

Abstract
Objectives:
Respiratory infections in the postacute phase of traumatic brain injury impede optimal recovery and contribute substantially to overall morbidity and mortality. This study investigated bidirectional innate immune responses between the injured brain and lung, using a controlled cortical impact model followed by secondary Streptococcus pneumoniae infection in mice.
Design:
Experimental study.
Setting:
Research laboratory.
Subjects:
Adult male C57BL/6J mice.
Interventions:
C57BL/6J mice were subjected to sham surgery or moderate-level controlled cortical impact and infected intranasally with S. pneumoniae (1,500 colony-forming units) or vehicle (phosphate-buffered saline) at 3 or 60 days post-injury.
Main Results:
At 3 days post-injury, S. pneumoniae-infected traumatic brain injury mice (TBI + Sp) had a 25% mortality rate, in contrast to no mortality in S. pneumoniae-infected sham (Sham + Sp) animals. TBI + Sp mice infected 60 days post-injury had a 60% mortality compared with 5% mortality in Sham + Sp mice. In both studies, TBI + Sp mice had poorer motor function recovery compared with TBI + PBS mice. There was increased expression of pro-inflammatory markers in cortex of TBI + Sp compared with TBI + PBS mice after both early and late infection, indicating enhanced post-traumatic neuroinflammation. In addition, monocytes from lungs of TBI + Sp mice were immunosuppressed acutely after traumatic brain injury and could not produce interleukin-1β, tumor necrosis factor-α, or reactive oxygen species. In contrast, after delayed infection monocytes from TBI + Sp mice had higher levels of interleukin-1β, tumor necrosis factor-α, and reactive oxygen species when compared with Sham + Sp mice. Increased bacterial burden and pathology was also found in lungs of TBI + Sp mice.
Conclusions:
Traumatic brain injury causes monocyte functional impairments that may affect the host’s susceptibility to respiratory infections. Chronically injured mice had greater mortality following S. pneumoniae infection, which suggests that respiratory infections even late after traumatic brain injury may pose a more serious threat than is currently appreciated.
Keywords: immunosuppression, lung infection, monocyte function, neuroinflammation, Streptococcus pneumoniae, traumatic brain injury
Traumatic brain injury (TBI) is the leading cause of mortality in young adults and a major cause of death and disability across all ages in all countries (1). A major risk factor for poorer outcome after severe TBI is bacterial infection (2). Given the frequent need for mechanical ventilation, ventilator-acquired pneumonia (VAP) represents 23–74% of nosocomial infections in severe TBI patients (3-7) and is a major contributor to post-TBI com-plications (8-11). Further, at 5–20 years after brain trauma, survivors are 2.3–4.3 times more likely to die than the general population, with increased risk of death from pneumonia or sepsis (12).
Streptococcus pneumoniae is a gram-positive bacterium and a common cause of community-acquired pneumonia (13). Neutrophils are the first innate immune cells to respond to S. pneumoniae, heavily recruited to the lung and nasopharynx (13). Neutrophils phagocytose bacteria and produce reactive oxygen species (ROS); this can result in epithelial cell signaling, and infiltration of monocytes into lung (13). Alveolar macrophages are activated after infection; however, it is the infiltrating monocytes that induce critical immune responses. They are activated by damage-associated molecular patterns, such as high mobility group box protein 1 (HMGB1), upregulating nuclear factor kappa B signaling, and interleukin (IL)–1β production (13). Whereas neutrophils are necessary for the initial response to infection, monocytes and IL-1β are essential for bacterial clearance and immune resolution (14).
Clinical studies report marked cellular immunosuppression acutely after TBI (15, 16), which results in alterations in monocyte gene expression and increased susceptibility to infection (17, 18). Experimental studies confirm acute post-traumatic immunosuppression (19, 20). Furthermore, we recently reported that monocytes are hyper-responsive chronically after TBI in mice, producing high levels of IL-1β and ROS (19). However, whether long-lasting alterations in monocyte function have implications for TBI clinically is unknown. This study investigates the immunological and neurologic impact of TBI on the host’s response to S. pneumoniae at 3 (acute) or 60 (chronic) days post-injury to model early- and late-onset pneumonia following severe brain trauma, respectively. Male C57BL/6J mice were subjected to moderate-level controlled cortical impact (CCI) followed by S. pneumoniae infection, and molecular and cellular analyses were used to examine bidirectional effects of brain injury and lung infection.
MATERIALS AND METHODS
Detailed methods are available in the Supplementary File (Supplemental Digital Content 1, http://links.lww.com/CCM/F349).
Animals
Adult male C57BL/6J mice (12-wk-old; Jackson Laboratories, Bar Harbor, ME) were housed in a specific pathogen-free facility (12 hr light/dark cycle). All mice had access to sterilized chow and hyperchlorinated water ad libitum. Animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine.
Acute TBI + Sp model: Sham surgery or CCI was performed on 20–25 mice/surgical condition. At 3 days post-injury, mice were infected intranasally with S. pneumoniae or vehicle (phosphate-buffered saline) (n = 10–15/group). Mice used for flow cytometry, histology, and reverse transcriptasepolymerase chain reaction (RT-PCR) analyses were euthanized at 3 days post-infection. Mice evaluated for mortality were euthanized at 7 days post-infection.
Chronic TBI + Sp model: Sham surgery or CCI was performed on 20–25 mice/surgical condition. At 60 days post-injury, mice were administered S. pneumoniae or PBS (n = 10–15/group). Mice used for flow cytometry, histology, RT-PCR, were euthanized at 3 days post-infection, and mice used for mortality were euthanized 7 days post-infection.
Controlled Cortical Impact
Moderate-level CCI was induced using an impactor velocity of 6 m/s and deformation depth of 2 mm, as previously described (21). Sham mice underwent the same procedure as CCI except for craniectomy and cortical impact.
Infection
S. pneumoniae was prepared as detailed in the Supplementary File (Supplemental Digital Content 1, http://links.lww.com/CCM/F349). Prior to infection, frozen stocks were diluted to 1,500 colony-forming units (CFUs), as previously described (22). Mice were anesthetized with isoflurane prior to administration of 25 μL of S. pneumoniae inoculum to each nare for a total of 50 μL of S. pneumoniae. Mice were not mechanically ventilated at any point during the study. Mice were housed in a Biosafety Level 2 facility until euthanasia. Mice were examined daily for weight loss and mortality. If a mouse lost greater than or equal to 20% body weight, the animal was humanely euthanized.
