

Acinetobacter baumannii remains a leading cause of hospital-acquired infections. Widespread multidrug resistance in this species has prompted the WHO to name carbapenem-resistant A. baumannii as its top priority for research and development of new antibiotics. Many strains of A. baumannii undergo a high-frequency virulence switch, which is an attractive target for new therapeutics targeting this pathogen. This study reports a novel mechanism controlling the frequency of switching in strain AB5075. The rate of switching from the virulent opaque (VIR-O) to the avirulent translucent (AV-T) variant is positively influenced by the copy number of an antibiotic resistance locus encoded on a plasmid-borne composite integron. Our data suggest that this locus encodes a small RNA that regulates opacity switching. Low-switching opaque variants, which harbor a single copy of this locus, also exhibit decreased virulence. This study increases our understanding of this critical phenotypic switch, while also identifying potential targets for virulence-based A. baumannii treatments.
KEYWORDS: Acinetobacter, phenotypic switching, integron, sRNA, virulence regulation
ABSTRACT
We describe a novel genetic mechanism in which tandem amplification of a plasmid-borne integron regulates virulence, opacity variation, and global gene expression by altering levels of a putative small RNA (sRNA) in Acinetobacter baumannii AB5075. Copy number of this amplified locus correlated with the rate of switching between virulent opaque (VIR-O) and avirulent translucent (AV-T) cells. We found that prototypical VIR-O colonies, which exhibit high levels of switching and visible sectoring with AV-T cells by 24 h of growth, harbor two copies of this locus. However, a subset of opaque colonies that did not form AV-T sectors within 24 h were found to harbor only one copy. The colonies with decreased sectoring to AV-T were designated low-switching opaque (LSO) variants and were found to exhibit a 3-log decrease in switching relative to that of the VIR-O. Overexpression studies revealed that the element regulating switching was localized to the 5′ end of the aadB gene within the amplified locus. Northern blotting indicated that an sRNA of approximately 300 nucleotides (nt) is encoded in this region and is likely responsible for regulating switching to AV-T. Copy number of the ∼300-nt sRNA was also found to affect virulence, as the LSO variant exhibited decreased virulence during murine lung infections. Global transcriptional profiling revealed that >100 genes were differentially expressed between VIR-O and LSO variants, suggesting that the ∼300-nt sRNA may act as a global regulator. Several virulence genes exhibited decreased expression in LSO cells, potentially explaining their decreased virulence.
INTRODUCTION
Acinetobacter baumannii is a Gram-negative nosocomial pathogen. This bacterium causes a range of opportunistic infections, including pneumonia, urinary tract infections, meningitis, bloodstream infections, and wound infections (1). A. baumannii infections are frequently difficult to treat due to widespread antimicrobial resistance in this species, which has prompted the WHO to name carbapenem-resistant A. baumannii as its top priority for research and development of new antimicrobials (2). This highlights the need for the identification of new targets for virulence-focused therapeutics for A. baumannii. Although a number of virulence factors have been identified in A. baumannii (reviewed in reference 3), a complete understanding of the regulation of the pathogenesis of this organism is lacking. Many strains of A. baumannii, including isolate AB5075, are capable of undergoing a high-frequency switch between virulent opaque (VIR-O) variants and avirulent translucent (AV-T) variants (4–8). Of these, only the VIR-O variant is capable of causing disease in mice and Galleria mellonella waxworms, and this variant is selected for in vivo (4, 6). This virulence switch represents an attractive target for pathogenesis-focused therapeutics in A. baumannii, but currently, the mechanisms underlying switching are incompletely understood.
Opacity variation in A. baumannii is influenced by a number of gene products. The OmpR/EnvZ two-component system negatively regulates VIR-O to AV-T switching (7), whereas this switch is positively affected by the ArpAB efflux pump (6). The TetR-type transcriptional regulator ABUW_1645 appears to be involved in maintenance of the AV-T state, as this gene is upregulated in the AV-T variant, stimulates conversion of VIR-O cells to the AV-T form when overexpressed, and slightly increases the rate of AV-T-to-VIR-O switching when disrupted (4). ABUW_1645 does not transcriptionally regulate ompR-envZ or arpAB and the hyperswitching phenotype of an ompR mutant does not require ABUW_1645 (4). The VIR-O-to-AV-T switch also requires relA, encoding (p)ppGpp synthetase (9).
p1AB5075 is the largest plasmid carried by A. baumannii AB5075. It is nearly 84 kb in size and harbors resistance island 2 (RI-2) (10). RI-2 consists of two miniature inverted-repeat transposable elements (MITEs) flanking a composite integron (10). This composite integron is composed of two fused class 1 integrons and therefore encodes two highly homologous copies of the integrase gene intI (one of which is actually a pseudogene) (10). The two intI alleles flank the resistance genes aadB, cmlA, aadA1, and strAB (10). We previously showed that the genes between the two copies of intI undergo spontaneous, RecA-dependent high-copy amplification, which results in increased resistance to tobramycin and gentamicin due to increased expression of the aminoglycoside adenylyltransferase gene aadB (11). These resistance genes are presumably expressed from the integron cassette promoter Pc, which is upstream of aadB (12–15). Class 1 integron cassette promoter regions also harbor binding sites for the DNA binding proteins FIS, LexA, IHF, and H-NS, a small open reading frame encoding ORF-11, a nonfunctional peptide whose translation enhances translation of cassette genes, and the integron attI site (12, 16, 17). These promoter characteristics appear to be conserved in RI-2.
In bacteria, small RNAs (sRNAs) are generally 50 to 500 nucleotides (nt) in length and act as posttranscriptional regulators by base pairing to mRNA targets (18). sRNAs typically base pair with their targets over fairly short sequences, usually between 7 and 12 nt (19). sRNAs can act as either positive or negative regulators of mRNA activity (18, 20, 21). These transcripts can be derived from intergenic regions or 5′ or 3′ untranslated regions (UTRs) of coding genes and can be independently transcribed or generated by RNase cleavage of larger transcripts (18, 22–25).
In this paper, we report that A. baumannii produces a third opacity variant along with AV-T and VIR-O. Designated the low-switching opaque (LSO) variant, this third subpopulation exhibits dramatically reduced levels of switching to AV-T relative to that for the VIR-O. Switching frequencies in the LSO and VIR-O variants are controlled by the copy number of a region between the two copies of intI on RI-2 within p1AB5075. We demonstrate that the element controlling switching is encoded at the 5′ end of the aadB gene within RI-2 and that this element is likely an sRNA. Finally, we also show that the LSO variant exhibits decreased virulence relative to that of the VIR-O, which, based on transcriptional profiling, may be due to decreased expression of virulence factors in this variant.
RESULTS
Wild-type AB5075 forms two opaque subpopulations.
Colonies of the VIR-O variant formed distinct sectors after 24 h of growth, which were composed of cells that have switched to the translucent AV-T variant (Fig. 1A). However, we occasionally observed that opaque colonies in our stocks did not sector by 24 h (Fig. 1B). As this low-frequency sectoring phenotype was stable upon restreaking, we concluded that these colonies represented a distinct subpopulation, which was designated low-switching opaque (LSO). Comparison of the switching frequencies between VIR-O and LSO colonies revealed a nearly 3-log difference in switching (Fig. 1C). LSO and VIR-O populations exhibited similar growth rates in LB broth, suggesting that the defect in switching in the LSO variant is not due to decreased growth (see Fig. S1 in the supplemental material). To determine whether the LSO subpopulation was formed due to random mutation, the rates of interconversion between VIR-O and LSO variants were measured in 24-h-old colonies. Interestingly, the rate of conversion from VIR-O to LSO was 4% (Fig. 1D), which was higher than the 0.2% frequency found for the conversion from LSO to VIR-O (Fig. 1E). We also measured the frequency of LSO colonies in stationary-phase overnight cultures inoculated with the VIR-O variant, which yielded a frequency of 2% (Fig. 1F). These frequencies were much higher than would be expected if the conversion between variants was due to the generation of a random point mutation, suggesting that the LSO colonies do not arise by this mechanism.
FIG 1.

Wild-type AB5075 produces two subpopulations of opaque variants. (A) Colonies representative of the VIR-O subpopulation were photographed after 24 h of growth on 0.5× LB with 0.8% agar using a dissecting microscope. The translucent sectors forming around the colony edges are composed of AV-T cells. (B) Colonies representative of the LSO population were photographed after 24 h of growth on 0.5× LB with 0.8% agar using a dissecting microscope. (C) Switching to AV-T of VIR-O and LSO colonies was quantified after 24 h of growth. Data represent averages and standard errors of the means (SEMs) from six colonies collected in two independent experiments. ****, P ≤ 0.0001, unpaired two-tailed t test. (D) Serial dilutions of 24-h-old VIR-O colonies were plated, and the number of resulting LSO colonies was counted to determine the frequency of conversion. (E) Serial dilutions of 24-h-old LSO colonies were plated, and the number of resulting VIR-O colonies was counted to determine the frequency of conversion. For panels D and E, data represent averages and SEMs frim five colonies collected in three independent experiments. (F) Cultures of the VIR-O variant were grown overnight with shaking, serially diluted, and plated to enumerate the frequency of LSO variants. Data represent the average and SEM from three independent cultures.
The LSO and VIR-O subpopulations do not exhibit growth differences in LB broth. Data represent averages and SEMs from two independent experiments (the final two time points were collected in one experiment only). Download FIG S1, TIF file, 1.0 MB (1,013.5KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The rate of switching in different opaque subpopulations is correlated with copy number of an antibiotic resistance locus.
To determine the genetic differences between the VIR-O and LSO subpopulations, representatives from each were subjected to whole-genome sequencing (WGS) using PacBio technology. WGS revealed the VIR-O and LSO cells differed by an approximately 6-kb duplication present within a composite integron (RI-2) on the large plasmid p1AB5075 (10). The duplicated region consists of five genes flanked by two highly homologous copies of the integrase gene intI, which share 99% nucleic acid identity across 1,062 bp (Fig. 2A). In the VIR-O variant, a tandem duplication of the entire locus has occurred, presumably through recombination between the two copies of intI (Fig. 2A). The duplication in this region was confirmed through Southern blotting and quantitative PCR (qPCR) (Fig. 2B) (set A). For the Southern blot, genomic DNA (gDNA) was digested with ScaI, an enzyme that cuts outside the duplication, and the blot was treated with a probe specific for the duplicated region. Results were further confirmed by using qPCR to measure the copy number of aadB, the first gene within the duplication. Results for both of these assays confirmed that the VIR-O variant contains a duplication of this region. Aside from intI, all of the genes in the duplicated region mediate resistance to aminoglycosides or chloramphenicol. As expected, this duplication resulted in modest increases in resistance to some of these antimicrobials (see Table S1).
FIG 2.

Copy number of an antibiotic resistance locus positively correlates with switching frequency to AV-T. (A) PacBio whole-genome sequencing of the VIR-O and LSO isolates revealed a duplication in the VIR-O variant. This duplication comprises part of a composite integron encoded on the plasmid p1AB5075 and consists of five antibiotic resistance genes flanked by two highly homologous copies of intI. (B) Southern blotting and qPCR analysis of the duplicated region were performed with two sets of LSO and VIR-O isolates (set A samples were previously used for PacBio sequencing). For the Southern blot, gDNA was digested with ScaI and probed with digoxigenin-labeled DNA specific for aadB. The table indicates copy number of aadB in each sample as calculated by qPCR. Copy number was normalized to aacA4, a gene harbored on p1AB5075 outside the duplicated region. Data represent averages and standard deviations (SDs) from three independent samples. (C) Switching frequencies to AV-T were measured for set B VIR-O and LSO colonies after 16 h of growth (differences not significant by one-way analysis of variance [ANOVA] with Tukey’s posttest). (D) The AB5075 isolate hetR-O2 was previously reported to harbor 17 to 20 copies of the duplicated region (11). Switching to AV-T of hetR-O2 and VIR-O was measured for colonies after 16 h of growth. *, P ≤ 0.05, unpaired two-tailed t test. (E) An AV-T isolate harboring two copies of the duplicated region was used to derive two isolates harboring a single copy of the region, designated AV-T-56 and AV-T-80. Switching from AV-T to VIR-O was measured for colonies after 24 h of growth (differences not significant by one-way ANOVA with Dunnett’s posttest). In panels C to E, data represent the averages and SEMs from five or six colonies collected in two independent experiments.
