

Abstract
Purpose of review.
Senescent cells are now known to accumulate in multiple tissues with aging and through their inflammation (the senescence-associated secretory phenotype, SASP), contribute to aging and chronic diseases. Here we review the roles of senescent osteocytes in the context of bone loss.
Recent findings.
Numerous studies have established that senescent osteocytes accumulate in the bone microenvironment with aging in mice and in humans. Moreover, at least in mice, elimination of senescent cells results in attenuation of age-related bone loss. Osteocyte senescence also occurs in response to other cellular stressors, including radiotherapy, chemotherapy, and metabolic dysfunction where it appears to mediate skeletal deterioration.
Summary.
Osteocyte senescence is linked to bone loss associated with aging and other conditions. Senescent osteocytes are potential therapeutic targets to alleviate skeletal dysfunction. Additional studies better defining the underlying mechanisms as well as translating these exciting findings from mouse models to humans are needed.
Keywords: Osteocyte, Senescence, Aging, Radiotherapy, Chemotherapy, Type 2 Diabetes Mellitus
Introduction
Cellular senescence is a cell fate, like differentiation, proliferation, or apoptosis, that involves essentially irreversible proliferative arrest, tumor suppressor activation, altered chromatin organization, apoptosis resistance, and frequently is associated with increased protein synthesis [1]. The phenomenon was discovered in the early 1960s [2] by Leonard Hayflick who found that with prolonged serial culture, normal human embryonic fibroblasts eventually lost their capacity to divide, yet they remained viable. This growth arrest phase was coined “senescence” [2]. It is now well established that various types of stress converge to cause a cell to enter the cellular senescence program. Examples of senescence-inducing stressors include internal and external cell damaging insults such as DNA breaks, oncogenic stimuli, reactive metabolites and oxygen species, proteotoxic stress, and inflammation [1].
The pathways that drive a cell into senescence are largely centered on the cyclin-dependent kinase inhibitors, most notably p16Ink4a and p21Cip1 (p53 is upstream of p21Cip1), that in response to accumulating cell damage are upregulated as a protective mechanism to halt cell proliferation and prevent malignant transformation [1]. The resistance of senescent cells to apoptosis is acquired via activation of several senescent cell anti-apoptotic pathways (or SCAPs) [3]. Among the most common morphological features present with the proliferative arrest and apoptosis resistance of senescent cells, are the higher-order unfolding of satellite heterochromatin (termed senescence-associated distension of satellites [SADS] [4]) and the acquisition of DNA damage at sites of telomeres – i.e., telomere-associated foci [5].
Despite their growth arrest, senescent cells are highly metabolically active and can acquire a distinctive pro-inflammatory secretome, termed the senescence-associated secretory phenotype (SASP) [6]. The SASP varies by senescent cell type and based on the senescence-inducing stressor, but is typically comprised of cytokines, chemokines, matrix-degrading metalloproteinases (MMPs), and growth factors that can transmit senescence to neighboring, previously healthy cells via a senescence-induced bystander effect [7]. Thus, by spreading local and systemic inflammation over prolonged periods, the SASP can drive stem cell dysfunction, aberrant remodeling, and tissue deterioration [1].
It is now evident that senescent cells accumulate in vivo with aging and at pathological sites of multiple chronic diseases [1]. Likewise, the biological relevance and consequences of senescent cells in aging, cancer, and various etiological conditions are becoming increasingly apparent [1]. This conceptual framework has predominantly stemmed from the discovery of genetic and pharmacological approaches to eliminate senescent cells in vivo. For example, the development of a novel transgene, termed INK-ATTAC [8] – i.e., p16Ink4a-linked apoptosis through targeted activation of caspase, has enabled the elimination of p16Ink4a-positive senescent cells in mice. More recently, the identification of drugs, termed ‘senolytics’ [9], that target SCAPs to specifically kill senescent cells has led to a better understanding of the causal roles of senescent cells and has opened new opportunities to therapeutically target senescent cells to prevent or alleviate numerous comorbidities as a group, instead of one at a time [10], to thereby extend healthspan (i.e., the period of life free of chronic disease). In several different models of aging and chronic disease, multiple groups have demonstrated that genetic or pharmacological elimination of senescent cells prevents or alleviates numerous comorbidities [8, 11–36].
There is also recent mounting interest in the roles of senescent cells in bone [37–39]. Indeed, this is a rapidly emerging area as, to date, multiple studies have investigated the contributions of senescent cells to bone loss in various settings (as summarized in Table 1). The purpose of this review is to summarize the existing knowledge of skeletal cellular senescence in mice and humans with aging as well as in preclinical models of cancer therapy and type 2 diabetes mellitus (T2DM), with a focus on senescent osteocytes, given the immense biological importance of the osteocyte in bone biology (reviewed elsewhere [40, 41] and in this Issue of Current Osteoporosis Reports).
Table 1:
Summary of senescence biomarkers in bone and effects of senolytic (genetic or pharmacological) approaches on senescence biomarkers in models of aging, type 2 diabetes mellitus (T2DM), and cancer therapy.