Bronchoalveolar Lavage
Bronchoalveolar lavage (BAL) was performed as detailed in the Supplementary File (Supplemental Digital Content 1, http://links.lww.com/CCM/F349).
Evans Blue and Fluorescein Tracers
Tail vein injection of Evans blue or sodium fluorescein in mice were performed as detailed in the Supplementary File (Supplemental Digital Content 1, http://links.lww.com/CCM/F349).
Neurobehavior
Mice performed cylinder and grip strength tasks as previously described (23, 24).
Flow Cytometry
Flow cytometry was performed as detailed in the Supplementary File (Supplemental Digital Content 1, http://links.lww.com/CCM/F349).
Quantitative RT-PCR
Messenger RNA (mRNA) analysis was performed as previously described (22, 24).
HMGB1
Enzyme-linked immunosorbent assay (IBL International, Hamburg, Germany) was used to quantify HMGB1 in plasma.
Lung Histopathology
Lung sections were prepared as detailed in the Supplementary File (Supplemental Digital Content 1, http://links.lww.com/CCM/F349). The slides were read in a blind manner (J.C.B.) and examined for parameters of pulmonary inflammation, that is, peribronchiolitis, perivasculitis, interstitial pneumonia, and alveolitis, as previously described (25).
Statistical Analyses
Data were analyzed for normal distribution and are presented as arithmetic mean ± sem. Sham versus TBI alone comparisons were analyzed by Student t test. Mortality data were analyzed using Mantel-Cox analysis. Neurobehavior was analyzed by two-way analysis of variance (ANOVA) with repeated measures and Tukey post hoc analysis. All other studies were analyzed by two-way ANOVA with Tukey post hoc analysis. Statistics were performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA).
RESULTS
TBI Increases Lung Permeability and Infiltration of Myeloid Cells in Lung
To investigate early peripheral responses after TBI, C57BL/6J mice were subjected to sham surgery or moderate-level CCI and plasma was collected at 3, 24, 72 hours, and 7 days post-injury to measure HMGB1 levels. Circulating HMGB1 increased within 3 hours (Fig. 1A), and peaked at 24 hours. Flow cytometry was performed to assess myeloid function in lung. At 24 hours post-injury, there were increased numbers of myeloid cells in the lung of TBI compared with sham mice (Fig. 1B). There were also increased numbers of myeloid cells in BAL fluid of TBI mice (Fig. S2A, Supplemental Digital Content 3, http://links.lww.com/CCM/F351; legend, Supplemental Digital Content 1, http://links.lww.com/CCM/F349). When compared with sham levels, infiltrating Ly6C+ monocytes in lungs of TBI mice had higher transforming growth factor-β (TGFβ) expression (Fig. 1C) and deficits in phagocytic function (Fig. 1D) as demonstrated by reduced latex bead engulfment in an ex vivo phagocytosis assay. There was increased lung permeability after TBI, as shown by an increased permeability ratio of fluorescein in BAL versus plasma (Fig. S2B, Supplemental Digital Content 3, http://links.lww.com/CCM/F351; legend, Supplemental Digital Content 1, http://links.lww.com/CCM/F349) and increased Evans blue dye accumulation in lungs of TBI mice (Fig. S2C, Supplemental Digital Content 3, http://links.lww.com/CCM/F351; legend, Supplemental Digital Content 1, http://links.lww.com/CCM/F349).
Figure 1.
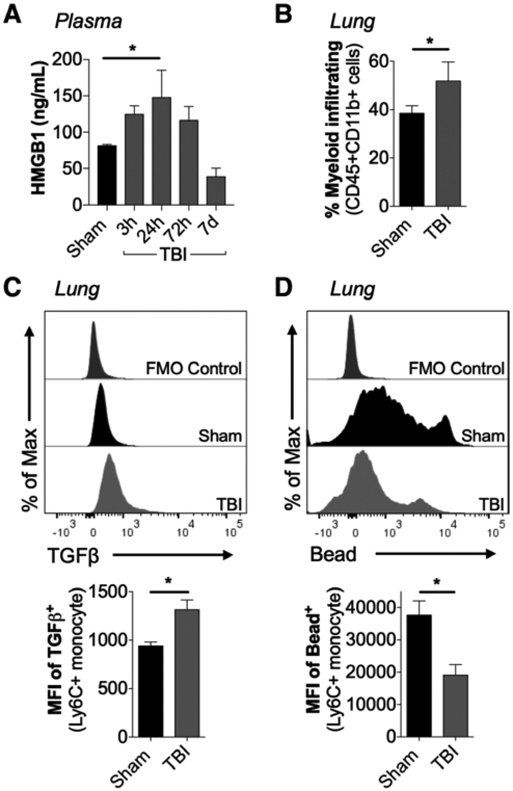
Experimental traumatic brain injury (TBI) alters myeloid cell function in lung. A, Adult male C57Bl/6J mice were subjected to sham surgery or moderate-level controlled cortical impact and plasma was collected at 3, 24, 72 hr and 7 d post-injury. When compared to levels in sham, high mobility group box protein 1 (HMGB1) was increased in plasma of TBI mice (p < 0.05 vs sham). B, Flow cytometric analysis of whole lung tissue collected at 24 hr post-injury demonstrated an increase in lung-infiltrating myeloid cells (CD45+CD11b+) in TBI mice (p < 0.05 vs sham). C, At 24 hr post-injury, TBI increased transforming growth factor-β (TGFβ) production in lung-infiltrating TBI monocytes (CD45+CD11b+Ly6C+Ly6G−) when compared to levels in sham mice (p < 0.05). D, At 24 hr post-injury, lung-infiltrating monocytes had decreased phagocytic activity in TBI mice compared with sham mice (p < 0.05), as measured by latex bead uptake assay. Statistical analyses. *p < 0.05 versus sham; one-way analysis of variance followed by post hoc Tukey test (A). *p < 0.05 versus sham; Student t test (B–D). Mean ± sem; n = 5–8/group. FMO = fluorescence minus one control, MFI = mean fluorescence intensity.