The LSO variant exhibits decreased antimicrobial resistance relative to the VIR-O. Resistance was evaluated to the antibiotic targets of resistance genes harbored within the duplicated region on p1AB5075. Strains were directly compared by inoculating the two variants on either side of an Etest strip. Data shown are representative results from a single experiment; experiments were performed twice to assess the reproducibility of trends. a MICs were measured after 16 h of growth at 37°C. b MICs were measured after 6 h of growth at 37°C. The two variants did not exhibit a reproducible difference in resistance to chloramphenicol after 6 h of growth (data not shown). TOB, tobramycin; GEN, gentamicin; STR, streptomycin. Download Table S1, DOCX file, 0.1 MB (64.9KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To determine whether copy number of this region is actually correlated with opacity switching frequency, we measured copy number of this region in a second independent set of interrelated LSO and VIR-O variants. Starting with a VIR-O variant that was independent from that used for WGS, we isolated and stocked a second independent LSO variant. This LSO variant was then used to isolate a new independent VIR-O (VIR-O2). These three related samples were designated “set B” samples to distinguish them from the “set A” samples used for WGS. Southern blot analysis with the set B LSO and starting VIR-O variants gave band sizes similar to those seen for the set A samples (Fig. 2B). Interestingly, set B VIR-O2 yielded a larger band than the other two VIR-O samples, suggesting further amplification of this region. qPCR analysis confirmed the Southern blot results for the set B VIR-O and LSO samples and indicated that set B VIR-O2 harbors four copies of the duplicated region (Fig. 2B). To determine whether VIR-O2 exhibited higher levels of switching due to this further increase in copy number, switching to AV-T of the set B samples was quantified. Switching was measured after 16 h of growth, a time point before VIR-O colonies typically exhibit high levels of switching. VIR-O2 colonies exhibited an 86-fold increase in switching relative to that of the original VIR-O, although the results were variable between samples and differences were not statistically significant (Fig. 2C). However, this trend toward increased switching with increased copy number suggests that amplification of this region does indeed affect switching rate from VIR-O to AV-T.
Our laboratories previously reported that high-level RecA-dependent amplifications of the same duplicated region are responsible for tobramycin heteroresistance in AB5075 (11). To determine whether colonies exhibiting high-level amplification of this region also exhibit hyperswitching from VIR-O to AV-T, we quantified switching in the previously characterized tobramycin-resistant isolate hetR-O2, which harbors 17 to 20 copies of the duplicated region (11). After 16 h of growth, hetR-O2 exhibited a significant 15-fold increase in switching relative to that of the control VIR-O (Fig. 2D). This result further suggests that the VIR-O to AV-T switching rate is controlled by the copy number of this region.
We further evaluated whether copy number of the duplicated region also correlated with an increased AV-T-to-VIR-O switching frequency. To identify AV-T isolates with one copy of the duplicated region, an AV-T isolate harboring two copies was screened for colonies with decreased tobramycin resistance. The resulting colonies (designated AV-T-56 and AV-T-80) were confirmed to have a single copy of aadB by qPCR (data not shown). Quantification of switching in these isolates and the original AV-T demonstrated that switching frequency from AV-T to VIR-O was not affected by copy number of the duplicated region (Fig. 2E). This result provides further evidence that AB5075 employs distinct mechanisms to switch from VIR-O to AV-T and vice versa, as suggested by previous research (6).
To more directly examine the influence of the duplicated region on VIR-O to AV-T switching, the LSO background was used to generate a strain that was cured of the p1AB5075 plasmid, designated LSO Δp1AB5075. LSO Δp1AB5075 exhibited similar levels of switching to that of its parent variant (Fig. 3A), indicating that the plasmid-encoded region is not required for basal levels of VIR-O-to-AV-T switching. To test whether p1AB5075 plasmids harboring two copies of the duplicated region are sufficient to stimulate increased levels of switching, LSO Δp1AB5075 was transformed with total DNA isolated from either the LSO or VIR-O variant to move p1AB5075 back into these strains. Transformants that had taken up p1AB5075 were selected for using tobramycin. Transformants generated using LSO DNA maintained low levels of switching (Fig. 3A). Transformation with VIR-O DNA yielded a mix of low-switching and normally switching colonies (Fig. 3A). When qPCR was used to measure aadB copy number in these transformants, it was determined that a low-switching transformant (transformant A) had taken up a plasmid containing one copy of the duplicated region, whereas a switching transformant (transformant B) had taken up a plasmid containing two copies (Fig. 3B). This experiment demonstrates that uptake of p1AB5075 harboring two copies of the duplicated region is sufficient to increase VIR-O-to-AV-T switching in AB5075.
FIG 3.

p1AB5075 carrying two copies of the duplicated region is sufficient to increase switching to AV-T. (A) Strain LSO Δp1AB5075, which has been cured of the p1AB5075 plasmid, was retransformed with DNA derived from the LSO or VIR-O variant to restore p1AB5075, and switching to AV-T was measured for parent variant and transformant colonies after 24 h of growth. Data represent averages and SEMs from six colonies collected in three independent experiments. ***, P ≤ 0.001; ****, P ≤ 0.0001 relative to LSO Δp1AB5075 by one-way ANOVA with Dunnett’s posttest. (B) Copy numbers of the aadB gene were measured for variants shown in panel A by qPCR. Data represent averages and SDs from two independent experiments. ND, not detectable.
Overexpression of the aadB gene leads to increased opacity switching, but AadB protein is dispensable for this phenotype.
To identify the gene(s) responsible for stimulating switching within the duplicated region, portions of this region were overexpressed in the LSO variant, and switching to AV-T was examined. We first used plasmid pWHaadB, consisting of the aminoglycoside resistance gene aadB and upstream DNA cloned into plasmid pWH1266; this plasmid exhibits an average copy number of 8.63 ± 5.73. This plasmid stimulated switching >800-fold in the LSO background (Fig. 4). A plasmid construct was also generated containing all five resistance genes in the duplicated region; this stimulated similar levels of switching in an LSO background as that of a construct harboring aadB alone, suggesting that all of the stimulatory activity in the duplicated region was derived from aadB (see Fig. S2).
FIG 4.
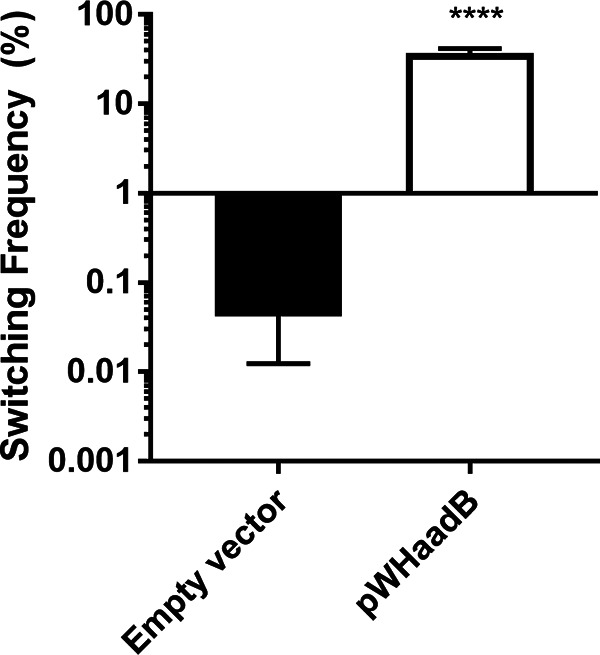
Overexpression of the aadB region is sufficient to stimulate switching. The aadB gene was cloned along with its native promoter into the vector pWH1266 (60) and transformed into the LSO variant. Switching to AV-T was measured for colonies after 24 h of growth. Data shown represent averages and SEMs from at least five colonies collected in two independent experiments. ****, P ≤ 0.0001 by unpaired two-tailed t test.
Overexpression of the entire duplicated region increases switching to the same level as overexpression of aadB alone. The aadB gene (paadB) and the duplicated region stretching from strB to aadB (pDR) were cloned into vector pQF1266.Blue and overexpressed in the LSO variant. Switching to AV-T was measured for colonies after 24 h of growth, indicating that pDR and paadB both stimulate switching similarly compared to the empty vector control. Data shown represent averages and SEMs from at least five colonies collected in two independent experiments. ****, P ≤ 0.0001, one-way ANOVA with Tukey’s posttest. Download FIG S2, TIF file, 0.3 MB (288KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
AadB mediates aminoglycoside resistance by adenylylating the antibiotic; this enzyme is not known to act on cellular substrates. Therefore, it was unexpected that overproduction of this protein would have an effect on VIR-O-to-AV-T switching. To determine if the AadB protein was mediating the effect on switching, a nonsense mutation was introduced at the fifth amino acid of aadB (pWHaadBstop). This allele was still capable of stimulating switching >800-fold when overexpressed in the LSO background (see Fig. S3), demonstrating that AadB is not responsible for regulating the rate of VIR-O-to-AV-T switching.
The AadB protein is not required for stimulation of switching by the aadB region. A stop codon was introduced into the fifth amino acid of aadB on plasmid pWHaadB, and the resulting plasmid (pWHaadBstop) was transformed into the LSO variant. Data shown represent averages and SEMs from six 24-h-old colonies collected in two independent experiments. **, P ≤ 0.01 by unpaired two-tailed t test. Download FIG S3, TIF file, 0.2 MB (211.8KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The element responsible for controlling switching rate is encoded at the 5′ end and upstream of aadB.
To determine where the element affecting switching was localized in the pWHaadB plasmid insert, we mutagenized this plasmid with the EZ-Tn5 <Kan-2> transposon. Mutant plasmids were transformed into the LSO variant, and two screens were performed. Insertions that blocked the ability of the plasmid to cause switching were identified by screening for transformants that no longer exhibited the highly sectored colony phenotype stimulated by pWHaadB. Insertions in aadB that did not abolish switching were also isolated by screening for plasmids that had lost the ability to confer tobramycin resistance in Escherichia coli followed by a screen for plasmids that still stimulated switching in the LSO variant. When both groups of insertions were mapped onto the aadB region, all of the insertions blocking switching localized to the 5′ end and upstream of aadB, while most of the insertions within the aadB open reading frame (ORF) did not affect switching (Fig. 5A). Insertions blocking switching were relatively evenly distributed between the 5′ portion of aadB and the Pc promoter, suggesting that the element affecting switching is encoded in this region.
FIG 5.

The region responsible for stimulating switching is encoded toward the 5′ end and upstream of aadB. (A) The insert in plasmid pWHaadB contains part of the 5′ end of the intI ORF (gray rectangle), as well as the promoters for intI (PintI, gray bent arrow) and aadB (Pc, green bent arrow). To determine which part of the insert was responsible for stimulating switching, pWHaadB was mutagenized with EZ-Tn5 <Kan-2>. Insertions that blocked the stimulation of switching are indicated by closed triangles, whereas insertions that did not affect sectoring are indicated by open triangles. Plasmids were then generated containing truncated inserts of different lengths to further test for the stimulation of switching in the LSO variant. The remaining length of each plasmid insert is indicated in parentheses. Plasmids pWHaadB-T (B), pWHaadB-TA (C), pWHaadB-TB (D), and pWHaadB-UP (E) were transformed into the LSO variant, and switching to AV-T was measured for 24-h-old colonies. Data shown represent averages, SEMs, and individual switching frequencies from six colonies collected from at least two independent experiments. ****, P ≤ 0.0001, one-way ANOVA with Tukey’s posttest.