| Reference | Animal or Human Characteristics | Senescence Biomarkers | Senolytic Approach | Δ Senescence Biomarkers |
|---|---|---|---|---|
| Farr et al., 2016 [20] | C57BL/6 mice ♀♂ Old (24m) vs. young (6m) |
↑p16Ink4a (OP, OB, OCY, B, T, Myeloid) - ♀ & ♂ ↑p21Cip1 (OCY) - ♂ ↑p53 (OCY, Myeloid) - ♀ ↑SASP, senescent OCY, TIF+ OCY |
- | - |
| Human bone biopsies♀ Old (72-87y) vs. young (23-30y) |
↑p16Ink4a, p21Cip1, SASP | - | - | |
| Farr et al., 2017 [42] | C57BL/6 mice ♀♂ Old (20m) vs. young (6-12m) |
↑p16Ink4a (OCY) ♀ & ♂ |
INK-ATTAC ♀, 20m AP20187 vs. VEH |
↓p16Ink4a, EGFP, ↓senescent OCY (SADS+) |
| C57BL/6 ♂ 20m D+Q vs. VEH |
↓p16Ink4a ↓senescent OCY(SADS+) |
|||
| Piemontese et al., 2017 [46] | C57BL/6 mice ♀♂ Old (20-21 m) vs. young (6-7m) |
↑p16Ink4a (OCY), DNA damage (γH2AX) | - | - |
| Chandra et al., 2020 [35] | C57BL/6 mice ♂ R vs. NR |
↑p16Ink4a, p21Cip1, SASP ↑SA-β-Gal+ and TIF+ osteoblasts ↑TIF+ OCY |
C57BL/6 ♂ 4m D+Q vs. VEH |
↓p16Ink4a, p21Cip1,SASP ↓TIF+ OCY |
| Eckhardt et al., 2020 [56] | C57BL/6 mice ♂ 7m HFD/STZ vs. control |
↑p16Ink4a, p21Cip1, SASP (OCY) ↑ senescent OCY (SADS+, TAF+) |
- | - |
| Yao et al., 2020 [36] | FVB/NJ mice ♀, 6w R vs. NR |
↑p16Ink4a, SASP ↓ HMGB1 |
- | - |
| FVB/NJ mice ↑, 6w DOXO vs. VEH |
↑p16Ink4a, SASP, SA-β-Gal+ ↓HMGB1 |
INK-ATTAC ♀ 16w AP20187 vs. VEH |
↑p16Ink4a, SASP ↓ HMGB1 |
Key: m, month; y, year; R, radiated; NR, non-radiated; HFD, high-fat diet; STZ, streptozotocin; DOXO, doxorubicin; OP, osteoprogenitor; OB, osteoblast; OCY, osteocytes; B, B-cells; T, T-cells; SASP, senescence-associated secretory phenotype; TIF, telomere dysfunction-induced foci; INK-ATTAC, p16Ink4a-linked apoptosis through targeted activation of caspase; EGFP, enhance green fluorescent protein (encodes for the INK-ATTAC transgene); SA-β-Gal, senescence-associated β-galactosidase; D, dasatinib; Q, quercetin. The upward (↑) and downward (↓) arrows represent the results for the study group in bold versus the contralateral group.
Identification of Senescent Osteocytes in Bone with Aging
In studies in mice, our group identified senescent cells in bone and characterized their SASP in vivo [42]. From young (6-month-old) and old (24-month-old) female and male mice, we developed approaches using magnetic-activated cell sorting (MACS) to rapidly isolate enriched in vivo populations of various cell types from the bone microenvironment [42]. From mouse bone marrow, these populations included myeloid cells (CD14+), B cells (CD19+), and T cells (CD3+) as well as osteoblast progenitors (Lin−/Lepr+) [42]. In addition, we subjected mouse bones to serial liberase digestions to release cells, and then used MACS to first deplete for hematopoietic/endothelial markers (CD31/34/45/54−), followed by a positive selection for alkaline phosphatase (AP) to enrich for osteoblasts (AP+/CD31/34/45/54−) [42]. We then demonstrated that the remaining liberase-digested bone samples (termed “osteocyte-enriched bone samples”) were highly enriched for several key osteocyte markers (i.e., Dmp1, Phex, Mepe, Sost, Fgf23) [42], consistent with methods developed by the Bonewald laboratory [43]. These approaches allow for the rapid (within 2-3 hrs of sacrificing the animal) isolation of various major cell populations from the bone microenvironment, and subsequent analysis of in vivo gene expression signatures.
In each of the isolated cell populations, we then compared the levels of the senescence markers, p16Ink4a and p21Cip1, between young and old mice. Our data established that p16Ink4a mRNA expression increased significantly (~5-10 fold) with aging in myeloid cells, B and T cells, osteoblast progenitors, osteoblasts, and osteocytes; results were similar in female and male mice [42]. By contrast, p21Cip1 mRNA levels did not change with aging in the majority of the isolated populations. Interestingly, however, p21Cip1 mRNA levels did increase significantly with aging in osteocytes from male mice [42]. Consistent with the higher expression of the senescence-related cyclin-dependent kinase inhibitors, p16Ink4a and p21Cip1, we found that a subset of osteocytes in bone cortices of old mice displayed altered chromatin organization (i.e., large-scale unraveling of peri-centromeric satellite heterochromatin or SADS), which is a consistent characteristic of senescent cells [4]. Using a threshold of ≥4 SADS per cell to define cellular senescence, we detected a significantly increased number of senescent osteocytes in bone cortices of old (~11% senescent) as compared to young (~2% senescent) mice [42]. To further confirm osteocyte senescence with aging, we isolated primary osteocytes from bones of young and old mice (using techniques developed by the Bonewald laboratory [44]) and measured DNA damage (53BP1) co-localized with telomeres (telomere-induced foci, TIF), another established biomarker of cellular senescence related to impaired DNA damage repair mechanisms [45]. With aging, our data demonstrated that the number of TIF+ osteocytes increased 6-fold, thus providing further evidence for an accumulation of senescent osteocytes in old bone [42]. The age-associated increase in the proportion of senescent osteocytes we observed in vivo was consistent with results from another group [46], which also found increased senescence markers (e.g., p16Ink4a mRNA expression and DNA damage) in osteocyte-enriched bone samples of old relative to young mice. In addition, Piemontese et al. [46] reported a potential connection in old mice between osteocyte senescence, increased production of RANKL, and endocortical resorption leading to thinning of the cortex and increased cortical porosity with aging. Taken together, these findings establish that at least a subset of osteocytes become senescent with old age in mice.