Lung Infection in TBI Mice Delays Motor Function Recovery, Exacerbates Lung Pathology, and Increases Mortality
To model post-traumatic respiratory infection, C57BL/6J mice were subjected to sham surgery or CCI and infected with S. pneumoniae (1,500 CFUs) or vehicle (PBS) at 3 days post-TBI (Fig. 2A). Immune function and lung pathology were assessed at 3 days post-infection, and mortality assessed at 7 days postinfection in a separate cohort of animals. Sp-infected TBI mice (TBI + Sp) exhibited increased mortality (25–30%) compared with Sp-infected sham mice (Sham + Sp, 0%; Fig. 2B). Sham and TBI mice inoculated with vehicle survived. Neurobehavioral testing to assess forelimb function demonstrated that all injured mice had equivalent levels of forelimb impairment in the cylinder task post-TBI (Fig. 2C). However, whereas impairments in TBI + PBS mice recovered, impairments persisted in TBI + Sp mice and were increased compared with TBI + PBS mice (Fig. 2C). Analysis of forelimb grip strength revealed that all mice had equivalent grip strength post-TBI, whereas after S. pneumoniae infection, TBI + Sp mice had reduced grip strength compared with TBI + PBS mice (Fig. 2D).
Figure 2.

Lung infection acutely after traumatic brain injury (TBI) delays motor function recovery and increases mortality and lung pathology in mice. A, Experimental design. Two cohorts of C57Bl/6J mice were subjected to sham surgery or controlled cortical impact on day (D) 0 and were infected with Streptococcus pneumoniae (Sp) (1,500 colony-forming units) or vehicle (phosphate-buffered saline [PBS]) at D3. All mice were tested on the cylinder test and grip strength task at D2 and D6. The first cohort was euthanized at D6 for flow cytometry, quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR), histology, and 16S ribosomal RNA (rRNA) polymerase chain reaction (PCR) analyses. The second cohort was assessed for mortality at D10 (7d post-infection). B, Mortality. In three separate experiments, TBI + Sp mice had increased mortality (25–30%) compared with Sp-infected sham (Sham + Sp) mice (0%; p < 0.05). C, On the cylinder test, Sp-infected TBI (TBI + Sp) mice and TBI + PBS mice had similar impairments and significant left-paw preference (p < 0.01 vs sham; TBI + Sp vs TBI + PBS p > 0.05). Following S. pneumoniae infection, TBI + Sp mice had increased left-paw preference compared with TBI + PBS mice (p < 0.05) on D6. D, On the grip strength task, TBI + Sp and TBI + PBS mice had similar grip strength at D2. Following S. pneumoniae infection, TBI + Sp mice had decreased grip strength as compared with TBI + PBS mice (p < 0.05) on D6. E, Representative hematoxylin and eosin lung images from TBI + Sp and Sham + Sp mice. Scale bar = 500 μm. F, Scoring of peribronchiolitis, perivasculitis, interstitial pneumonia, and alveolitis in the lung revealed an effect of S. pneumoniae infection (p < 0.01 vs Sham + PBS). TBI + Sp mice had significantly increased alveolitis compared with Sham + Sp mice (p < 0.05). Statistical analyses: mortality was analyzed by Mantel-Cox analysis. All other outcomes were analyzed by two-way analysis of variance followed by post hoc Tukey test. *p < 0.05 versus Sham + PBS or TBI + PBS. Mean ± sem; n = 8–15/group.
16S ribosomal RNA (rRNA) for S. pneumoniae was quantified in lung and cortex to assess bacterial burden. Lungs of TBI + Sp mice had lower cycle threshold values for 16S rRNA than Sham + Sp mice, indicating increased bacterial load in lung after TBI (Fig. S3, Supplemental Digital Content 4, http://links.lww.com/CCM/F352; legend, Supplemental Digital Content 1, http://links.lww.com/CCM/F349). Importantly, bacteria were not present in cortex following S. pneumoniae infection (Fig. S3, Supplemental Digital Content 4, http://links.lww.com/CCM/F352; legend, Supplemental Digital Content 1, http://links.lww.com/CCM/F349). Histopathological scoring revealed that TBI + Sp mice had increased alveolitis in lung compared with Sham + Sp mice (Fig. 2, E and F).
Lung Infection After Acute TBI Increases Circulating HMGB1, Brain and Lung Inflammation
At 3 days post-infection, HMGB1 was increased in plasma of TBI + Sp mice when compared with all other groups (Fig. 3A). To determine whether S. pneumoniae infection could alter neuroinflammation, microglial activation markers were assessed. Pro-inflammatory tumor necrosis factor (TNF)–α, IL-1β, and nicotinamide adenine dinucleotide phosphate oxidase (NOX2) mRNA were increased in cortex of TBI + Sp compared with TBI + PBS mice (Fig. 3B). In addition, alternative activation markers, Arginase-1 and chitinase-like protein 3 (YM1) mRNA, were also increased in cortex of TBI + Sp mice (Fig. 3B). There was no effect of infection on other alternative activation (TGFβ, CD206, Fizz1) or inflammasome (NLR family pyrin domain containing 3 [NLRP3], Caspase-1) markers. Analysis of lung focused on mice challenged with S. pneumoniae because TBI alone had no effect on lung inflammation at this time point. When compared with Sham + PBS levels, there was increased expression of pro-inflammatory IL-1β, TNFα, and NOX2 mRNA in lung of Sham + Sp and TBI + Sp mice (Fig. 3C). Notably, there was a reduction in TNFα in TBI + Sp compared with Sham + Sp mice (Fig. 3C). TBI + Sp mice also had increased expression of M2 alveolar macrophage markers, YM1, Arginase-1, and Fizz1 mRNA, compared with levels in Sham + Sp mice (Fig. 3C). Expression levels of other M2 (TGFβ, CD206) and inflammasome (NLRP3, Caspase-1) markers were unchanged by S. pneumoniae.
Figure 3.

