

Abstract
Enhancer of Zester Homolog 2 (EZH2), a histone lysine methyltransferase and the catalytic component of Polycomb Repressive Complex 2, has been extensively investigated as a chromatin regulator and a transcriptional suppressor by methylating H3 at lysine 27 (H3K27). EZH2 is upregulated or mutated in most cancers, and its expression levels are negatively associated with clinical outcomes. However, the current developed small molecule inhibitors targeting EZH2 enzymatic activities could not inhibit the growth and progression of solid tumors. Here, we discovered an anti-histamine drug, ebastine, as a novel EZH2 inhibitor by targeting EZH2 transcription and subsequently downregulating EZH2 protein level and H3K27 tri-methylation in multiple cancer cell lines at concentrations below 10μM. The inhibition of EZH2 by ebastine further impaired the progression, migration, and invasiveness of these cancer cells. Overexpression of EZH2 wild-type and its mutant, H689A (lacking methyltransferase activity), rescued the neoplastic properties of these cancer cells after ebastine treatment, suggesting that EZH2 targeted by ebastine is independent of its enzymatic function. Next-generation RNA-sequencing analysis also revealed that C4-2 cells treated with 8μM ebastine showed a gene profiling pattern similar to EZH2-knockdown C4-2 cells, which was distinctively different from cells treated with GSK126, an EZH2 enzyme inhibitor. Additionally, ebastine treatment effectively reduced tumor growth and progression, and enhanced progression-free survival in triple-negative breast cancer (TNBC) and drug-resistant castration-resistant prostate cancer (CRPC) patient-derived xenograft (PDX) mice. Our data demonstrated that ebastine is a novel, safe, and potent anticancer agent for patients with advanced cancer by targeting the oncoprotein EZH2.
Keywords: EZH2, Ebastine, Prostate cancer, Breast cancer, Drug Re-purposing
Introduction
The Polycomb group (PcG) proteins form repressive complexes (PRCs) with diverse and conserved proteins that perform their functions as crucial epigenetic modifiers and transcriptional regulators in many cellular processes, including cell differentiation, cell cycle regulation, DNA damage repair, stem cell self-renewal, and disease development and progression (1). EZH2 forms the core of PRC2 along with EED and SUZ12, and methylates histone H3 at lysine 27 (H3K27) to suppress the downstream targets (1). Many studies have reported that EZH2 overexpression enhances the proliferative properties of cancer cells while knocking down or inhibiting EZH2 could induce apoptosis and autophagy in cancers (2). Importantly, recent evidence reveals that H3K27 plays a critical role in epigenetic regulation and cancer initiation/progression. A point mutant of lysine 27 (H3K27M) could induce pediatric high-grade gliomas (pHGGs) (3). Nevertheless, EZH2 inhibition can still effectively reduce the growth of tumors harboring the H3K27M mutant (4), suggesting that EZH2 may perform its oncogenic functions beyond its methyltransferase activities. EZH2 crosstalks with diverse epigenetic machinery in the course of chromatin compaction and gene expression regulation (5).
We and many other groups have reported that EZH2 is a biomarker of aggressive prostate and breast cancer (6,7). The expression levels of EZH2 are profoundly involved in the progression of prostate cancer, especially in the lethal castration-resistant status (6). While EZH2 expression is very low or undetectable in benign cells, EZH2 mRNA and protein levels are elevated in advanced cancers. EZH2 overexpression in prostate and breast cancer represents a high possibility of metastasis, high Gleason Score, and adverse clinical prognosis (6,8). Additionally, many studies demonstrated a strong correlation between EZH2 expression levels and the progression of many other cancer types, including but not limited to lung cancer (9,10), bladder cancer (11), endometrial cancer (12), melanoma (13), as well as non-solid neoplasms from hematopoietic and lymphoid origins (14,15).
In search of new treatment strategies for advanced cancers, EZH2 presents to be a promising therapeutic target. Tremendous efforts have been made to develop small molecule inhibitors against EZH2. 3-deazaneplanocin A (DZNep) is the first identified EZH2 inhibitor that targets S-adenosyl-L-homocysteine hydrolase (SAH), a cofactor known to be required by EZH2-dependent methylation (16). DZNep decreases EZH2 protein level, but it is not an ideal EZH2 inhibitor due to its non-specific inhibition to histone methylation and excessive toxicity in animal models (17-19). Other EZH2 inhibitors such as GSK126, EPZ5687, EPZ6438, and EI1, specifically target the lysine methyltransferase activity of EZH2 but do not alter EZH2 expression levels (15,20-22). Disappointingly, these EZH2 enzyme inhibitors alone only suppress the growth of lymphomas harboring EZH2 gain-of-function mutations, but not that of the solid tumors in which EZH2 are upregulated. Hereby, we report that ebastine (23), a second-generation antagonist of the histamine H1 receptor, which has been extensively evaluated for its safety and toxicity and approved for anti-allergy therapy in many European countries, could be repurposed for cancer therapy by targeting EZH2 in cancer cells.
Materials and methods
Cell lines
MDA-MB-231, PC3, DU145, MCF7, H82, H146, L1236, H526, VCaP, Jeko-1, SUM159, HDLM2, and HEK293T cells were purchased from ATCC in 2016. C4-2 was a gift from Dr. Leland W. Chang. All cell lines were tested and authenticated at the University of Arizona Genetic Core (Tucson, AZ) in 2017 and 2019. PC3, and MDA-MB-231 were cultured in DMEM (GenDEPOT) supplemented with 10% FBS (GenDEPOT). DU145, C4-2, MCF7, H82, H146, L1236, H526, Jeko-1 and HDLM2 were cultured in RPMI 1640 (GenDEPOT) supplemented with 10% FBS (GenDEPOT). SUM159 was cultured in Ham’s F-12 (Gibco) with 10% FBS. All cells were frozen at passage 2-3 and used within 20 passages after each thawing. The cells were cultured in a 37°C incubator and a humidified atmosphere with 5% CO2. All cell lines were authenticated by the University of Arizona Genetics Core using short tandem repeat (STR) profiling. Cell lines were mycoplasma negative as reported by routine lab tests.