In our young (6-month) versus old (24-month) mouse study [42], we next aimed to examine the in vivo age-related changes in the SASP produced by various cells within the bone microenvironment by measuring the mRNA expression levels of a panel of 36 previously identified SASP factors [6]. Using Gene Set Enrichment Analysis (GSEA), we found relatively few SASP changes with aging in osteoblast progenitors, osteoblasts, B cells, and T cells [42]. By contrast, there was a profound age-associated SASP signature identified in both myeloid cells and in osteocytes as 23-26 of the 36 queried SASP factors significantly increased with aging in these cell populations [42]. Thus, although only a relatively small proportion of cells within bone marrow and bone itself become senescent with aging (e.g., ~11% of osteocytes), it appears that these cell populations, particularly senescent myeloid cells and osteocytes, are likely capable of contributing to the development of a pro-inflammatory local bone microenvironment that contributes to skeletal dysfunction, at least in mice.
To establish whether our findings of increased cellular senescence in bone with aging in mice extended to humans, we isolated small needle bone biopsies from the posterior iliac crest of younger (27±3 years) versus older postmenopausal (78±5 years) women and examined senescence markers and SASP factors by rt-qPCR [42]. The bone biopsies contained bone marrow elements as well as both trabecular and cortical bone comprised predominantly of osteocytes. Despite their heterogeneity, consistent with our data in mice, bone biopsies from older women had significantly higher mRNA expression levels of the senescence markers, p16INK4a and P21Cip1, as well as several SASP factors (12 of the 36 queried) in comparison to biopsies from younger women [42]. These data thus suggest parallel findings between mice and humans, and prompted us to next ask the question of whether eliminating senescent cells prevents age-related bone loss. To address this question, we utilized both genetic and pharmacological approaches to eliminate senescent cells.
Eliminating Senescent Cells Using Genetic Versus Pharmacological Approaches
As noted earlier, global clearance of senescent cells can be achieved using INK-ATTAC [8] as this mouse model contains a “suicide” transgene driven by the p16Ink4a promoter that permits the inducible elimination of senescent cells by activating an apoptosis cascade specifically in p16Ink4a-positive cells resulting in their ablation in response to administration of a synthetic drug (AP20187). Several studies in models of aging and various chronic diseases have demonstrated that clearing senescent cells using INK-ATTAC prevents or delays multiple major chronic diseases of aging, including cardiovascular disease, frailty, hepatic steatosis, pulmonary fibrotic disease, metabolic dysfunction, neurodegenerative diseases, renal disease, osteoarthritis, and osteoporosis [8, 11–25]. Alternatively, systemic senescent cell clearance can be achieved pharmacologically by periodically administering senolytics that, as noted earlier, target SCAPs to specifically kill senescent cells [3]. For example, the combination of dasatinib (D; a tyrosine kinase inhibitor) plus quercetin (Q; a natural flavonoid present in many fruits and vegetables) (D+Q) was identified as among the first generation senolytics with established in vivo efficacy [26]. The combination D+Q was more senolytic than either alone and remained effective in eliminating senescent cells when delivered intermittently [26], thereby avoiding potential off-target, non-senolytic actions as these compounds have relatively short in vivo half-lives (<12 hrs). Consistent with the genetic INK-ATTAC approach, intermittent senolytic therapy has been shown to improve several aspects of healthspan in preclinical models of aging and chronic disease [26–36].
Effects of Senescent Cell Clearance on Age-Related Bone Loss
Based on the collective data demonstrating that senescent cells are present at the time and location of osteoporosis with aging in humans [42] as in mice [42, 46], we hypothesized that cellular senescence, including the age-associated accumulation of senescent osteocytes, has a causal role in mediating the pathogenesis of osteoporosis. In order to test this hypothesis, we utilized both genetic (INK-ATTAC) and pharmacological (senolytics D+Q) approaches to target senescent cells [20]. These interventional studies were initiated in mice at 20-months of age as our analysis of changes in p16Ink4a throughout the murine lifespan indicated that expression of this senescence marker in osteocyte-enriched bone samples begins to increase significantly in both males and females around 18 months of age [20], which coincides with the timing of age-related bone loss in both sexes. Over the course of four months, old INK-ATTAC mice were treated twice weekly with AP20187, whereas old wild-type mice were treated once monthly with D+Q. Both approaches resulted in at least partial elimination of senescent osteocytes and improved bone mass, microarchitecture, and strength by suppressing bone resorption (reducing osteoclast numbers) and either maintaining (trabecular skeletal sites) or increasing (cortical skeletal sites) osteoblast numbers as well as indices of bone formation [20]. These data thus implicate cellular senescence at the nexus of skeletal aging. Furthermore, the absence of any effects of these interventions on the skeleton in young mice [20] points to their specificity in eliminating senescent cells that accumulate with aging, and the intermittent senolytic dosing strategy employed, along with the short in vivo half-lives of D+Q (<12 hrs), reduces the likelihood of potential off-target effects and undesired side effects on non-senescent cells. These findings provide “proof-of-concept” evidence in mice for selectively eliminating senescent cells, including senescent osteocytes, to potentially alleviate age-related bone loss. It is noteworthy, however, that clearance of senescent osteoclast progenitors was not enough to prevent bone loss in old mice [47], which further implicates senescent osteocytes as perhaps the key drivers of age-related bone loss. In addition to age-related osteoporosis, recent emerging data suggest that targeting cellular senescence in mice also has beneficial skeletal effects in the settings of radiotherapy- and chemotherapy-driven bone loss [35, 36], thus expanding the potential applications of this strategy to treat other conditions of skeletal dysfunction.