Lung infection after acute traumatic brain injury (TBI) increases circulating high mobility group box protein 1 (HMGB1), inflammation in brain and lung, and impairs monocyte functional responses. A, There was a significant increase in circulating HMGB1 in plasma of Streptococcus pneumoniae (Sp)-infected TBI (TBI + Sp) mice compared with all other groups (p < 0.05). B, Analysis of neuroinflammation demonstrated an effect of S. pneumoniae infection on tumor necrosis factor-α (TNFα), interleukin (IL)–1β, and nicotinamide adenine dinucleotide phosphate oxidase (NOX2) messenger RNA (mRNA) expression in cortex of TBI + Sp mice compared with TBI + phosphate buffered saline (PBS) mice (p < 0.05), but no change in NLR family pyrin domain containing 3 (NLRP3) and Caspase-1 mRNA. TBI + Sp mice had increased cortical expression of alternatively activated markers, chitinase-like protein 3 (YM1), and Arginase-1 mRNA (p < 0.05). Other markers (transforming-growth factor-β [TGFβ], CD206, and Fizz1 mRNA) were unaffected by S. pneumoniae infection. Dotted line represents Sham + PBS mRNA expression levels. C, In the lung, TNFα mRNA in Sp-infected sham (Sham + Sp) mice was significantly increased as compared with TBI + Sp (p < 0.05), but IL-1β and NOX2 mRNA expression was not different between infected groups (p > 0.05). Analysis of M2 macrophage markers demonstrated significantly increased expression of YM1, Arginase-1, and Fizz1 mRNA in TBI + Sp as compared with Sham + Sp (p < 0.05). TGFβ, CD206, NLRP3, and Caspase-1 mRNA expression was unchanged with S. pneumoniae infection. Dotted line represents Sham + PBS mRNA expression levels. D, Flow cytometry analysis of lung tissue. Representative dot plots show the gating strategy used to identify myeloid cell populations (CD45+CD11b+) in lung, including neutrophils (Ly6C−Ly6G+) and monocytes (Ly6C+Ly6G−). Sp infection increased the percentage of lung-infiltrating neutrophils in Sham + Sp when compared with Sham + PBS mice (p < 0.05). Neutrophil infiltration was significantly reduced in lungs of TBI + Sp mice (p < 0.05 vs Sham + Sp). E, Analysis of lung-infiltrating monocytes demonstrated that Sham + Sp monocytes produced higher levels of IL-1β (Ei) and TNFα (Eii) than Sham + PBS monocytes (p < 0.05 vs Sham + PBS). IL-1β and TNFα production was significantly reduced in monocytes of TBI + Sp mice (p < 0.05 vs Sham + Sp). Ly6C+ monocytes from TBI + Sp and Sham + Sp mice produced higher levels of reactive oxygen species (ROS) when compared with Sham + PBS (Eiii) (p < 0.05 vs Sham + PBS). There was no difference in ROS levels between monocytes from TBI + PBS and TBI + Sp mice. Statistical analysis: Two-way analysis of variance followed by Tukey post hoc test, *p < 0.05 versus Sham + PBS or TBI + PBS. Mean ± sem; n = 8–15/group. FMO = fluorescence-minus one control, MFI = mean fluorescence intensity, ns = not significant.
S. pneumoniae Infection Impairs Monocyte Functional Responses After TBI
Myeloid function in lung was then assessed by flow cytometry. S. pneumoniae infection increased infiltration of Ly6G+ neutrophils into lungs of sham mice (Fig. 3D); however, TBI mitigated this response such that TBI + Sp mice had reduced numbers of neutrophils when compared with levels in Sham + Sp mice. Analysis of lung-infiltrating Ly6C+ monocytes demonstrated that S. pneumoniae increased IL-1β, TNFα, and ROS production in sham mice (Fig. 3Ei-iii). In contrast, TBI suppressed IL-1β, TNFα, and ROS levels in monocytes of TBI + Sp mice.
Delayed Lung Infection Increases Motor Function Impairments, Exacerbates Lung Pathology, and Increases Mortality in Chronically Injured Mice
To investigate the immunological consequences of a delayed respiratory infection months after TBI, sham and CCI mice were infected with S. pneumoniae (1,500 CFUs) or vehicle (PBS) at 60 days post-injury (Fig. 4A). Immune function and lung pathology were assessed at 3 days post-infection, and mortality assessed at 7 days post-infection in a separate cohort of animals. Chronic TBI mice that received S. pneumoniae (TBI + Sp) had increased mortality (50–60%) when compared with Sham + Sp mice (5–10%; Fig. 4B). Sham and TBI mice inoculated with vehicle survived. In the cylinder task, forelimb impairments persisted in TBI + Sp mice when compared with recovery levels in TBI + PBS mice (Fig. 4C). Similarly, following S. pneumoniae infection, chronic TBI + Sp mice had reduced forelimb grip strength when compared with TBI + PBS mice (Fig. 4D).
Figure 4.

Chronic traumatic brain injury (TBI) mice that are challenged by a delayed Streptococcus pneumoniae (Sp) infection have persistent motor function impairments and increased lung pathology and mortality. A, Experimental design. Two cohorts of C57Bl/6J mice were subjected to sham surgery or controlled cortical impact on day (D) 0 and were infected with S. pneumoniae (1,500 colony-forming units) or vehicle (phosphate-buffered saline [PBS]) at D60. All mice were tested on the cylinder test and grip strength task at D59 and D63. The first cohort was euthanized at D63 for flow cytometry, quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR), histology, and 16S ribosomal RNA (rRNA) polymerase chain reaction (PCR) analyses. The second cohort was assessed for mortality at D67 (7d post-infection). B, Mortality. In three separate experiments, TBI + Sp mice had increased 7-d mortality (50–60%) compared with Sp-infected sham (Sham + Sp) mice (5–10%). C, On the cylinder test, all chronic TBI mice had significant forelimb impairments and showed similar left-paw preferences at D59. Following S. pneumoniae infection, TBI + Sp mice had significantly increased left-paw preference compared with TBI + PBS mice (p < 0.05 vs TBI + PBS) on D63. D, On the grip strength task, all TBI mice had similar grip strength at D59. Following S. pneumoniae infection, TBI + Sp mice had significantly decreased grip strength compared with TBI + PBS mice (p < 0.05 vs TBI + PBS) on D63. E, Representative hematoxylin and eosin lung images from TBI + Sp and Sham + Sp mice. Scale bar = 500 μm. F, Scoring of peribronchiolitis, perivasculitis, interstitial pneumonia, and alveolitis in the lung revealed an effect of S. pneumoniae infection (p < 0.01 vs Sham + PBS). TBI + Sp mice had significantly increased interstitial pneumonia and alveolitis compared with Sham + Sp mice (p < 0.05 vs Sham + Sp). Statistical analysis: mortality was analyzed by Mantel-Cox analysis. All other outcomes were analyzed by two-way analysis of variance followed by Tukey post hoc test. *p < 0.05 versus Sham + PBS or TBI + PBS; ns = not significant. Mean ± sem; n = 8–15/group. D = day, Sham + Sp = Sp-infected sham mice, TBI + Sp = Sp-infected TBI mice.