To confirm that the element responsible for stimulating switching is encoded toward the 5′ end and upstream of aadB, truncated fragments of the aadB region were overexpressed in the LSO variant. As depicted in Fig. 5A, truncated insertions extended by differing lengths into the aadB gene. Plasmids pWHaadB-T (harboring the longest truncated insert), pWHaadB-TA, and pWHaadB-TB all appeared to stimulate switching in the LSO variant, although not to the same degree as the full-length pWHaadB plasmid (Fig. 5B to D). For unknown reasons, colonies overexpressing these plasmids exhibited high variability in their levels of switching, resulting in differences in switching between these transformants not being significantly higher than those of the empty vector controls. However, these plasmids were still classified as being biologically active, as transformants clearly exhibited more sectoring/switching than empty vector controls on average. The shortest truncated insert, pWHaadB-UP, did not stimulate sectoring and appeared similar to the empty vector control in quantitative switching assays (Fig. 5E). The results of these truncation studies confirm those obtained through transposon mutagenesis of pWHaadB. The fragment encoded by pWHaadB-UP terminates upstream of some of the transposon insertions that blocked switching by pWHaadB, whereas the shortest truncated plasmid to still stimulate switching, pWHaadB-TB, includes the insertion sites for all transposons that blocked switching. Taken together, these results indicate that the element responsible for stimulating switching is encoded at the 5′ end and upstream of aadB.
Possible role for an sRNA in VIR-O-to-AV-T switching.
We hypothesized that the element affecting switching was likely a regulatory sRNA or a small peptide encoded upstream of aadB, but our data could also be explained by the presence of a protein binding site titrating a regulator required for controlling switching. To discriminate between these possibilities, site-directed mutagenesis was used to introduce a two-base pair change in the −35 region of Pc (TTGACA→TTGAGT) in plasmid pWHaadB. This mutation decreased transcription 25-fold from that of the Pc promoter (see Fig. S4A) and the mutated plasmid was no longer able to stimulate switching when overexpressed in the LSO background (Fig. S4B), indicating that transcription from Pc was required for the upstream element to increase switching. This result indicates that the upstream element is likely an sRNA or a small peptide. While there are a number of small putative ORFs encoded within the upstream region of aadB, transposon insertions affecting switching stimulated by pWHaadB do not localize to any of these potential peptides, suggesting that none of them are responsible for controlling the switching rate (see Fig. S5).
Transcription from Pc is required for the stimulation of switching. (A) Two single base changes were introduced to disrupt the −35 region of Pc in pWHaadB (TTGACA→TTGAGT). The resulting plasmid (pWHaadB-Q35) exhibited a 25-fold decrease in promoter activity as measured by qRT-PCR. Data represent averages and SEMs from three independent samples. ***, P ≤ 0.001 relative to empty vector control, one-way ANOVA with Dunnett’s posttest. (B) Plasmid pWHaadB-Q35 did not stimulate switching in the LSO background relative to that of an empty vector control at 24 h of growth. Data represent averages and SEMs from six colonies collected in two independent experiments (differences are not significant by unpaired two-tailed t test). Download FIG S4, TIF file, 0.8 MB (795.5KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Transposon insertions blocking switching do not localize to any predicted ORFs encoded in the aadB 5′ region. The insert in pWHaadB-TB, the shortest plasmid insert that appears to stimulate switching, is shown. Sequence features are as indicated in Fig. 5A. Predicted ORFs in this region were identified using NCBI ORFfinder using both ATG and alternative start codons. ORFs are represented by black arrows; only ORFs completely contained within the pWHaadB-TB fragment are shown. The transposon insertions blocking switching do not align specifically with any of these ORFs, suggesting that none of these ORFs are responsible for the switching phenotype stimulated by this construct. Download FIG S5, TIF file, 0.2 MB (217.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Based on the above-described results, the possibility that an sRNA controlled the rate of switching was investigated. To determine whether any small transcripts were produced from the Pc promoter, Northern blotting was performed on RNA separated on a 10% acrylamide urea gel. Initial Northern blots were conducted with probe oSA134 (see Table S2; Fig. S6A), a digoxigenin-labeled single-stranded DNA oligonucleotide probe that only binds transcripts derived from the Pc promoter. This probe hybridizes starting 40 nt downstream of the reported transcriptional start site for Pc (26). Northern blotting was performed using LSO Δp1AB5075 overexpressing pWHaadB, pWHaadB-TA, pWHaadB-TB, and pWHaadB-UP along with an empty vector control (Fig. 6). Strains containing the three longer inserts all exhibited the same five major RNA products, which were roughly 300, 150, 110, 80, and 70 nt in size. All of these transcripts are too small for the full aadB gene (the aadB ORF is 534 bp). While a larger transcript encoding the aadB gene is not evident on this blot, transcripts in excess of 300 nt are present on other blots using another probe for this region (Fig. S6). Interestingly, in the strain overexpressing pWHaadB-UP, the ∼300-nt RNA was largely absent. Since this plasmid was not capable of stimulating switching in the LSO background, this result suggests that the ∼300-nt RNA may be involved in stimulating switching.
FIG 6.
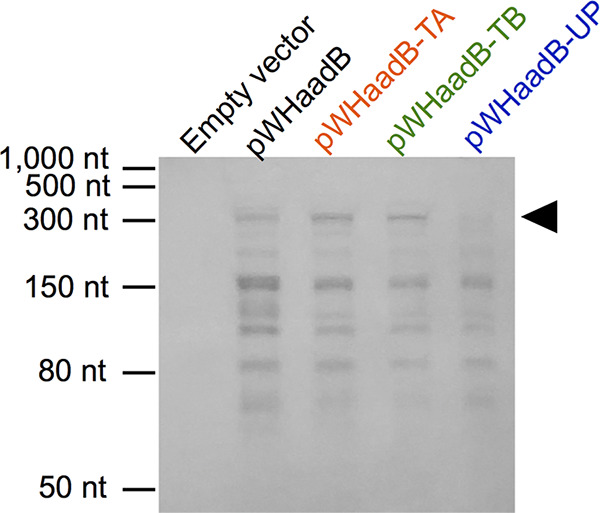
Small RNA species are produced from the 5′ end and upstream of aadB. Northern blotting was performed using oSA134 to examine RNAs produced from the Pc promoter on overexpression plasmids transformed into LSO Δp1AB5075. An ∼300-nt RNA appears to be missing or reduced in sample pWHaadB-UP (black arrowhead).
The 3′ end of the ∼300-nt sRNA was identified by Northern blotting. (A) Three additional probes (oSA190, oSA191, and oSA197) were used for Northern blotting. All probes are 30-nt single-stranded oligonucleotides that should only bind transcripts produced from the same strand as aadB; probes hybridize to the region indicated. Other sequence features are shown as in Fig. 5A. (B) Northern blotting was performed using oSA190, oSA191, and oSA197 to examine RNAs produced from pWHaadB transformed into LSO Δp1AB5075. The ∼300-nt RNA was only weakly bound by oSA197 (white arrowhead). (C) A dot blot was performed using 2-fold serial dilutions of AB5075 gDNA and oSA197 to assess the ability of this probe to bind nucleic acids. Probe oSA191 was used as a positive control. Download FIG S6, TIF file, 1.5 MB (1.5MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Oligonucleotides used in this study. DIG, digoxigenin. Download Table S2, DOCX file, 0.1 MB (90.8KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To identify the endpoint of the ∼300-nt sRNA, additional Northern blotting was performed using probes that bind toward the 3′ end of this transcript. Probe oSA190 was designed to bind 209 nt downstream of the end of oSA134 (Table S2; Fig. S6A). Probe oSA191 binds immediately 3′ of oSA190, and oSA197 binds immediately 3′ of oSA191 (Table S2; Fig. S6A). Northern blotting of LSO Δp1AB5075 pWHaadB with these probes revealed that both oSA190 and oSA191 bind strongly to the ∼300-nt RNA (as evidenced by the production of dark bands on the Northern blot), whereas oSA197 binds this band only weakly (Fig. S6B). Dot blotting of gDNA with oSA197 confirmed that this probe is not defective for binding in general (Fig. S6C), suggesting that the ∼300-nt RNA terminates within the region bound by oSA197. This would make the ∼300-nt RNA between 339 and 368 nt in length, assuming that the transcript begins at the reported start site for Pc (26).
To determine whether the ∼300-nt RNA was produced via RNase processing, we overexpressed aadB in single mutants defective for all nonessential ribonucleases obtained from the AB5075 transposon library (10). VIR-O mutants were qualitatively evaluated for their degree of switching to AV-T when aadB was overexpressed. Mutants that did not exhibit clearly increased switching were examined by Northern blotting. None of the evaluated mutants showed consistent differences in production of the ∼300-nt band, while minor differences in the intensities of some other bands were observed for some ribonucleases (see Fig. S7). The results of these experiments are summarized in Table 1.
TABLE 1.
Effects of aadB overexpression and transcript accumulation in RNase mutantsa
| Accession no. | Gene name | Result of aadB overexpression | aadB transcript accumulation |
|---|---|---|---|
| ABUW_0038 | NAb | Increased switchingc | NDd |
| ABUW_0444 | rnr | Switching not increased | Minor changes in band intensity |
| ABUW_0456 | NA | Increased switchingc | ND |
| ABUW_0466 | NA | Increased switching | ND |
| ABUW_0719 | cafA | Switching not increased | No change |
| ABUW_1032 | rnc | Inconsistent switching phenotype | No change |
| ABUW_1369 | rnz | Increased switching | ND |
| ABUW_2146 | NA | Increased switching | ND |
| ABUW_2751 | rnhB | Increased switching | ND |
| ABUW_2826 | rnt | Increased switching | ND |
| ABUW_3316 | NA | Increased switching | ND |
| ABUW_3367 | NA | Increased switching | ND |
| ABUW_3478 | rne | Too mucoid to see switching | No change |
| ABUW_3518 | pnp | Increased switching | ND |
| ABUW_3841 | rph | Inconsistent switching phenotype | No change |
| ABUW_5006 | NA | Switching not increased | No change |
VIR-O mutants were transformed with aadB overexpression plasmids and were qualitatively assessed for increased switching based on the degree of colony sectoring on 0.5× LB with 0.8% agar. Mutants that did not clearly exhibit increased switching due to aadB overexpression were further evaluated by Northern blotting with oSA134.
NA, not available.
aadB was overexpressed using plasmid pWHaadB (tetracycline resistant), as transposon mutations conferred hygromycin resistance. All other mutations conferred tetracycline resistance, and so aadB was overexpressed using plasmid paadB (hygromycin resistant).
ND, not determined.
Northern blotting does not indicate a role for ribonucleases in the synthesis of the 300-nt sRNA. (A) T26 insertions in rnr (ABUW_0444), cafA (ABUW_0719), rne (ABUW_3478), and ABUW_5006 were moved into a VIR-O variant and electroporated with paadB or an empty vector control. Northern blotting was performed for all samples alongside WT VIR-O paadB and an empty vector control using probe oSA134. (B) T26 insertions in rph (ABUW_3841) and rnc (ABUW_1032) were moved into an LSO Δp1AB5075 strain and electroporated with paadB. Northern blotting was performed for samples alongside an LSO Δp1AB5075 paadB control using oSA134. Download FIG S7, TIF file, 0.9 MB (986.5KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
VIR-O and LSO subpopulations exhibit differences in virulence and global gene expression.
We hypothesized that because the VIR-O variant is more virulent than the AV-T variant, and because the LSO variant switches to AV-T at a lower frequency, that the LSO variant would exhibit increased virulence relative to that of VIR-O. To evaluate this possibility, mice were intranasally inoculated with either the VIR-O or LSO variant, and CFU were enumerated in the lungs, spleen, and liver at 24 h postinfection (hpi) (Fig. 7). Surprisingly, the LSO variant exhibited at least a 3-log decrease in the geometric mean of CFU in all three organs, demonstrating that this variant is less virulent than VIR-O.
FIG 7.