Radiotherapy-Induced Bone Loss
In addition to old age, other skeletal conditions have been identified in which senescent cells accumulate in bone and thus patients with such conditions may benefit from senolytic therapy. For example, mounting evidence has linked radiation therapy in cancer patients to chronic bone loss and cell senescence [48–50]. Radiation induces senescence by causing cell damage, including DNA breaks and chromatin disruption [49]. While radiation therapy is effective in slowing cancer, it consequently becomes detrimental to bone, as senescence and skeletal dysfunction ensue. Indeed, Chandra and colleagues [35] recently showed in young adult mice that senescent osteoblasts and osteocytes (characterized by increased senescence-associated β-galactosidase [SA-β-Gal] staining and TIFs) accumulate prematurely in the setting of focal radiation therapy (FRT)-induced bone loss. Consistent with another study [36], whole-bone mRNA analysis revealed that radiation caused increased expression of senescence (p16Ink4a and p21Cip1) as well as several SASP genes [35]. Following FRT, mice treated with the senolytic cocktail, D+Q, had reduced TIF+ osteocytes, preserved bone formation, and attenuated bone loss as compared to vehicle-treated mice [35]. These results are consistent with previous findings in old mice [20] demonstrating that senolytic therapy eliminates senescent osteocytes and has beneficial effects on bone turnover.
Chemotherapy-Induced Bone Loss
Alongside radiation, chemotherapy is another strategy to induce growth arrest of cancer cells. However, activated cellular senescence is a common byproduct, as observed with numerous chemotherapeutic drugs, including bleomycin [51], cisplatin [52] and hydroxyurea [53], among others. Recently, Yao and colleagues [36] hypothesized that bone loss resulting from chemotherapy may be a consequence of increased skeletal senescence. Similar to radiation therapy, mouse long-bone samples enriched for osteocytes (flushed by centrifugation) from young mice treated with the chemotherapeutic, doxorubicin (DOXO), displayed upregulated p16Ink4a mRNA expression relative to osteocyte-enriched bones from vehicle-treated mice [36]. In addition, bone marrow cells depleted of hematopoietic/endothelial marker also exhibited robust elevated mRNA levels of p16Ink4a and p21Cip1, as well as increased SASP (e.g., IL6) and reduced levels of the apoptotic marker, Hmgb1 [36]. Following DOXO-therapy, treatment of INK-ATTAC mice with AP20187 was sufficient to rescue chemotherapy-induced bone loss [36]. At the cellular level, DOXO-treated mice exhibited increased osteoclast number and reduced bone formation rates; these phenotypes were normalized after clearance of senescent cells with AP20187 [36]. These results that are thus consistent with findings in old INK-ATTAC mice treated with AP20187 [20]. Therefore, cellular senescence may represent a therapeutic target to preserve bone in patients with cancer following chemotherapy [36].
Accelerated Osteocyte Senescence and Skeletal Fragility in Type 2 Diabetes Mellitus (T2DM)
Patients with T2DM are at higher risk for fractures despite normal or higher bone mineral density (BMD) [54, 55]; thus, fracture risk in these individuals may stem from poor bone quality [56]. We hypothesized that cellular senescence may represent a mechanistic link between skeletal fragility and T2DM [57] given that: i) bone cell senescence increases in old age [42, 46]; ii) eliminating senescent cells prevents skeletal fragility with aging [20]; and iii) emerging data showing that obesity prior to aging causes the premature accumulation of senescent cells at etiological sites including adipose tissue [24], liver [18], brain [22], and pancreatic β cells [25], contributing to metabolic dysfunction [58].
To examine whether T2DM causes accelerated osteocyte senescence and skeletal dysfunction [57], we recently leveraged a non-genetic wild-type mouse model that closely mimics human T2DM – i.e., the high-fat diet (HFD)/streptozotocin (STZ) mouse of T2DM [59–61] that develops overt hyperglycemia, dysfunctional insulin secretion, insulin resistance, and β-cell deterioration. Additional advantages of this T2DM model of human metabolic dysfunction include both environmental (in obesity) and temporal (after skeletal maturity) control of T2DM onset [59–61]. After 3-4 months of metabolic dysfunction in young adulthood, we found that HFD/STZ (i.e., T2DM) mice develop poor bone quality, similar to that which manifests in humans [62, 63], including significantly deteriorated trabecular and cortical bone microarchitecture, impaired bone material properties, and diminished biomechanical strength [57]. Mice with T2DM also displayed dysfunctional bone remodeling, including higher bone resorption and defective bone formation, based on bone histomorphometric analyses and circulating bone turnover markers [57]. Using mass spectrometry, we also found that T2DM mice had increased skeletal and circulating levels of the advanced glycation endproduct (AGE), Nε-( 1-carboxymethyl)-L-Lysine (CML), an established activator of the receptor for AGE (RAGE) pathway [64]. Finally, we demonstrated that T2DM led to the accelerated accumulation of senescent osteocytes, characterized by increased expression of the cyclin-dependent kinase inhibitors, p16Ink4a and p21Cip1, in osteocyte-enriched bone samples, a greater proportion of SADS+ [4] osteocytes, and significantly more telomere-associated foci – TAF+ [5] osteocytes as compared to age-matched mice on low-fat control chow [57]. Interestingly, senescent osteocytes in mice with T2DM developed a unique pro-inflammatory SASP signature comprised predominantly of significantly upregulated levels of MMPs (i.e., Mmp3, Mmp9, Mmp12, and Mmp13) and Nfkb1 (NF-κB), a downstream target of the RAGE signaling pathway activated by CML [64]. Collectively, these observations point to the RAGE pathway and senescent cells as potential therapeutic targets to alleviate diabetic skeletal fragility [57]. However, whether there is a direct causal relationship between cellular senescence, RAGE signaling, and skeletal fragility in T2DM has not been established. This hypothesis needs to be tested.