Chronic TBI + Sp mice had increased bacterial load in the lung following infection (Fig. S4, Supplemental Digital Content 5, http://links.lww.com/CCM/F353; legend, Supplemental Digital Content 1, http://links.lww.com/CCM/F349); there was no evidence of bacteria in the cortex (Fig. S4, Supplemental Digital Content 5, http://links.lww.com/CCM/F353; legend, Supplemental Digital Content 1, http://links.lww.com/CCM/F349). Chronic TBI + Sp mice also had increased interstitial pneumonitis and alveolitis in lung when compared with Sham + Sp mice (Fig. 4, E and F).
Lung Infection After Chronic TBI Increases Circulating HMGB1, Brain and Lung Inflammation
Analysis of HMGB1 in plasma demonstrated that chronic TBI + Sp mice had increased circulating HMGB1 compared with all other groups (Fig. 5A). In the cortex, TNFα and IL-1β mRNA were increased in TBI + Sp compared with TBI + PBS mice (Fig. 5B). YM1 mRNA was also increased in TBI + Sp mice. Expression levels of other alternative activation and inflammasome markers were not changed by S. pneumoniae.
Figure 5.

Lung infection after chronic traumatic brain injury (TBI) increases circulating high mobility group box protein 1 (HMGB1), inflammation in lung, and dysregulates monocyte functional responses. A, Circulating HMGB1 in plasma was significantly increased in chronic Streptococcus pneumoniae (Sp)-infected TBI (TBI + Sp) mice compared with all other groups (p < 0.05). B, S. pneumoniae significantly increased tumor necrosis factor-α (TNFα) and interleukin (IL)–1β messenger RNA (mRNA) in cortex of TBI + Sp mice compared with TBI + phosphate-buffered saline (PBS) mice (p < 0.05). There was no change in nicotinamide adenine dinucleotide phosphate oxidase (NOX2), NLR family pyrin domain containing 3 (NLRP3), and Caspase-1 mRNA. TBI + Sp mice had increased expression of alternative activation marker, chitinase-like protein 3 (YM1) (p < 0.05), but not other markers (Fizz1, transforming-growth factor-β [TGFβ], CD206, Arginase-1). Dotted line represents TBI + PBS mRNA expression levels. C, In the lung, IL-1β and TNFα mRNA in TBI + Sp was significantly increased as compared with Sp-infected sham (Sham + Sp) mice (p < 0.05). NOX2 mRNA was not changed by S. pneumoniae infection. There was significantly increased expression of M2 macrophage markers, YM1 Arginase-1 mRNA in TBI + Sp as compared with Sham + Sp (p < 0.05). Fizz1, TGFβ, CD206 mRNA expression was not changed by S. pneumoniae infection. There was increased expression of NLRP3 and Caspase-1 mRNA in TBI + Sp as compared with Sham + Sp (p < 0.05). Dotted line represents Sham + PBS mRNA expression levels. D, Flow cytometry analysis of infiltrating Ly6G+ neutrophils in lung. S. pneumoniae infection increased the percentage of lung-infiltrating neutrophils in Sham + Sp when compared with levels in Sham +PBS mice (p < 0.05). The effect of S. pneumoniae infection on neutrophil infiltration in lung was significantly increased in chronic TBI + Sp mice (p < 0.05 vs Sham + Sp). E, Flow cytometry analysis of Ly6C+ monocytes in lung. Sham + Sp monocytes produced significantly higher levels of IL-1β (Ei), TNFα (Eii), and reactive oxygen species (ROS) (Eiii) than Sham + PBS monocytes (p < 0.05 vs Sham + PBS). The effect of S. pneumoniae infection on monocyte IL-1β, TNFα, and ROS production was significantly increased in lungs of chronic TBI + Sp mice (p < 0.05 vs Sham + Sp). Statistical analysis: Two-way analysis of variance followed by Tukey post hoc test, *p < 0.05 versus Sham + PBS or TBI + PBS. Mean ± sem; n = 8–15/group. FMO = florescence minus one control, MFI = mean fluorescence intensity, ns = not significant.
In the lung, there was a robust increase in IL-1β mRNA in TBI + Sp compared with Sham + Sp mice (Fig. 5C). In contrast to the acute model (Fig. 3C), TNFα mRNA was increased in chronic TBI + Sp compared with Sham + Sp mice (Fig. 5C). Arginase-1 and YM1 mRNA were also increased in lungs of TBI + Sp compared with Sham + Sp mice. Notably, expression of NLRP3, apoptosis-associated speck-like protein containing C-terminal caspase recruitment domain, and cleaved Caspase-1 protein were also increased in lung of chronic TBI + Sp mice (Fig. 5C; and Fig. S5, Supplemental Digital Content 6, http://links.lww.com/CCM/F354; legend, Supplemental Digital Content 1, http://links.lww.com/CCM/F349).
Delayed S. pneumoniae Infection Increases Monocyte Cytokine and ROS Levels After Chronic TBI
In contrast to the acute model (Fig. 2D), there was increased neutrophil infiltration in lung of chronic TBI + Sp mice when compared with Sham + Sp levels (Fig. 5D). Furthermore, lung-infiltrating monocytes in chronic TBI + Sp mice produced higher levels of IL-1β, TNFα, and ROS when compared with levels in Sham + Sp mice (Fig. 5Ei-iii).