Reagents and antibodies
GSK126 (406228, MedKoo), ebastine (ab141928, Abcam), and carebastine (23076, Cayman Chemical) were dissolved in 100% ethanol or DMSO for cell treatment. Lipofectamine 3000 (Thermo Fisher Scientific) was used to perform the transfection of EZH2 shRNA (Sigma). The following antibodies were used for western blot: EZH2 (5246, Cell Signaling), GAPDH (sc-32233, Santa Cruz), H3K27me3 (9733, Cell Signaling), H3 (9715, Cell signaling), β-Actin (A2228, Sigma), and LC3-A/B (12741, Cell Signaling).
Western blotting
For protein extraction, lysates were added to 1X reducing SDS-sample buffer prepared by lysis buffer and 4X reducing SDS-sample buffer (BP-110R, Boston BioProducts) and heated to 95 °C for 10 min. Protein levels were assessed by standard SDS–polyacrylamide gel electrophoresis and transferred to PVDF membranes (162-0177, BIO-RAD). Images were captured using the ChemiDoc XRS+ Molecular Imager system (BIO-RAD). Primary antibodies used in western blotting are listed above. Blots were incubated overnight with primary antibodies at 4 °C, followed by detection with Clean-Blot IP Detection Reagent (HRP) (21230, Thermo Fisher Scientific), goat anti-mouse IgG (H+L)-HRP (SA001-500, GenDEPOT), or goat anti-rabbit IgG (H+L)-HRP (SA002-500, GenDEPOT) secondary antibody.
Lentiviral constructs
Lentivirus was packaged by co-transfection of constructs with third-generation packaging plasmids pMD2.G, pRRE, and pRSV/REV with Fugene HD (Roche) into HEK293T cells. The transfection mixture was replaced with growth medium 24 h after transfection (2 μg of MDLG, 1 μg of VSVG, 1 μg of Rev, and 4 μg of target plasmid). The supernatant was collected at 72 h and 96 h after transfection and centrifuged to remove HEK293T cells. Lentiviral titers were determined by p24 assay, in addition to functional titration to determine an MOI of 1 for each initial batch of virus.
Reporter Luciferase Assays
The EZH2 reporter, the void vector control, and SEAP plasmid construct were purchased from GeneCopoeia (Rockville, MD). Upon cultivation of C4-2 cells in 96-well plates for 24 h, the EZH2 reporter or the void vector control was co-transfected together with the SEAP plasmid construct, which was used as an indicator of transfection internal control, into the cells. After 24 h of incubation, the transfection system was removed and the cells were treated with 4 μM and 8 μM of ebastine. Cells were then lysed after 24 h, 48 h, and 72 h of incubation, and the bioluminescence test was conducted using the Secrete-Pair Dual Luminescence Assay Kit (LF061, GeneCopoeia). The bioluminescence was read on Synergy 2 Multi-Mode Reader (BioTek). EZH2 promoter luciferase activity was normalized with the void vector and SEAP luciferase activity. Each experiment was performed in quadruplicate.
RNA-sequencing analysis
The vector control, GSK126, and EZH2KD#1 and EZH2KD#2 RNA-sequencing data were obtained from previous publication (24). Ebastine treated samples were prepared together with control, GSK126 treated and EZH2 KD samples at the same batch and sequenced at same batch. The RNA-seq reads were mapped to the human reference genome version hg19 using TopHat (version 2.0.12) default parameters (25). The human reference gene set (RefSeq gene) was downloaded from https://www.ncbi.nlm.nih.gov/refseq/rsg/. Cuffdiff (v2.0.12) was used to calculate gene expression levels and the significance of differential expression based on the classic-FPKM using default parameters (26). Differentially expressed gene threshold was set for P<0.05. For clustering analysis, we used a hierarchical clustering method with Spearman correlation distance to cluster samples based on the log scaled FPKM, and used MORPHEUS (https://software.broadinstitute.org/morpheus/) to plot the heat map. The significance of overlapping genes between the two groups was calculated using Fisher’s exact test. Gene set enrichment analysis (GSEA) was applied to assess the significance of associations between AR target genes and genes affected by ebastine treatment or EZH2 knockdown (27).
Cell growth assay
Cells were seeded in 96-well plates and treated with different concentrations of ebastine for 72 h. Bioluminescence was measured to quantify cell viability using CellTiter-Glo® Luminescent Cell Viability Assay Kit (Promega) and was read on Synergy 2 Multi-Mode Reader (BioTek). The cell proliferation curve was drawn and fit by the bioluminescence to drug concentration. Half-maximum inhibitory concentration (IC50) was calculated with non-linear fitting.
Wound-healing assay
Cell migrating capabilities were detected using wound-healing assay. C4-2 cells were plated with 80-90% confluence in 6-well plates. Wounds were created across the monolayer of cell culture using a Bioclean pipette tip. The cells were incubated in serum-free medium supplemented with DMSO, 4 μM or 8 μM of ebastine after rinsed with PBS. Wound closures were captured at 0, 24, and 72 h.
Boyden chamber invasion assay
Polycarbonate membrane cell culture inserts (CLS3422, Corning) were applied with Basement Membrane Matrix (Trevigen). After the matrix condensed at 37°C in cell incubator, each 24-well insert was seeded with 1×105 of C4-2 cells in RPMI-1640 without FBS. RPMI-1640 with FBS was added to each well containing the insert. Ebastine or vehicle (ethanol or DMSO) was added to keep the same concentration inside and outside of the inserts. Each insert was fixed with methanol, and cells that permeated through the membrane were stained with 0.5% crystal violet. Images were captured, and the cell count was calculated using ImageJ (1.8.0).