Evidence for Cellular Senescence of Post-Mitotic Cells
In addition to senescent osteocytes, multiple other post-mitotic, terminally differentiated, tissue-resident cells develop senescent-like features, including SASP acquisition, and have been shown to mediate deleterious effects on neighboring cells in their respective tissues [65]. As early as the 1950’s, lipofuscin – the “age pigment” – was reported to accumulate in long-lived post-mitotic cells such as neurons [66], cardiomyocytes [67], and osteocytes [68]. While most studies examining cellular senescence have focused on the growth arrest of proliferative-competent cells, further investigation of long-lived post-mitotic cells has led to exciting recent insights. For example, senescent neurons have been a focus in the context of major age-related cognitive diseases [21, 22, 34]. Interestingly, up to 80% of Purkinje and 40% of cortical, hippocampal, and peripheral post-mitotic neurons in the myenteric plexus of aged mice show markers of senescence [69]. Cardiomyocytes, another major post-mitotic cell type, have also been shown to take on a profound senescence phenotype with advancing age in both mice and humans [23]. Clearance of senescent cells in aged mice using both genetic and pharmacological approaches reduced cardiomyocyte senescence and alleviated cardiac hypertrophy and fibrosis [23]. Findings of senescence-like features in additional post-mitotic cells, including adipocytes [70] and hepatocytes [18], have led to interest in how these cells become senescent in the setting of little to no active replication to thereby induce DNA damage and telomere shortening. Although still unclear, it has been proposed that senescence of post-mitotic cells arises at least in part from mitochondrial dysfunction [23], therefore acting independent of cell division. Additional underling mechanisms are likely shared among post-mitotic senescent cells, including senescent osteocytes, a better understanding of which could provide novel insights into the pathophysiology of numerous conditions, including osteoporosis.
Senolytics in Human Clinical Trials
Senolytics are currently in clinical trials, and results from the first studies were recently reported. For example, the first-in-human pilot clinical trial of D+Q provided initial evidence that senolytic therapy may improve aspects of physical function as nine doses of D+Q over three weeks led to better walking distance and speed as well as ability to rise from a chair by five days after the final dose in 14 human subjects with idiopathic pulmonary fibrosis (IPF), a debilitating fibrotic disease associated with senescent cell accumulation in the lungs [71]. More recently, early interim findings from a phase 1, open-label, study of a 3-day oral course of D+Q in nine subjects with diabetic kidney disease showed that senolytics reduced the burden of senescent cells in adipose tissue by eleven days after the final dose [72]. As an extension to the aforementioned preliminary studies, placebo-controlled, human trials are currently underway as more information about safety, side effects, tolerability, and efficacy is needed. Furthermore, additional carefully monitored trials in human subjects with other conditions, including age-related bone loss, are initiating in the near future.
Conclusions
It is now clear that in mice and in humans, senescent cells, including senescent osteocytes, accumulate in the bone microenvironment with aging and in response to other cellular stressors (e.g., radiotherapy, chemotherapy, T2DM; see Fig. 1). These senescent osteocytes contribute to the pro-inflammatory SASP in the bone microenvironment (and perhaps systemically), leading to an increase in bone resorption and decrease in bone formation. Whether the specific elimination of senescent osteocytes is sufficient to prevent age-related bone loss remains to be seen. Nevertheless, even a partial elimination of these senescent cells or inhibition of their SASP results, at least in mice, in preserved bone mass through favorable effects on bone metabolism. These pre-clinical studies have now set the stage for early-stage human trials, currently underway, examining whether reducing the burden of senescent cells in humans using senolytics can reduce bone resorption/increase bone formation and thereby prevent or ameliorate age-related bone loss, with the added advantage of potentially also delaying frailty [33] and reductions in muscle mass that may also contribute to fracture risk.
Fig. 1. Osteocyte cellular senescence in bone and effects of senolytics on bone metabolism.
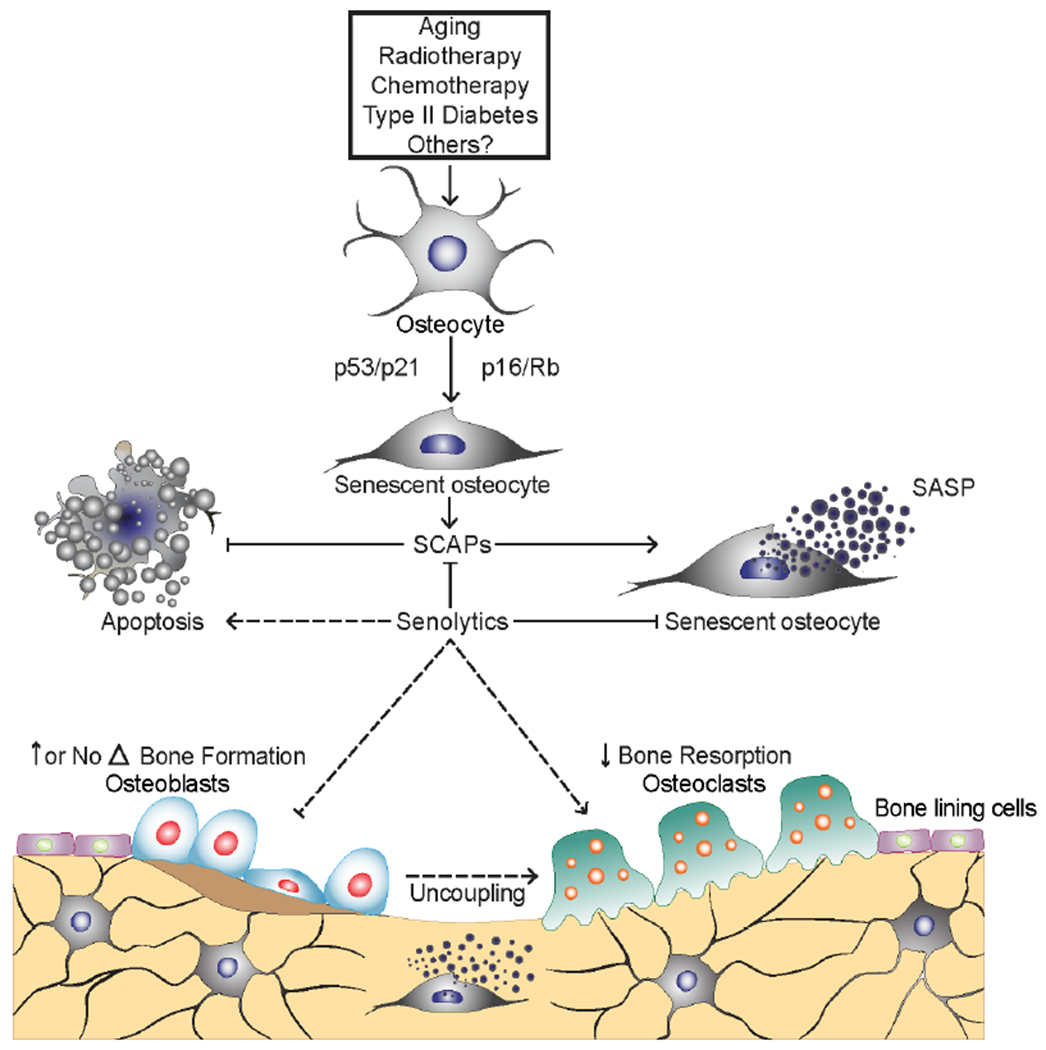