DISCUSSION
Lung infection following severe TBI is a serious complication that increases mortality (8, 9). Our experimental studies confirmed this relationship; TBI mice infected with S. pneumoniae at 3 days post-TBI exhibited 25% mortality compared with 0% in Sham + Sp mice. Furthermore, a delayed S. pneumoniae infection at 60 days post-TBI caused even higher mortality rates (50–60%). Importantly, respiratory infection also exacerbated motor function impairments after TBI. Chronic post-traumatic neuroinflammation was significantly increased following S. pneumoniae infection. In the lung, time-dependent immunosuppression and dysfunctional cellular responses to S. pneumoniae occurred after TBI. Acutely after TBI, lung-infiltrating monocytes had deficits in innate immune functions critical for bacterial clearance. They produced lower levels of pro-inflammatory cytokines and ROS in response to S. pneumoniae, which suggests an immunosuppressed phenotype. In contrast, chronically after TBI monocytes responded more vigorously to bacterial infection and produced high levels of TNFα, IL-1β, and ROS. M2 macrophage markers were also upregulated in lung, perhaps as a host response to counteract the damaging effects of pro-inflammatory agents. However, with marked mortality differences and increased bacterial burden in lung, TBI mice at either acute or chronic time points were unable to respond to S. pneumoniae and control inflammation. These preclinical studies suggest that bidirectional brain-lung interactions influence key innate immune functions in monocytes, which may affect physiologic and neurologic outcomes in severely injured patients.
To our knowledge, this is the first preclinical study to use “living” gram-positive bacteria (S. pneumoniae) that cause respiratory infections commonly contracted by TBI patients (9, 26). Unfortunately, due to safety precautions surrounding the use of living bacteria in our facility, we were unable to comprehensively assess cognitive function using established tests (27). Despite these limitations, we identified differences in sensorimotor function between TBI + Sp and TBI + PBS (Vehicle) mice. It is unclear whether S. pneumoniae-induced neurologic impairments are sustained after TBI or resolve once S. pneumoniae-induced lung inflammation subsides. Future studies to include evaluations of chronic time points post-infection will be required to address this open question. Nevertheless, neurologic differences were associated with increased pro-inflammatory neuroinflammation (TNFα, IL-1β, and NOX2); such markers are expressed by reactive microglia during chronic neuroinflammation (28, 29). Stroke and spinal cord injury studies also reported exacerbated neuroinflammatory responses following secondary challenge with S. pneumoniae or influenza infections (30, 31).
The increased lung pathology and bacterial burden in TBI + Sp may be due to acute immunosuppression. Following TBI, monocytes in the lung had impaired phagocytic activity and produced high levels of TGFβ. Furthermore, monocytes in lung were unresponsive to S. pneumoniae and failed to upregulate TNFα and IL-1β. Other CNS injuries elicit similar levels of peripheral immunosuppression. In experimental stroke studies, acetylcholine release in the spleen causes monocyte functional deficits, leading to increased infection rates (32, 33). Norepinephrine and epinephrine also impact peripheral immunosuppression (31, 34), such that β-receptor blockers inhibit cytokine production and cell migration after CNS injury (35). Further, the effects of glucocorticoids and signaling through the hypothalamus-pituitary-adrenal axis can cause immune paralysis following TBI (36).
TBI patients have increased HMGB1 in plasma within hours of brain trauma (37). Mechanistically, experimental studies indicate that acute lung injury after TBI is caused, in part, by HMGB1 release from damaged endothelial cells in the blood-brain barrier, and subsequent Toll-like receptor 4/receptor for advanced glycation end products signaling in lung—leading to inflammasome activation and inflammation (38-40). In lung injury models, pharmacological blockade of HMGB1 preserves lung function (41), and early HMGB1 inhibition also prevents neurologic deficits following experimental TBI (42, 43). Our “two-hit” model of moderate-severe TBI plus secondary lung infection increased circulating HMGB1 in TBI + Sp mice compared with all other groups. Mice that were infected at 60 days post-injury had the highest levels of HMGB1. Inflammasome components were also robustly increased in lung at 60 days post-injury, indicating that TBI may prime alveolar macrophages/monocytes to produce more IL-1β upon secondary infection by upregulating NLRP3 inflammasome. Cellular analyses confirmed excessive IL-1β production in lung-infiltrating monocytes of chronic TBI + Sp mice. Therefore, if respiratory infections occur during the chronic phase of TBI when monocyte innate immune function (e.g., respiratory burst and phagocytic activity) is impaired (19), excessive HMGB1 release may exacerbate an aggressive immune reaction capable of inducing bystander injury (44).
Because monocytes repopulate within days of a TBI (45), chronic impairments in monocyte function may reflect inherent changes in bone marrow (BM) progenitor cells. Prior immunophenotyping of BM from chronically brain-injured mice revealed long-lived deficits in monocyte function (19). In stroke models, myeloid progenitor stem cells are altered early after the ischemic brain injury, perhaps through epigenetic mechanisms that regulate myelopoiesis (46, 47). The innate immune system can develop memory through epigenetic reprogramming (48), such that re-stimulated BM-derived monocytes may produce a more robust response following secondary challenge. Alternatively, an initial stress response to trauma may epigenetically modify inflammatory genes causing them to become refractory to a subsequent challenge (49, 50). Additional studies are needed to investigate the mechanisms that drive persistent systemic immune impairments chronically after TBI.
Although we identified TBI-induced impairments in systemic immune function that may affect the host’s susceptibility to respiratory infections, our preclinical study has several limitations. In the hospital setting, severe TBI patients often develop VAP acutely after brain trauma (7). Here, we sought to compare early versus late respiratory infection post-TBI. Chronically injured patients are extubated and are more likely to develop community-acquired pneumonia caused by gram-positive bacteria spread by inhalation of aerosols (13). Thus, we developed a model of delayed community-acquired pneumonia that was induced via nasopharynx administration of S. pneumoniae (gram-positive bacteria) rather than through direct tracheal infection with gram-negative bacteria, which would mimic VAP. However, S. pneumoniae is also recognized as a nosocomial pathogen in ICU patient populations (4, 51), including the elderly who are known to have poorer outcomes after TBI. Further, our histopathological assessments of lung revealed a diffuse multi-lobe pneumonia in TBI + Sp mice, which is commonly seen in VAP. Another limitation of this study was that bacterial load in lung post-infection was not assayed by CFU assay but was determined by 16S rRNA analysis. This approach may artificially inflate bacterial load readout due to presence of 16S rRNA from dead bacteria.
