 (B6; Thy1.2/1.2) mice were purchased from the National Cancer Institute (Frederick, MD) and maintained in the animal facilities at the University of Iowa at the appropriate biosafety level. P14 (obtained from Michael J. Bevan) and OT-I (Jackson Labs) T cell receptor-transgenic (TCR-Tg) mice (Thy1.1/1.1) were bred and maintained at the University of Iowa (Iowa City, IA). OT-I mice were crossed with B6 eGFP mice (Jackson Labs) at the University of Iowa to yield OT-I-eGFP Thy1.1 B6 mice. LCMV-Armstrong virus was injected i.p. (2×105 PFU). Attenuated (ActA-deficient) or virulent Listeria monocytogenes expressing chicken ovalbumin (Lm-Ova) (13) was injected i.v.
Generating memory CD8 T cells and enrichment
To generate primary memory CD8 T cells, 5 × 104 naïve CD8 T cells enriched from the spleen of naïve P14 or OT-I eGFP mice (CD90.1) and were adoptively transferred into B6 recipients (CD90.2) i.v., followed by infection with LCMV-Armstrong or Lm-Ova. At the indicated time point, recipient spleens were harvested, homogenized, and single-cell suspensions were prepared. The CD90.1 CD8 T cells were negatively enriched using the mouse CD8a T cell isolation kit and CD90.2 microbeads (Miltenyi Biotec), following the manufacturer’s protocols with >98 % purity.
Plasmid vectors
CD8 T cells transfected with the pX458 plasmid that encodes rCas9 from S. pyogenes with a 2A-EGFP tag (Addgene, #48138). This pX458 plasmid was cloned with the indicated target-gene specific sgRNAs (please see below for sgRNA sequences). Transfections employing the “nickase mutant” used the pX461 plasmid that encodes Cas9n (D10A nickase mutant) from S. pyogenes with 2A-EGFP (Addgene, #48140) (14) and cloned with the indicated target-gene specific sgRNAs.
In vitro transcription and RNP complex formation
In vitro synthesis of sgRNAs was performed using the manufacturer’s instruction for the MEGAscript T7 kit (ThermoFisher). The reaction products were purified using alcohol-precipitation, resuspended in water and quantified using a spectrometer. The ribonucleotide-protein (RNP) complexes were generated and used as described in detail before(3). In short, RNP complexes were generated in under 10ul or RNP reaction buffer by co-incubating 30 ug of sgRNA with10 ug of rCas9 protein (IDT) at RT for 15 min.
Nucleofection
Nucleofection of CD8 T cells were performed following the manufacturer’s (Lonza) protocol, and as described in detail elsewhere (3). In short, 1×106 CD8 T cells were resuspended in 100uL of Nucleofector buffer (Lonza) along with the RNP complex, with or without the p53siRNA (Santa Cruz, sc-29436) in a glass cuvette (Lonza). Nucleofector 2b device (Lonza) with Nucleofector program X001 was used. Following nucleofection, cell suspension was removed from cuvette, placed in Eppendorf tube with 100 uL of RPMI 1640 (Gibco) containing 10% fetal calf serum (Gibco). After incubation at 37°C for 10 min, the CD8 T cells were then placed in culture containing the T cell growth media (Lonza) or transferred intravenously into recipient mouse.
PMA/ionomycin stimulation
Blood was collected from the mice and the RBC lysed using Vitalyse (cmdg). The cells were then washed in RPMI with 10% fetal calf serum and stimulated for 3.5 h in the presence of Brefeldin A (Biolegend), 5 ng/ml PMA (Sigma) and 500 ng/ml ionomycin (Sigma) as described in detailed before(15). The cells were subsequently stained for surface markers and intracellular cytokines after membrane permeabilization (Fix Perm kit, BD Biosciences) before analysis by flow cytometry.
Flow cytometry
The cells were stained with the following antibodies: CD8 (clone 53–6.7, eBioscience), CD4 (clone GK1.5, eBioscience), CD90.1 (clone OX-7, eBioscience), CD90.2 (clone 53–2.1, eBioscience), PD-1 (clone J43, Biolegend), KLRG1 (clone 2F1, Biolegend), p53 (clone pAb 240, Novus Biologicals) TNF (Clone MP6-XT22, Biolegend) and IFNγ (clone XMG1.2, Biolegend) at the appropriate dilution and with compatible fluorochromes. The lymphocyte gates in the samples were plotted directly to examine CD8 T cell populations depicted in the flow plots.
In vitro T cell culture and proliferation assay
In vitro proliferation assay in CD8 T cells was performed as described in detail previously (4) In short, CD8 T cells were stained with CellTrace Violet (Invitrogen) as per the manufacturers protocol, before transferring them to a 96 well U-bottom plate in T cell growth media (Lonza). The indicated doses of the IL-7 (Peprotech, Cat #: 200–07) and IL-15 (Peprotech, Cat #: 210–15) were added into the culture media and T cell proliferation was determined at various time points by dye dilution using flow-cytometry.
DNA sequencing
CD8 T cells were harvested from mouse periphery at 530 dpi. CD8 T cell single cell clones were obtained by limiting dilution in 96 well plate as described in detail before (3). Following proliferation of the clones, the CD8 T cells were harvested and DNA extracted and the indicated gene sequenced at the Iowa Institute of Human Genetics (IIHG).
Assessing tissue bacterial burden
Naïve IFNgKO mice (Jackson Labs) were adoptively transferred with Cas9-IfngsgRNA transfected 6X105 OT-I CD8 T cells i.v. Control mice either received no cells or cells transfected with only rCas9 or rCas9-CtrlsgRNA. The next day, the recipient mice were challenged with 4X103 CFUs of virulent Listeria monocytogenes expressing Ova (Lm-Ova), i.v. Four days after the challenge, liver and spleen were harvested and assessed for bacterial burden (4).
Colony Counting
L. monocytogenes infection burdens in mice were determine as described in detail before(16). In short, the livers and spleens obtained from the Lm-Ova challenged mice were weighed, homogenized in antibiotic-free RPMI1640 media (Gibco) containing 10%FCS (Gibco), with a mechanical disruptor. Serial dilutions were performed and aliquots were placed on 5 cm petri dishes containing LB-Ampicillin agar. The Lm-Ova bacteria are transgenically ampicillin resistant. Plates were incubated at 37C, 5% CO2 overnight and colonies were counted by hand the next morning, to calculate the bacterial burden per gram of tissue.
Quantitative RT-PCR (qRT-PCR)
L. monocytogenes tissue burdens were determined as described in detail elsewhere(17). Total tissue DNA was extracted using the phenol/ chloroform and quantitative PCR was performed with the following primers: hlyA-177-F (TGCAAGTCCTAAGACGCCA) and hlyA-177-R (CACTGCATCTCCGTGGTATACTAA). The CT Ratios were normalized against the housekeeping gene for mouse Tumor Necrosis factor as the reference, using the following primers: TNF-5241 (TCCCTCTCATCAGTTCTATGGCCCA, TNF-5411 (CAGCAAGCATCTATGCACTTAGACCCC)
List of sgRNAs used
Klrg1 sgRNA: CCTTACATTTCCGGACAACC (crispr.mit.edu)
eGFP sgRNA: GGTGGTGCAGATGAACTTCA (18)
p53 sgRNA: AGGAGCTCCTGACACTCGGA (crispr.mit.edu)
Pdcd1 sgRNA: GACACACGGCGCAATGACAG (3)
IFNg sgRNA: GGCTTTCAATGACTGTGCCG, GGCTTTGCAGCTCTTCCTCA (crispr.mit.edu)
Results
CRISPR/Cas9 mediated targeted gene disruption prevents proliferation of memory CD8 T cells in vivo.