Senescent cells, including senescent osteocytes, accumulate with aging, radiotherapy, chemotherapy, type 2 diabetes mellitus (T2DM) in bone, where their senescence-associated secretory phenotype (SASP) increases bone resorption by osteoclasts and reduces bone formation by osteoblasts. In old mice, senolytics reduce the burden of senescent cells, which leads to a reduction in bone resorption with either increased (cortical bone) or maintained (trabecular bone) bone formation, resulting in a beneficial ‘uncoupling’ between bone resorption and bone formation. Dashed lines indicate beneficial effects of senescent cells on osteoclasts, osteoblasts, and the coupling between osteoclasts and osteoblasts. Adapted and reproduced with permission from [20], SpringerNature.
Acknowledgements:
This work was supported by National Institutes of Health (NIH) grants P01 AG062413 (to J.N.F., S.K.), R21 AG065868 (to J.N.F, S.K.), K01 AR070241 (to J.N.F.) as well as a High-Risk Pilot Award (to J.N.F.) and Career Development Award (J.N.F.) from the Mayo Clinic Robert and Arlene Kogod Center on Aging, and the Richard F. Emslander Career Development Award in Endocrinology (to J.N.F.). All authors state that they do not have a relevant conflict of interest.
Funding: This work was supported by National Institutes of Health (NIH) grants P01 AG062413 (J.N.F., S.K.), R21 AG065868 (J.N.F, S.K.), K01 AR070241 (J.N.F.), as well as a High-Risk Pilot Award (J.N.F.) and Career Development Award (J.N.F.) from the Mayo Clinic Robert and Arlene Kogod Center on Aging, and the Richard F. Emslander Career Development Award in Endocrinology (J.N.F.).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest
Joshua Farr, Japneet Kaur, Madison Doolittle and Sundeep Khosla declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References:
Papers of particular interest, published recently, have been highlighted as:
• Of importance
••Of major importance
- [1].Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961;25: 585–621. [DOI] [PubMed] [Google Scholar]
- [3].Wissler Gerdes EO, Zhu Y, Tchkonia T, Kirkland JL. Discovery, development, and future application of senolytics: theories and predictions. FEBS J 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Swanson EC, Manning B, Zhang H, Lawrence JB. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. The Journal of cell biology 2013;203: 929–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nature communications 2012;3: 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual review of pathology 2010;5: 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. A senescent cell bystander effect: senescence-induced senescence. Aging cell 2012; 11: 345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deuersen JM. Clearance of pl6Ink4a-positive senescent cells delay aging-associated disorders. Nature 2011;479: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kirkland JL, Tchkonia T, Zhu Y, Niedemhofer LJ, Robbins PD. The Clinical Potential of Senolytic Drugs. Journal of the American Geriatrics Society 2017;65: 2297–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. The Journal of clinical investigation 2013; 123: 966–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Laberge RM, Adler D, DeMaria M, Mechtouf N, Teachenor R, Cardin GB, Desprez PY, Campisi J, Rodier F. Mitochondrial DNA damage induces apoptosis in senescent cells. Cell death & disease 2013;4: e727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, Hoeijmakers JH, de Bruin A, Hara E, Campisi J. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Developmental cell 2014;31: 722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, Sepe A, Johnson KO, Stout MB, Giorgadze N, Jensen MD, LeBrasseur NK, Tchkonia T, Kirkland JL. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 2015;4: e12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, Kirkland JL, Miller JD. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging cell 2016; 15: 973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring pl6(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530: 184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016;354: 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, van Deursen JM, Campisi J, Elisseeff JH. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nature medicine 2017;23: 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, Grellscheid SN, Hoeijmakers JHJ, Bamhoom S, Mann DA, Bird TG, Vermeij WP, Kirkland JL, Passos JF, von Zglinicki T, Jurk D. Cellular senescence drives age-dependent hepatic steatosis. Nature communications 2017;8: 15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, KitaH, LeBrasseur NK. Cellular senescence mediates fibrotic pulmonary disease. Nature communications 2017;8: 14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]••.Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ, Pirtskhalava T, Tchkonia T, Oursler MJ, Kirkland JL, Khosla S. Targeting cellular senescence prevents age-related bone loss in mice. Nature medicine 2017;23: 1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first demonstration that senescent cells have a causal role in mediating age-related bone loss in naturally-aged mice.