The LSO variant exhibits decreased virulence in a mouse lung infection model. Mice were inoculated intranasally with 5 × 107 CFU of VIR-O or LSO bacteria, and CFU were enumerated at 24 hpi. The LSO variant showed decreased colonization of the lungs (A) and dissemination to the spleen (B) and liver (C) compared to that of the VIR-O. Data represent the geometric mean of CFU counts from 10 mice. ***, P ≤ 0.001 by two-tailed Mann-Whitney U test.
To obtain a global view of gene expression differences between VIR-O and LSO variants, and to determine why the LSO variant might be less virulent, RNA sequencing (RNA-seq) was performed on each subpopulation in triplicates. Results of the RNA-seq are detailed in Data Set S1. Using a fold change cutoff of 1.5 (log2-fold 0.7) and a P value cutoff of 0.05, we identified 55 upregulated genes and 87 downregulated genes in the LSO variant. As expected, the genes within the duplicated region were all downregulated in the LSO variant. Interestingly, several genes involved in pathways that were previously implicated in A. baumannii virulence were found to be downregulated in the LSO variant, including the phospholipase C gene plc1 (27), genes involved in cysteine metabolism/sulfur assimilation (cysW, cysT, and cysN) (28), and genes involved in the phenylacetic acid catabolic pathway (paaK, paaJ, and paaI2) (29, 30). The LSO variant also showed decreased expression of genes from the csu operon, which is involved in the production of pili important for surface adherence and biofilm formation (30–32). These differences in gene expression may underlie the decreased virulence observed in the LSO strain.
RNA-seq results comparing the VIR-O and LSO. Triplicate cultures of the VIR-O and LSO variants were grown to mid-log phase and subjected to RNA-seq. Data sets indicate genes that were up- or downregulated at least 1.5-fold (log2-fold of 0.7) in the LSO variant. Only genes that exhibited P values of 0.05 or less are shown. Download Data Set S1, XLSX file, 0.4 MB (372.6KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
This study establishes that A. baumannii AB5075 produces two distinct subpopulations of opaque variants, designated VIR-O and LSO. The two subpopulations differ not only in their rates of switching to AV-T but also in their gene expression and virulence. The switching frequencies of these opaque variants were positively correlated with copy number of a locus in RI-2 on plasmid p1AB5075, with VIR-O variants exhibiting two or more copies of this locus. Copy number of this locus appears to vary through homologous recombination between two alleles of intI, leading to the interconversion of the LSO and VIR-O subpopulations. The VIR-O variant gives rise to the LSO variant at a higher frequency than the reverse; this is likely due to the inherent instability of tandem duplications, which are easily lost through recombination between their large regions of homology (33). High-level amplification of this locus can also occur, leading to hyperswitching to AV-T and increased resistance to tobramycin (11). The element controlling switching is encoded toward the 5′ end and upstream of the aadB gene in RI-2, and our evidence suggests that this element is likely an sRNA. Switching stimulated by overexpression of aadB required transcription from the integron cassette promoter Pc but did not appear to require any of the small ORFs predicted upstream of aadB. Northern blotting revealed multiple small transcripts produced downstream of Pc, providing further evidence that one or more sRNAs encoded in this region are responsible for controlling switching.
While our results suggest that an sRNA produced from the 5′ end of the aadB transcript is responsible for stimulating switching, there are many open questions about this model that remain to be answered. Our Northern blotting results have specifically implicated a transcript of ∼300 nt in switching, as an sRNA of this length was found to be produced from overexpression plasmids that stimulated switching but was reduced or absent from a plasmid lacking stimulatory activity (Fig. 6). Future studies should determine the precise sequence of this sRNA as well as the mechanism by which it is produced. Disruption of nonessential ribonucleases did not reveal any differences in production of the ∼300-nt sRNA (Table 1), suggesting that this sRNA is either generated by transcriptional termination or via processing by an essential RNase not examined here. Finally, the target(s) of this sRNA, as well as any chaperones involved in target binding, remains to be identified. Our data indicate that the target(s) involved in switching is not encoded on p1AB5075, as LSO Δp1AB5075 transformed with pWHaadB still exhibits increased switching (data not shown). Genes previously implicated in switching such as ompR, envZ, arpAB, ABUW_1645, and relA were not differentially expressed between LSO and VIR-O variants based on the RNA-seq analysis. However, since sRNAs act at the posttranscriptional level, regulation of these genes by the sRNA may be missed in this analysis. Identification of the chromosomal target(s) of this sRNA will be extremely valuable for understanding the regulation of switching in A. baumannii.
The putative sRNA described here exhibits a number of unexpected genetic and regulatory features. First, it is interesting that the putative sRNA is encoded on a transmissible plasmid. Although this is unusual, there is precedence for horizontally acquired sRNAs being produced in other bacterial species. For example, Salmonella enterica serovar Typhimurium has been shown to produce sRNAs from horizontally acquired genetic elements (34, 35). One of these, TnpA, is produced from the 5′ UTR of a transposase gene and functions to regulate expression of virulence genes by directly binding to mRNA of the pathogenicity regulator invF (35). A plasmid-derived sRNA has also been shown to regulate genetic competence in Legionella pneumophila (36). sRNAs associated with mobile elements have also been identified in Staphylococcus aureus (37), Xanthomonas campestris (38), and Coxiella burnetii (39). Taken together, these studies and ours suggest that horizontally transferred elements may be an underappreciated source of sRNAs.
It is also unusual that two or more copies of the sRNA-encoding locus are required to stimulate significant levels of opacity switching. Figure 3 demonstrates that the sRNA is not required for baseline levels of switching, as strain LSO Δp1AB5075, which does not harbor the sRNA, exhibits the same level of opacity switching as the wild-type LSO. Changes in switching to AV-T are only observed when copy number of the sRNA-encoding locus is increased, either through spontaneous amplification (Fig. 2 and 3) or overexpression on a multicopy plasmid (Fig. 4). This suggests that the sRNA must be expressed past a certain threshold before switching can be activated. This could potentially occur if its mRNA target(s) involved in switching is produced in great excess to the sRNA; perhaps when the sRNA is encoded in single copy, it is produced to insufficient levels to affect switching.
It is also interesting that the truncated plasmids pWHaadB-T, pWHaadB-TA, and pWHaadB-TB all caused much more variable switching phenotypes than the full-length plasmid pWHaadB. Although LSO colonies overexpressing the truncated plasmids exhibited increased average switching compared to that of an empty vector control, the differences were not found to be statistically significant due to the wide variability. The reason for this variability is unclear but could be due to variable levels of sRNA production from these plasmids. Even if the entire sequence for the active sRNA is harbored by each of these plasmids, it is possible that truncation of the inserts could affect the efficiency of posttranscriptional processing or transcriptional termination, which could potentially result in variable levels of transcripts between cells. If the sRNA only causes an observable phenotype when expressed past a certain threshold, then slight variations in transcript levels between colonies could result in dramatic differences in switching phenotypes. However, more research is needed to confirm this hypothesis.
This work is not the first report of a regulatory RNA element encoded in the promoter region of class 1 integrons. The promoter regions of aminoglycoside resistance integrons have also been reported to encode an aminoglycoside-responsive riboswitch, although the presence of this element is controversial (40–42). In 2013, Jia et al. characterized a putative 76-nt riboswitch that exhibits 97% sequence identity across 61 bp to the 5′ UTR of aadB (40). Although the putative sRNA described here extends over a larger sequence than this riboswitch, it is interesting that the RI-2 promoter region may encode both a riboswitch and an sRNA. A riboswitch was recently reported to affect stability of an upstream sRNA harbored by Vibrio cholerae (43). In Listeria monocytogenes, a riboswitch has also been shown to act as an sRNA in trans (23), and a riboswitch-containing sRNA has also been identified in Enterococcus faecalis (44). More work will be needed to determine the interplay in regulation between the putative integron riboswitch and the sRNA described here.
It remains to be seen whether the mechanism controlling switching frequency described here is widespread in A. baumannii. A BLAST search with the first 300 nt of the aadB transcript yields hits to >100 A. baumannii genomes, with coverage ranging from 73% to 100%. Integrons encoding aminoglycoside modifying enzymes, including aadB, are also widely reported in the A. baumannii literature (45–50). As the ∼300-nt sRNA appears to be encoded mostly within the integron promoter region, it is possible that other integrons, even those not harboring aadB, still contain this transcript. Together, these pieces of evidence suggest that the active sRNA is likely to be widespread in this species. However, the data presented here demonstrate that in AB5075, the presence of this sRNA in single copy is not enough to stimulate switching, as duplications of the sRNA-encoding region are also required. Our results suggest that in AB5075, amplification likely occurs through homologous recombination between flanking intI alleles within the RI-2 integron (11). As of this writing, RI-2 has only been identified in two other A. baumannii strains besides AB5075 (51, 52), suggesting that this mechanism of copy number variation may be restricted to certain A. baumannii strains. However, in other strains, amplifications of this locus could occur by other mechanisms, including transposition or increases in plasmid copy number. Indeed, amplification of a different aminoglycoside resistance-encoding integron has been reported in a clinical isolate of A. baumannii, even though this strain lacks the flanking intI alleles (53). Any A. baumannii strains that do not harbor the active sRNA (or are incapable of varying its copy number or expression) would be expected to behave as LSO variants. However, these strains could still have the ability to acquire the active sRNA, since RI-2 is carried on a transmissible plasmid (10, 51, 54). As VIR-O variants harboring duplications within RI-2 exhibit increased virulence, dissemination of this plasmid could be of public health concern.
It was unexpected that the LSO variant exhibited decreased levels of virulence compared to that of the VIR-O variant. Previous studies comparing virulence of VIR-O and AV-T isolates suggested that differences in virulence between the two variants are due to levels of capsule, which is a virulence factor that contributes to A. baumannii resistance to host lysozyme (4, 55). Increased capsule levels partly underlie the appearance of opaque A. baumannii colonies, as AB5075 VIR-O mutants lacking capsule appear more translucent than the wild type (55). However, the in vivo results observed for the LSO variant demonstrate that an opaque colony appearance does not necessarily translate to virulence. While one could hypothesize that the decreased virulence of the LSO variant indicates a role for the AV-T form in infection, preliminary infection experiments with mixtures of LSO and AV-T variants indicate that this is not the case (data not shown). This suggests that other intrinsic factors in addition to capsule contribute to the increased virulence of the VIR-O variant compared to LSO. We hypothesize that the LSO variant is less virulent due to its decreased expression of multiple virulence factors, including ones involved in metabolism and pili production (see Data Set S1 in the supplemental material).
Our data suggest that AB5075 should maintain a low number of duplications of the sRNA-encoding locus to ensure maximal virulence. While Fig. 7 demonstrates that a single duplication of this locus results in a dramatic increase in virulence, Fig. 2D demonstrates that high-level amplification of this locus results in hyperswitching to the AV-T state, which should result in decreased virulence in vivo. Variant hetR-O2 characterized in Fig. 2D was first isolated using selection for increased tobramycin resistance (11), demonstrating that exposure to high levels of tobramycin can select for high-copy amplifications of RI-2 and hyperswitching to AV-T. This suggests that tobramycin treatment may lead to decreased virulence of AB5075 by increasing switching to AV-T; however, this hypothesis remains to be experimentally verified. A recent study demonstrated that pretreatment with the aminoglycoside kanamycin results in decreased virulence in AB5075, although the mechanism behind this appears to involve aminoglycoside-induced changes and not changes selected for by aminoglycosides (56).