EZH2 Rescue Assay
Ezh2 wild-type and Ezh2 mutant H689A cloned into MigR1 (GFP) retroviral vector were provided by Dr. Yi Zhang (Temple University). Retrovirus was packaged by co-transfection of constructs with second-generation packaging plasmids pUMVC and pCMV-VSVG with Lipofectamine (Fisher) into HEK293T cells. The transfection mixture was replaced with growth medium 24 h after transfection (1 μg of pCMV-VSVG, 3 μg of pUMVC, and 4 μg of target plasmid). The supernatant was collected at 72 h and 96 h after transfection and centrifuged to remove the cells. For retroviral infection, retrovirus supernatant was added to C4-2 cells in the presence of 10 μg/ml polybrene (Sigma). Cells were incubated at 37°C for 6-7 h, then change to fresh growth medium. The same retroviral infection procedure was repeated on the next day. The proliferative, invasive and migrating properties of these cells in response to ebastine treatment were evaluated as described in previous sections.
Autophagy and Apoptosis assays
C4-2 cells treated with ebastine at dose gradients for 72 h and lysed for western blotting to detect LC3-A/B. Densitometry measurements of bands were quantitated and calculated in ImageJ (1.8.0). Autophagosome activity was detected with specific dye using an autophagy assay kit (MAK138, Sigma). The pictures were captured under fluorescence microscopy, and bioluminescence was read on Synergy 2 Multi-Mode Reader (BioTek).
Apoptosis was detected using FITC annexin V apoptosis detection kit (556547, BD Biosciences) in C4-2 cells treated with ebastine for 72 h. The staining was analyzed by flow cytometry (LX200 Luminex Multiplexing Assay system).
Murine prostate and breast tumor xenograft models
CB17SCID mice were purchased from Charles River. Animal care and conditions were based on the institutional and National Institutes of Health protocols and guidelines, and all studies were approved by Houston Methodist Institution Animal Care and Use Committee. Tumor xenograft model was induced as previously described (28).
For VCaP castration-resistant prostate tumor model, male mice were anesthetized using 2% isoflurane (inhalation), and 2 × 106 of VCaP prostate cancer cells suspended in 100 μl of PBS with 50% Basement Membrane Matrix (Trevigen) were implanted subcutaneously into the dorsal flank on the right side of each mouse. Tumor volumes were measured by length (a), width (b), and calculated as tumor volume = MIN (a)2 × MAX(b)/2. For VCaP castration-resistant prostate tumor model, VCaP tumor-bearing mice were castrated when tumors were approximately 200-300mm3 in size (approximately 5 weeks after implantation of tumor cells) and once tumors started to relapse, mice were randomized and treated with vehicle (n=13) or ebastine (10 mg·kg−1 per day (n=10), 5 days per week) orally. All animals were euthanized after 28 days of treatment. The body weight of the mice was monitored during the study.
For SUM159 triple-negative breast tumor model, female mice were anesthetized using 2% isoflurane (inhalation), and 2 × 106 of SUM159 prostate cancer cells suspended in 100 μl of PBS with 50% Basement Membrane Matrix (Trevigen) were implanted subcutaneously into the mammary fat pad (MFP). Tumor volumes were measured by length (a), width (b), and calculated as tumor volume = MIN(a)2 × MAX(b)/2. Mice were randomized and treated with vehicle (n=11) or ebastine (30 mg·kg−1 per day (n=8), 5 days per week) via oral gavage, and terminated about 28 days later. Body weight of mice was monitored throughout the entire study.
Murine prostate and breast PDX models
Mice with LuCaP 35CR castration-resistant prostate cancer patient-derived xenograft (PDX) (gift from Dr. Eva Corey) or BCM3887 triple-negative breast cancer PDX (gift from Dr. Jenny Chang) were euthanized when the tumor size over 1000mm3. All cells used in this model were tested for murine-associated pathogens at IDEXX BioResearch (Columbia, MO). The tumors were excised, minced in wash medium (DMEM with 5% FBS, 50ug/ml Gentamicin). One piece of minced PDX tumor (about 8mm3) were implanted subcutaneously into the dorsal flank on the right side of the 5-week-old pre-castrated male mice (LuCaP 35CR model), or 5-week-old female mice (BCM3887). For the BCM3887 group, when the tumors were approximately 100mm3 in size, mice were randomized and treated with vehicle (n=10) or ebastine (10 mg·kg−1 per day (n=5) or 30 mg·kg−1 per day (n=5), 5 days per week). For the LuCaP 35CR group, when the tumors were approximately 100mm3 in size, mice were randomized and treated with enzalutamide (10 mg·kg−1 per day (n=12), 5 days per week) only or ebastine + enzalutamide (30 mg·kg−1 and 10 mg·kg−1 per day, respectively (n=13), 5 days per week). Tumor diameters and body weight were recorded twice per week. Mice were euthanized about 28 days later.
Quantification of ebastine and its metabolite, carebastine in mouse tumor
Ebastine and carebastine in mouse tumors were extracted and measured based on a previously established method with slight modifications (23). An internal standard, methaqualone (10 μM in acetonitrile), and external standard, terfenadine (2.5 μg/mL in acetonitrile), were obtained from Sigma (M-015, St. Louis, MO) and Tocris Bioscience (3948/50, Minneapolis, MN), respectively. Briefly, each tumor sample was weighed and homogenized in five times its volume of phosphate buffer solution (0.5M, pH 7.4) on ice using a sonicator ultrasonic homogenizer (EpiShear™ Probe Sonicator, Active Motif®, Carlsbed, CA). Acetonitrile with methaqualone was added to the homogenate, vortexed vigorous for 1 min, and centrifuged at 15,700 ×g for 10 min to precipitate proteins from the homogenate. The supernatant was collected for quantification.
