

Abstract
Camptothecins are potent topoisomerase I inhibitors used to treat high-risk pediatric solid tumors, but they often show poor efficacy due to intrinsic or acquired chemoresistance. Here we developed a multivalent, polymer-based prodrug of a structurally optimized camptothecin (SN22) designed to overcome key chemoresistance mechanisms. The ability of SN22 vs. SN38 (the active form of irinotecan/CPT-11) to overcome efflux pump-driven drug resistance was tested. Tumor uptake and biodistribution of SN22 as a polymer-based prodrug (PEG-[SN22]4) compared to SN38 was determined. The therapeutic efficacy of PEG-[SN22]4 to CPT-11 was compared in: a) spontaneous neuroblastomas (NB) in transgenic TH-MYCN mice; b) orthotopic xenografts of a drug-resistant NB line SK-N-BE(2)C (mutated TP53); c) flank xenografts of a drug-resistant NB-PDX; and d) xenografts of Ewing sarcoma and rhabdomyosarcoma. Unlike SN38, SN22 inhibited NB cell growth regardless of ABCG2 expression levels. SN22 prodrug delivery resulted in sustained intratumoral drug concentrations, dramatically higher than those of SN38 at all time points. CPT-11/SN38 treatment had only marginal effects on tumors in transgenic mice, but PEG-[SN22]4 treatment caused complete tumor regression lasting over 6 months (tumor-free at necropsy). PEG-[SN22]4 also markedly extended survival of mice with drug-resistant, orthotopic NB and it caused long-term (6+ month) remissions in 80-100% of NB and sarcoma xenografts. SN22 administered as a multivalent polymeric prodrug resulted in increased and protracted tumor drug exposure compared to CPT-11, leading to long-term “cures” in NB models of intrinsic or acquired drug resistance, and models of high-risk sarcomas, warranting its further development for clinical trials.
Keywords: neuroblastoma, nanoparticle, camptothecin, irinotecan, SN38, SN22, drug resistance, ABCG2
Introduction
Despite intensive multimodality therapy, less than half of high-risk solid tumors like neuroblastoma (NB), Ewing sarcoma (EWS) and rhabdomyosarcoma (RMS) can be cured, and many of the survivors experience serious short- and long-term toxicities. Irinotecan (CPT-11) is a topoisomerase 1 inhibitor of the camptothecin family that stabilizes the topo-1/DNA complex, causing double-strand DNA breaks and cell death (1), and it is one of the most potent and commonly used drugs to treat these patients. However, CPT-11 is often ineffective against high-risk tumors due to intrinsic or acquired drug resistance. CPT-11 is a prodrug that requires enzymatic activation, primarily by hepatic carboxylesterases, and only a small fraction of its bioactive metabolite (SN38) reaches the tumor, limiting its efficiency as a cancer therapeutic (1). Furthermore, it causes significant adverse effects in many patients, including bone marrow suppression, and intractable diarrhea as serious, dose-limiting toxicities (2).
We have developed a multivalent, polymer-based prodrug of a pharmacologically enhanced camptothecin (SN22) (3), designed to achieve sustained intratumoral drug levels, overcome ABC transporter-mediated drug efflux, and avoid enzymatic inactivation by glucuronosyltransferases, which are key mechanisms driving intrinsic or acquired drug resistance (4–6). Unlike SN38 or topotecan, SN22 lacks a phenolic hydroxyl at position 10 of the A ring. This structural difference results in stronger and more sustained anti-tumor activity by protecting the molecule against both glucuronidation and cell efflux by ABC transporters - two processes that severely limit the performance of the clinically used camptothecins. The multivalent macromolecule evaluated here incorporates 4 residues of SN22 linked through a cleavable bond to a 4-arm polyethylene glycol (PEG) scaffold to form a hydrolytically activatable prodrug, PEG-[SN22]4. This single-component formulation of SN22 was designed to further improve therapeutic efficacy and safety by extending circulation time, increasing intratumoral accumulation, and reducing systemic exposure.
In this study, we tested the efficacy of PEG-[SN22]4 compared to free and/or liposomal CPT-11 to treat NB, EWS, and RMS in experimental mouse models. We used the TH-MYCN transgenic model of spontaneous NB, as these tumors exhibit intrinsic drug resistance due to the high expression of ABCG2, an ABC transporter associated with particularly aggressive disease (7). We also tested a chemo-resistant, patient-derived xenograft (PDX) of NB (with nonfunctional TP53), as well as a chemo-resistant orthotopic xenograft of the cell line SK-N-BE(2)C (with a TP53 mutation) (8). To demonstrate broad therapeutic applicability of this experimental treatment strategy integrating structural optimization with prodrug-based delivery, we examined the performance of PEG-[SN22]4 in two aggressive sarcoma models, using TC71, a chemo-resistant EWS cell line with nonfunctional p53 derived from a recurrent tumor; and RH30, an RMS cell line derived from a fusion-positive (alveolar) tumor. The results of our studies show that PEG-[SN22]4 is a cancer therapeutic that is highly effective in treating at least three different types of high-risk pediatric solid tumors with intrinsic or acquired drug resistance, and warrants further development for clinical trials.
Materials and Methods
Drug and prodrug formulations
CPT-11 (Camptosar; Pfizer) and liposomal CPT-11 (MM-398, Onivyde; Ipsen) (9) were obtained from the pharmacy at Children’s Hospital of Philadelphia–CHOP (Philadelphia, PA) and diluted in 0.9% normal saline before IV administration. The hydrolytically cleavable prodrug PEG-[SN22]4 with an alpha-alkoxy ester-based design (10–12) was assembled through direct coupling of carboxylated 4-arm-PEG (JenKem Technology, Mn = 20,553 Da) with SN22 using 1,3-dicyclohexylcarbodiimide as an activating agent for the carboxylic groups, 4-dimethylaminopyridine tosylate (DPTS) as a catalyst and dichloromethane (CH2Cl2) as a solvent. The polymeric conjugate contained 0.17 mmol/g of the drug. 1H NMR confirmed that SN22 was covalently bound to the carboxylated polymer by ester bonds between the carboxylic groups and 20-OH of SN22. A similar approach was used to form a control construct, PEG-[SN38]4. The phenolic 10-OH of SN38 (AstaTech , Bristol, PA) was first protected with tert-butyldiphenylsilyl (TBDPS) group by action of tert-butyl(chloro)diphenylsilane (TBDPS-Cl) in the presence of imidazole in N-methylpyrrolidone. The resulting 10-TBDPS-protected SN38 was then reacted as above with carboxylated 4-arm PEG. The conjugate was deprotected with pyridinium fluoride in methylene chloride, showing 0.17 mmol/g of SN38 covalently bound by the 20-OH to the polymer via ester bonds. PEG-[SN38]4 served as a technical control for PEG-[SN22]4, allowing to delineate the relative contributions of the macromolecule-based prodrug design, and the delivered therapeutic (SN22 vs. SN38).