Finally, the discovery of LSO variants has important implications for future studies using A. baumannii AB5075. As LSO, VIR-O, and AV-T variants all exhibit distinct phenotypes in terms of colony morphology, virulence, and global gene expression (4, 5), it is critical that future studies of AB5075 take opacity variants into account when comparing phenotypes between mutant and control strains. This study demonstrates that opaque isolates in particular should be assessed for their degree of sectoring to AV-T prior to further experimentation to avoid spurious results in virulence and other studies.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Stocks of bacteria were maintained at −80°C in 15% glycerol. Pure stocks of A. baumannii VIR-O isolates were generated as described previously (57). Liquid cultures were prepared in sterile LB broth, supplemented with tetracycline (Sigma-Aldrich, St. Louis, MO), hygromycin (Invitrogen, Carlsbad, CA), tobramycin (Sigma-Aldrich), or kanamycin (Sigma-Aldrich) as appropriate. To prevent opacity switching, which occurs at high culture density, cultures were grown overnight at room temperature or 37°C without shaking followed by growth at 37°C with shaking to the desired optical density at 600 nm (OD600). MIC and cloning experiments were performed on LB medium supplemented with 1.5% agar. For switching assays and qualitative examination of switching, 0.5× LB supplemented with 0.8% agar was used. Plates were supplemented with tetracycline, hygromycin, tobramycin, or kanamycin as indicated in the text and legends.
Wild-type LSO variants were isolated by plating serial dilutions of pure wild-type VIR-O stocks on 0.5× LB with 0.8% agar. Rare colonies that lacked AV-T sectors at 24 h were passaged onto 0.5× LB with 0.8% agar to confirm phenotypic stability; single colonies from restreaks were cultured and stocked as described above. The wild-type isolate designated set B VIR-O2 was derived by plating serial dilutions of an isolated LSO colony onto 0.5× LB with 0.8% agar. Colonies were screened for sectoring at approximately 24 h of growth; a sectoring colony was passaged onto 0.5× LB with 0.8% agar to confirm the phenotype. A single colony was then cultured and stocked as described above.
AV-T isolates carrying a single copy of the duplicated region were screened for by plating serial dilutions of an AV-T strain harboring two copies on 0.5× LB with 0.8% agar. Colonies were then patched onto 0.5× LB with 0.8% agar with and without 32 μg/ml of tobramycin. Patches that grew poorly on tobramycin were struck from the plate without antibiotic onto 0.5× LB with 0.8% agar, and single colonies were cultured and stocked as described above. Copy numbers of the duplicated region for the resulting isolates AV-T-56 and AV-T-80, as well as the parental AV-T isolate, were determined by qPCR as described below.
An LSO variant cured of plasmid p1AB5075 was serendipitously generated in an attempt to knockout aadB. A strain containing a T26 insertion in aadB was obtained from the AB5075 transposon library established at the University of Washington (10). Genomic DNA (gDNA) was prepared from an overnight culture of this strain grown at 37°C with shaking as described below. Diluted gDNA was used as a template for PCR using Phusion Hot Start II DNA polymerase (Thermo Scientific, Waltham, MA) with primers oSA65 and oSA66 (see Table S2 in the supplemental material) to amplify the aadB gene and the transposon disruption. The resulting fragment was ligated into the SmaI site of the counterselectable suicide plasmid pEX100 (58); the ligation was transformed into TransforMax EC100 electrocompetent E. coli (Lucigen, Middleton, WI), and transformants were selected by plating on 10 μg/ml tetracycline. The resulting plasmid was designated pEX100-aadBTnS. A concentrated preparation of pEX100-aadBTnS was made by purifying plasmid from 4 ml of E. coli culture using the Qiaprep Miniprep kit (Qiagen, Germantown, MD) followed by concentration through ethanol precipitation. This concentrated DNA was electroporated into an LSO culture of AB5075 as described below. Transformants were selected using 5 μg/ml tetracycline. Colonies were then screened for sensitivity to 10% sucrose to identify transformants that had incorporated the suicide plasmid by homologous recombination. Cells that had undergone a second crossover to lose the plasmid backbone were selected for by culturing for several hours without antibiotic at 37°C with shaking followed by plating serial dilutions on no salt LB with 10% sucrose. Double-crossover colonies were then screened for tetracycline resistance to determine whether they had retained the T26 marker. The colony designated LSO Δp1AB5075 was determined to be tetracycline sensitive and was originally assumed to have reverted to the wild-type genotype upon the second crossover. We determined that LSO Δp1AB5075 had actually lost the p1AB5075 plasmid by attempting to amplify fragments of DNA from this plasmid using primer sets oSA69/70, oSA67/68, oSA86/87, and oSA104/105. All of these PCRs were unsuccessful, while control PCRs using primers in the chromosomal mutS gene (osA7 and oSA8) still amplified the expected band.
Strains in which p1AB5075 had been reintroduced into LSO Δp1AB5075 were generated by electroporation. gDNA was prepared from VIR-O and LSO cultures grown to an OD of 0.5, as described below. gDNA from each strain was transformed into LSO Δp1AB5075 as described below. Transformants that had recovered the p1AB5075 plasmid were selected for by plating on 0.5× LB with 0.8% agar supplemented with 2 μg/ml tobramycin. Switching phenotypes were assessed qualitatively by observing transformants under a dissecting microscope with oblique illumination; colonies were passaged onto 0.5× LB with 0.8% agar with 2 μg/ml tobramycin and then cultured in LB to generate stocks as described above.
T26 insertions in RNase genes were obtained from the transposon mutant library prepared by Gallagher et al. (10). Initial screening was performed by electroporating the indicated plasmids directly into VIR-O colonies obtained from the library, using the method described below. Strains for Northern blotting were reconstructed in either the LSO Δp1AB5075 strain (rph::Tc, rnc::Tc) or a VIR-O isolate (remaining mutants) by transduction. Briefly, supernatants from stationary-phase cultures containing the desired library mutation were filtered and mixed with the appropriate AB5075 isolate grown to early-log phase. Mixtures were spotted onto LB plates and incubated for 4 h at 37°C. Spots were resuspended in LB, and transductants were selected for using 10 μg/ml tetracycline. Transductants were stocked as described above, and insertions were confirmed by PCR with Taq DNA polymerase (New England Biolabs, Ipswich, MA) using primers given in Table S2.
Growth curves.
Cultures were grown overnight without shaking to exponential phase (0.05 < OD < 0.5). Cultures were normalized to an OD of 0.05 and grown at 37°C with shaking to early stationary phase (OD > 1.0).
Electroporation of AB5075.
Electroporations were conducted with cultures of AB5075 isolates grown at 37°C with shaking to late log phase. Cells were pelleted by centrifugation and washed three times with 10% glycerol to induce electrocompetence. Cells were mixed with either gDNA or plasmid DNA in 2-mm cuvettes and electroporated at 2.50 kV. Transformants were recovered in 1 ml LB for 30 min at 37°C without shaking followed by 1 h at 37°C with shaking. Transformants were then selected for by plating on the appropriate antibiotic.
Opacity switching assays.
Switching frequencies were calculated as previously described (57). Switching assays were conducted on plates containing 20 ml of 0.5× LB with 0.8% agar supplemented with 100 μg/ml hygromycin or 5 μg/ml tetracycline, as appropriate to ensure maintenance of plasmids. Assays were performed using six 24-h-old colonies from at least two independent experiments, unless otherwise noted.
VIR-O and LSO interconversion assays.
To estimate the rate at which VIR-O and LSO interconvert, suspensions of 24-h-old colonies (both variants) or cultures (VIR-O only) were serially diluted on 0.5× LB with 0.8% agar to determine CFU; colony suspensions and cultures were then stored at 4°C. Following enumeration of CFU, suspensions and cultures were plated to obtain approximately 50 colonies per plate across several plates in order to screen several hundred colonies. After 48 h of growth, colonies were screened for sectoring using a dissecting microscope with oblique illumination. Colonies that appeared to exhibit the phenotype of the other variant were passaged onto 0.5× LB with 0.8% agar alongside a VIR-O positive control to confirm the phenotype. The frequency of conversion was calculated by dividing the number of confirmed colonies of the other phenotype by the total number of colonies screened.
gDNA isolation.
Unless otherwise noted, cultures used for gDNA preparations were grown with shaking at 37°C to an OD600 of 0.5. One milliliter of cells was pelleted through centrifugation; pellets were then resuspended in Tris-EDTA. Cells were lysed by incubation for 1 h at 37°C with 0.5% SDS and 400 μg/ml proteinase K. Sodium chloride was added to a final concentration of 0.7 M, and DNA was extracted twice using equal volumes of phenol-chloroform and isoamyl alcohol. DNA was isolated by mixing with equal volumes of 95% ethanol until a precipitate was visible. DNA pellets were collected by centrifugation, washed twice with 75% ethanol, dried, and resuspended in molecular-grade water.
Whole-genome sequencing.
PacBio WGS was performed at the University of Maryland Genomics Resource Center.
MIC assays.
MICs were determined using a modified Etest assay as previously described (11). Briefly, two strains at an OD600 of 0.1 were inoculated onto either side of an Etest strip (bioMérieux, Marcy-l’Étoile, France) by pipetting 10 μl of culture next to the bottom of the strip, tilting the plate to spread the culture up the side of the strip, and pipetting away excess culture at the top of the strip. MICs were assessed after 6 or 16 h, as noted in the text and legends. Experiments were performed two independent times to confirm the reproducibility of trends.
Southern blotting.
A DNA probe specific for aadB was generated by PCR using primers oSA65 and oSA66 (Table S2) and Phusion Hot Start II DNA polymerase, with LSO gDNA as a template. The resulting fragment was gel purified using the UltraClean DNA purification kit (Mo Bio Laboratories, Carlsbad, CA). The PCR product was then diluted in Tris-EDTA, boiled for 10 min to denature the DNA, and incubated in an ice water bath for 5 min. The DNA was then labeled with digoxigenin (DIG) by incubating with DIG labeling mix, hexanucleotide mix, and Klenow polymerase (Roche, Mannheim, Germany) at 37°C for at least 4 h. The labeled probe was diluted 1:500 in prehybridization solution, which consists of 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 0.1% N-lauroylsarcosine, 0.02% SDS, and 5 mg/ml blocking reagent (Roche), and stored at −20°C. A DNA probe specific for the Gene Ruler High Range DNA ladder (Thermo Scientific) was also generated by the above-described protocol using ladder diluted 1:2,500 in Tris-EDTA.
gDNA samples for Southern blotting were generated as described above. Concentrations of gDNA samples were determined using a NanoDrop ND-100 spectrophotometer; between 3.4 and 4.6 μg of each sample was digested with ScaI (New England Biolabs) for 6 h at 37°C. Samples were electrophoresed alongside the Gene Ruler High Range DNA ladder on a 0.5% Tris-acetate-EDTA (TAE)-agarose gel overnight at 25 V. Gels were poststained with ethidium bromide (EtBr) and photographed. Gels were washed for 10 min in 0.2 N HCl, rinsed three times with distilled water, washed twice for 15 min with denaturation solution (0.5 M NaOH, 1.5 M NaCl), and washed for 30 min with neutralization solution (0.5 M Tris-HCl, 1.5 M NaCl, pH 7.5). Nucleic acids were transferred onto a 0.22-μm nylon membrane via capillary transfer using a TurboBlotter (Schleicher & Schuell, Keene, NH) overnight at room temperature. Transfer was performed using 20× SSC as a buffer. The membrane was then baked in a vacuum oven at 80°C with 330.2 mm Hg for 2 h. The membrane was cut into separate lanes containing gDNA samples from lanes containing ladder; the two sections were treated separately for subsequent steps. Membranes were incubated in prehybridization solution for 2 h at 65°C. Membranes were then incubated with the appropriate probe (aadB probe for gDNA lanes, ladder probe for ladder lanes) overnight at 65°C. Probes were boiled for 10 min prior to addition to the membrane. Membranes were washed twice for 15 min with 2× SSC with 0.1% SDS and then washed twice for 15 min with 0.1× SSC with 0.1% SDS at 65°C. Membranes were then incubated for 2 min in buffer 1, which is 0.1 M Tris (pH 7.5) with 0.15 M NaCl. Membranes were then blocked for 1 h in buffer 2, which is buffer 1 with 10 mg/ml blocking reagent. Membranes were incubated for 30 min in buffer 1 containing a 1:5,000 dilution of α-DIG alkaline phosphatase (Roche). Membranes were then washed twice for 15 min in buffer 1 followed by a 2-min incubation in buffer 3 (0.1 M Tris, 0.1 M NaCl, pH 9.5). Finally, membranes were developed in color substrate solution, which was prepared by adding 105 μl 5-bromo-4-chloro-3-indolyl-phosphate (BCIP) (Roche) and 135 μl 4-nitroblue tetrazolium chloride (NBT) (Roche) to 30 ml of buffer 3. Once bands were clearly visible, development was stopped by washing membranes for 5 min in distilled water. Following development, the two sections of the membrane were realigned and photographed.