Standards and samples were both spiked with an equal amount of internal standards and analyzed by High-Performance Liquid Chromatography and Triple-quadruple Mass Spectrometry and Tandem Mass Spectrometry (HPLC-MS/MS). Specifically, the system consisted of a Thermo TSQ in line with an electrospray source and a Vanquish (Thermo) UHPLC consisting of a binary pump, degasser, and auto-sampler outfitted with a Kinetex EVO C18 column (Phenomenex, dimensions of 2.1 mm × 50 mm and a 5 μm particle size). Elution is at isocratic mode with a mobile phase containing 5 mM ammonium acetate in water and acetonitrile (50%:50%, v/v) at 0.4 mL/min. In positive mode, the capillary of ESI source was set to 350 °C, with sheath gas at 50 arbitrary units, auxiliary gas at 10 arbitrary units and the spray voltage at 3.5 kV. A selective reaction monitoring (SRM) of the protonated precursor ion and the related product ions for ebastine, carebastine, terfenadine and methaqualone (m/z 470.2 167.1, 500.2 191.1, 472.3 436.1 and 251.4 132, respectively) was monitored. Calibration graphs were derived from the peak area ratio of targets to internal standard with linear regression. Data acquisition and analysis were carried out by Xcalibur 4.1 software and Tracefinder 4.1 software, respectively (both from Thermo Fisher Scientific).
Results
Ebastine decreases EZH2 and H3K27 methylation levels in cancer.
Recently, several anti-histamine drugs have been reported to effectively inhibit tumor growth (29). Among these drugs, astemizole has been shown to disrupt EZH2-EED interaction and induce EZH2 degradation (30). Unfortunately, the use of astemizole, particularly overdose, may cause ventricular arrhythmias (31). Other anti-histamine drugs were screened to determine the potential to decrease EZH2 protein levels in cancer cells. Besides astemizole, ebastine markedly reduced EZH2 and H3K27 tri-methylation levels at concentrations below 20μM as compared to other anti-histamine drugs, including chlorpheniramine, fexofenadine, haloperidol, mizolastine, or terbinafine, in MDA-MB-231 (Fig. 1A) and MCF7 (Fig. S1A) breast cancer cells. To further investigate whether ebastine can reduce EZH2 in other cancer cells, we treated prostate cancer cells (DU145, VCaP), small cell lung cancer (H146, H82, H526), and lymphoma (L1236, Jeko-1, HDLM2) with ebastine at various concentrations. As shown in Fig. 1B and Fig. S1B, ebastine dose-dependently reduces the protein level of EZH2 and H3K27me3 in these cancer cell lines.
Figure 1. Ebastine decreases EZH2 protein and H3K27 methylation in cancer.
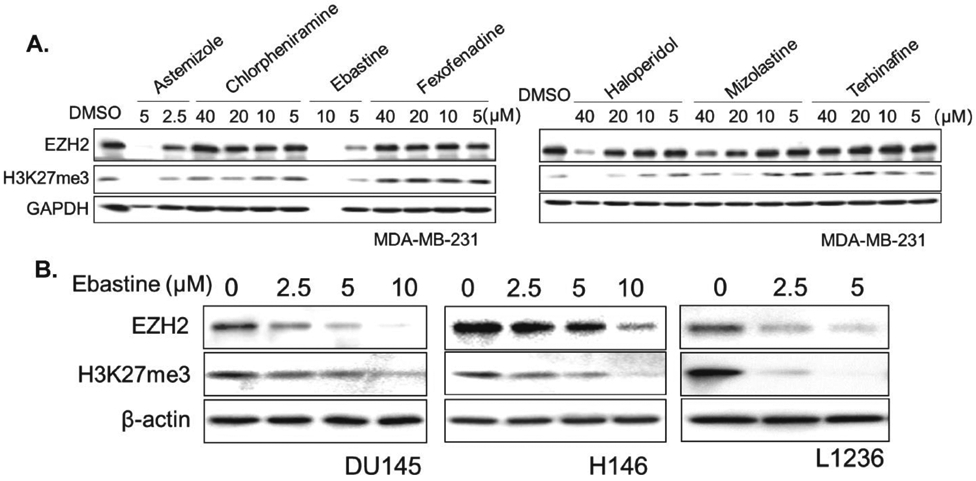
(A) MDA-MB-231 cells were treated with astemizole, chlorpheniramine, ebastine, fexofenadine, haloperidol at different doses. After 72 h, total cell lysates were blotted for EZH2 and H3K27me3, with GAPDH served as a loading control. (B) DU145, H146, L126 cells were treated with ebastine at different doses for 72 h and lysed for immunoblot analysis. β-actin served as a loading control.
Ebastine decreased EZH2 transcript levels by regulating its promoter activities.
The decrease of EZH2 protein may be a result of either increase EZH2 degradation or inhibit transcription or translation. Real-time quantitative PCR (qPCR) analysis demonstrates that ebastine markedly reduced the mRNA level of EZH2 dose-dependently along with the reduction of EZH2 protein level in PC3 (Fig. 2A, C), C4-2 (Fig. S2A, B) and SUM159 (Fig. 2B, D). Interestingly, the protein level of EED, but not SUZ12, decreases along with EZH2 after treated with ebastine (Fig. S2C), which is consistent with our previous finding (24). We then hypothesized that ebastine may repress EZH2 transcription by inhibiting its promoter activities. To test this hypothesis, we co-transfected a secreted Gaussia luciferase reporter containing EZH2 upstream promoter, along with a secreted embryonic alkaline phosphatase (SEAP) reporter as an internal control for transfection efficiency into C4-2 cells. We observed that the promoter activities of EZH2 significantly declined with ebastine treatment in a dose-dependent manner (Fig. 2E). Moreover, we conducted a next-generation RNA-sequencing in C4-2 cells treated with ebastine (8μM), GSK126 (8μM, EZH2 enzymatic inhibitor), and EZH2 shRNA (EZH2-knockdown). Intriguingly, we observed a similar gene expression pattern (Fig. S3A) and a considerable overlapping of upregulated or downregulated genes in ebastine-treated cells and EZH2-knockdown cells, whereas the GSK126-treated group showed a distinct gene profiling pattern as compared to EZH2 knockdown (Fig. 2F-G). Gene Set Enrichment Analysis (GSEA) further confirmed that ebastine-treatment-increased genes were significantly enriched (Q value <0.001) in the signature of EZH2-knockdown upregulated genes (Fig. 2G), and ebastine-treatment-decreased genes were significantly enriched (Q value <0.001) in the signature of EZH2-knockdown down-regulated genes (Fig. 2H). Among all of the cancer-related pathways, cell cycle is significantly down-regulated after ebastine treatment (Fig. S3B-C). Our results demonstrate that ebastine treatment decreases EZH2 transcript in cancer cells, and exhibits similar effect as EZH2-knockdown in C4-2 cells, suggesting that ebastine may be a better inhibitor for EZH2 as compared to GSK126.