Cell lines and in vitro proliferation inhibition
We used the NB cell lines NLF and SK-N-BE(2)C, and the patient-derived xenograft (PDX) COG-415x, for in vitro or in vivo studies focusing on NB. NLF is available from Kerafast (ECP008; Boston, MA); SK-N-BE(2)C was purchased from ATCC (ATCC CRL-2268; Manassas, VA); and COG-415x is a PDX available from the Children’s Oncology Group Childhood Cancer Repository (https://www.cccells.org/cellreqs-nbl.php). For cell growth inhibition studies, NLF cells transfected to stably express ABCG2 were plated in a 96-well plate and treated with various concentrations of SN38 or SN22. Confluency was measured over 3 days using the IncuCyte S3 Live-Cell Analysis System (Sartorius, #4647). SK-N-BE(2)C cells were transfected with luciferase for bioluminescent monitoring. We also used the Ewing’s sarcoma (EWS) cell line TC71, which was obtained from a recurrent tumor with nonfunctional p53, and the rhabdomyosarcoma (RMS) cell line Rh30, which is a fusion-positive “alveolar” RMS from an untreated patient. The EWS and RMS lines were obtained from the Childhood Cancer Repository of the Children’s Oncology Group (Lubbock, Tx). Integrity and authenticity of all cells were validated with mycoplasma testing and STR profiling on an annual basis at Cell Center Services at the University of Pennsylvania (Philadelphia, PA). Cells were maintained in a humidified atmosphere of 95% air and 5% CO2.
Mouse models of NB
Transgenic mouse model.
TH-MYCN mice were originally obtained as a gift from Dr. William Weiss (13). They have been maintained in our breeding colony and the genotype has been confirmed. Only mice with two copies of the TH-MYCN transgene were used in the studies, as they reliably develop spontaneous tumors in the paraspinal ganglia or adrenal glands at 4-6 weeks of age. A cell line was also established from a spontaneous tumor in a TH-MYCN transgenic mouse. Homozygous TH-MYCN mice received treatment as described below when they had clearly palpable tumors (day 0). Mice were treated according to the following groups: a) vehicle (saline) (n=8), b) free CPT-11 (n=7; 15 mg/kg), c) liposomal CPT-11 (n=6; 15 mg/kg), d) PEG-[SN38]4 (n=6; 10 mg/kg), or e) PEG-[SN22]4 (n=6; 10 mg/kg). Each treatment was given intravenously by tail vein 1x/week for 4 weeks. All doses are expressed as equivalent amounts of corresponding drugs (SN22, SN38 or CPT-11) in each formulation. The doses of free CPT-11 and PEG-[SN38]4 were chosen based on equivalency to that of SN22 in PEG-[SN22]4. We used the dose and schedule for liposomal CPT-11 as described previously (9). Tumor size was assessed by palpation, and body weight was recorded 2x/week. Mice were removed from study when signs of distress, discomfort, lethargy, or weight loss were apparent. The presence or absence of a tumor was confirmed by necropsy. Mice were maintained under humidity- and temperature-controlled conditions in a light/dark cycle that was set at 12-hr intervals. The Institutional Animal Care and Use Committee (IACUC) at Children’s Hospital of Philadelphia Research Institute approved all animal studies described in this report.
Orthotopic/adrenal xenograft model.
We used 6-week-old female athymic nude (CrTac:NCr-Foxn1nu) mice from Taconic Laboratory (Model #NCRNU-F). Luciferase-expressing SK-N-BE(2)C cells suspended in 20 μL of Basement Membrane Extract (Cultrex) were implanted at 106 cells per mouse into the suprarenal fat pad of anesthetized athymic nude mice. After reaching a tumor size of 1 cm3 (~ 14 days after inoculation), tumor-bearing mice were randomized into groups of 5 mice per treatment arm and administered treatments by tail vein 1x/week for 4 weeks as described above for the TH-MYCN mice. Body weights were obtained 2x/week. Therapeutic response and tumor progression were determined by measuring bioluminescent signal using D-luciferin potassium salt (PerkinElmer, Bridgeville, PA, USA) as a substrate.
Flank xenograft models.
We used 6-wk-old female athymic nude (Foxn1nu/Foxn1/nu) mice from Jackson Laboratory (Jax stock #007850). For the NB PDX study, we used COG-N-415x as a flank xenograft. PDX tissue was maintained by serial transplantation in nude mice. This line was derived from a relapse tumor that had both MYCN amplification and TP53 mutation, so it represented a very high-risk tumor. PDX studies were initiated by injecting approximately 2x107 cells of homogenized PDX tissue suspended in 100 μL Matrigel subcutaneously in the right flank of nude mice. For the sarcoma xenograft studies, a suspension of 107 TC-71 or Rh30 cells in 0.1 mL Matrigel (Corning) was implanted subcutaneously in the right flank of nude mice. The length (l) and width (w) of tumors were measured twice per week using a caliper. The volume (cm3) was calculated as (0.523 × l × w2)/1000, where l > w. Treatment started when the average tumor volume was 0.2 cm3. Mice with the COG-415x xenografts were treated with vehicle (saline), liposomal CPT-11, or PEG-[SN22]4 1x/week for 4 weeks. Mice with TC-71 or Rh30 xenografts (n=10/arm) received vehicle (saline), free CPT-11 (15 mg/kg), or PEG-[SN22]4 (10 mg/kg) 1x/week for 4 weeks. Body weights were obtained 2x/week, and treatment doses adjusted if there was a >10% change. Mice were removed from the study when tumor volumes reached 3.0 cm3.
Biodistribution and toxicity study of formulations
Homozygous TH-MYCN mice with easily palpable tumors (n=4/arm, each time point) were given a single dose by tail vein of either PEG-[SN22]4 (10 mg/kg), free CPT-11 (15 mg/kg) (14), liposomal CPT-11 (15 mg/kg) (9), or PEG-[SN38]4 (10 mg/kg) as controls. Blood was obtained by cardiac puncture and collected into 2-mL collection tubes containing sodium heparin (BD). Tissue (tumor, liver, spleen, intestine) was collected post-euthanasia after systemic perfusion with cold saline at 4, 24, and 72 hours. Samples were stored at −80°C until analyzed by the Bioanalytical Core Laboratory at CHOP Research Institute, as described previously (15–17). Total SN38 and SN22 levels were analyzed by an HPLC-MS assay, adapted from a prior publication (17). Complete blood count (CBC) was carried out using the HEMAVET®950FS (Drew Scientific) at 4, 24, and 72 hours (and 168 hours for PEG-[SN22]4). Liver enzymes ALT and AST were measured by the Translational Core laboratory of the CHOP Research Institute.
Western blot analysis
Tumor cell lysate (50-100 μg) was separated by SDS-PAGE through a 4-12% gradient gel (Invitrogen). Proteins were transferred to a nitrocellulose membrane (GE Healthcare Life Sciences), and the membrane was blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20. The membrane was incubated with an anti-ABCG2 primary antibody (Santa Cruz Biotechnology, sc-377176) overnight at 4°C, followed by HRP-linked anti-mouse IgG secondary antibody (GE Healthcare, NA931V).
Statistical analysis
Cell growth inhibition was compared using two-way ANOVA, and the Bonferroni-Holm step-down method was applied to adjust for the raw p values from linear mixed-effects model. Event-free survival (EFS) was estimated using the Kaplan-Meier method and compared using a log-rank (Mantel-Cox) test. Toxicities were summarized using descriptive statistics for each group.
Results
Strong SN22-mediated NB cell growth inhibitory effect independent of ABCG2 expression levels
To assess the effect of ABCG2 expression on sensitivity to SN38 or SN22, we identified an ABCG2-null NB cell line (NLF), transfected it with an ABCG2 expression vector, and then selected single-cell clones with low (NLF-R1) or intermediate (NLF-R2) levels of ABCG2 expression (Fig. 1A). Next, we assessed the sensitivity of parent NLF and ABCG2-expressing NLF clones to different concentrations of SN38 or SN22 by comparing their growth monitored continuously using IncuCyte® S3 Live-Cell Analysis System. NB cells derived from a spontaneous tumor in the TH-MYCN mouse (TH-MYCN-G1) were exposed in vitro to different concentrations of SN38 or SN22. The IC50 at 48 hr for SN38 (80 nM) was about 11 times higher than the IC50 at 48 hr for SN22 (7 nm). We showed that clones with higher expression levels of ABCG2 were increasingly resistant to SN38, whereas they remained fully sensitive to SN22 (Fig. 1B). This agrees with previous studies showing that endogenous ABCG2 expression, which characterizes aggressive NBs and NB stem cells (4, 18, 19), contributes to drug resistance to CPT-11/SN38 and other chemotherapeutics. It also demonstrates that, in analogy to the intrinsically resistant form of human disease, this mechanism also drives differential responsiveness to structurally distinct camptothecin drugs in the genetically engineered TH-MYCN mouse model.