Quantitative PCR.
qPCR was performed using gDNA samples prepared as described above. qPCR primers were designed to amplify approximately 150-bp fragments from each gene of interest and were generated using Primer-BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast). qPCR of aadB was performed using primers oSA69 and oSA70 (Table S2); qPCR primers for the control genes aacA4 and clpX are denoted in Table S2. qPCR was performed using iQ SYBR green Supermix (Bio-Rad, Hercules, CA) and a Bio-Rad CFX Connect cycler. Cycle parameters were 95°C for 3 min, followed by 40 cycles of 95°C for 10 s, 55°C for 10 s, and 72°C for 20 s. Melt curves were then collected to confirm primer specificity. For Fig. 2 and 3, copy number was calculated relative to a control strain (LSO), and a control gene (aacA4) using the comparative cycle threshold (2−ΔCT) method (59). To determine the copy number of the pWHaadB plasmid, aadB copy number in the strain LSO Δp1AB5075 pWHaadB was normalized to the chromosomal gene clpX by the 2−ΔΔCT method.
Overexpression of duplicated region fragments.
Fragments for overexpression were generated by PCR using Phusion Hot Start II polymerase and gDNA generated from a stationary-phase culture of AB5075 (pWHaadB, pWHaadB-T, pWHaadB-TA, pWHaadB-TB, pWHaadB-UP, and paadB) or gDNA from a mid-log LSO culture (pDR) as a template. All fragments were generated using oSA66 as a forward primer; reverse primers are indicated in Table S2. Fragments were ligated into pWH1266 (60) cut with ScaI (pWHaadB, pWHaadB-T, pWHaadB-TA, pWHaadB-TB, and pWHaadB-UP) or pQF1266.Blue (see below) cut with SmaI (paadB and pDR). Plasmids were isolated using TransforMax EC100 electrocompetent E. coli plated on 5 μg/ml tobramycin (pWHaadB), 10 μg/ml tetracycline (other pWH1266 derivatives), or 100 μg/ml hygromycin (pQF1266.Blue derivatives). Inserts were confirmed by restriction digestion and Sanger sequencing; plasmids were chosen with the inserts in the same orientation relative to the plasmid backbone. For the pWH1266 derivatives, inserts were all oriented with Pc on the opposite strand from the blaTEM-1 promoter. Plasmids were introduced into the LSO, LSO Δp1AB5075, and RNase mutant isolates, as indicated in the text and legends, by electroporation.
Plasmid pQF1266.Blue was constructed from pQF50 (61) by first inserting a hygromycin resistance gene from pMQ310 (62) into the ScaI site within the beta-lactamase gene. This resulted in plasmid pQF50.hyg. Next, a 1.4-kb fragment containing the origin of replication from pWH1266 was generated by PCR using the primers ATATCCATGGGATCGTGAAATATCTATGA and ATATCCATGGGGATTTTATTTTGCGTTACA, each containing a site for the restriction enzyme NcoI. The resulting PCR product was digested with NcoI and ligated to NcoI-digested pQF50.hyg. Plasmids containing the 1266 ori region in each orientation resulted in either blue or white colonies on plates containing 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal), indicating a promoter was present in one orientation that drove beta-galactosidase expression. This plasmid was designated pQF1266.Blue.
Site-directed mutagenesis.
Mutagenized plasmids pWHaadBstop and pWHaadB-Q35 were generated using a QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Primers used for mutagenesis are listed in Table S2. Briefly, pWHaadB was amplified using mutagenesis primers according to the manufacturer’s instructions. The parental plasmids were digested with DpnI, and the remaining mutagenized plasmids were transformed into TransforMax EC100 electrocompetent E. coli. Transformants were selected for using 10 μg/ml tetracycline, and mutations were confirmed by Sanger sequencing. Mutagenized plasmids were then electroporated into the LSO variant as described above.
Screen for transposon insertions in pWHaadB.
Plasmid pWHaadB was mutagenized using the EZ-Tn5 <Kan-2> transposon kit (Lucigen) according to the manufacturer’s instructions. The mutagenized plasmid was transformed into TransforMax EC100 electrocompetent E. coli, and transformants were selected for by plating on LB with 50 μg/ml of kanamycin. Transformants were then pooled and plasmids were purified. To obtain insertions that blocked switching stimulated by pWHaadB, pooled plasmids were transformed into the LSO as described above, and transformants were selected by plating on 10 μg/ml tetracycline. Transformants were pooled and serial dilutions were plated on 0.5× LB with 0.8% agar to screen for sectoring. Colonies that exhibited decreased sectoring were passaged onto 0.5× LB with 0.8% agar alongside positive controls to confirm phenotypes. Colonies were then cultured, plasmids were purified, and transposon insertions were mapped by Sanger sequencing using the primers Kan-2 FP-1 and RP-1 (Lucigen).
To screen for insertions in aadB that did not block switching stimulated by pWHaadB, E. coli transformants were individually patched onto LB containing 10 μg/ml tetracycline and LB containing 10 μg/ml tobramycin. Patches that failed to grow on tobramycin were cultured, and then plasmids were individually purified and transformed into the LSO variant. Transformants were screened for their ability to switch; plasmids that still stimulated switching were sequenced with primer Kan-2 FP-1 to map the insertion.
RNA purification.
RNA was prepared from cultures grown in LB medium to an OD600 of 0.5. Cultures were supplemented with 5 μg/ml tetracycline or 100 μg/ml hygromycin, as appropriate. Samples for Northern blotting were collected using RNAprotect Bacteria reagent (Qiagen), according to the manufacturer’s protocol. Cell pellets were flash frozen and stored at −80°C. RNA was isolated using the MasterPure RNA purification kit (Epicentre, Madison, WI) according to the manufacturer’s instructions.
Quantitative reverse transcriptase PCR.
RNA purification was performed as outlined above, after which, samples were treated twice with Turbo DNA-free (Thermo Fisher) according to the manufacturer’s instructions. To confirm that samples were not contaminated with DNA following DNase treatment, PCR of RNA samples was performed. Total RNA (1 μg) was used to prepare cDNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) with random primers. Reaction mixtures lacking reverse transcriptase were also performed as a control for the presence of contaminating DNA. Incubation conditions for cDNA synthesis were 25°C for 5 min, 42°C for 45 min, and 85°C for 5 min. cDNA reactions and controls were then diluted 1:10 with sterile water and used as a template for reverse transcriptase quantitative PCR (qRT-PCR). Oligonucleotide primer pairs for qRT-PCR were designed as for qPCR. Primer sequences are indicated in Table S2. qRT-PCR conditions were identical to those used for qPCR. Data were generated using cDNA prepared from three independent RNA isolations, and qRT-PCRs were performed in technical triplicates to ensure accuracy. Fold changes in gene expression relative to the control strain (LSO pWH1266) and a control gene (clpX) were determined using the 2−ΔΔCT method (59).
Northern blotting.
Concentrations of RNA samples were determined using a NanoDrop ND-100 spectrophotometer. Either 2.7 μg (Fig. S7A, left blot) or 3.6 μg (other blots) of RNA was diluted in an equal volumes of 2× RNA loading dye and incubated at 65°C for 10 min. Samples were then electrophoresed on a prerun 10% Mini-PROTEAN Tris-borate-EDTA (TBE)-urea gel (Bio-Rad) at 200 V. Samples were run alongside Low Range ssRNA ladder (New England Biolabs), which was denatured by incubating at 90°C for 5 min followed by a 2-min incubation on ice prior to electrophoresis. Gels were poststained with EtBr in 0.1% diethyl pyrocarbonate (DEPC)-treated distilled water for 30 min and photographed. Gels were washed again with 0.1% DEPC distilled water for 30 min. Nucleic acids were transferred onto a 0.22-μm nylon membrane via capillary transfer overnight at room temperature, as described above for Southern blotting. Membranes were visualized with a UV lamp, and ladder bands were marked with a ball point pen; membranes were then baked in a vacuum oven at 80°C with 330.2 mm Hg for 2 h. Membranes were incubated with DIG Easy Hyb (Roche) for 2 h at 39°C. Membranes were then incubated overnight at 39°C with the appropriate probe (Table S2). Probes were single-stranded oligonucleotides labeled at the 3′ and 5′ ends with DIG. Probes were diluted to 100 ng/ml in 0.1% DEPC prehybridization solution (see “Southern blotting”) and boiled for 10 min before use. After probing, membranes were washed for 15 min in 0.1% DEPC, 2× SSC, and 0.1% SDS, followed by two 15-min washes in 0.1% DEPC, 0.2× SSC, and 0.1% SDS at 65°C. Membranes were washed for 1 min in maleate buffer (100 mM maleic acid, 150 mM NaCl, 0.1% DEPC, pH 7.5). Membranes were then incubated for 1 h in maleate buffer containing 2% blocking reagent (Roche). Membranes were incubated for 30 min in maleate buffer containing 2% blocking reagent and a 1:5,000 dilution of α-DIG alkaline phosphatase (Roche). Membranes were then washed twice for 15 min with maleate buffer followed by a 5-min incubation in 0.1% DEPC buffer 3 (see “Southern blotting”). Finally, membranes were developed as described above for the Southern blots. Blots were photographed after developing for 2 h (Fig. 6) or overnight (Fig. S5 and S6).
Dot blotting.
Twofold serial dilutions of concentrated AB5075 gDNA were spotted onto a 0.22-μm nylon membrane in duplicates. The membrane was incubated on filter paper soaked in denaturation solution (see “Southern blotting”) for 5 min followed by two 5-min incubations on filter paper soaked in neutralization solution (see “Southern blotting”). The membrane was then incubated on filter paper soaked in 20× SSC for 5 min. The membrane was baked for 2 h in a vacuum oven at 80°C with 330.2 mm Hg and subsequently treated according to the Southern blotting protocol. The membrane was cut in half to separate duplicate spots; one set was probed with oSA197, while the other set was probed with oSA191 as a control (Table S2). The blot was photographed following development overnight.
Mice.
Wild-type (WT) C57BL/6J female mice were purchased from Jackson Laboratories and used at age 8 to 10 weeks. Mice were housed under specific-pathogen-free conditions at Yerkes National Primate Center, Emory University. Experimental studies were performed in accordance with the Institutional Animal Care and Use Committee guidelines.
Mouse pulmonary infection models.
Approximately 5 × 107 CFU were administered per mouse for infections to quantify the bacterial load. Overnight standing bacteria at room temperature were subcultured in LB broth and grown at 37°C with shaking to an OD600 of ∼0.15, washed, and resuspended in phosphate-buffered saline (PBS). Fifty microliters of bacterial inocula was inoculated intranasally (i.n.) to each mouse. Mice were anesthetized with isoflurane immediately prior to i.n. inoculation. At 24 h postinfection, the mice were sacrificed, and the lungs, spleen, and liver were harvested, homogenized, and plated for CFU on 0.5× LB plates.
RNA sequencing.
RNA-seq was performed at the Yerkes Genomics Core at Emory University. RNA-seq was performed in triplicates using samples generated from three independent cultures of the LSO and VIR-O variants. Upon sample collection, cultures were struck onto a 0.5× LB and 0.8% agar plate and checked for the presence of contaminating AV-T variants; only samples with greater than ∼95% opaque variants were used for RNA-seq. RNA purification was performed as outlined above, after which, samples were treated twice with Turbo DNA-free according to the manufacturer’s instructions. To confirm that samples were not contaminated with DNA following DNase treatment, PCR of RNA samples was performed using primers abaR-qPCR-for and abaR-qPCR-rev (Table S2). Samples were then processed for RNA-sequencing at the Yerkes National Primate Research Center Genomic core.