Figure 2. Ebastine decreases EZH2 transcript levels by regulating EZH2 promoter activities.

(A) PC3 cells and (B) SUM159 treated with ebastine for 72 h were lysed and blotted for EZH2 and H3K27me3, with H3 and GAPDH served as a loading control. RNA was extracted from PC3 cells (C) and SUM159 (D) treated with ebastine for 72 h and quantified using RT-qPCR. The relative expression of EZH2 was normalized by GAPDH. * P<0.05 vs vehicle, mean ± SEM. (E) C4-2 cells were co-transfected with a secreted Gaussia luciferase reporter containing EZH2 upstream promoter and a secreted embryonic alkaline phosphatase (SEAP) reporter. The SEAP group acted as an internal control. Ebastine treatment was performed for 24, 48 or 72 h. (F) Heat map for the expression level of genes down- or upregulated by EZH2 knockdown, GSK126 (8 μM) or ebastine (8 μM) treatment. (G) The number of overlapped differential genes between EZH2-KD and ebastine-treated group is significantly larger than the number of genes overlapped by GSK126-treated vs EZH2-KD or Ebastine-treated. (H-I) GSEA analysis of ebastine targeted genes that are significantly enriched by genes upregulated after EZH2 knockdown (Q value <0.001; H) or down-regulated after EZH2 knockdown (Q value <0.001; I).
Ebastine impaired malignant neoplasm and induced cell cycle arrest and autophagy
The mRNA and protein levels of EZH2 are associated with the neoplastic properties in many cancers (6,8-14). Consistently, we found that ebastine treatment significantly inhibits the growth of various cancer cell lines with IC50 below 12μM (Fig. 3A and Fig. S4A-G). Additionally, ebastine treatment dose-dependently abolished the ability to migrate and invade in C4-2 cells, as demonstrated by wound-healing assay (Fig. 3B) and Boyden chamber invasion assay (Fig. 3C), respectively. Similar results were observed in MDA-MB-231 breast cancer cells (Fig. S4H). To confirm the anti-neoplastic activity of ebastine is specifically targeting EZH2, we overexpressed EZH2 wild-type and its H689A mutant (lacking methyltransferase activity) driven by a MSCV promoter in C4-2 cells and NIH3T3 cells. We found that wild-type EZH2 restored EZH2 and H3K27 protein levels, while H689A mutant only restored EZH2 protein levels, but not H3K27me levels in C4-2 cells (Fig 3D-E) and NIH3T3 cells (Fig. S5A). Interestingly, both EZH2 wild-type and H689A mutant could significantly rescue back ebastine treatment-reduced neoplastic properties of C4-2 prostate cancer cells (Fig. 3F-H) as well as NIH3T3 cells (Fig. S5B), suggesting the inhibition of cancerous properties by ebastine is through targeting EZH2 regardless of its enzymatic function.
Figure 3. Ebastine has potent therapeutic effects on prostate cancer.

(A) Growth curve of C4-2 cells treated with ebastine at different concentrations to determine the half-maximal inhibitory concentration (IC50). (B) Migration images of C4-2 cells treated with ebastine and vehicle at 0, 24, and 72 h. The percentage of closure was calculated by dividing the width of 0 h. * P<0.05 vs vehicle, mean ± SEM. (C) Invasion assay using transwell insert and matrigel to assess the invasiveness of C4-2 cells after ebastine treatment. Cell count was analyzed in the lower panel. **P<0.01 vs Vehicle, mean ± SEM. (D-H) EZH2 rescue assay in C4-2 cell for protein level (D), mRNA level (E), growth (F), invasion (G) and migration (H). (I) C4-2 treated with ebastine for 48 h were lysed and blotted with anti-LC3-A/B antibody, and presented as the ratio of LC3-A/B-II/I to GAPDH. (J) Cell cycle distribution of C4-2 cells treated with ebastine and vehicle.
EZH2 is involved in many physiological and pathological cellular processes as an epigenetic regulator (32). The previous study reported that knocking down EZH2 may induce cell cycle arrest and autophagy (32,33). Consistently, we found that ebastine treatment-induced autophagy dose-dependently as indicated by the elevated ratio of LC3-A/B-II to LC3-A/B-I using western blotting (Fig. 3I) and increased staining of proprietary fluorescent autophagosome (Fig. S4I) in C4-2 cells. Furthermore, ebastine induced G2/M cell cycle arrest in a dose-dependent manner (Fig. 3J). Our data clearly demonstrate that ebastine inhibits the oncogenic phenotypes of cancer cells in vitro by targeting EZH2 and induces cell cycle arrest and autophagy.
To compare the effects of ebastine and EZH2/PRC2 enzymatic-inhibitors in inhibiting cancer cell growth, migration and invasion, we treated Du145 and C4-2 cells with 4 μM of Ebastine, GSK126, EPZ6438 and EED226 in parallel for 72 h. Immunoblot analysis revealed that all drugs decreased H3K27me3 levels, but only Ebastine decreased EZH2 protein levels (Fig. S6A, B). While the GSK126, EPZ6438 and EED226 inhibited Du145 cell growth with IC50 of 20.18 μM, 77.58 μM, and 24.62 μM, respectively, ebastine showed a better inhibition in cell growth with IC50 of 11.15 μM (Fig. S6C). A similar effect was observed in C4-2 cells (Fig. S6D). In addition, the Boyden chamber invasion assay and transwell migration assays revealed that Ebastine significantly and markedly reduced the migration and invasiveness properties of Du145 and C4-2 (Fig. S6E, F), while GSK126, EPZ6438 and EED226 only marginally reduced these properties of cancer cells.