- [21].Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018;562: 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Kruger P, Fielder E, Victorelli S, Ruswhandi RA, Giorgadze N, Pirtskhalava T, Podgomi O, Enikolopov G, Johnson KO, Xu M, Inman C, Palmer AK, Schafer M, Weigl M, Ikeno Y, Bums TC, Passos JF, von Zglinicki T, Kirkland JL, Jurk D. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab 2019;29: 1061–1077 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, Birch J, Salmonowicz H, Ogrodnik M, Jurk D, Proctor C, Correia-Melo C, Victorelli S, Fielder E, Berlinguer-Palmini R, Owens A, Greaves LC, Kolsky KL, Parini A, Douin-Echinard V, LeBrasseur NK, Arthur HM, Tual-Chalot S, Schafer MJ, Roos CM, Miller JD, Robertson N, Mann J, Adams PD, Tchkonia T, Kirkland JL, Mialet-Perez J, Richardson GD, Passos JF. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J 2019;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, Praia LG, van Dijk TH, Verkade E, Casaclang-Verzosa G, Johnson KO, Cubro H, Doomebal EJ, Ogrodnik M, Jurk D, Jensen MD, Chini EN, Miller JD, Matveyenko A, Stout MB, Schafer MJ, White TA, Hickson LJ, Demaria M, Garovic V, Grande J, Arriaga EA, Kuipers F, von Zglinicki T, LeBrasseur NK, Campisi J, Tchkonia T, Kirkland JL. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019; 18: e12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Aguayo-Mazzucato C, Andie J, Lee TB Jr, Midha A, Talemal L, Chipashvili V, Hollister-Lock, van Deursen J, Weir G, Bonner-Weir S. Acceleration of beta Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]••.Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O’Hara SP, LaRusso NF, Miller JD, Roos CM, Verzosa GC, LeBrasseur NK, Wren JD, Farr JN, Khosla S, Stout MB, McGowan SJ, Fuhrmann-Stroissnigg H, Gurkar AU, Zhao J, Colangelo D, Dorronsoro A, Ling YY, Barghouthy AS, Navarro DC, Sano T, Robbins PD, Niedemhofer LJ, Kirkland JL. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging cell 2015;14: 644–58. [DOI] [PMC free article] [PubMed] [Google Scholar]; First demonstration that pharmacologically targeting senescent cell anti-apoptotic pathways (SCAPs) enables senescent cell killing (senolytic) activity.
- [27].Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, Ben-Porath I, Krizhanovsky V. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 2016;7: 11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Bums N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016;22: 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhu Y, Doomebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedemhofer LJ, Robbins PD, Tchkonia T, Kirkland JL. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017;9: 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, Grassi D, Gregg SQ, Stripay JL, Dorronsoro A, Corbo L, Tang P, Bukata C, Ring N, Giacca M, Li X, Tchkonia T, Kirkland JL, Niedemhofer LJ, Robbins PD. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun 2017; 8: 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, van IWF, Houtsmuller AB, Pothof J, de Bruin RWF, Madl T, Hoeijmakers JHJ, Campisi J, de Keizer PLJ. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017;169: 132–147 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, McGuckian C, Wade EA, Kato JI, Grassi D, Wentworth M, Burd CE, Arriaga EA, Ladiges WL, Tchkonia T, Kirkland JL, Robbins PD, Niedemhofer LJ. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018;36: 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, Langhi LGP, Weigl M, Giorgadze N, LeBrasseur NK, Miller JD, Jurk D, Singh RJ, Allison DB, Ejima K, Hubbard GB, Ikeno Y, Cubro H, Garovic VD, Hou X, Weroha SJ, Robbins PD, Niedemhofer LJ, Khosla S, Tchkonia T, Kirkland JL. Senolytics improve physical function and increase lifespan in old age. Nature medicine 2018;24: 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, Orr ME. Tau protein aggregation is associated with cellular senescence in the brain. Aging cell 2018: e12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chandra A, Lagnado AB, Farr JN, Monroe DG, Park S, Hachfeld C, Tchkonia T, Kirkland JL, Khosla S, Passos JF, Pignolo RJ. Targeted Reduction of Senescent Cell Burden Alleviates Focal Radiotherapy-Related Bone Loss. J Bone Miner Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yao Z, Murali B, Ren Q, Luo X, Faget DV, Cole T, Ricci B, Thotala D, Monahan J, van Deursen JM, Baker D, Faccio R, Schwarz JK, Stewart SA. Therapy-Induced Senescence Drives Bone Loss. Cancer Res 2020;80: 1171–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Khosla S, Farr JN, Kirkland JL. Inhibiting Cellular Senescence: A New Therapeutic Paradigm for Age-Related Osteoporosis. The Journal of clinical endocrinology and metabolism 2018; 103: 1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Farr JN, Almeida M. The Spectrum of Fundamental Basic Science Discoveries Contributing to Organismal Aging. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2018;33: 1568–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Farr JN, Khosla S. Cellular senescence in bone. Bone 2019; 121: 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dallas SL, Prideaux M, Bonewald LF. The osteocyte: an endocrine cell … and more. Endocrine reviews 2013;34: 658–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Robling AG, Bonewald LF. The Osteocyte: New Insights. Annu Rev Physiol 2020;82: 485–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42]••.Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, Drake MT, Tchkonia T, LeBrasseur NK, Kirkland JL, Bonewald LF, Pignolo RJ, Monroe DG, Khosla S. Identification of Senescent Cells in the Bone Microenvironment. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2016;31: 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was among the first to systimatically identify senescent cells in the bone mircoenvironment with aging, including senescent osteocytes, and characerized their SASP.