Statistical analyses.
Statistical analyses were performed with Prism 5 and 7 (GraphPad Software, Inc., La Jolla, CA).
Data availability.
RNA sequencing information and files have been deposited to the GEO database under the accession numbers GSE156206, GSM4726730, GSM4726731, GSM4726732, GSM4726733, GSM4726734, and GSM4726735.
ACKNOWLEDGMENTS
This work was supported by the following grants to P.N.R.: R21AI142489 from the National Institutes of Health and I01BX001725 and IK6BX004470, both from the U.S. Department of Veterans Affairs. D.S.W. is supported by a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease award, VA Merit award I01 BX002788 and NIH grant AI141883. S.E.A. was supported by T32 training grant AI106699 from the NIH. The Yerkes NHP Genomics core is supported in part by NIH P51 OD11132. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Department of Veterans Affairs.
We have no conflicts of interest to disclose.
Footnotes
This article is a direct contribution from Philip N. Rather, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Colin Manoil, University of Washington, and Melissa Brown, Flinders University.
Citation Anderson SE, Chin CY, Weiss DS, Rather PN. 2020. Copy number of an integron-encoded antibiotic resistance locus regulates a virulence and opacity switch in Acinetobacter baumannii AB5075. mBio 11:e02338-20. https://doi.org/10.1128/mBio.02338-20.
REFERENCES
- 1.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. 2017. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. http://who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf.
- 3.Wong D, Nielsen TB, Bonomo RA, Pantapalangkoor P, Luna B, Spellberg B. 2017. Clinical and pathophysiological overview of Acinetobacter infections: a century of challenges. Clin Microbiol Rev 30:409–447. doi: 10.1128/CMR.00058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chin CY, Tipton KA, Farokhyfar M, Burd EM, Weiss DS, Rather PN. 2018. A high-frequency phenotypic switch links bacterial virulence and environmental survival in Acinetobacter baumannii. Nat Microbiol 3:563–569. doi: 10.1038/s41564-018-0151-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tipton KA, Dimitrova D, Rather PN. 2015. Phase-variable control of multiple phenotypes in Acinetobacter baumannii strain AB5075. J Bacteriol 197:2593–2599. doi: 10.1128/JB.00188-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tipton KA, Farokhyfar M, Rather PN. 2017. Multiple roles for a novel RND-type efflux system in Acinetobacter baumannii AB5075. Microbiologyopen 6:e00418. doi: 10.1002/mbo3.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tipton KA, Rather PN. 2017. An ompR/envZ two-component system ortholog regulates phase variation, osmotic tolerance, motility, and virulence in Acinetobacter baumannii strain AB5075. J Bacteriol 199:e00705-16. doi: 10.1128/JB.00705-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad I, Karah N, Nadeem A, Wai SN, Uhlin BE. 2019. Analysis of colony phase variation switch in Acinetobacter baumannii clinical isolates. PLoS One 14:e0210082. doi: 10.1371/journal.pone.0210082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pérez-Varela M, Tierney ARP, Kim JS, Vázquez-Torres A, Rather P. 2020. Characterization of RelA in Acinetobacter baumannii. J Bacteriol 202:e00045-20. doi: 10.1128/JB.00045-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallagher LA, Ramage E, Weiss EJ, Radey M, Hayden HS, Held KG, Huse HK, Zurawski DV, Brittnacher MJ, Manoil C. 2015. Resources for genetic and genomic analysis of emerging pathogen Acinetobacter baumannii. J Bacteriol 197:2027–2035. doi: 10.1128/JB.00131-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson SE, Sherman EX, Weiss DS, Rather PN. 2018. Aminoglycoside heteroresistance in Acinetobacter baumannii AB5075. mSphere 3:e00271-20. doi: 10.1128/mSphere.00271-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cagle CA, Shearer JE, Summers AO. 2011. Regulation of the integrase and cassette promoters of the class 1 integron by nucleoid-associated proteins. Microbiology (Reading) 157:2841–2853. doi: 10.1099/mic.0.046987-0. [DOI] [PubMed] [Google Scholar]
- 13.Levesque C, Brassard S, Lapointe J, Roy PH. 1994. Diversity and relative strength of tandem promoters for the antibiotic-resistance genes of several integrons. Gene 142:49–54. doi: 10.1016/0378-1119(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 14.Papagiannitsis CC, Tzouvelekis LS, Miriagou V. 2009. Relative strengths of the class 1 integron promoter hybrid 2 and the combinations of strong and hybrid 1 with an active p2 promoter. Antimicrob Agents Chemother 53:277–280. doi: 10.1128/AAC.00912-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collis CM, Hall RM. 1995. Expression of antibiotic resistance genes in the integrated cassettes of integrons. Antimicrob Agents Chemother 39:155–162. doi: 10.1128/aac.39.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanau-Bercot B, Podglajen I, Casin I, Collatz E. 2002. An intrinsic control element for translational initiation in class 1 integrons. Mol Microbiol 44:119–130. doi: 10.1046/j.1365-2958.2002.02843.x. [DOI] [PubMed] [Google Scholar]
- 17.Partridge SR, Recchia GD, Scaramuzzi C, Collis CM, Stokes HW, Hall RM. 2000. Definition of the attI1 site of class 1 integrons. Microbiology 146:2855–2864. doi: 10.1099/00221287-146-11-2855. [DOI] [PubMed] [Google Scholar]
- 18.Carrier MC, Lalaouna D, Masse E. 2018. Broadening the definition of bacterial small RNAs: characteristics and mechanisms of action. Annu Rev Microbiol 72:141–161. doi: 10.1146/annurev-micro-090817-062607. [DOI] [PubMed] [Google Scholar]
- 19.Kunne T, Swarts DC, Brouns SJ. 2014. Planting the seed: target recognition of short guide RNAs. Trends Microbiol 22:74–83. doi: 10.1016/j.tim.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Frohlich KS, Papenfort K, Fekete A, Vogel J. 2013. A small RNA activates CFA synthase by isoform-specific mRNA stabilization. EMBO J 32:2963–2979. doi: 10.1038/emboj.2013.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat Rev Microbiol 9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim S, Reyes D, Beaume M, Francois P, Cheung A. 2014. Contribution of teg49 small RNA in the 5' upstream transcriptional region of sarA to virulence in Staphylococcus aureus. Infect Immun 82:4369–4379. doi: 10.1128/IAI.02002-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. 2009. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139:770–779. doi: 10.1016/j.cell.2009.08.046. [DOI] [PubMed] [Google Scholar]
- 24.Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. 2012. An atlas of Hfq-bound transcripts reveals 3' UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 31:4005–4019. doi: 10.1038/emboj.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chao Y, Vogel J. 2016. A 3' UTR-derived small RNA provides the regulatory noncoding arm of the inner membrane stress response. Mol Cell 61:352–363. doi: 10.1016/j.molcel.2015.12.023. [DOI] [PubMed] [Google Scholar]
- 26.Guerin E, Jove T, Tabesse A, Mazel D, Ploy MC. 2011. High-level gene cassette transcription prevents integrase expression in class 1 integrons. J Bacteriol 193:5675–5682. doi: 10.1128/JB.05246-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiester SE, Arivett BA, Schmidt RE, Beckett AC, Ticak T, Carrier MV, Ghosh R, Ohneck EJ, Metz ML, Sellin Jeffries MK, Actis LA. 2016. Iron-regulated phospholipase C activity contributes to the cytolytic activity and virulence of Acinetobacter baumannii. PLoS One 11:e0167068. doi: 10.1371/journal.pone.0167068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gebhardt MJ, Gallagher LA, Jacobson RK, Usacheva EA, Peterson LR, Zurawski DV, Shuman HA. 2015. Joint transcriptional control of virulence and resistance to antibiotic and environmental stress in Acinetobacter baumannii. mBio 6:e01660-15. doi: 10.1128/mBio.01660-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhuiyan MS, Ellett F, Murray GL, Kostoulias X, Cerqueira GM, Schulze KE, Mahamad Maifiah MH, Li J, Creek DJ, Lieschke GJ, Peleg AY. 2016. Acinetobacter baumannii phenylacetic acid metabolism influences infection outcome through a direct effect on neutrophil chemotaxis. Proc Natl Acad Sci U S A 113:9599–9604. doi: 10.1073/pnas.1523116113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerqueira GM, Kostoulias X, Khoo C, Aibinu I, Qu Y, Traven A, Peleg AY. 2014. A global virulence regulator in Acinetobacter baumannii and its control of the phenylacetic acid catabolic pathway. J Infect Dis 210:46–55. doi: 10.1093/infdis/jiu024. [DOI] [PubMed] [Google Scholar]
- 31.Tomaras AP, Dorsey CW, Edelmann RE, Actis LA. 2003. Attachment to and biofilm formation on abiotic surfaces by Acinetobacter baumannii: involvement of a novel chaperone-usher pili assembly system. Microbiology (Reading) 149:3473–3484. doi: 10.1099/mic.0.26541-0. [DOI] [PubMed] [Google Scholar]
- 32.Pakharukova N, Tuittila M, Paavilainen S, Malmi H, Parilova O, Teneberg S, Knight SD, Zavialov AV. 2018. Structural basis for Acinetobacter baumannii biofilm formation. Proc Natl Acad Sci U S A 115:5558–5563. doi: 10.1073/pnas.1800961115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersson DI, Hughes D. 2009. Gene amplification and adaptive evolution in bacteria. Annu Rev Genet 43:167–195. doi: 10.1146/annurev-genet-102108-134805. [DOI] [PubMed] [Google Scholar]
- 34.Padalon-Brauch G, Hershberg R, Elgrably-Weiss M, Baruch K, Rosenshine I, Margalit H, Altuvia S. 2008. Small RNAs encoded within genetic islands of Salmonella Typhimurium show host-induced expression and role in virulence. Nucleic Acids Res 36:1913–1927. doi: 10.1093/nar/gkn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellis MJ, Trussler RS, Charles O, Haniford DB. 2017. A transposon-derived small RNA regulates gene expression in Salmonella Typhimurium. Nucleic Acids Res 45:5470–5486. doi: 10.1093/nar/gkx094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durieux I, Ginevra C, Attaiech L, Picq K, Juan PA, Jarraud S, Charpentier X. 2019. Diverse conjugative elements silence natural transformation in Legionella species. Proc Natl Acad Sci U S A 116:18613–18618. doi: 10.1073/pnas.1909374116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pichon C, Felden B. 2005. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc Natl Acad Sci U S A 102:14249–14254. doi: 10.1073/pnas.0503838102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen XL, Tang DJ, Jiang RP, He YQ, Jiang BL, Lu GT, Tang JL. 2011. sRNA-Xcc1, an integron-encoded transposon- and plasmid-transferred trans-acting sRNA, is under the positive control of the key virulence regulators HrpG and HrpX of Xanthomonas campestris pathovar campestris. RNA Biol 8:947–953. doi: 10.4161/rna.8.6.16690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wachter S, Raghavan R, Wachter J, Minnick MF. 2018. Identification of novel MITEs (miniature inverted-repeat transposable elements) in Coxiella burnetii: implications for protein and small RNA evolution. BMC Genomics 19:247. doi: 10.1186/s12864-018-4608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jia X, Zhang J, Sun W, He W, Jiang H, Chen D, Murchie AI. 2013. Riboswitch control of aminoglycoside antibiotic resistance. Cell 152:68–81. doi: 10.1016/j.cell.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 41.Roth A, Breaker RR. 2013. Integron attI1 sites, not riboswitches, associate with antibiotic resistance genes. Cell 153:1417–1418. doi: 10.1016/j.cell.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S, He W, Sun W, Zhang J, Chang Y, Chen D, Murchie AIH. 2019. Integron-derived aminoglycoside-sensing riboswitches control aminoglycoside acetyltransferase resistance gene expression. Antimicrob Agents Chemother 63:e00236-19. doi: 10.1128/AAC.00236-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pursley BR, Fernandez NL, Severin GB, Waters CM. 2019. The Vc2 cyclic di-GMP dependent riboswitch of Vibrio cholerae regulates expression of an upstream putative small RNA by controlling RNA stability. J Bacteriol 201:e00293-19. doi: 10.1128/JB.00293-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DebRoy S, Gebbie M, Ramesh A, Goodson JR, Cruz MR, van Hoof A, Winkler WC, Garsin DA. 2014. Riboswitches. A riboswitch-containing sRNA controls gene expression by sequestration of a response regulator. Science 345:937–940. doi: 10.1126/science.1255091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nemec A, Dolzani L, Brisse S, van den Broek P, Dijkshoorn L. 2004. Diversity of aminoglycoside-resistance genes and their association with class 1 integrons among strains of pan-European Acinetobacter baumannii clones. J Med Microbiol 53:1233–1240. doi: 10.1099/jmm.0.45716-0. [DOI] [PubMed] [Google Scholar]