Ebastine inhibits tumor progression in vivo
We then evaluated the therapeutic potential of ebastine against triple-negative breast cancer (TNBC) in vivo using murine xenograft models with TNBC patient-derived xenograft (PDX) BCM3887 and TNBC cell line SUM159 xenograft. Once tumor size reached to 100mm3, mice were randomized to 3 groups (vehicle, ebastine 10mg/kg/day, ebastine 30mg/kg/day). As shown in Fig. 4A, ebastine treatment significantly and dose-dependently reduced tumor growth and progression as indicated by tumor weight. Kaplan-Meier survival plot indicated that a higher dose of ebastine enhanced the survival of TNBC-PDX mice (Fig. 4B). Similarly, ebastine treatment reduced tumor growth and progression in SUM159 xenograft mice, as indicated by tumor volume (Fig. 4C) and tumor weight (Fig. 4D). More importantly, immunoblot analysis using tumor lysate from the ebastine-treated and control mice showed that EZH2 protein levels were decreased in vivo upon ebastine treatment (Fig. 4E). No significant change in body weight of these mice was observed among different treatments (Fig. S7A-B).
Figure 4. Therapeutic efficacy of ebastine in triple-negative breast cancer murine models.

BCM3887 PDX mouse model was generated as described in the “Methods” section. Mice carrying BCM3887 PDX received vehicle or ebastine (10mg/kg, 30mg/kg per day) 5 days per week orally. (A) Caliper measurements were taken every 4 days to determine tumor volume. * P<0.05 , vs vehicle, mean tumor volume ± SEM. (B) Kaplan-Meier survival plot comparing progression-free survival of BCM3887 PDX model. Significance was tested using unpaired t-test and presented as * P<0.05. (C) Caliper measurements were taken every 4 days to determine the tumor volume of SUM159 xenograft mice. (D) Tumor weight was measured after sacrifice. (E) Tumor samples (n=7) were ground and lysed for western blot analysis. Proteins were blotted and quantitated to compare the level of EZH2 between the ebastine-treated group and the vehicle-treated group. * P<0.05, vs vehicle, Mean ± SEM.
Androgen-deprivation therapy (ADT) is the first line of treatment against early-stage prostate cancer. However, most patients who received ADT will inevitably develop castration resistance, which poses a huge challenge to clinical treatment. In the context of prostate cancer, EZH2 oncogenic activity is correlated with tumor progression and castration-resistance. To evaluate the therapeutic effects of ebastine in prostate cancer, we first utilized the CRPC VCaP xenograft mouse model. As expected, ebastine treatment significantly and markedly reduced the VCaP CRPC xenograft tumor growth (Fig. 5A). We also observed better survival of mice with ebastine treatment as compared to those with vehicle control, according to Kaplan-Meier analysis (Fig. 5B). We then evaluated the therapeutic efficacy of ebastine in LuCaP 35CR, an aggressive CRPC, enzalutamide-resistant, and abiraterone-resistant PDX model (34). The combination of ebastine with enzalutamide significantly enhanced the anti-cancer activity of enzalutamide in the LuCaP 35CR PDX model as indicated by the inhibition of tumor growth (Fig. 5C), the reduction of overall tumor weight (Fig. 5D), and enhanced progression-free survival (Fig. 5E). Noteworthy, the protein and mRNA levels of EZH2 and its downstream targets were significantly reduced in the tumor lysate of LuCaP 35CR xenograft mice after ebastine treatment as compared to those of control (Fig. 5F-G). The levels of ebastine and its metabolite, carebastine, were measured using HPLC-MS/MS and found that ebastine and carebastine levels significantly elevated 20-fold after ebastine treatment in the tumor lysate of mice (Fig. 5H-I). Again, no significant changes in body weight were observed in mice among different treatment groups (Fig. S7C-D).
Figure 5. Therapeutic efficacy of ebastine in castration-resistant prostate cancer murine models.

Castration-resistant VCaP xenograft mouse models were generated as described in the “Methods” section. Castrated mice carrying CRPC xenograft received vehicle or ebastine treatment (10mg/kg, 30mg/kg) five days per week. (A) Caliper measurements were taken every 4 days to obtain tumor volume. Mean tumor volume ± SEM, * P<0.05 vs. Vehicle. (B) Kaplan-Meier survival plot comparing progression-free survival and significance was tested using unpaired t-test. LuCap 35CR PDX mouse models were established. Mice received Enzalutamide (10mg/kg per day) or enzalutamide (10mg/kg per day) + ebastine (30mg/kg per day) orally. (C) Tumor volume was obtained using caliper every 4 days. Mean tumor volume ± SEM. (D) Tumors were dissected and weighted after 35 days of treatment. Significance was tested using unpaired t-test. * P<0.05, vs vehicle, Mean ± SEM. (E) Kaplan-Meier survival plot compares progression-free survival. Significance was tested using unpaired t-test. (F) Tumor samples (control n=6, ebastine n=7) were ground and lysed for western blot analysis. Proteins were blotted and quantitated to compare the level of EZH2, AR, and PSA between the ebastine-treated group and the vehicle-treated group. (G) RNA was extracted to compare the level of EZH2, AR, and PSA. The relative expression was standardized with GAPDH. (H-I) The concentration of carebastine, metabolites of ebastine, and ebastine using HPLC-MS/MS from tumor lysates. * P<0.05, *** P<0.0001 vs vehicle, Mean ± SEM.