Figure 1. ABCG2 expression and growth inhibition of NLF and transfected clones.
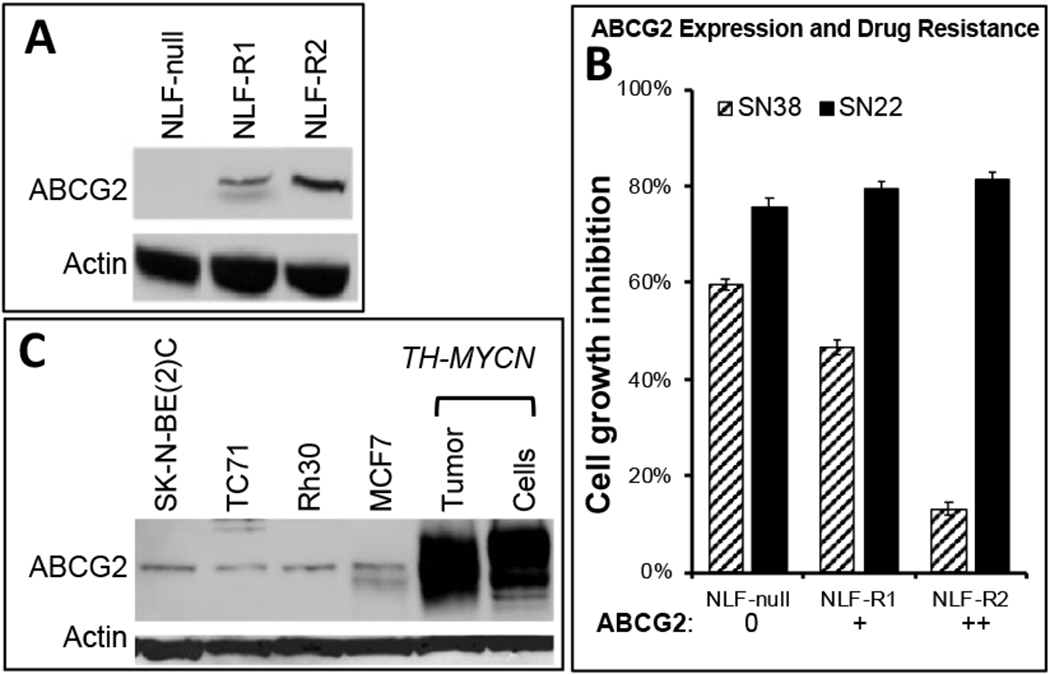
A. Western analysis was performed on NLF and two single-cell clones with an ABCG2 antibody (Santa Cruz), compared to an actin control. Clones R1 and R2 had low and intermediate levels of ABCG2 expression, respectively. B. Growth of NLF and ABCG2-expressing clones (panel A) in the presence of SN38 or SN22 (50 nM). Growth shown at day 4. C. Western analysis of other cell lines used in this study (SK-N-BE(2)C, TC71, Rh30). MCF7 was included as a positive control. Tissue from a TH-MYCN primary (1°) NB and a cell line derived from a TH-MYCN primary tumor (G1) showed very high endogenous ABCG2 expression.
Efficacy of camptothecin formulations in NB mouse models with intrinsic or acquired drug resistance
TH-MYCN transgenic mouse model.
PEG-[SN22]4 was comparatively tested against intrinsically resistant de novo NBs in a TH-MYCN transgenic mouse model (13). Consistent with the markedly stronger sensitivity of TH-MYCN-derived tumor cells to SN22 vs. SN38, NB tumors in TH-MYCN mice express high levels of ABCG2 (Fig. 1A, 1C). Mice were divided into 5 groups once tumors became palpable (4-5 weeks): vehicle (saline), free CPT-11, liposomal CPT-11, PEG-[SN38]4, or PEG-[SN22]4 at comparable doses, and treated 1x/week for 4 weeks. Tumors progressed rapidly in untreated mice, and tumor growth was only slightly delayed by CPT-11 treatment. Mice were sacrificed when they became symptomatic from tumor burden. Interestingly, some mice with growing tumors treated with liposomal CPT-11 developed a progressive neurological syndrome that included ataxia and rolling behavior, and they were removed from the study. However, no neurological symptoms were observed, and all tumors regressed with PEG-[SN22]4 treatment. They became non-palpable within 1-2 weeks of treatment (Fig. 2), and remained undetectable in all mice for over 180 days after the initiation of the treatment. Representative mice underwent necropsy, and the site of the tumor was examined grossly and microscopically, but no evidence of tumor was found in any animal. These results suggest that PEG-[SN22]4 is extremely effective against intrinsically resistant, ABCG2-overexpressing tumors arising in immunocompetent mice, as evidenced by long-term remissions and cures in all the animals treated with only 4 weekly doses of the prodrug.
Figure 2. Efficacy of PEG-[SN22]4 in a TH-MYCN transgenic mouse model.
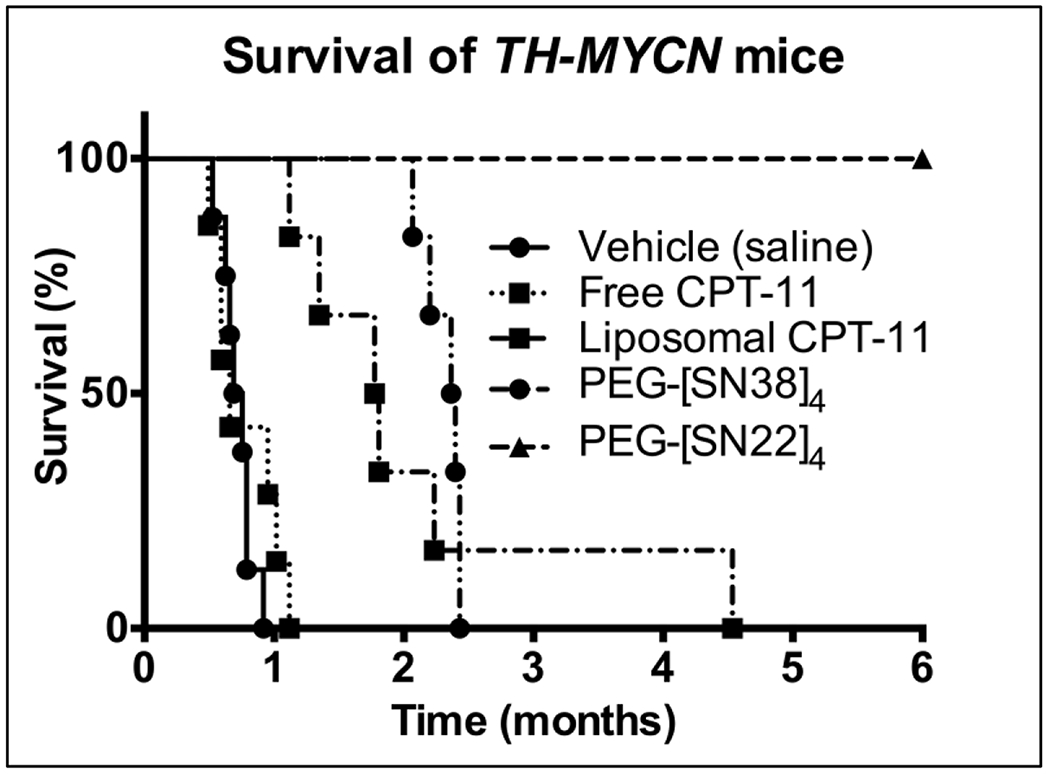
Homozygous TH-MYCN mice with palpable tumors were treated with vehicle (saline) (n=8), Free CPT-11 (15 mg/kg/dose; n=7), Liposomal CPT-11 (15 mg/kg/dose; n=6), PEG-[SN38]4 (10 mg/kg/dose; n=6) or PEG-[SN22]4 (10 mg/kg/dose; n=6) IV by tail vein 1x/week x 4 weeks. Treatments started when the mice were about 5 weeks old with a tumor volume of about 1-2 cm3. Mice were removed from the study when they showed signs of distress due to tumor burden. PEG-[SN22]4 had rapid tumor regression, and no tumor was found at necropsy after 6 months. Log-rank (Mantel-Cox) Test: SN22 vs. Vehicle: p=0.0003; SN22 vs. Free CPT-11: p=0.0004; SN22 vs. Liposomal CPT-11: p=0.0005; SN22 vs. PEG-[SN38]4: p=0.0008.