- 46.Liu CC, Tang CY, Chang KC, Kuo HY, Liou ML. 2014. A comparative study of class 1 integrons in Acinetobacter baumannii. Gene 544:75–82. doi: 10.1016/j.gene.2014.04.047. [DOI] [PubMed] [Google Scholar]
- 47.Lin MF, Liou ML, Tu CC, Yeh HW, Lan CY. 2013. Molecular epidemiology of integron-associated antimicrobial gene cassettes in the clinical isolates of Acinetobacter baumannii from northern Taiwan. Ann Lab Med 33:242–247. doi: 10.3343/alm.2013.33.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin MF, Chang KC, Yang CY, Yang CM, Xiao CC, Kuo HY, Liou ML. 2010. Role of integrons in antimicrobial susceptibility patterns of Acinetobacter baumannii. Jpn J Infect Dis 63:440–443. [PubMed] [Google Scholar]
- 49.Azizi O, Shakibaie MR, Badmasti F, Modarresi F, Ramazanzadeh R, Mansouri S, Shahcheraghi F. 2016. Class 1 integrons in non-clonal multidrug-resistant Acinetobacter baumannii from Iran, description of the new blaIMP-55 allele in In1243. J Med Microbiol 65:928–936. doi: 10.1099/jmm.0.000315. [DOI] [PubMed] [Google Scholar]
- 50.Mirshekar M, Shahcheraghi F, Azizi O, Solgi H, Badmasti F. 2018. Diversity of class 1 integrons, and disruption of carO and dacD by insertion sequences among Acinetobacter baumannii isolates in Tehran, Iran. Microb Drug Resist 24:359–366. doi: 10.1089/mdr.2017.0152. [DOI] [PubMed] [Google Scholar]
- 51.Mabrouk A, Grosso F, Botelho J, Achour W, Ben Hassen A, Peixe L. 2017. GES-14-producing Acinetobacter baumannii isolates in a neonatal intensive care unit in Tunisia are associated with a typical Middle East clone and a transferable plasmid. Antimicrob Agents Chemother 61:e00142-17. doi: 10.1128/AAC.00142-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonnin RA, Nordmann P, Potron A, Lecuyer H, Zahar J-R, Poirel L. 2011. Carbapenem-hydrolyzing GES-type extended-spectrum β-lactamase in Acinetobacter baumannii. Antimicrob Agents Chemother 55:349–354. doi: 10.1128/AAC.00773-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGann P, Courvalin P, Snesrud E, Clifford RJ, Yoon EJ, Onmus-Leone F, Ong AC, Kwak YI, Grillot-Courvalin C, Lesho E, Waterman PE. 2014. Amplification of aminoglycoside resistance gene aphA1 in Acinetobacter baumannii results in tobramycin therapy failure. mBio 5:e00915-14. doi: 10.1128/mBio.00915-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamidian M, Holt KE, Pickard D, Dougan G, Hall RM. 2014. A GC1 Acinetobacter baumannii isolate carrying AbaR3 and the aminoglycoside resistance transposon TnaphA6 in a conjugative plasmid. J Antimicrob Chemother 69:955–958. doi: 10.1093/jac/dkt454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tipton KA, Chin CY, Farokhyfar M, Weiss DS, Rather PN. 2018. Role of capsule in resistance to disinfectants, host antimicrobials, and desiccation in Acinetobacter baumannii. Antimicrob Agents Chemother 62:e01188-18. doi: 10.1128/AAC.01188-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hood-Pishchany MI, Pham L, Wijers CD, Burns WJ, Boyd KL, Palmer LD, Skaar EP, Noto MJ. 2020. Broad-spectrum suppression of bacterial pneumonia by aminoglycoside-propagated Acinetobacter baumannii. PLoS Pathog 16:e1008374. doi: 10.1371/journal.ppat.1008374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anderson SE, Rather PN. 2019. Distinguishing colony opacity variants and measuring opacity variation in Acinetobacter baumannii. Methods Mol Biol 1946:151–157. doi: 10.1007/978-1-4939-9118-1_14. [DOI] [PubMed] [Google Scholar]
- 58.Schweizer HP, Hoang TT. 1995. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene 158:15–22. doi: 10.1016/0378-1119(95)00055-b. [DOI] [PubMed] [Google Scholar]
- 59.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 60.Hunger M, Schmucker R, Kishan V, Hillen W. 1990. Analysis and nucleotide sequence of an origin of DNA replication in Acinetobacter calcoaceticus and its use for Escherichia coli shuttle plasmids. Gene 87:45–51. doi: 10.1016/0378-1119(90)90494-c. [DOI] [PubMed] [Google Scholar]
- 61.Farinha MA, Kropinski AM. 1990. Construction of broad-host-range plasmid vectors for easy visible selection and analysis of promoters. J Bacteriol 172:3496–3499. doi: 10.1128/jb.172.6.3496-3499.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalivoda EJ, Horzempa J, Stella NA, Sadaf A, Kowalski RP, Nau GJ, Shanks RMQ. 2011. New vector tools with a hygromycin resistance marker for use with opportunistic pathogens. Mol Biotechnol 48:7–14. doi: 10.1007/s12033-010-9342-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The LSO and VIR-O subpopulations do not exhibit growth differences in LB broth. Data represent averages and SEMs from two independent experiments (the final two time points were collected in one experiment only). Download FIG S1, TIF file, 1.0 MB (1,013.5KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The LSO variant exhibits decreased antimicrobial resistance relative to the VIR-O. Resistance was evaluated to the antibiotic targets of resistance genes harbored within the duplicated region on p1AB5075. Strains were directly compared by inoculating the two variants on either side of an Etest strip. Data shown are representative results from a single experiment; experiments were performed twice to assess the reproducibility of trends. a MICs were measured after 16 h of growth at 37°C. b MICs were measured after 6 h of growth at 37°C. The two variants did not exhibit a reproducible difference in resistance to chloramphenicol after 6 h of growth (data not shown). TOB, tobramycin; GEN, gentamicin; STR, streptomycin. Download Table S1, DOCX file, 0.1 MB (64.9KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Overexpression of the entire duplicated region increases switching to the same level as overexpression of aadB alone. The aadB gene (paadB) and the duplicated region stretching from strB to aadB (pDR) were cloned into vector pQF1266.Blue and overexpressed in the LSO variant. Switching to AV-T was measured for colonies after 24 h of growth, indicating that pDR and paadB both stimulate switching similarly compared to the empty vector control. Data shown represent averages and SEMs from at least five colonies collected in two independent experiments. ****, P ≤ 0.0001, one-way ANOVA with Tukey’s posttest. Download FIG S2, TIF file, 0.3 MB (288KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The AadB protein is not required for stimulation of switching by the aadB region. A stop codon was introduced into the fifth amino acid of aadB on plasmid pWHaadB, and the resulting plasmid (pWHaadBstop) was transformed into the LSO variant. Data shown represent averages and SEMs from six 24-h-old colonies collected in two independent experiments. **, P ≤ 0.01 by unpaired two-tailed t test. Download FIG S3, TIF file, 0.2 MB (211.8KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Transcription from Pc is required for the stimulation of switching. (A) Two single base changes were introduced to disrupt the −35 region of Pc in pWHaadB (TTGACA→TTGAGT). The resulting plasmid (pWHaadB-Q35) exhibited a 25-fold decrease in promoter activity as measured by qRT-PCR. Data represent averages and SEMs from three independent samples. ***, P ≤ 0.001 relative to empty vector control, one-way ANOVA with Dunnett’s posttest. (B) Plasmid pWHaadB-Q35 did not stimulate switching in the LSO background relative to that of an empty vector control at 24 h of growth. Data represent averages and SEMs from six colonies collected in two independent experiments (differences are not significant by unpaired two-tailed t test). Download FIG S4, TIF file, 0.8 MB (795.5KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Transposon insertions blocking switching do not localize to any predicted ORFs encoded in the aadB 5′ region. The insert in pWHaadB-TB, the shortest plasmid insert that appears to stimulate switching, is shown. Sequence features are as indicated in Fig. 5A. Predicted ORFs in this region were identified using NCBI ORFfinder using both ATG and alternative start codons. ORFs are represented by black arrows; only ORFs completely contained within the pWHaadB-TB fragment are shown. The transposon insertions blocking switching do not align specifically with any of these ORFs, suggesting that none of these ORFs are responsible for the switching phenotype stimulated by this construct. Download FIG S5, TIF file, 0.2 MB (217.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The 3′ end of the ∼300-nt sRNA was identified by Northern blotting. (A) Three additional probes (oSA190, oSA191, and oSA197) were used for Northern blotting. All probes are 30-nt single-stranded oligonucleotides that should only bind transcripts produced from the same strand as aadB; probes hybridize to the region indicated. Other sequence features are shown as in Fig. 5A. (B) Northern blotting was performed using oSA190, oSA191, and oSA197 to examine RNAs produced from pWHaadB transformed into LSO Δp1AB5075. The ∼300-nt RNA was only weakly bound by oSA197 (white arrowhead). (C) A dot blot was performed using 2-fold serial dilutions of AB5075 gDNA and oSA197 to assess the ability of this probe to bind nucleic acids. Probe oSA191 was used as a positive control. Download FIG S6, TIF file, 1.5 MB (1.5MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Oligonucleotides used in this study. DIG, digoxigenin. Download Table S2, DOCX file, 0.1 MB (90.8KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Northern blotting does not indicate a role for ribonucleases in the synthesis of the 300-nt sRNA. (A) T26 insertions in rnr (ABUW_0444), cafA (ABUW_0719), rne (ABUW_3478), and ABUW_5006 were moved into a VIR-O variant and electroporated with paadB or an empty vector control. Northern blotting was performed for all samples alongside WT VIR-O paadB and an empty vector control using probe oSA134. (B) T26 insertions in rph (ABUW_3841) and rnc (ABUW_1032) were moved into an LSO Δp1AB5075 strain and electroporated with paadB. Northern blotting was performed for samples alongside an LSO Δp1AB5075 paadB control using oSA134. Download FIG S7, TIF file, 0.9 MB (986.5KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
RNA-seq results comparing the VIR-O and LSO. Triplicate cultures of the VIR-O and LSO variants were grown to mid-log phase and subjected to RNA-seq. Data sets indicate genes that were up- or downregulated at least 1.5-fold (log2-fold of 0.7) in the LSO variant. Only genes that exhibited P values of 0.05 or less are shown. Download Data Set S1, XLSX file, 0.4 MB (372.6KB, xlsx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data Availability Statement
RNA sequencing information and files have been deposited to the GEO database under the accession numbers GSE156206, GSM4726730, GSM4726731, GSM4726732, GSM4726733, GSM4726734, and GSM4726735.