Discussion
EZH2 has been reported as an oncogene for over 18 years (6,7). The emerging role of EZH2 in cancer progression and metastasis prompt researchers and pharmaceutical companies to develop EZH2 inhibitors for cancer therapy. A number of small molecular EZH2 inhibitors have recently been identified. Among these inhibitors, GSK126, EPZ005687, EPZ006438, and EI1 are remarkable for their high selectivity to EZH2, improved pharmacokinetic properties, and fewer side effects (15,20,35). Despite their high therapeutic and clinical translation potential, the use of these EZH2 inhibitors is limited to sarcomas or lymphomas harboring EZH2 gain-of-function mutants (15,35-38). Several studies reported that these EZH2 enzymatic inhibitors alone are not sufficient to suppress the growth and progression of solid tumors with high wild-type EZH2 expression levels unless these EZH2 inhibitors are combined with other inhibitors (39-41), or in tumors harboring other mutations such as ARID1A, EGFR or BRG1 mutants (42,43).
Mounting evidence suggests that EZH2 can perform its oncogenic functions independently of its methyltransferase activity (44,45). EZH2 could act as a transcriptional activator to activate AR expression by binding to enhancer regions of AR to promote prostate cancer progression (40). EZH2 can also regulate NF-κB signaling independently of its C-term enzymatic SET domain by interacting with REL-A and REL-B or activating REL-B in breast cancer (46,47). Therefore, targeting the transcript or protein levels of EZH2 could be a better therapeutic option by inhibiting both methyltransferase-dependent and -independent oncogenic functions of EZH2.
In this study, we first discovered ebastine, an anti-histamine drug for allergy treatment, as a potent EZH2 inhibitor by targeting the promoter of EZH2, which downregulates both mRNA and protein levels of EZH2. As previously reported that the mRNA and protein levels of EZH2 are associated with the neoplastic properties in many cancers (6,8-14). We consistently demonstrated that ebastine effectively inhibits the proliferation, migration, and invasion of various breast and prostate cells in vitro. When compared with GSK126 and other EZH2 enzyme inhibitors EPZ6438 and EED226, ebastine showed better inhibition against EZH2 and demonstrated more robust anti-cancer activities. The RNA-sequencing analysis also demonstrated that cells treated with ebastine exhibited gene regulation signature as similar to EZH2 knockdown, which was distinct from those treated with GSK126. Most importantly, this inhibition of EZH2 by ebastine is independent of EZH2 enzymatic activity, as demonstrated by the rescue using the H689A mutant. Additionally, the in vivo studies in murine xenograft models and PDX models further supported that ebastine could effectively inhibit tumor growth and progression of advanced cancers and drug-resistant cancers. In addition to ebastine, its metabolite, carebastine, also found to be elevated in the tumor of mice treated with ebastine and demonstrated to reduce EZH2 protein level and exhibit anti-cancer activities in C4-2 and DU145 prostate cancer cells (Fig. S8).
Altogether, our results revealed that ebastine could be a promising therapeutic intervention for cancers bearing EZH2 overexpression or mutations. A retrospective study reported that breast cancer patients taking ebastine for allergy treatment had a better overall and breast cancer-specific survival rate compared to those not taking anti-histamine drugs (48-50), further suggesting the safety and the potential of using ebastine in clinical settings. With the present data, ebastine is a safe drug for daily use and should be implemented in cancer treatment intervention either as a preventive agent for people with allergy syndrome or therapeutic options for patients with advanced cancers after careful and systematic evaluation.
Supplementary Material
Acknowledgment
We would like to acknowledge Dr. Peng Gao at the Metabolomics Developing Core Facility of Northwestern University for the HPLC-MS/MS support. We also wish to acknowledge the following individuals for their kind gifts: Dr. Leland W. Chang for the C4-2 cells, Dr. Yi Zhang for the MigR1 (GFP) retroviral vectors of Ezh2 wild-type and Ezh2 mutant H689A, and Dr. Eva Corey for LuCaP 35CR PDX.
Financial support: This work is supported in part by grants from Prostate Cancer Foundation (13YOUN007 to Q.C.), U.S. Department of Defense (W81XWH-15-1-0639, W81XWH-17-1-0357 and W81XWH-19-1-0563 to Q.C.), American Cancer Society (RSG-15-192-01 to Q.C.), and NIH/NCI (R01CA208257 to Q.C. and Prostate SPORE P50CA180995 DRP) and Northwestern Univ. Polsky Urologic Cancer Institute (to Q.C.); K.C. is supported in part by grants from NIH/NHLBI (R01CA208257, HL100397, and HL099997) and Department of Defense (W81XWH-17-1-0357 and W81XWH-19-1-0563).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Reference
- 1.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. [DOI] [PubMed] [Google Scholar]
- 2.Yao Y, Hu H, Yang Y, Zhou G, Shang Z, Yang X, et al. Downregulation of Enhancer of Zeste Homolog 2 (EZH2) is essential for the Induction of Autophagy and Apoptosis in Colorectal Cancer Cells. Genes (Basel). 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pengelly AR, Copur O, Jackle H, Herzig A, Muller J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science. 2013;339:698–9. [DOI] [PubMed] [Google Scholar]
- 4.Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23:483–92. [DOI] [PubMed] [Google Scholar]
- 5.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9. [DOI] [PubMed] [Google Scholar]
- 7.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Leenders GJ, Dukers D, Hessels D, van den Kieboom SW, Hulsbergen CA, Witjes JA, et al. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur Urol. 2007;52:455–63. [DOI] [PubMed] [Google Scholar]
- 9.Poirier JT, Gardner EE, Connis N, Moreira AL, de Stanchina E, Hann CL, et al. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene. 2015;34:5869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kikuchi J, Kinoshita I, Shimizu Y, Kikuchi E, Konishi J, Oizumi S, et al. Distinctive expression of the polycomb group proteins Bmi1 polycomb ring finger oncogene and enhancer of zeste homolog 2 in nonsmall cell lung cancers and their clinical and clinicopathologic significance. Cancer. 2010;116:3015–24. [DOI] [PubMed] [Google Scholar]