Pharmacokinetics and biodistribution.
We determined the levels of SN22 (for PEG-[SN22]4) or SN38 as well as CPT-11 where appropriate (for free or liposomal CPT-11, or PEG-[SN38]4) in blood, tumor, liver, spleen, and intestine at 4, 24 and 72 hr after giving a single dose of each agent as given in the above preclinical trials. Tissue levels were expressed as both drug concentration (in μg/g) and total drug per tissue or organ tested (in total μg) (Table 1, Supplemental Table 1). The blood concentration of SN22 at 4 hr was 227, 11 and 3 times greater than the level of SN38 from an equivalent dose of free CPT-11, liposomal CPT-11, and PEG-[SN38]4, respectively. Although the SN22 concentration in blood decreased over time (7-fold in the next 20 hr), SN22 administered as the prodrug was present in circulation at all time points at levels markedly exceeding those of SN38 delivered by all control compounds. In contrast, the drug delivered as free or liposomal CPT-11 was fully eliminated from blood by 24 and 72 hr, respectively. Consistent with its longevity in the circulation enabling prolonged accumulation in the tumor tissue, intratumoral level of SN22 at 24 h was 731, 6.8, and 2.5 greater than that of SN38 from equivalent doses of free CPT-11, liposomal CPT-11, and PEG-[SN38]4, respectively. Furthermore, superior dwell time in the circulation translated into superior drug concentration in the tumor at all time points, compared to all controls, including the liposomal CPT-11 formulation (9) and the prodrug construct analogously designed with SN38 (PEG-[SN38]4). With PEG-[SN22]4, slightly higher drug levels were seen in the liver, spleen, and intestine, but the tumor/liver ratio of all agents still strongly favored PEG-[SN22]4.
Table 1.
Concentration of SN38 or SN22 in Blood and Tumor Over Time.
| BLOOD | Drug concentration (μg/g) | ||
|---|---|---|---|
| 4 h | 24 h | 72 h | |
| Free CPT-11 (SN38) | 0.13 ± 0.07 | < 0.01 | < 0.01 |
| Liposomal CPT-11 (SN38) | 2.62 ± 0.35 | 1.06 ± 0.58 | < 0.01 |
| PEG-[SN38]4 (SN38) | 8.98 ± 0.47 | 1.05 ± 0.15 | 0.30 ± 0.05 |
| PEG-[SN22]4 (SN22) | 29.50 ± 1.01 | 4.34 ± 0.29 | 0.53 ± 0.11 |
| TUMOR | Drug concentration (μg/g) | ||
| 4 h | 24 h | 72 h | |
| Free CPT-11 (SN38) | 0.14 ± 0.03 | < 0.01 | < 0.01 |
| Liposomal CPT-11 (SN38) | 0.72 ± 0.17 | 1.07 ± 0.22 | 0.27 ± 0.05 |
| PEG-[SN38]4 (SN38) | 3.11 ± 0.33 | 2.99 ± 0.22 | 3.47 ± 0.46 |
| PEG-[SN22]4 (SN22) | 6.67 ± 0.86 | 7.31 ± 0.52 | 26.48 ± 7.40 |
Orthotopic xenograft NB model.
Next, we compared PEG-[SN22]4 to free CPT-11 and to PEG-[SN38]4 in an orthotopic xenograft mouse model of NB using the chemoresistant NB line SK-N-BE(2)C with mutated TP53. To allow for bioluminescent imaging, cells stably expressing luciferase were used in this study. Mice were treated 1x/week for 4 weeks with either PEG-[SN22]4 (10 mg/kg/dose), PEG-[SN38]4 (10mg/kg/dose) or free CPT-11 (15 mg/kg/dose). In this model of acquired chemoresistance, CPT-11 had no effect, whereas both PEG-[SN22]4 and PEG-[SN38]4 were initially effective at shrinking the tumor (Fig. 3). However, tumors rapidly regrew right after treatment cessation in the PEG-[SN38]4 treated mice, but PEG-[SN22]4 treatment resulted in complete disappearance of the tumor-associated signal and lasting inhibition of tumor growth for a number of weeks beyond the treatment period. The median survival with vehicle or CPT-11 treatment was 20 days, whereas the median survival with PEG-[SN22]4 was 72 days. This suggests that PEG-[SN22]4 had superior efficacy against recurrent, refractory disease with acquired drug resistance stemming from dysfunctional TP53.
Figure 3. Efficacy of PEG-[SN22]4 in an orthotopic mouse model.