Successful CRISPR/Cas9 editing and subsequent evaluation of bona fide memory CD8 T cells remains an unrealized, yet potentially fruitful experimental and translational goal. To achieve this goal, we generated memory T cell receptor (TCR) transgenic CD8 T cells specific to the lymphocytic choriomeningitis virus (LCMV) GP33–41 antigen (P14 cells, CD90.1) in CD90.2 B6 mice, as depicted in Fig 1A. The memory P14 CD8 T cells were negatively enriched, transfected with the plasmid (pX458) encoding Cas9, eGFP (to identify transfected cells) and the indicated target-gene specific sgRNA (14) before adoptively transferring them to congenically distinct recipient mice. The recipient mice were subsequently challenged with LCMV to induce recall responses from the transfected P14 cells (Supplemental Fig 1A). The adoptively transferred memory P14 cell population expanded and expressed vector-derived eGFP when transfected with the ‘empty’ (no sgRNA template encoded) pX458 plasmid (Supplemental Fig 1A). As a proof of concept, we chose to disrupt the Klrg1 gene in memory P14 cells, since KLRG1 deficiency does not influence CD8 T cell proliferation or function in mice (19). Based on this, we expected the Klrg1sgRNA encoded pX458 (pX458-Klrg1sgRNA) transfected P14 cells to proliferate like those with the control pX458 empty plasmid. To our surprise, pX458-Klrg1sgRNA transfected memory P14 cells failed to undergo secondary expansion and accumulate in the recipient mice, after challenge with LCMV (Supplemental Fig 1B). We next targeted the Pdcd1 gene (encoding PD-1) for disruption in memory P14 cells, following the same approach. PD-1 deficiency does not disrupt cognate-antigen mediated proliferation of memory CD8 T cells (20). Nevertheless, we failed to detect secondary expansion and accumulation of the pX458-Pdcd1sgRNA transfected P14 CD8 cell population (Supplemental Fig 1B). Thus, while transfection of memory P14 cells with pX458 lacking sgRNA did not hinder their secondary expansion and accumulation, merely co-transfecting them with pX458 and Klrg1 or Pdcd1 targeting sgRNAs (but not scrambled sgRNA; data not shown) prevented their proliferation in the recipient mice challenged with LCMV (Supplemental Fig 1C). Cas9 nuclease functions by inducing double-strand breaks in the genomic DNA(14). To determine if the double-strand breaks (DSBs) in genomic DNA itself may have been responsible for the failure of CD8 T cells proliferation, we induced single strand breaks in the P14 CD8 T cell DNA using a ‘nickase’ mutant Cas9 (nCas9) encoding pX461 plasmid (14) and the Klrg1sgRNA. The nCas9-Klrg1sgRNA transfected P14 CD8 T cells also failed to proliferate in the recipient mice (Supplemental Fig 1D).
Figure 1. CRISPR/Cas9-mediated p53 gene disruption enables expansion of transfected memory CD8 T cells in mice.
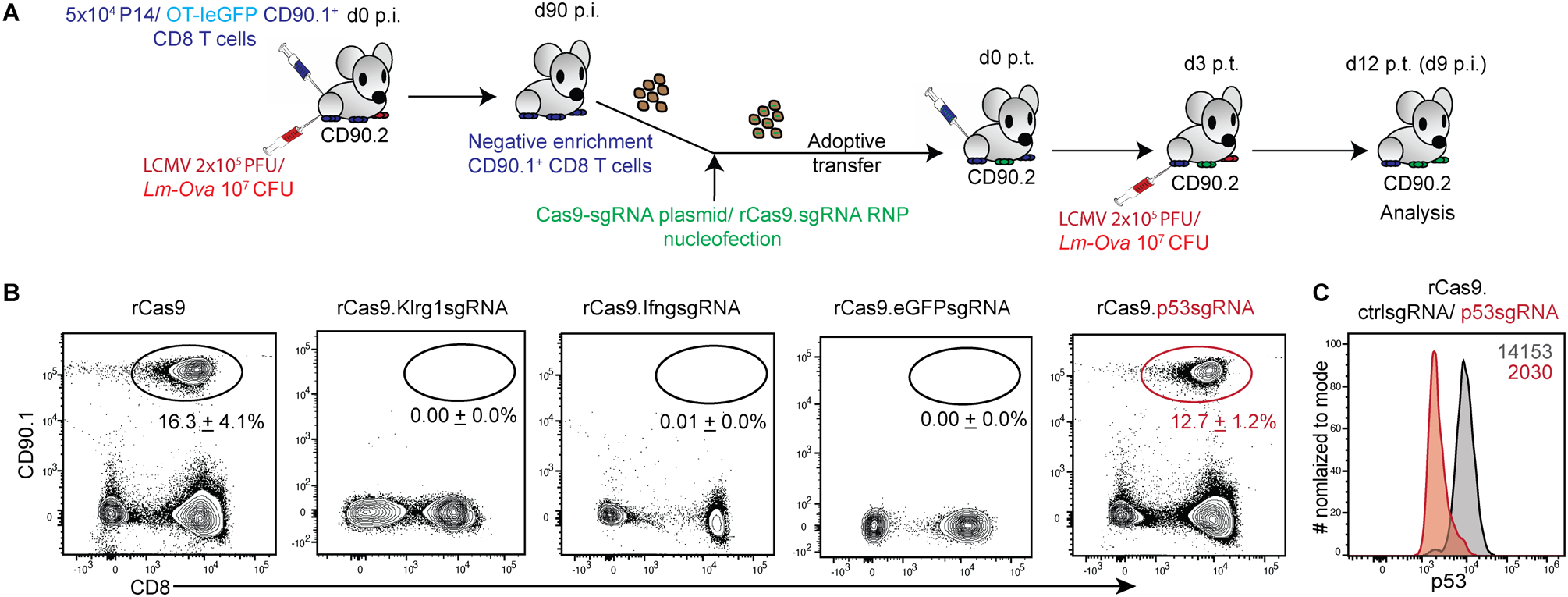
(A) Schematic depicting the experimental model used to generate genetically modified, congenically distinct, defined antigen-specific, primary memory CD8 T cells. Memory P14 or OT-IeGFP TCR transgenic (CD90.1+) CD8 T cells generated in donor (CD90.2+) mice (d90 post infection, p.i.) were selectively enriched, transfected by nucleofection with either plasmid encoding recombinant Cas9 (rCas9) and separate sgRNA or recombinant Cas9/sgRNA complexes and adoptively transferred immediately into recipient (CD90.2+) mice. The recipients were challenged with LCMV-Armstrong or Lm-Ova on d3 post-transfer (p.t.) of cells. Transferred (CD90.1+) CD8 T cells were assessed in blood by flow cytometry at d12p.t. (B) Representative flow-plots depicting frequencies of OT-IeGFP TCR transgenic (CD90.1+) CD8 T cells, transfected with rCas9 and the indicated sgRNAs before adoptive transfer to (CD90.2+) recipient mice and challenged with Lm-Ova, and examined at d9 p.i. Numbers inset represent the frequencies of the CD90.1 gated OT-I population presented as mean ± SEM from at least 2 independent experiments with ≥ 3 recipient mice per group. (C) Representative histograms of p53 expression levels in CD8 T cells co-transfected with rCas9 and ctrl sgRNA (grey) or p53sgRNA (red) and expanded in vivo as shown in 1A. Numbers inset represent the gMFI values of p53 expression. Data representative of 2 separate experiments.