Clinically, TBI survivors are at increased risk of death by aspiration pneumonia and sepsis between 5 and 20 years after trauma (12). Here, we showed that chronically injured mice infected with S. pneumoniae had increased mortality, exacerbated motor function impairments and neuroinflammation, dysfunctional monocyte responses, and increased pathology in lung. Although neuroinflammation is known to continue for years following experimental TBI (29), long-term changes in peripheral immunity have only recently been reported (19, 52). In fact, systemic injection of lipopolysaccharide following diffuse TBI in mice exaggerates glial activation and worsens cognitive outcomes (53, 54). Therefore, long-term TBI survivors may be at higher risk of overall worsened neurologic and immunological outcomes following respiratory infection, which has further implications on morbidity and mortality.
Supplementary Material
ACKNOWLEDGMENTS
We thank Xiaoxuan Fan, PhD and Karen Underwood, BSc of the University of Maryland Greenebaum Comprehensive Cancer Center Flow Cytometry Facility for support with flow cytometry studies.
Supported, in part, by grant from the National Institutes of Health grants R01NS082308 (to Dr. Loane), R01NS037313 (to Dr. Faden), F32NS105355 (to Dr. Ritzel), and R01125215 (to Dr. Vogel), and Science Foundation Ireland grant 17/FRL/4860 (to Dr. Loane). Support for Dr. Doran was provided by T32AI095190 (to Dr. Vogel).
Drs. Ritzel, Faden, Vogel, and Loane’s institution received funding from National Institutes of Health (NIH) R01, NIH F32 fellowship, and NIH T32 Training grants. Dr. Loane received support for article research from Science Foundation Ireland (17/FRL/4860). Dr. Blanco disclosed he is an employee of Sigmovir Biosystems, Rockville, MD. The remaining authors have disclosed that they do not have any potential conflicts of interest.
Footnotes
All procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website (http://journals.lww.com/ccmjournal).
REFERENCES
- 1.Maas AIR, Menon DK, Adelson PD, et al. ; InTBIR Participants and Investigators: Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol 2017; 16:987–1048 [DOI] [PubMed] [Google Scholar]
- 2.Sharma R, Shultz SR, Robinson MJ, et al. : Infections after a traumatic brain injury: The complex interplay between the immune and neurological systems. Brain Behav Immun 2019; 79:63–74 [DOI] [PubMed] [Google Scholar]
- 3.Bronchard R, Albaladejo P, Brezac G, et al. : Early onset pneumonia: Risk factors and consequences in head trauma patients. Anesthesiology 2004; 100:234–239 [DOI] [PubMed] [Google Scholar]
- 4.Cazzadori A, Di Perri G, Vento S, et al. : Aetiology of pneumonia following isolated closed head injury. Respir Med 1997; 91:193–199 [DOI] [PubMed] [Google Scholar]
- 5.Kourbeti IS, Vakis AF, Papadakis JA, et al. : Infections in traumatic brain injury patients. Clin Microbiol Infect 2012; 18:359–364 [DOI] [PubMed] [Google Scholar]
- 6.Zygun DA, Zuege DJ, Boiteau PJ, et al. : Ventilator-associated pneumonia in severe traumatic brain injury. Neurocrit Care 2006; 5:108–114 [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Liu C, Xiao W, et al. : Incidence, risk factors, and outcomes of ventilator-associated pneumonia in traumatic brain injury: A meta-analysis. Neurocrit Care 2019. July 12. [online ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbier F, Sonneville R, Boulain T: Prevention of pneumonia after severe traumatic brain injury. Lancet Respir Med 2014; 2:674–675 [DOI] [PubMed] [Google Scholar]
- 9.Esnault P, Nguyen C, Bordes J, et al. : Early-onset ventilator-associated pneumonia in patients with severe traumatic brain injury: Incidence, risk factors, and consequences in cerebral oxygenation and outcome. Neurocrit Care 2017; 27:187–198 [DOI] [PubMed] [Google Scholar]
- 10.Kesinger MR, Kumar RG, Wagner AK, et al. : Hospital-acquired pneumonia is an independent predictor of poor global outcome in severe traumatic brain injury up to 5 years after discharge. J Trauma Acute Care Surg 2015; 78:396–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott BN, Roberts DJ, Robertson HL, et al. : Incidence, prevalence, and occurrence rate of infection among adults hospitalized after traumatic brain injury: Study protocol for a systematic review and meta-analysis. Syst Rev 2013; 2:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison-Felix C, Kolakowsky-Hayner SA, Hammond FM, et al. : Mortality after surviving traumatic brain injury: Risks based on age groups. J Head Trauma Rehabil 2012; 27:E45–E56 [DOI] [PubMed] [Google Scholar]
- 13.Weiser JN, Ferreira DM, Paton JC: Streptococcus pneumoniae: Transmission, colonization and invasion. Nat Rev Microbiol 2018; 16:355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kafka D, Ling E, Feldman G, et al. : Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int Immunol 2008; 20:1139–1146 [DOI] [PubMed] [Google Scholar]
- 15.Quattrocchi KB, Frank EH, Miller CH, et al. : Suppression of cellular immune activity following severe head injury. J Neurotrauma 1990; 7:77–87 [DOI] [PubMed] [Google Scholar]
- 16.Quattrocchi KB, Miller CH, Wagner FC Jr, et al. : Cell-mediated immunity in severely head-injured patients: The role of suppressor lymphocytes and serum factors. J Neurosurg 1992; 77:694–699 [DOI] [PubMed] [Google Scholar]
- 17.Gouel-Cheron A, Allaouchiche B, Guignant C, et al. ; AzuRea Group: Early interleukin-6 and slope of monocyte human leukocyte antigen-DR: A powerful association to predict the development of sepsis after major trauma. PLoS One 2012; 7:e33095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang Y, Xu H, Du X, et al. : Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: A microarray study. J Cereb Blood Flow Metab 2006; 26:1089–1102 [DOI] [PubMed] [Google Scholar]
- 19.Ritzel RM, Doran SJ, Barrett JP, et al. : Chronic alterations in systemic immune function after traumatic brain injury. J Neurotrauma 2018; 35:1419–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vermeij JD, Aslami H, Fluiter K, et al. : Traumatic brain injury in rats induces lung injury and systemic immune suppression. J Neurotrauma 2013; 30:2073–2079 [DOI] [PubMed] [Google Scholar]