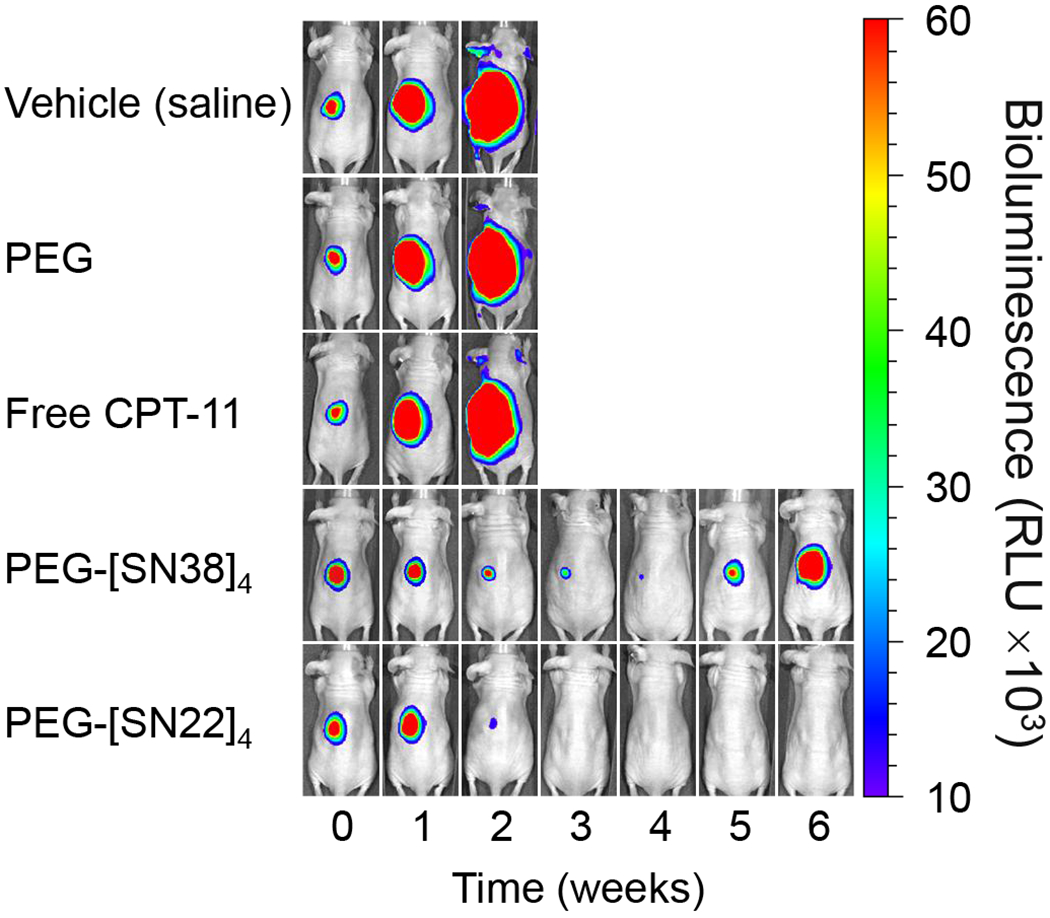
Chemoresistant, luciferase-transfected, SK-N-BE(2)C cells (107) were injected in the perirenal fat pad of nude mice. Mice were divided into five groups: vehicle (saline), or treatment with blank PEG, Free CPT-11, PEG-[SN38]4 or PEG-[SN22]4 (5 mice per group). Treatment started when tumors reached about 0.2 cm3. Treatment with either PEG-[SN38]4 or PEG-[SN22]4 produced tumor regression, but only PEG-[SN22]4 treatment caused complete tumor disappearance.
Flank xenograft PDX NB model.
For an NB PDX study, we used COG-N-415x as a flank xenograft. PDX tissue was maintained by serial transplantation in nude mice. PDX studies were initiated by injecting approximately 2x107 cells of homogenized PDX tissue suspended in 100 μL. Treatment started when the average tumor volume was 0.2 cm3. Mice received vehicle (saline; n=5), liposomal CPT-11 (15 mg/kg; n=5), or PEG-[SN22]4 (10 mg/kg; n=4) 1x/week for 4 weeks. Untreated tumors progressed rapidly. Tumors in mice treated with liposomal CPT-11 showed regression but began progressing within weeks after stopping treatment, and all had progressed and had to be sacrificed by 3 mo. However, all the tumors in mice treated with PEG-[SN22]4 regressed completely, and no recurrence was observed up to 6+ months (Fig. 4).
Figure 4. Treatment of patient derived xenograft (PDX), COG-N-415x, with PEG[SN22]4.
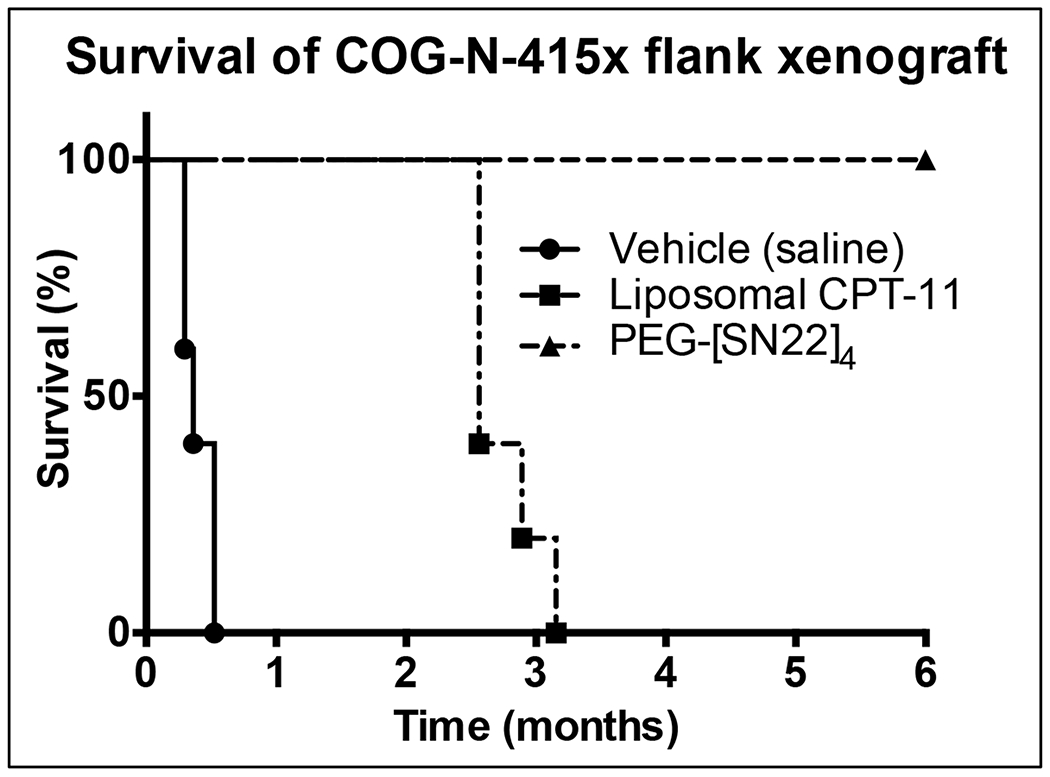
Nude mice with tumors were treated with vehicle (saline; n=5), Liposomal CPT-11 (15 mg/kg/dose; n=5), or PEG-[SN22]4 (10 mg/kg/dose; n=4), by tail vein 1x/week for 4 weeks. Mice treated with PEG-[SN22]4 were tumor-free for over 6 months. Log-rank (Mantel-Cox) test: PEG-[SN22]4 vs Liposomal CPT-11: p=0.0038
Efficacy of PEG-[SN22]4 in mouse models of aggressive sarcomas
To demonstrate the efficacy of PEG-[SN22]4 to treat other high-risk childhood solid tumors, we treated two representative high-risk sarcomas growing as orthotopic/flank xenografts in mice inoculated either with TC-71, a chemoresistant EWS with nonfunctional p53 (20), or an alveolar (fusion-positive) RMS line Rh30 with mutated TP53. After 180 days, all EWS mice treated with PEG-[SN22]4 had complete tumor regression with no palpable tumors (Fig. 5A). Complete regression was also achieved with the RMS xenografts treated with PEG-[SN22]4, but slow tumor regrowth occurred in two out of ten mice at around 150 days (Fig. 5B), whereas eight mice remained tumor-free beyond 180 days. In contrast, free CPT-11 had no significant effect on tumor progression in either aggressive sarcoma model.
Figure 5. Survival of EWS and RMS xenografts treated with PEG[SN22]4.