Target gene disruption and transfection efficiencies in cells can be greatly enhanced by substituting Cas9 encoding plasmids with ribonucleoprotein complexes (RNPs) consisting of recombinant Cas9 (rCas9) and the target-gene specific sgRNA(3, 5). We thus co-transfected memory P14 cells with RNPs consisting of rCas9 and in vitro transcribed Klrg1sgRNA or the scrambled control sgRNA, before adoptively transferring them into recipient mice and challenging them with LCMV (Fig 1A). Here also, the adoptively transferred P14 cells failed to expand and accumulate following Klrg1 targeted gene disruption (Supplemental Fig 1E). Taken together, these data show that CRISPR/Cas9 mediated DNA breaks in memory P14 cells prevents their expansion and accumulation in response to LCMV infection in mice.
CRISPR/Cas9 mediated genomic DNA damage may hinder memory CD8 T cell proliferation.
To rule out the possibility that T cells with a particular transgenic TCR might influence memory CD8 T cell proliferation in CRISPR/Cas9 mediated targeted gene disruption, we next used chicken ovalbumin antigen specific TCR transgenic OT-I CD8 T cells (that also co-expressed eGFP, OT-IeGFP) for targeted gene disruption (Fig 1A). As with the P14 CD8 T cells, Klrg1 gene disrupted memory OT-IeGFP CD8 T cells failed to expand and accumulate in the recipient mice when challenged with Listeria monocytogenes expressing the ovalbumin protein (Lm-Ova) (Fig 1B). Similar results were obtained when we targeted genes that are known to serve specific functions in CD8 T cells- the Ifng gene for cytotoxicity (Fig 1B) or the Pdcd1 gene for co-inhibition (data not shown). Neither of these gene products are known to directly influence the proliferative abilities of memory CD8 T cells. Notably, disrupting the exogenous and non-essential eGFP gene using CRIPSR/Cas9 also prevented the expansion and accumulation memory OT-IeGFP CD8 T cells in the recipient mice (Fig 1B), indicating that genomic DNA damage itself may be responsible for this defect rather than the targeting of any specific gene. It is noteworthy that CRISPR/Cas9 mediated gene disruption also hinders the cytokine driven proliferation of the memory CD8 T cells in vitro. rCas9.Klrg1sgRNA RNP, but not rCas9 itself (data not shown) or the rCas9.Ctrlg1sgRNA RNP transfected memory OT-IeGFP cells failed to proliferate in response to IL-7 and IL-15 (Supplemental Fig 2A). Additionally, letting the adoptively transferred memory OT-IeGFP CD8 T cells ‘rest’ in vitro (or in vivo in the recipient mice; data not shown) to undergo DNA repair for up to 10 days after transfection with rCas9.Klrg1sgRNA RNP also did not rescue their inability to proliferate on cognate antigen encounter (Supplemental Fig 2B). These data suggested that targeted gene disruption using CRISPR/Cas9 hinders the proliferation of memory CD8 T cells in response to cytokines or antigen-stimulation.
P53 hinders proliferation of memory CD8 T cells after CRISPR/Cas9 mediated targeted gene disruption.
DNA damage is known to elicit cell-cycle arrest and apoptosis in cells (21, 22). While there are many molecular components in the DNA damage response pathway, the pro-apoptotic transcription factor p53 is an integral part of this response (22, 23). T cells provisionally downregulate p53 expression to facilitate antigen specific-proliferative responses in vitro (24), as well as to cope with double-strand breaks introduced by T cell receptor recombination during development (25). It is noteworthy that proliferating T cells are very sensitive to DNA damage responses and undergo rapid cell-death on detecting induced DNA damages (26, 27). We thus hypothesized that memory CD8 T cells that undergo CRISPR/Cas9 mediated DNA breaks might undergo apoptotic death in response to proliferative cues in vivo, possibly mediated by p53 (28). It is known that human pluripotent stem cells that express lower p53 levels are more amenable to CRISPR/Cas9 mediated targeted gene disruption(29, 30). Considering these notions, we chose to undertake CRISPR/Cas9 mediated targeted disruption of the p53 gene itself in memory CD8 T cells, to possibly rescue the proliferating cells from undergoing apoptotic death in response to the DNA damage introduced by editing. Indeed, memory OT-IeGFP cells transfected with rCas9.p53sgRNA RNP expanded and accumulated in the recipient mice after challenge with Lm-Ova (Fig 1B). The rCas9.p53sgRNA RNP transfected memory OT-I CD8 T cell population that expanded in the recipient mice also showed a concurrent loss of p53 expression (Fig 1C). Taken together, these data indicated that the p53 mediated apoptotic response to DNA damage may be responsible for our inability to perform CRISPR/Cas9 mediated targeted gene disruption and amplify memory CD8 T cell responses by antigen restimulation in vivo.
Targeted gene disruption in memory CD8 T cells under the aegis of p53siRNA
The above data also suggested that we could achieve targeted gene disruption via CRISPR/Cas9 in memory CD8 T cells, if the p53 function was compromised. However, p53 is a critical tumor suppressor gene, and is associated with tumor development (31). In addition, T cells lacking the p53 gene gave rise to spontaneous T cell lymphomas in mice (32). Considering the risk, concurrent p53 gene disruption may not be a feasible approach to achieving other specific gene deletions in memory CD8 T cells. Nevertheless, we surmised that a temporary loss of p53 function would present the window of opportunity to repair the CRISPR/Cas9 induced DNA breaks in memory CD8 T cells, without the long-term risks associated with the perpetual loss of p53 function. Hence, we tested if we could achieve targeted gene disruption in memory CD8 T cells, under the protection of a temporary knock-down of p53 transcription factor using p53siRNA.
To test this hypothesis, we first assessed whether knockdown of p53 could rescue proliferation of transgenic T cells in which the non-essential eGFP gene had been disrupted. As depicted in Fig 2A, we enriched memory OT-IeGFP CD8 T cells generated in donor mice, co-transfected them with rCas9.eGFPsgRNA, rCas9 or rCas9.ctrlsgRNA (data not shown) and p53siRNA before adoptively transferring them to congenically distinct recipient mice. The transferred OT-IeGFP CD8 T cells expanded and accumulated after challenge with Lm-Ova and established long-term (530 day) persistence in the recipient mice (Fig 2B). A substantial proportion of rCas9.eGFPsgRNA transfected memory OT-IeGFP CD8 T cells also exhibited loss of eGFP expression and this loss of eGFP was sustained in their memory phase (Fig 2C). The gene disruption was also retained on secondary expansion of these rCas9-eGFPsgRNA memory OT-IeGFP CD8 T cells (Fig 2B–C). These data suggested that CRISPR/Cas9 mediated targeted gene disruption can be achieved in antigen experienced CD8 T cells in vivo, under temporarily subdued p53 function.