- 21.Loane DJ, Pocivavsek A, Moussa CE, et al. : Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med 2009; 15:377–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen WH, Toapanta FR, Shirey KA, et al. : Potential role for alternatively activated macrophages in the secondary bacterial infection during recovery from influenza. Immunol Lett 2012; 141:227–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doran SJ, Ritzel RM, Glaser EP, et al. : Sex differences in acute neuroinflammation after experimental traumatic brain injury are mediated by infiltrating myeloid cells. J Neurotrauma 2019; 36:1040–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritzel RM, Doran SJ, Glaser EP, et al. : Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol Aging 2019; 77:194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanco JC, Core S, Pletneva LM, et al. : Prophylactic antibody treatment and intramuscular immunization reduce infectious human rhinovirus 16 load in the lower respiratory tract of challenged cotton rats. Trials Vaccinol 2014; 3:52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Principi N, Esposito S: Prevention of community-acquired pneumonia with available pneumococcal vaccines. Int J Mol Sci 2016; 18:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Z, Loane DJ, Murray MG 2nd, et al. : Comparing the predictive value of multiple cognitive, affective, and motor tasks after rodent traumatic brain injury. J Neurotrauma 2012; 29:2475–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar A, Barrett JP, Alvarez-Croda DM, et al. : NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav Immun 2016; 58:291–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loane DJ, Kumar A, Stoica BA, et al. : Progressive neurodegeneration after experimental brain trauma: Association with chronic microglial activation. J Neuropathol Exp Neurol 2014; 73:14–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dénes Á, Pradillo JM, Drake C, et al. : Streptococcus pneumoniae worsens cerebral ischemia via interleukin 1 and platelet glycoprotein Ibα. Ann Neurol 2014; 75:670–683 [DOI] [PubMed] [Google Scholar]
- 31.Norden DM, Bethea JR, Jiang J: Impaired CD8 T cell antiviral immunity following acute spinal cord injury. J Neuroinflammation 2018; 15:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lafargue M, Xu L, Carlès M, et al. : Stroke-induced activation of the α7 nicotinic receptor increases Pseudomonas aeruginosa lung injury. FASEB J 2012; 26:2919–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prass K, Meisel C, Höflich C, et al. : Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med 2003; 198:725–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prüss H, Tedeschi A, Thiriot A, et al. : Spinal cord injury-induced immunodeficiency is mediated by a sympathetic-neuroendocrine adrenal reflex. Nat Neurosci 2017; 20:1549–1559 [DOI] [PubMed] [Google Scholar]
- 35.Griffin GD: Stroke, mTBI, infection, antibiotics and beta blockade: Connecting the dots. Med Hypotheses 2015; 85:224–229 [DOI] [PubMed] [Google Scholar]
- 36.Dong T, Zhi L, Bhayana B, et al. : Cortisol-induced immune suppression by a blockade of lymphocyte egress in traumatic brain injury. J Neuroinflammation 2016; 13:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang KY, Yu GF, Zhang ZY, et al. : Plasma high-mobility group box 1 levels and prediction of outcome in patients with traumatic brain injury. Clin Chim Acta 2012; 413:1737–1741 [DOI] [PubMed] [Google Scholar]
- 38.Kerr NA, de Rivero Vaccari JP, Abbassi S, et al. : Traumatic brain injury-induced acute lung injury: Evidence for activation and inhibition of a neural-respiratory-inflammasome axis. J Neurotrauma 2018; 35:2067–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopez-Aguilar J, Blanch L: Brain injury requires lung protection. Ann Transl Med 2015; 3:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicolls MR, Laubach VE: Traumatic brain injury: Lungs in a RAGE. Sci Transl Med 2014; 6:252fs34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Zhang J, Wang B, et al. : Blocking HMGB1 signal pathway protects early radiation-induced lung injury. Int J Clin Exp Pathol 2015; 8:4815–4822 [PMC free article] [PubMed] [Google Scholar]
- 42.Okuma Y, Liu K, Wake H, et al. : Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann Neurol 2012; 72:373–384 [DOI] [PubMed] [Google Scholar]
- 43.Webster KM, Shultz SR, Ozturk E, et al. : Targeting high-mobility group box protein 1 (HMGB1) in pediatric traumatic brain injury: Chronic neuroinflammatory, behavioral, and epileptogenic consequences. Exp Neurol 2019; 320:112979. [DOI] [PubMed] [Google Scholar]
- 44.Hazeldine J, Lord JM, Belli A: Traumatic brain injury and peripheral immune suppression: Primer and prospectus. Front Neurol 2015; 6:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patel AA, Zhang Y, Fullerton JN, et al. : The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med 2017; 214:1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Courties G, Herisson F, Sager HB, et al. : Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res 2015; 116:407–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Denes A, McColl BW, Leow-Dyke SF, et al. : Experimental stroke-induced changes in the bone marrow reveal complex regulation of leukocyte responses. J Cereb Blood Flow Metab 2011; 31:1036–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Netea MG, Joosten LA, Latz E, et al. Trained immunity: A program of innate immune memory in health and disease. Science 2016; 352:aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hotchkiss RS, Monneret G, Payen D: Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat Rev Immunol 2013; 13:862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shirey KA, Perkins DJ, Lai W, et al. : Influenza “Trains” the host for enhanced susceptibility to secondary bacterial infection. mBio 2019; 10:e00810–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paradisi F, Corti G, Cinelli R: Streptococcus pneumoniae as an agent of nosocomial infection: Treatment in the era of penicillin-resistant strains. Clin Microbiol Infect 2001; 7(Suppl 4):34–42 [DOI] [PubMed] [Google Scholar]
- 52.Schwulst SJ, Trahanas DM, Saber R, et al. : Traumatic brain injury-induced alterations in peripheral immunity. J Trauma Acute Care Surg 2013; 75:780–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fenn AM, Gensel JC, Huang Y, et al. : Immune activation promotes depression 1 month after diffuse brain injury: A role for primed microglia. Biol Psychiatry 2014; 76:575–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muccigrosso MM, Ford J, Benner B, et al. : Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun 2016; 54:95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.