- [43].Stem AR, Stem MM, Van Dyke ME, Jahn K, Prideaux M, Bonewald LF. Isolation and culture of primary osteocytes from the long bones of skeletally mature and aged mice. BioTechniques 2012;52: 361–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jahn K, Kato S, Wysolmerski J, Bonewald LF. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2012;27: 1018–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kenyon J, Gerson SL. The role of DNA damage repair in aging of adult stem cells. Nucleic acids research 2007;35: 7557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Piemontese M, Almeida M, Robling AG, Kim HN, Xiong J, Thostenson JD, Weinstein RS, Manolagas SC, O'Brien CA, Jilka RL. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kim HN, Chang J, Iyer S, Han L, Campisi J, Manolagas SC, Zhou D, Almeida M. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 2019;18: el2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pacheco R, Stock H. Effects of radiation on bone. Curr Osteoporos Rep 2013; 11: 299–304. [DOI] [PubMed] [Google Scholar]
- [49].Zhang J, Qiu X, Xi K, Hu W, Pei H, Nie J, Wang Z, Ding J, Shang P, Li B, Zhou G. Therapeutic ionizing radiation induced bone loss: a review of in vivo and in vitro findings. Connect Tissue Res 2018;59: 509–522. [DOI] [PubMed] [Google Scholar]
- [50].Yaprak G, Gemici C, Temizkan S, Ozdemir S, Dogan BC, Seseogullari OO. Osteoporosis development and vertebral fractures after abdominal irradiation in patients with gastric cancer. BMC Cancer 2018;18: 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 1998;16: 1113–23. [DOI] [PubMed] [Google Scholar]
- [52].Wang X, Wong SC, Pan J, Tsao SW, Fung KH, Kwong DL, Sham JS, Nicholls JM. Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res 1998;58: 5019–22. [PubMed] [Google Scholar]
- [53].Park JI, Jeong JS, Han JY, Kim DI, Gao YH, Park SC, Rodgers GP, Kim IH. Hydroxyurea induces a senescence-like change of K562 human erythroleukemia cell. J Cancer Res Clin Oncol 2000;126: 455–60. [PubMed] [Google Scholar]
- [54].Ma L, Oei L, Jiang L, Estrada K, Chen H, Wang Z, Yu Q, Zillikens MC, Gao X, Rivadeneira F. Association between bone mineral density and type 2 diabetes mellitus: a meta-analysis of observational studies. Eur J Epidemiol 2012;27: 319–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bonds DE, Larson JC, Schwartz AV, Strotmeyer ES, Robbins J, Rodriguez BL, Johnson KC, Margolis KL. Risk of fracture in women with type 2 diabetes: the Women’s Health Initiative Observational Study. The Journal of clinical endocrinology and metabolism 2006;91: 3404–10. [DOI] [PubMed] [Google Scholar]
- [56].Farr JN, Khosla S. Determinants of bone strength and quality in diabetes mellitus in humans. Bone 2016;82: 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Eckhardt BA, Rowsey JL, Thicke BS, Fraser DG, O’Grady KL, Bondar OP, Hines JM, Singh RJ, Thomson AR, Rakshit K, Vella A, Matveyenko AV, Khosla S, Monroe DG, and Farr JN. Accelerated osteocyte senescence and skeletal fragility in mice with type 2 diabetes. JCI Insight 2020 (In Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Palmer AK, Gustafson B, Kirkland JL, Smith U. Cellular senescence: at the nexus between ageing and diabetes. Diabetologia2019;62: 1835–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Luo J, Quan J, Tsai J, Hobensack CK, Sullivan C, Hector R, Reaven GM. Nongenetic mouse models of non-insulin-dependent diabetes mellitus. Metabolism 1998;47: 663–8. [DOI] [PubMed] [Google Scholar]
- [60].Mu J, Woods J, Zhou YP, Roy RS, Li Z, Zycband E, Feng Y, Zhu L, Li C, Howard AD, Moller DE, Thomberry NA, Zhang BB. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 2006;55: 1695–704. [DOI] [PubMed] [Google Scholar]
- [61].Mu J, Petrov A, Eiermann GJ, Woods J, Zhou YP, Li Z, Zycband E, Feng Y, Zhu L, Roy RS, Howard AD, Li C, Thornberry NA, Zhang BB. Inhibition of DPP-4 with sitagliptin improves glycemic control and restores islet cell mass and function in a rodent model of type 2 diabetes. Eur J Pharmacol 2009;623: 148–54. [DOI] [PubMed] [Google Scholar]
- [62].Burghardt AJ, Issever AS, Schwartz AV, Davis KA, Masharani U, Majumdar S, Ling TM. High-resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 2010;95: 5045–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Farr JN, Drake MT, Amin S, Melton LJ 3rd, McCready LK, Khosla S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2014;29: 787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Litwinoff E, Hurtado Del Pozo C, Ramasamy R, Schmidt AM. Emerging Targets for Therapeutic Development in Diabetes and Its Complications: The RAGE Signaling Pathway. Clin Pharmacol Ther 2015;98: 135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sapieha P, Mallette FA. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol 2018;28: 595–607. [DOI] [PubMed] [Google Scholar]
- [66].Reichel W, Hollander J, Clark JH, Strehler BL. Lipofuscin pigment accumulation as a function of age and distribution in rodent brain. J Gerontol 1968;23: 71–8. [DOI] [PubMed] [Google Scholar]
- [67].Strehler BL, Mark DD, Mildvan AS. GEE MV: Rate and magnitude of age pigment accumulation in the human myocardium. J Gerontol 1959; 14: 430–9. [DOI] [PubMed] [Google Scholar]
- [68].Tonna EA. Accumulation of lipofuscin (age pigment) in aging skeletal connective tissues as revealed by electron microscopy. J Gerontol 1975;30: 3–8. [DOI] [PubMed] [Google Scholar]
- [69].Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Saffrey MJ, Cameron K, von Zglinicki T. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging cell 2012; 11: 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F, Komuro I. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nature medicine 2009;15: 1082–7. [DOI] [PubMed] [Google Scholar]
- [71].Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashimi SK, Prata L, Masternak MM, Kritchevsky S, Musi N, Kirkland JL. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;pii:S2352-3964(18)30629-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, Kellogg TA, Khosla S, Koerber DM, Lagnado AB, Lawson DK, LeBrasseur NK, Lerman LO, McDonald KM, McKenzie TJ, Passos JF, Pignolo RJ, Pirtskhalava T, Saadiq IM, Schaefer KK, Textor SC, Victorelli SG, Volkman TL, Xue A, Wentworth MA, Wissler Gerdes EO, Zhu Y, Tchkonia T, Kirkland JL. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019;47: 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]