Figure. 2. CRISPR/Cas9 mediated ablation of eGFP expression in CD8 T cells.
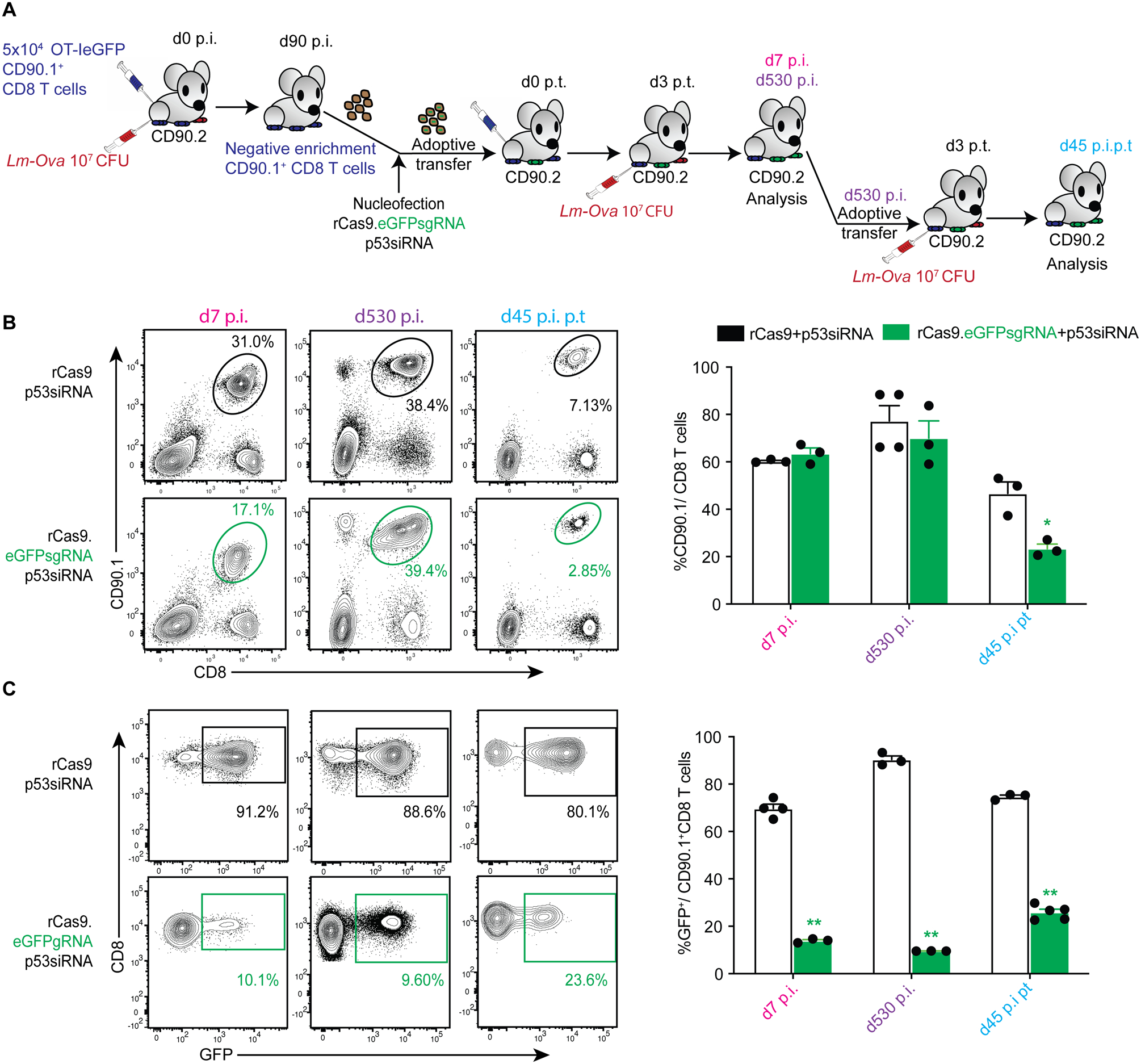
(A) Schematic depicting the experimental model to ablate eGFP from primary OT-IeGFP TCR transgenic (CD90.1+) CD8 T cells. Memory OT-IeGFP TCR transgenic (CD90.1+) CD8 T cells generated in donor (CD90.2+) mice (d90 post infection, p.i.) were enriched, transfected with rCas9/sgRNA complexes by nucleofection and adoptively transferred immediately into recipient (CD90.2+) mice. The recipients were subsequently challenged with Lm-Ova on d3 post-transfer (p.t.) and the OT-I TCR transgenic (CD90.1+) CD8 T cells in circulation examined by flow cytometry on d7 and d530 p.i. The OT-I TCR transgenic (CD90.1+) CD8 T cells were subsequently adoptively transferred to secondary recipients (CD90.2+) on d530 p.i and challenged with Lm-Ova on d3 p.t. OT-I (CD90.1+) CD8 T cells in the secondary recipients were analyzed on d45 p.i. (B) Representative flow-plots depicting frequencies of OT-IeGFP TCR transgenic (CD90.1+) CD8 T cells and (C) frequencies of GFP expressing OT-I in groups transfected with the indicated reagents, in primary and secondary recipient B6 mice challenged with Lm-ova, analyzed at the indicated time-points. Data summarized as bar graphs in the right panel with each data point representing an individual sample, represents 1 of 3 independent experiments with ≥ 3 mice per group and analyzed with t-tests comparing the eGFPsgRNA and control transfected groups at each time point. *p ≤ 0.05, **p ≤ 0.01.
Functional alteration of memory CD8 T cells by CRISPR/Cas9 mediated targeted disruption
To test if we could employ CRISPR/Cas9 to functionally alter memory CD8 T cells, we next targeted the Ifng gene, which is critical for the ability of CD8 T cells to protect against many infections, including Listeria monocytogenes infection (33, 34). As with the targeted gene disruption of the eGFP gene, we adoptively transferred memory OT-I cells obtained from donor mice into recipient mice after co-transfection with rCas9.IfngsgRNA, rCas9 or rCas9.CtrlsgRNA (data not shown) and p53siRNA, before challenging with Lm-Ova to drive their cognate antigen mediated expansion. While the rCas9.IfngsgRNA transfected memory OT-I CD8 T cells expanded and accumulated (Fig 3A) in response to Lm-Ova infection, a substantial proportion of these cells showed loss of IFNγ production in response to ex vivo stimulation. In contrast, TNF, a non-targeted cytokine produced by CD8 T cells after stimulation, was not reduced in rCas9.IfngsgRNA transfected memory OT-I CD8 T cells indicating that these cells were viable and responsive to stimulation (Supplemental Fig 3). As with the eGFP gene disruption, we observed that the rCas9.IfngsgRNA transfected memory OT-I CD8 T cells underwent secondary memory expansion after being adoptively transferred to recipient mice which were subsequently challenged with Lm-Ova (Fig 3A), and remained unable to produce IFNγ (Fig 3B).