- 11.Weikert S, Christoph F, Kollermann J, Muller M, Schrader M, Miller K, et al. Expression levels of the EZH2 polycomb transcriptional repressor correlate with aggressiveness and invasive potential of bladder carcinomas. Int J Mol Med. 2005;16:349–53. [PubMed] [Google Scholar]
- 12.Eskander RN, Ji T, Huynh B, Wardeh R, Randall LM, Hoang B. Inhibition of enhancer of zeste homolog 2 (EZH2) expression is associated with decreased tumor cell proliferation, migration, and invasion in endometrial cancer cell lines. Int J Gynecol Cancer. 2013;23:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6:6051. [DOI] [PubMed] [Google Scholar]
- 14.Simon C, Chagraoui J, Krosl J, Gendron P, Wilhelm B, Lemieux S, et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 2012;26:651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12. [DOI] [PubMed] [Google Scholar]
- 16.Glazer RI, Hartman KD, Knode MC, Richard MM, Chiang PK, Tseng CK, et al. 3-Deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem Biophys Res Commun. 1986;135:688–94. [DOI] [PubMed] [Google Scholar]
- 17.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun F, Lee L, Zhang Z, Wang X, Yu Q, Duan X, et al. Preclinical pharmacokinetic studies of 3-deazaneplanocin A, a potent epigenetic anticancer agent, and its human pharmacokinetic prediction using GastroPlus. Eur J Pharm Sci. 2015;77:290–302. [DOI] [PubMed] [Google Scholar]
- 20.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6. [DOI] [PubMed] [Google Scholar]
- 21.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. P Natl Acad Sci USA. 2013;110:7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi W, Chan HM, Teng L, Li L, Chuai SN, Zhang RP, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. P Natl Acad Sci USA. 2012;109:21360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang W, Liu KH, Ryu JY, Shin JG. Simultaneous determination of ebastine and its three metabolites in plasma using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;813:75–80. [DOI] [PubMed] [Google Scholar]
- 24.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Morley M, Brandimarto J, Hannenhalli S, Hu Y, Ashley EA, et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 2015;105:83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellegaard AM, Dehlendorff C, Vind AC, Anand A, Cederkvist L, Petersen NHT, et al. Repurposing Cationic Amphiphilic Antihistamines for Cancer Treatment. EBioMedicine. 2016;9:130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kong X, Chen L, Jiao L, Jiang X, Lian F, Lu J, et al. Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2. J Med Chem. 2014;57:9512–21. [DOI] [PubMed] [Google Scholar]
- 30.Clark A, Love H. Astemizole-induced ventricular arrhythmias: an unexpected cause of convulsions. International Journal of Cardiology. 1991;33:165–7. [DOI] [PubMed] [Google Scholar]
- 31.Liu Q, Wang G, Li Q, Jiang W, Kim JS, Wang R, et al. Polycomb group proteins EZH2 and EED directly regulate androgen receptor in advanced prostate cancer. Int J Cancer. 2019;145:415–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei FZ, Cao ZY, Wang X, Wang H, Cai MY, Li TT, et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy. 2015;11:2309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang R, Wang R, Chang H, Wu F, Liu C, Deng D, et al. Downregulation of Ezh2 expression by RNA interference induces cell cycle arrest in the G0/G1 phase and apoptosis in U87 human glioma cells. Oncol Rep. 2012;28:2278–84. [DOI] [PubMed] [Google Scholar]
- 34.Lam HM, McMullin R, Nguyen HM, Coleman I, Gormley M, Gulati R, et al. Characterization of an Abiraterone Ultraresponsive Phenotype in Castration-Resistant Prostate Cancer Patient-Derived Xenografts. Clin Cancer Res. 2017;23:2301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13:842–54. [DOI] [PubMed] [Google Scholar]
- 36.Shen JK, Cote GM, Gao Y, Choy E, Mankin HJ, Hornicek FJ, et al. Targeting EZH2-mediated methylation of H3K27 inhibits proliferation and migration of Synovial Sarcoma in vitro. Sci Rep. 2016;6:25239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu B, Shen X, Zhang L, Liu D, Zhang C, Cao J, et al. Discovery of EBI-2511: A Highly Potent and Orally Active EZH2 Inhibitor for the Treatment of Non-Hodgkin's Lymphoma. ACS Med Chem Lett. 2018;9:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stacchiotti S, Zuco V, Tortoreto M, Cominetti D, Frezza AM, Percio S, et al. Comparative Assessment of Antitumor Effects and Autophagy Induction as a Resistance Mechanism by Cytotoxics and EZH2 Inhibition in INI1-Negative Epithelioid Sarcoma Patient-Derived Xenograft. Cancers (Basel). 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell. 2017;31:286–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Lee Y, Lu X, Song B, Fong K-W, Cao Q, et al. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018;25:2808–20.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamaguchi H, Du Y, Nakai K, Ding M, Chang SS, Hsu JL, et al. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene. 2018;37:208–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21:231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015;520:239–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan J, Ng SB, Tay JL, Lin B, Koh TL, Tan J, et al. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood. 2013;121:4512–20. [DOI] [PubMed] [Google Scholar]
- 45.Karantanos T, Boussiotis VA. JAK3-mediated phosphorylation of EZH2: a novel mechanism of non-canonical EZH2 activation and oncogenic function. Transl Cancer Res. 2016;5:S1208–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43:798–810. [DOI] [PubMed] [Google Scholar]
- 47.Lawrence CL, Baldwin AS. Non-Canonical EZH2 Transcriptionally Activates RelB in Triple Negative Breast Cancer. PLoS One. 2016;11:e0165005.27764181 [Google Scholar]
- 48.Olsson HL. Use of certain antihistamines among breast cancer patients confers survival benefit. Cancer Res. 2018;78:5695. [Google Scholar]
- 49.Olsson HL, Einefors R, Broberg P. Second generation antihistamines after breast cancer diagnosis to improve prognosis both in patients with ER+ and ER− breast cancer. Journal of Clinical Oncology. 2015;33:3062-. [Google Scholar]
- 50.Olsson HL, Broberg P, Einefors R, Fritz I. Effects of antihistamine use on survival in breast cancer. Journal of Clinical Oncology. 2018;36:e12527–e. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.