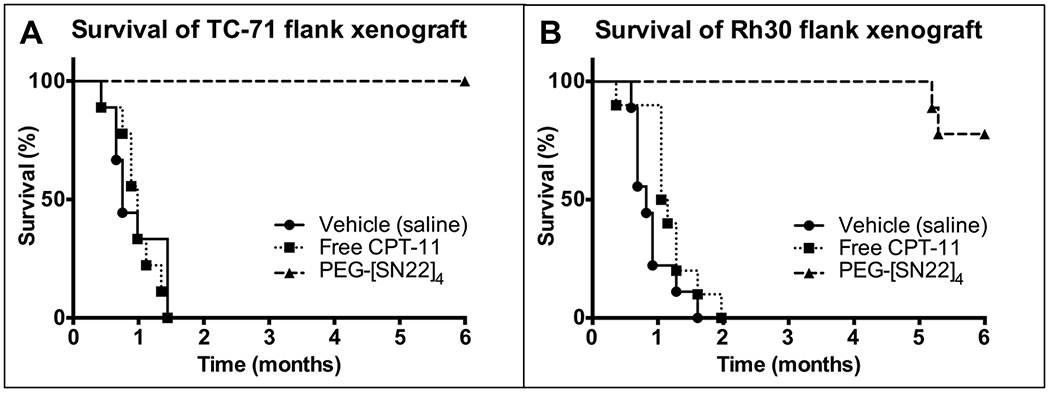
Mice were treated with vehicle (saline), Free CPT-11 (15 mg/kg/dose), or PEG-[SN22]4 (10 mg/kg/dose) (n=10 for all) IV by tail vein once a week x 4 weeks. 5A. Survival of animals with flank xenografts of the EWS cell line TC-71 (chemo-resistant) after 4 weeks of treatment. None of the PEG-[SN22]4 animals recurred. Log-rank (Mantel-Cox) Test: SN22 vs. vehicle: p<0.0001; SN22 vs. free CPT-11: p<0.0001. 5B. Survival of animals with flank xenografts of the alveolar (fusion-positive) RMS cell line Rh30 after 4 weeks of treatment. Two of the animals treated with PEG-[SN22]4 had a late recurrence, and one had a small, slow-growing tumor, but the 7 others were tumor free after 6 months. Log-rank (Mantel-Cox) Test: SN22 vs. vehicle: p<0.0001; SN22 vs. free CPT-11: p<0.0001.
Drug toxicity
In addition to pharmacokinetics and biodistribution, we monitored mouse weight and behavior, as well as complete blood counts (CBCs) and liver function tests (LFTs) after a single injection of free or liposomal CPT-11, as well as PEG-[SN38]4 and PEG-[SN22]4 in 129SvJ mice without tumors. In general, there were only modest if any changes in the CBCs or LFTs. Complete blood counts, including total red and white blood count (RBC, WBC) were analyzed, and there were no significant changes in RBCs or platelets. Also, WBCs were all within the range of 4-10 x103/μl at 4, 24 and 72 hr after a single dose of each agent, and after 168 hr for treatment with PEG-[SN22]4 (Supplemental Fig. 1). Thus, all counts either remained in the normal range or returned to normal within a week. We also monitored liver transaminases, and the ALT level was about 3 times normal at 4 hr after free CPT-11 and about 2 times normal at 72 hr for liposomal CPT-11, with minimal changes after other treatments at all times after a single dose, with similar findings for AST levels (Supplemental Fig. 2). Thus, there were no significant hepatic or hematological toxicities with PEG-[SN22]4 compared to other related treatments. The only significant untoward effect observed in these studies was the development of ataxia and a rolling behavior in four of the six TH-MYCN (129SvJ) mice treated with liposomal CPT-11. Remarkably, we did not observe this behavior in mice treated with other regimens, in the immunocompromised mice bearing human xenografts, or in immunocompetent wild type 129SvJ mice treated with liposomal CPT-11 but not bearing tumors.
Discussion
High-risk solid tumors like NB, EWS, and RMS are treated with intensive, multimodality therapy, but it is ineffective in over half the cases due to intrinsic or acquired drug resistance. With conventional delivery of chemotherapeutic agents by vein or by mouth, only a small fraction of these agents reach the tumor, which contributes significantly both to treatment failure and to systemic toxicity. As a result, survivors often experience serious short-and long-term side effects, including second malignancies. Therefore, more effective and less toxic therapy is needed for these high-risk patients.
The multivalent macromolecular prodrug evaluated in this study is long-circulating and designed to deliver four molecules of SN22, a highly potent topoisomerase I inhibitor. SN22 has several distinct advantages over SN38 conventionally administered as a precursor (CPT-11). First, CPT-11 is an inactive prodrug that requires conversion to SN38, primarily by hepatic carboxylesterases. As a result, only 3-4% of CPT-11 is converted to SN38, and only a small fraction of this reaches the tumor, severely limiting delivery and on-target activity. In comparison, the macromolecular prodrug of SN22 is designed to generate an active topoisomerase 1 inhibitor directly in the tumor via a nonenzymatic, hydrolytic activation mechanism not requiring carboxylesterases. Additional advantages of our delivery approach stem from the distinct chemistry of SN22. Unlike SN38, SN22 lacks a phenolic hydroxyl at position 10 of the A ring (21). This hydroxyl makes SN38 a substrate for ABC transporters, like ABCG2 (Fig. 1), and it is also the site for glucuronidation, which inactivates SN38 and targets it for removal. Because SN22 lacks this hydroxyl, it cannot be easily exported from cancer cells or inactivated by glucuronosyltransferases. As a result, SN22 levels persist in the tumor for much longer, accounting for its greater efficacy. Furthermore, SN22 delivery as a macromolecular prodrug prevents widespread systemic tissue and organ exposure, as well as rapid renal clearance of the active compound, which are key to its greater accumulation in the tumor. This phenomenon is reflected in the dramatically higher blood and tumor drug levels we observed in our mouse model (Table 1, supplemental Table 1).
Initially, we used the TH-MYCN mouse model of MYCN-driven aggressive disease, where spontaneous tumors develop in locations similar to NB in children. Although these tumors have not been exposed to drugs, they have intrinsic drug resistance due to high expression of ABCG2—a characteristic of aggressive NBs and NB stem cells (4, 18, 19), and this contributes to drug resistance to CPT-11/SN38 and other chemotherapeutics vulnerable to ABCG2-mediated efflux. Consistent with this mechanism driving primary drug resistance in high-risk tumors, therapeutics delivering SN38 had limited efficacy, whereas four weekly doses of PEG-[SN22]4 fully eradicated established tumors and led to complete remission for at least 6 months (Fig. 2), as confirmed by necropsy and pathological examination.
Next, we treated orthotopic xenografts established with SK-N-BE(2)C, a NB cell line, and PDX xenografts of COG-415x, both with MYCN amplification, derived post-therapy, and with TP53 inactivation. Mutant TP53 usually represent an acquired drug resistance mechanism in recurrent NBs (8, 22). Thus, these two models recapitulate the essential features of recurrent neuroblastoma characterized by acquired drug resistance driven by one of the most common mechanisms. Although free CPT-11 had no effect in these two models, PEG-[SN22]4 caused rapid tumor shrinkage and produced durable remissions (Figs. 3 and 4). This demonstrates that prodrug-based delivery of SN22 was highly effective in treating drug-resistant disease driven by loss of TP53 function, likely due to extended pharmacological activity and reduced clearance rate of SN22, which is not susceptible to glucuronidation. Remarkably, these preliminary results demonstrate that the superior efficacy of PEG-[SN22]4 extends to other chemoresistance mechanisms, including those present in recurrent tumors, and is not restricted to de novo drug-resistant tumors characterized by ABC transporter overexpression.