Figure. 3. CRISPR/Cas9 mediated loss of IFNγ in murine primary memory CD8 T cells.
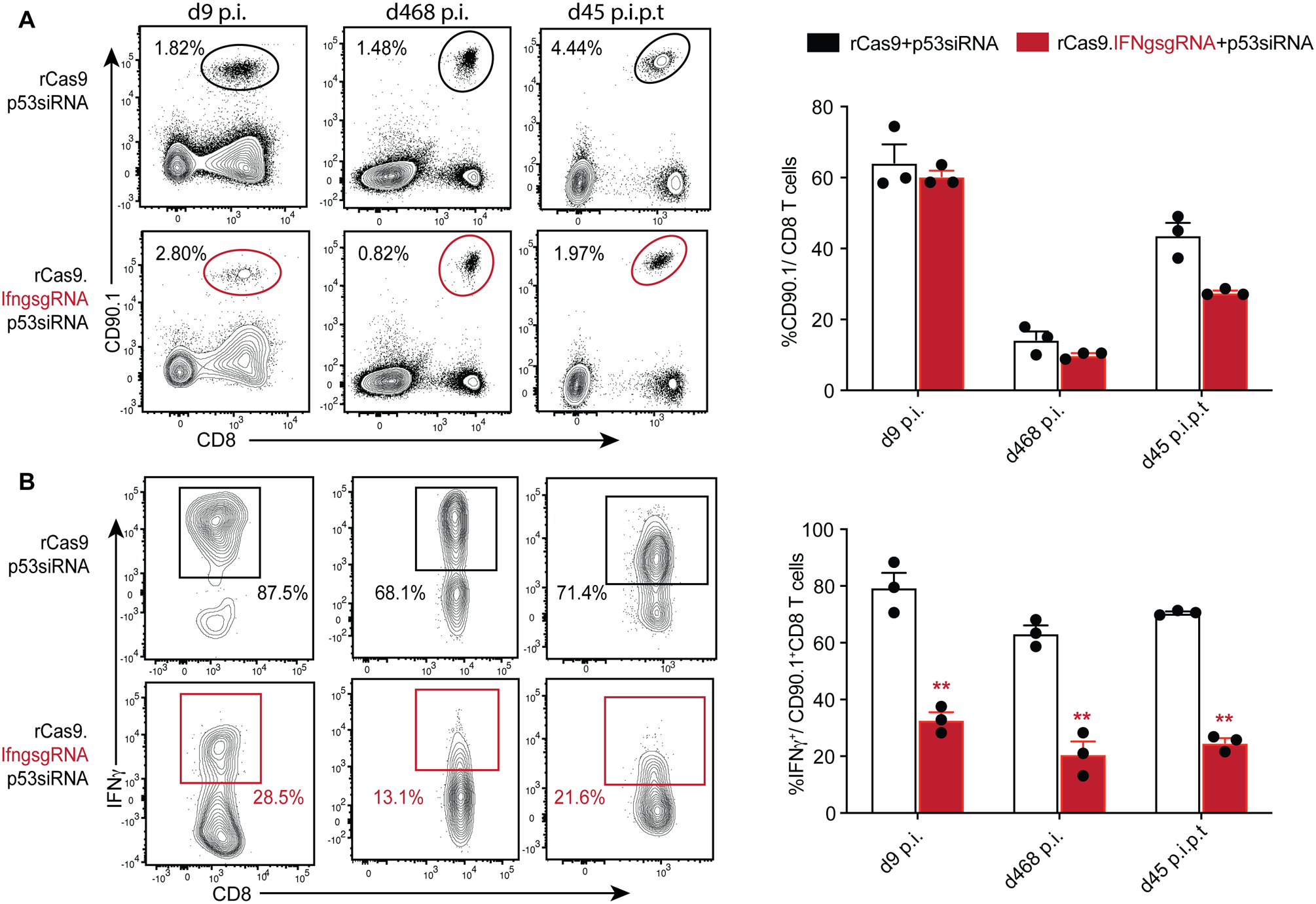
(A) Representative flow plots displaying the frequencies of OT-I TCR transgenic (CD90.1+) CD8 T cells transfected with indicated reagents, in recipient B6 mice challenged with Lm-ova, observed at the indicated effector and memory time-points, as well as after adoptive-transfer to secondary recipients, similar to in Fig 2A. Data summarized as bar graphs in the right panel. (B) Representative flow plots showing IFNγ expression at the indicated time points in response to PMA/ionomycin stimulation, in the corresponding gated populations depicted in Fig 3A. Data summarized as bar graphs in the right panel with each data point representing an individual sample, represents one of three independent experiments (n > 3 mice per group) and analyzed with t-tests comparing the IFNgsgRNA or control transfected groups at each time point ** = p < 0.01.
To determine if the CRISPR/Cas9 mediated loss of IFNγ in the OT-I CD8 T cells resulted in a perceivable functional deficiency, we determined the ability of the gene-edited OT-I CD8 T cells to protect from a challenge with virulent Lm-Ova in a model that is absolutely dependent on memory CD8 T cell-derived IFNγ (34). Memory OT-I CD8 T cells co-transfected with rCas9.IfngsgRNA or rCas9, and p53siRNA were adoptively transferred into IFNγ-deficient (IfngKO) recipient mice. These mice and control IfngKO mice were subsequently challenged with Lm-Ova infection (Fig 4A) and the resulting bacterial loads determined in the spleen and liver. Mice that received rCas9 + p53siRNA transfected OT-I (that maintained IFNγ expression) exhibited reduced bacterial numbers in both the spleen and livers. In sharp contrast, the mice that received OT-I CD8 T cells transfected with rCas9.IfngsgRNA and p53siRNA failed to control the Lm-Ova infection, yielding similar bacterial numbers as the infected naïve controls (Fig 4B). These data indicated that CRISPR/Cas9 mediated targeted gene disruption can be used to functionally alter memory CD8 T cells.
Figure. 4. CRISPR/Cas9 mediated loss of IFNγ in murine primary memory CD8 T cells limits the ability to control L. monocytogenes infection.
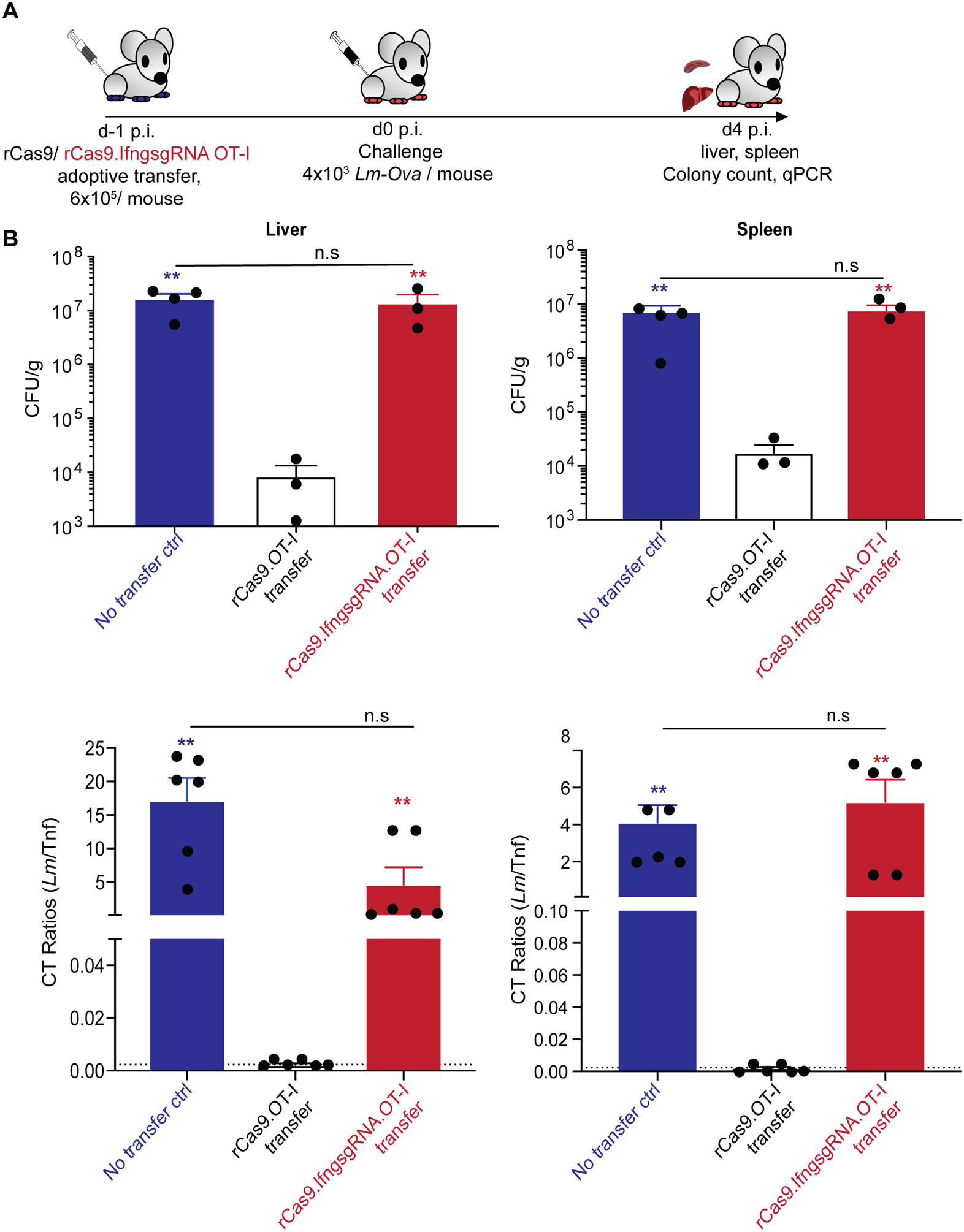
(A) Schematic depicting the experimental model for the challenge of IfngKO B6 recipient mice who received memory OT-I TCR transgenic (CD90.1+) CD8 T cells transfected with the indicated reagents. The IfngKO B6 recipient mice were challenged with virulent Lm-Ova the next day, sacrificed and organs harvested on 4 days later to determine control of bacterial infection. (B) Bacterial burden determined from the indicated groups by colony counts (top) and qPCR (bottom) for L. monocytogenes (hly gene) from the indicated organ homogenates. Dashed line indicates the limit of detection. Data representative of two independent experiments (n ≥ 3 mice per group) with each data point representing an individual sample, analyzed using one-way ANOVA comparing the indicated groups. ** = p < 0.01, n.s = p > 0.05
Discussion
In this study, we demonstrate that antigen experienced CD8 T cells fail to proliferate in response to cognate antigen encounter in vivo, after undergoing CRISPR/Cas9 mediated targeted gene disruption. We show that this defect is dependent on the pro-apoptotic transcription factor p53, which possibly drives the DNA damage response resulting from the DNA strand breaks introduced by the CRISPR/Cas9 system. We also show that disengaging the pro-apoptotic pathway either by ablating the p53 gene itself or temporarily subduing p53 function using p53siRNA may help create a window of opportunity for the CD8 T cells to undertake DNA damage repair without undergoing programmed cell-death. This approach makes CRISPR/Cas9 mediated targeted gene disruption possible in vivo, in memory CD8 T cells. Our approach also circumvents the necessity to expose the primary memory CD8 T cells obtained from a subject to non-specific activation or lengthy in vitro culture and cytokine induced non-specific expansion spanning multiple days, before returning back by adoptive transfer(10). The CD8 T cells obtained from mice were adoptively transferred to recipient mice after CD8 T cell enrichment and Cas9-sgRNA+p53siRNA transfection in under 20 minutes. Of note, naïve CD8 T cells were recently genetically modified with CRISPR/Cas9 in vivo, without the need for altering their p53 expression (12). The underlying differences in the responses of naïve and memory CD8 T cells to CRISPR/Cas9 gene editing remain to be elucidated, but our results suggest that subduing p53 function may be a requirement specific to memory CD8 T cells. The minimal in vitro manipulation coupled with the rapid re-transfusion time in our approach makes it a more biologically pertinent means to investigate CD8 T cell memory, and offers an opportunity to significantly improve upon the current CRISPR/Cas9 based immunotherapeutic approaches.
The ability to undertake single gene or gene family ablation in primary cells may be one of the most significant contributions of the CRISPR/cas9 system to basic biomedical sciences(1). Targeted gene disruption in T cells would help us investigate the genetic and transcriptional bases of T cell mediated immunity, establishment and maintenance of long-term memory, localization, tissue residence, recall responses etc., and help design better prophylactic and immuno-therapeutic approaches using it (35–37). Although whole gene knock-out mice and the Cre-lox system have been used to investigate the relevance of various genes in T cell memory (38–41), the ability to ablate genes after memory formation would help segregate their roles in the various aspects of memory formation, maintenance, recall etc. While inducible Cre-lox systems have partially filled this gap, the practical and technical limitations of the methodology (42, 43) have hindered a high-throughput investigation of the genetic basis of T cell memory. It is well established that T cell memory function is reflective of its transcriptional signature(44) and these transcriptional signatures vary between various iterations of memory CD8 T cells, depending on the frequency of antigen exposures(45). In addition to helping achieve single gene disruption in memory CD8 T cells, the CRISPR/Cas9 technology can be harnessed to alter transcriptional signatures of memory CD8 T cells by undertaking multi-gene knock-outs(10, 18, 46–48), at any stage of memory formation or maintenance. The genetically altered cells can be specifically identified by their targeted phenotypic changes or by single cell sequencing. Co-expression of fluorescent markers with CRISPR/Cas9 and target gene specific sgRNA in plasmid transfection-based strategies (eg. using pX458) can help identify the transfected cells using flow cytometry. Alternatively, disruption of a fluorescent protein or inert surface marker gene (eg. CD90.1) in CD8 T cells using specific sgRNAs encoded in plasmids with CRISPR/Cas9 and target gene specific sgRNA may also be used. Thus, enabling CRISPR/Cas9 gene editing of memory CD8 T cells by transient p53 silencing should open new avenues to explore and exploit the protective capacity of these cells.