To evaluate the applicability of this delivery strategy to other aggressive pediatric solid tumors, we also treated two therapeutically challenging sarcomas established as orthotopic/flank xenografts. TC71 is a drug-resistant EWS line with mutated, nonfunctional p53, and although free CPT-11 had no effect, PEG-[SN22]4 again achieved complete remissions and “cures” in all 10 mice, as demonstrated by 6 months without recurrence and confirmed by necropsy (Fig. 5A). CPT-11 had a modest and transient effect against xenografts established with Rh30, a fusion-positive, alveolar RMS line, but PEG-[SN22]4 produced complete and durable remissions after four weekly doses, with only 2 of 10 late recurrences after 6 months (Fig. 5B). Thus, PEG-[SN22]4 provides a highly promising approach for treating both NBs exhibiting intrinsic and acquired resistance, as well as high-risk EWS and RMS based on the results of our preclinical model studies.
Although much of the efficacy of PEG-[SN22]4 can be attributed to the unique chemical properties of SN22, the long-circulating aspect of the multivalent macromolecule resulted in sustained dwell time and significantly more accumulation in the tumor than was achieved with CPT-11 (free or liposomal) or an analogously made SN38 prodrug construct. Dramatically higher blood and tumor drug concentrations of SN22 were consistently achieved with PEG-[SN22]4 at all examined time points (Table 1, Supplemental Table 1). The higher and more prolonged on-target drug levels translated into remarkably greater efficacy in preclinical therapeutic trials, clearly indicating the superiority of the delivery approach combining structural enhancement of the pharmacophore with macromolecule-based prodrug design. Interestingly, the concentration of SN22 increased over time from 4 hr to 72 hr (Table 1, Supplemental Table 1). This was presumably due to the high levels of SN22 delivered and accumulated in the tumor as the PEG-[SN22]4 prodrug, the cytotoxicity and tumor shrinkage from released drug, and the persistence of SN22 levels because SN22 is not a substrate for glucuronosyltransferases. Thus, the ongoing cytotoxic activity of SN22 causing a rapid reduction in tumor mass outpaces the drug clearancefrom the tumor tissue, and results in a seemingly paradoxical increase in tumor tissue concentration. This effect is not seen appreciably in normal tissues because the action of SN22, a topoisomerase inhibitor, is highly specific to cells in cycle, like tumor cells, but not to primarily quiescent liver or other normal cells.
Thus, there are three major advantages of PEG-[SN22]4 as a therapeutic: 1) this multivalent macromolecular prodrug is long-circulating and delivers 50-100 times as much drug to the tumor initially; 2) SN22 cannot be glucuronidated, inactivated and eliminated, so there is sustained and even increased drug concentration over time, compared to conventional agents that dissipate quickly; and 3) unlike SN38, SN22 is not susceptible to efflux by ABCG2 (and potentially other) transporters. Most mechanisms of drug resistance, including mutated TP53, can be overcome by dramatically increasing the drug concentration and duration of drug exposure in the tumor, and this is an important advantage of our formulation employing SN22, due in large part to its resistance to glucuronidation. The therapeutic itself (SN22) has the added advantage that, once within the cell, it cannot be effluxed by ABCG2, a marker of high-risk disease, including aggressive variants of NB and NB stem cells.
The neurological syndrome that developed in four of the six TH-MYCN (129SvJ) mice treated with liposomal CPT-11 was unanticipated and is noteworthy. This side effect of ataxia and rolling behavior has not been reported previously in mice (9), nor have neurological side effects with ataxia been reported in human clinical trials (23, 24). The mechanism responsible for this adverse effect is unclear, but we did not see this side effect in immunocompetent mice treated with free CPT-11, in immunosuppressed mice, or in athymic nude (Foxn1nu/Foxn1/nu) mice with NB, EWS or RMS treated with liposomal CPT-11, nor in 129SvJ mice without tumors treated with 4 weekly doses of liposomal CPT-11. One possibility is that these mice developed an opsoclonus/myoclonus/ataxia syndrome similar to that experienced by some patients with NBs (25, 26). This is thought to be an autoimmune condition with antibodies directed against NBs that cross-react with normal neural antigens. This adverse reaction and its pathophysiological mechanism may warrant further investigation.
The profound antitumor efficacy of PEG-[SN22]4 in models of NB and other solid tumors was not accompanied by any significant toxicity. CPT-11 treatment caused transient elevations of transaminases ALT and AST, and a modest effect on blood counts, whereas PEG-[SN22]4 did not. None of the mice experienced significant weight loss, skin changes, or diarrhea. The chemical design of PEG-[SN22]4 compared to that of CPT-11 probably played an important role in reducing the likelihood of side effects. The dipiperidino moiety attached to SN38 to form CPT-11 as a water-soluble and enzymatically activatable precursor of SN38 contributes to the intractable diarrhea due to increased cholinergic activity, which is a dose-limiting side effect of CPT-11 (27–30). Furthermore, the glucuronidated form of SN38 in the GI tract also contributes to the GI toxicity, whereas the likelihood of intractable diarrhea is markedly reduced with SN22, which is not susceptible to glucuronidation (31–33) and does not release a dipiperidino moiety upon activation. Additionally, systemic exposure to free drug is dramatically reduced when using a long-circulating prodrug that is retained in the intravascular compartment, and the greater potency and prolonged half-life of SN22 administered in the prodrug form may help reduce the overall amount of drug required to treat aggressive disease. Taken together, the superior efficacy and improved biocompatibility of PEG-[SN22]4 demonstrated in clinically relevant models of high-risk solid tumors make it a highly promising new therapeutic capable of addressing the considerable limitations of clinically used camptothecins. Phase 1 clinical trials are warranted to further evaluate its therapeutic potential against aggressive malignancies, either alone or as part of combination therapy with temozolomide and other solid tumor therapeutics.
Supplementary Material
Significance:
SN22 is an effective and curative multivalent macromolecular agent in multiple solid tumor mouse models, overcoming common mechanisms of drug resistance with the potential to elicit fewer toxicities than most cancer therapeutics.
Acknowledgments
These studies were made possible based on support from the following Foundations: Solving Kids Cancer Foundation–US, Alex’s Lemonade Stand Foundation, the CURE Childhood Cancer Foundation, Make Some Noise: Cure Kids Cancer Foundation, Arms Wide Open Childhood Cancer Foundation, Pierce Phillips Charity, and Fishin’ for the Cure (G. Brodeur, M. Chorny), and NIH/NCI R01-CA251883 (M. Chorny, G. Brodeur). This work was also supported by the Audrey E. Evans Endowed Chair (G. Brodeur). We are grateful to Dr. Joshua Schiffman, Chairman and CEO of PEEL Therapeutics (our commercial partner collaborating with our group on the development of PEG-[SN22]4) for helpful discussions and careful reading of the manuscript.