CD8 T cells are also at the core of modern-day cancer therapy(49). CRIPSR/Cas9 technology has helped alter the CD8 T cell genome to ablate or express specific genes, to enhance their ability to proliferate, detect and kill tumor cells(10). However, these approaches are currently limited to utilizing CRISPR/Cas9 technology to undertake genetic alteration of antigen inexperienced (naïve) CD8 T cells. This approach also requires transgenic replacement of native TCRs of autologous T cells, with known tumor antigen specific TCRs, to promote tumor cell targeting after transfusion into a patient. While these CD8 T cells can be concurrently targeted with CRISPR/Cas9 mediated disruption of genes promoting T cell exhaustion (eg. Pdcd1), adequate expansion of these cells to generate transfusable numbers requires prolonged in vitro maintenance and non-specific, cytokine driven propagation (10). The ability to undertake targeted gene disruption in antigen experienced memory CD8 T cells will simplify this process greatly. Hypothetically, enriched antigen experienced, tumor specific memory CD8 T cells from a patient can be used for CRISPR/Cas9 mediated targeted gene disruption to restore their function, under the aegis of p53siRNA before transfusing it back into the patient. The memory CD8 T cells would undergo antigen re-encounter driven proliferation in vivo, and target the tumor cells. While this application needs to be tested rigorously in humans, our study in mice provides the proof of principle for the ability to collect antigen specific memory CD8 T cells, functionally alter them by targeted gene disruption using CRISPR/Cas9 and transfer it back into a recipient, with tangible functional consequences as intended.
In conclusion, we here demonstrate in vivo CRIPSR/Cas9 mediated targeted gene disruption in antigen-experienced, antigen-specific memory CD8 T cells. While DNA damage produced by targeted gene disruption precipitates apoptotic cell-death, preventing antigen-driven proliferation of these CD8 T cells in vivo, temporarily inhibiting p53-mediated apoptotic responses helps rescue these CD8 T cells. This facilitates reliable targeted gene disruption and functional transformation of memory CD8 T cells in vivo. We believe this is the first demonstration of CRISPR/Cas9 mediated gene editing in memory CD8 T cells.
Supplementary Material
Key points.
P53 precludes Cas9 mediated gene-disruption in memory CD8 T cells
Temporarily subduing p53 enables gene-editing in memory CD8 T cells in vivo.
Cas9 mediated gene ablations in memory CD8 T cells are retained through recall.
Acknowledgements
We would like to thank Dr. Duo Peng (Harvard University) for his insights and suggestions during the course of this study. We thank Drs. Vladimir Badovinac and Scott Anthony (University of Iowa), Kim Klonowski and Rick Tarleton (University of Georgia) for helpful suggestions on the manuscript, the University of Iowa vivarium staff and the University of Georgia Center for Tropical and Emerging Global Diseases Flow cytometry core facility for their help.
This work was supported by the United States National Institutes for Health grants AI42767, AI085515, AI114543, AI100527 to JTH and the University of Georgia Research Foundation startup grant to SPK.
Abbreviations used in this article:
- CRISPR
clustered regularly interspaced short palindromic repeats
- CD
Cluster of differentiation
- TCR
T-cell receptor
- sgRNA
Single guide ribonucleic acid
- Ctrl
control
- IFN
Interferon
- GFP
Green fluorescent protein
- LCMV
Lymphocytic choriomeningitis virus
References
- 1.Pickar-Oliver A, and Gersbach CA. 2019. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol 20: 490–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh DA, Borges da Silva H, Beura LK, Peng C, Hamilton SE, Masopust D, and Jameson SC. 2019. The Functional Requirement for CD69 in Establishment of Resident Memory CD8(+) T Cells Varies with Tissue Location. J Immunol 203: 946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seki A, and Rutz S. 2018. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med 215: 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hultquist JF, Hiatt J, Schumann K, McGregor MJ, Roth TL, Haas P, Doudna JA, Marson A, and Krogan NJ. 2019. CRISPR-Cas9 genome engineering of primary CD4(+) T cells for the interrogation of HIV-host factor interactions. Nat Protoc 14: 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, Haliburton GE, Ye CJ, Bluestone JA, Doudna JA, and Marson A. 2015. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A 112: 10437–10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salas-Mckee J, Kong W, Gladney WL, Jadlowsky JK, Plesa G, Davis MM, and Fraietta JA. 2019. CRISPR/Cas9-based genome editing in the era of CAR T cell immunotherapy. Hum Vaccin Immunother 15: 1126–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao Q, Dong X, Xu Q, Zhu L, Wang F, Hou Y, and Chao CC. 2019. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T-cell therapy. Cancer Med 8: 4254–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, Larson RC, Scarfo I, Bailey SR, Gerhard GM, Frigault MJ, Leick MB, Schmidts A, Sagert JG, Curry WT, Carter BS, and Maus MV. 2019. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer 7: 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee J, Bayarsaikhan D, Bayarsaikhan G, Kim JS, Schwarzbach E, and Lee B. 2020. Recent advances in genome editing of stem cells for drug discovery and therapeutic application. Pharmacol Ther: 107501. [DOI] [PubMed] [Google Scholar]
- 10.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, Tian L, Gonzalez VE, Xu J, Jung IY, Melenhorst JJ, Plesa G, Shea J, Matlawski T, Cervini A, Gaymon AL, Desjardins S, Lamontagne A, Salas-Mckee J, Fesnak A, Siegel DL, Levine BL, Jadlowsky JK, Young RM, Chew A, Hwang WT, Hexner EO, Carreno BM, Nobles CL, Bushman FD, Parker KR, Qi Y, Satpathy AT, Chang HY, Zhao Y, Lacey SF, and June CH. 2020. CRISPR-engineered T cells in patients with refractory cancer. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X, Kozhaya L, Tastan C, Placek L, Dogan M, Horne M, Abblett R, Karhan E, Vaeth M, Feske S, and Unutmaz D. 2018. Functional Interrogation of Primary Human T Cells via CRISPR Genetic Editing. J Immunol 201: 1586–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nussing S, House IG, Kearney CJ, Chen AXY, Vervoort SJ, Beavis PA, Oliaro J, Johnstone RW, Trapani JA, and Parish IA. 2020. Efficient CRISPR/Cas9 Gene Editing in Uncultured Naive Mouse T Cells for In Vivo Studies. J Immunol. [DOI] [PubMed] [Google Scholar]
- 13.Haring JS, Corbin GA, and Harty JT. 2005. Dynamic regulation of IFN-gamma signaling in antigen-specific CD8+ T cells responding to infection. J Immunol 174: 6791–6802. [DOI] [PubMed] [Google Scholar]
- 14.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, Waldschmidt TJ, Crompton PD, and Harty JT. 2012. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol 13: 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Badovinac VP, Porter BB, and Harty JT. 2004. CD8+ T cell contraction is controlled by early inflammation. Nat Immunol 5: 809–817. [DOI] [PubMed] [Google Scholar]