Abbreviations
- NB
neuroblastoma
- CPT-11
irinotecan
- PDX
patient-derived xenograft
Footnotes
Drugs: Camptosar (free irinotecan); Onivyde (liposomal irinotecan)
References
- 1.Mathijssen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, et al. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin Cancer Res. 2001;7(8):2182–94. [PubMed] [Google Scholar]
- 2.Blaney S, Berg SL, Pratt C, Weitman S, Sullivan J, Luchtman-Jones L, et al. A phase I study of irinotecan in pediatric patients: a pediatric oncology group study. Clin Cancer Res. 2001;7(1):32–7. [PubMed] [Google Scholar]
- 3.Nagata H, Kaneda N, Furuta T, Sawada S, Yokokura T, Miyasaka T, et al. Action of 7-ethylcamptothecin on tumor cells and its disposition in mice. Cancer Treat Rep. 1987;71(4):341–8. [PubMed] [Google Scholar]
- 4.Bates SE, Medina-Perez WY, Kohlhagen G, Antony S, Nadjem T, Robey RW, et al. ABCG2 mediates differential resistance to SN-38 (7-ethyl-10-hydroxycamptothecin) and homocamptothecins. J Pharmacol Exp Ther. 2004;310(2):836–42. [DOI] [PubMed] [Google Scholar]
- 5.Henderson MJ, Haber M, Porro A, Munoz MA, Iraci N, Xue C, et al. ABCC multidrug transporters in childhood neuroblastoma: clinical and biological effects independent of cytotoxic drug efflux. J Natl Cancer Inst. 2011;103(16):1236–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi T, Fujiwara Y, Yamakido M, Katoh O, Watanabe H, Mackenzie PI. The role of glucuronidation in 7-ethyl-10-hydroxycamptothecin resistance in vitro. Jpn J Cancer Res. 1997;88(12):1211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aravindan N, Jain D, Somasundaram DB, Herman TS, Aravindan S. Cancer stem cells in neuroblastoma therapy resistance. Cancer Drug Resist. 2019;2:948–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keshelava N, Zuo JJ, Chen P, Waidyaratne SN, Luna MC, Gomer CJ, et al. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001;61(16):6185–93. [PubMed] [Google Scholar]
- 9.Kang MH, Wang J, Makena MR, Lee JS, Paz N, Hall CP, et al. Activity of MM-398, nanoliposomal irinotecan (nal-IRI), in Ewing’s family tumor xenografts is associated with high exposure of tumor to drug and high SLFN11 expression. Clin Cancer Res. 2015;21(5):1139–50. [DOI] [PubMed] [Google Scholar]
- 10.Conover CD, Greenwald RB, Pendri A, Shum KL. Camptothecin delivery systems: the utility of amino acid spacers for the conjugation of camptothecin with polyethylene glycol to create prodrugs. Anticancer Drug Des. 1999;14(6):499–506. [PubMed] [Google Scholar]
- 11.Greenwald RB, Choe YH, McGuire J, Conover CD. Effective drug delivery by PEGylated drug conjugates. Adv Drug Deliv Rev. 2003;55(2):217–50. [DOI] [PubMed] [Google Scholar]
- 12.Greenwald RB, Pendri A, Conover CD, Lee C, Choe YH, Gilbert C, et al. Camptothecin-20-PEG ester transport forms: the effect of spacer groups on antitumor activity. Bioorg Med Chem. 1998;6(5):551–62. [DOI] [PubMed] [Google Scholar]
- 13.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16(11):2985–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson J, Zamboni WC, Cheshire PJ, Lutz L, Luo X, Li Y, et al. Efficacy of systemic administration of irinotecan against neuroblastoma xenografts. Clin Cancer Res. 1997;3(3):423–31. [PubMed] [Google Scholar]
- 15.Goldwirt L, Lemaitre F, Zahr N, Farinotti R, Fernandez C. A new UPLC-MS/MS method for the determination of irinotecan and 7-ethyl-10-hydroxycamptothecin (SN-38) in mice: application to plasma and brain pharmacokinetics. J Pharm Biomed Anal. 2012;66:325–33. [DOI] [PubMed] [Google Scholar]
- 16.Iyer R, Croucher JL, Chorny M, Mangino JL, Alferiev IS, Levy RJ, et al. Nanoparticle delivery of an SN38 conjugate is more effective than irinotecan in a mouse model of neuroblastoma. Cancer Lett. 2015;360(2):205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen F, Alferiev IS, Guan P, Guerrero DT, Kolla V, Moorthy GS, et al. Enhanced Intratumoral Delivery of SN38 as a Tocopherol Oxyacetate Prodrug Using Nanoparticles in a Neuroblastoma Xenograft Model. Clin Cancer Res. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandian V, Ramraj S, Khan FH, Azim T, Aravindan N. Metastatic neuroblastoma cancer stem cells exhibit flexible plasticity and adaptive stemness signaling. Stem Cell Res Ther. 2015;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acosta S, Lavarino C, Paris R, Garcia I, de Torres C, Rodriguez E, et al. Comprehensive characterization of neuroblastoma cell line subtypes reveals bilineage potential similar to neural crest stem cells. BMC Dev Biol. 2009;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.May WA, Grigoryan RS, Keshelava N, Cabral DJ, Christensen LL, Jenabi J, et al. Characterization and Drug Resistance Patterns of Ewing’s Sarcoma Family Tumor Cell Lines. PLoS ONE. 2013;8(12):e80060, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunimoto T, Nitta K, Tanaka T, Uehara N, Baba H, Takeuchi M, et al. Antitumor activity of a new camptothecin derivative, SN-22, against various murine tumors. J Pharmacobiodyn. 1987;10(3):148–51. [DOI] [PubMed] [Google Scholar]
- 22.Keshelava N, Zuo JJ, Waidyaratne NS, Triche TJ, Reynolds CP. p53 mutations and loss of p53 function confer multidrug resistance in neuroblastoma. Med Pediatr Oncol. 2000;35(6):563–8. [DOI] [PubMed] [Google Scholar]
- 23.Carnevale J, Ko AH. MM-398 (nanoliposomal irinotecan): emergence of a novel therapy for the treatment of advanced pancreatic cancer. Future Oncol. 2016;12(4):453–64. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H. Onivyde for the therapy of multiple solid tumors. Onco Targets Ther. 2016;9:3001–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antunes NL, Khakoo Y, Matthay KK, Seeger RC, Stram DO, Gerstner E, et al. Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J Pediatr Hematol Oncol. 2000;22(4):315–20. [DOI] [PubMed] [Google Scholar]
- 26.Rudnick E, Khakoo Y, Antunes NL, Seeger RC, Brodeur GM, Shimada H, et al. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies-a report from the Children’s Cancer Group Study. Med Pediatr Oncol. 2001;36(6):612–22. [DOI] [PubMed] [Google Scholar]
- 27.Dodds HM, Rivory LP. The mechanism for the inhibition of acetylcholinesterases by irinotecan (CPT-11). Mol Pharmacol. 1999;56(6):1346–53. [DOI] [PubMed] [Google Scholar]
- 28.Harel M, Hyatt JL, Brumshtein B, Morton CL, Yoon KJ, Wadkins RM, et al. The crystal structure of the complex of the anticancer prodrug 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothecin (CPT-11) with Torpedo californica acetylcholinesterase provides a molecular explanation for its cholinergic action. Mol Pharmacol. 2005;67(6):1874–81. [DOI] [PubMed] [Google Scholar]
- 29.Hyatt JL, Tsurkan L, Morton CL, Yoon KJ, Harel M, Brumshtein B, et al. Inhibition of acetylcholinesterase by the anticancer prodrug CPT-11. Chem Biol Interact. 2005;157-158:247–52. [DOI] [PubMed] [Google Scholar]
- 30.Zhou M, Liu M, He X, Yu H, Wu D, Yao Y, et al. Synthesis and biological evaluation of novel 10-substituted-7-ethyl-10-hydroxycamptothecin (SN-38) prodrugs. Molecules. 2014;19(12):19718–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta E, Lestingi TM, Mick R, Ramirez J, Vokes EE, Ratain MJ. Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhea. Cancer Res. 1994;54(14):3723–5. [PubMed] [Google Scholar]
- 32.Richardson G, Dobish R. Chemotherapy induced diarrhea. J Oncol Pharm Pract. 2007;13(4):181–98. [DOI] [PubMed] [Google Scholar]
- 33.Stein A, Voigt W, Jordan K. Chemotherapy-induced diarrhea: pathophysiology, frequency and guideline-based management. Ther Adv Med Oncol. 2010;2(1):51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.