- 17.Barbau-Piednoir E, Botteldoorn N, Yde M, Mahillon J, and Roosens NH. 2013. Development and validation of qualitative SYBR(R)Green real-time PCR for detection and discrimination of Listeria spp. and Listeria monocytogenes. Appl Microbiol Biotechnol 97: 4021–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng D, Kurup SP, Yao PY, Minning TA, and Tarleton RL. 2014. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio 6: e02097–02014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grundemann C, Schwartzkopff S, Koschella M, Schweier O, Peters C, Voehringer D, and Pircher H. 2010. The NK receptor KLRG1 is dispensable for virus-induced NK and CD8+ T-cell differentiation and function in vivo. European journal of immunology 40: 1303–1314. [DOI] [PubMed] [Google Scholar]
- 20.Charlton JJ, Chatzidakis I, Tsoukatou D, Boumpas DT, Garinis GA, and Mamalaki C. 2013. Programmed death-1 shapes memory phenotype CD8 T cell subsets in a cell-intrinsic manner. J Immunol 190: 6104–6114. [DOI] [PubMed] [Google Scholar]
- 21.Norbury CJ, and Zhivotovsky B. 2004. DNA damage-induced apoptosis. Oncogene 23: 2797–2808. [DOI] [PubMed] [Google Scholar]
- 22.Zhang XP, Liu F, and Wang W. 2011. Two-phase dynamics of p53 in the DNA damage response. Proc Natl Acad Sci U S A 108: 8990–8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aubrey BJ, Kelly GL, Janic A, Herold MJ, and Strasser A. 2018. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 25: 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe M, Moon KD, Vacchio MS, Hathcock KS, and Hodes RJ. 2014. Downmodulation of tumor suppressor p53 by T cell receptor signaling is critical for antigen-specific CD4(+) T cell responses. Immunity 40: 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haks MC, Krimpenfort P, van den Brakel JH, and Kruisbeek AM. 1999. Pre-TCR signaling and inactivation of p53 induces crucial cell survival pathways in pre-T cells. Immunity 11: 91–101. [DOI] [PubMed] [Google Scholar]
- 26.Heylmann D, Rodel F, Kindler T, and Kaina B. 2014. Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim Biophys Acta 1846: 121–129. [DOI] [PubMed] [Google Scholar]
- 27.McNally JP, Millen SH, Chaturvedi V, Lakes N, Terrell CE, Elfers EE, Carroll KR, Hogan SP, Andreassen PR, Kanter J, Allen CE, Henry MM, Greenberg JN, Ladisch S, Hermiston ML, Joyce M, Hildeman DA, Katz JD, and Jordan MB. 2017. Manipulating DNA damage-response signaling for the treatment of immune-mediated diseases. Proc Natl Acad Sci U S A 114: E4782–E4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haapaniemi E, Botla S, Persson J, Schmierer B, and Taipale J. 2018. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 24: 927–930. [DOI] [PubMed] [Google Scholar]
- 29.Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, Kommineni S, Chen J, Sondey M, Ye C, Randhawa R, Kulkarni T, Yang Z, McAllister G, Russ C, Reece-Hoyes J, Forrester W, Hoffman GR, Dolmetsch R, and Kaykas A. 2018. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 24: 939–946. [DOI] [PubMed] [Google Scholar]
- 30.Ferrarelli LK 2018. CRISPR, cancer, and p53. Science Signaling 11: eaau7344. [Google Scholar]
- 31.Ozaki T, and Nakagawara A. 2011. Role of p53 in Cell Death and Human Cancers. Cancers (Basel) 3: 994–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haines BB, Ryu CJ, Chang S, Protopopov A, Luch A, Kang YH, Draganov DD, Fragoso MF, Paik SG, Hong HJ, DePinho RA, and Chen J. 2006. Block of T cell development in P53-deficient mice accelerates development of lymphomas with characteristic RAG-dependent cytogenetic alterations. Cancer Cell 9: 109–120. [DOI] [PubMed] [Google Scholar]
- 33.Schoenborn JR, and Wilson CB. 2007. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 96: 41–101. [DOI] [PubMed] [Google Scholar]
- 34.Harty JT, and Pamer EG. 1995. CD8 T lymphocytes specific for the secreted p60 antigen protect against Listeria monocytogenes infection. J Immunol 154: 4642–4650. [PubMed] [Google Scholar]
- 35.Jameson SC, and Masopust D. 2018. Understanding Subset Diversity in T Cell Memory. Immunity 48: 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mueller SN, Gebhardt T, Carbone FR, and Heath WR. 2013. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol 31: 137–161. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Zhou G, Zhang L, and Zhao Q. 2019. Building Potent Chimeric Antigen Receptor T Cells With CRISPR Genome Editing. Front Immunol 10: 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, and Kedl RM. 2013. T cell responses: naive to memory and everything in between. Adv Physiol Educ 37: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, Lietzenmayer M, Kroehling L, Takumi A, Kometani K, Inoue T, Kluger Y, Kaech SM, Kurosaki T, Okada T, and Flavell RA. 2018. KLRG1(+) Effector CD8(+) T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity 48: 716–729 e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahman AH, Zhang R, Blosser CD, Hou B, Defranco AL, Maltzman JS, Wherry EJ, and Turka LA. 2011. Antiviral memory CD8 T-cell differentiation, maintenance, and secondary expansion occur independently of MyD88. Blood 117: 3123–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang N, and He YW. 2005. The antiapoptotic protein Bcl-xL is dispensable for the development of effector and memory T lymphocytes. J Immunol 174: 6967–6973. [DOI] [PubMed] [Google Scholar]
- 42.Kurachi M, Ngiow SF, Kurachi J, Chen Z, and Wherry EJ. 2019. Hidden Caveat of Inducible Cre Recombinase. Immunity 51: 591–592. [DOI] [PubMed] [Google Scholar]
- 43.Song AJ, and Palmiter RD. 2018. Detecting and Avoiding Problems When Using the Cre-lox System. Trends Genet 34: 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, Harty JT, and Badovinac VP. 2010. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity 33: 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Braeckel-Budimir N, Varga SM, Badovinac VP, and Harty JT. 2018. Repeated Antigen Exposure Extends the Durability of Influenza-Specific Lung-Resident Memory CD8(+) T Cells and Heterosubtypic Immunity. Cell Rep 24: 3374–3382 e3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khan FJ, Yuen G, and Luo J. 2019. Multiplexed CRISPR/Cas9 gene knockout with simple crRNA:tracrRNA co-transfection. Cell Biosci 9: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu H, Sui T, Liu D, Liu T, Chen M, Deng J, Xu Y, and Li Z. 2018. Multiple homologous genes knockout (KO) by CRISPR/Cas9 system in rabbit. Gene 647: 261–267. [DOI] [PubMed] [Google Scholar]
- 48.Sekine R, Kawata T, and Muramoto T. 2018. CRISPR/Cas9 mediated targeting of multiple genes in Dictyostelium. Sci Rep 8: 8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farhood B, Najafi M, and Mortezaee K. 2019. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol 234: 8509–8521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.