

Abstract
Chromodomain helicase DNA-binding protein 5 (CHD5) plays a crucial tumor suppressor role in multiple types of tumors. For this study, we investigated its clinical significance and the molecular mechanism(s) underlying tumorigenesis in renal cell carcinoma (RCC). Initially, CHD5 expression was assessed in primary tumor tissue and in tissue array. Correlations among CHD5 expression and clinicopathological characteristics were analyzed. Next, lentivirus-mediated CHD5 overexpression in the ACHN and 769-P cells was used to assess effects on proliferation, migration, invasion ability, and the regulation of the p14ARF/p53 and p16INK4a/RB signaling pathways. Finally, a xenograft mouse model was used to verify its impact on tumor growth in vivo. Results demonstrated that CHD5 was downregulated in tumor tissues and that low CHD5 expression was correlated with advanced TNM stage, high Fuhrman grade, lymph node metastasis, and poor survival. Overexpression of CHD5 inhibited proliferation, migration, and invasion in vitro; prompted cell cycle G1 phase arrest; induced apoptosis; and suppressed tumor growth in vivo. Furthermore, we confirmed that CHD5 activates the p53 and RB pathways to inhibit tumorigenesis in RCC. In summary, CHD5 is involved in the initiation and progression of RCC and may serve as a diagnostic biomarker and a potential therapeutic target for RCC.
1. Introduction
Renal cell carcinoma (RCC) is the third most common tumor in the genitourinary system, representing 3% of all cancers worldwide, and the incidence has increased annually by 2% during the past decades [1, 2]. However, RCC is the most lethal genitourinary cancer due to its high recurrence rate, metastasis rate, and resistance to radiotherapy and chemotherapy [3]. Therefore, discovering new tumor biomarkers and exploring the molecular mechanism(s) involved in the tumorigenesis of RCC would be beneficial for developing new diagnostic and therapeutic strategies.
Human 1p36 has been reported to be deleted in multiple types of tumors, including those of neurogenic, epithelial, and hematopoietic origins [4]. Bagchi et al. identified CHD5 as a tumor suppressor gene (TSG) mapped to 1p36 by a chromosome engineering approach in a mouse model and confirmed that CHD5 plays a tumor suppressor role in human neuroblastoma [5]. Subsequent studies demonstrated that CHD5 is downregulated in neuroblastoma and is correlated with unfavorable clinical features and poor survival; restoration of CHD5 expression could inhibit clonogenicity and tumorigenicity [6, 7]. Evidence soon emerged that CHD5 is downregulated by promoter hypermethylation and/or deletion in many other types of tumors, such as gastric cancer [8], colorectal cancer [9, 10], prostate cancer [11], laryngeal squamous cell carcinoma [12], lung cancer [13, 14], hepatocellular carcinoma [15], ovarian cancer [16], breast cancer [17], leukemia [18], pancreatic cancer [19], and glioma [20]. Moreover, low expression of CHD5 correlated with unfavorable clinical features and poor survival [21]. Therefore, downregulation of CHD5 may be a critical initiating molecular event in tumorigenesis and represent a prognostic biomarker of outcome in patients with cancer. However, only one previous study has reported that CHD5 is inactivated by promoter methylation in RCC [22]. The clinical significance of CHD5 expression in RCC and the molecular mechanism underlying tumorigenesis remain unclear.
In this study, we detected the expression of CHD5 in RCC tissues and cell lines and further assessed the correlation between CHD5 expression and clinicopathological characteristics. Subsequently, we used a gain-of-function assay to evaluate its effects on cell proliferation, migration, and invasion, as well as the key signaling pathways involved in the regulation of CHD5. Our study showed that downregulation of CHD5 expression was related to advanced TNM stage, high Fuhrman grade, lymph node metastasis, and short overall survival. Overexpression of CHD5 induced G1 phase arrest and apoptosis, thereby inhibiting proliferation in vivo and in vitro, and suppressed migration and invasion in vitro. CHD5 positively regulated the p14ARF/p53 and p16INK4a/RB pathways to exert tumor suppressor functions. In conclusion, we elucidated the role of CHD5 in the tumorigenesis of RCC, which might provide a new insight for the diagnosis and treatment of renal cancer.
2. Materials and Methods
2.1. Clinical Tissues and Cell Lines
Twenty-four pairs of fresh primary tumor tissues and adjacent tissues were obtained from ZhuJiang Hospital of Southern Medical University. Clinicopathological information was collected from hospital records. Informed consent was obtained from all patients with approval of the Ethics Committee of ZhuJiang Hospital of Southern Medical University.
Human RCC cell lines (786-0, 769-P, Caki-1, ACHN, and A498) and normal epithelial cell lines (HEK293 and HK-2) were ordered from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). RCC cell lines were grown in RPMI-1640 medium (Gibco, Grand Island, NY, USA) with 10% fetal bovine serum (FBS; Gibco). HEK293 and HK-2 were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) and DMEM/F12 (Gibco) with 10% FBS, respectively. All cells were incubated at 37°C with 5% CO2 atmosphere.
2.2. Immunohistochemistry Assay
The tissue array (Outdo Biotech, Shanghai, China) was applied for immunohistochemistry (IHC) staining to detect CHD5 expression in 90 RCC patients. Antigen unmasking was performed in citrate buffer by microwaving after deparaffinization and rehydration, followed by inactivation of endogenous peroxidase with 0.3% H2O2. Sections were blocked with goat serum (ZSGB-BIO, Beijing, China) and incubated with CHD5 (Cell Signaling Technology, Beverly, USA; 1 : 100 dilution) overnight at 4°C. Subsequently, sections were probed with anti-rabbit antibody and avidin-biotin peroxidase at room temperature and visualized using diaminobenzidine before counterstaining with hematoxylin. The percentage of positive cells (PP) were graded as follows: 0 (<1%), 1 (1-10%), 2 (11-50%), and 3 (>50%). Staining intensity (SI) was defined as follows: no staining, weak staining, moderate staining, and strong staining, corresponding to 0-3 points. PP multiplied by SI was identified as immunoreactivity scoring (IRS). IRS 0–1 was defined as low expression, and >1 was defined as high expression [19].
2.3. Lentivirus-Mediated Overexpression of CHD5
To generate CHD5 overexpression (CHD5-OE) cells using CRISPR/Cas9 gene editing technology, dCas9 vectors and sgRNA targeting the CHD5 gene were cloned into dCas-VP64-Puro and sgRNA-MS2-P65-HSF1-Neo, respectively. The LV-dCas9 and LV-sgRNA lentivirus were produced by GeneChem (Shanghai, China). We designed three pairs of sgRNA oligonucleotides, and the sequences were as follows: sgRNA1 (“CCTCGGCCGGCTGCGGGACT”), sgRNA2 (“CGGCGGCAGCGCCAGAGGCA”), and sgRNA3 (“GCCCGGGCTTTGCGGGGAGC”). The LV-dCas9-VP64 lentivirus was seeded in the ACHN and 769-P cells at a multiplicity of infection (MOI) of 20 and screened with puromycin (Beyotime, Shanghai, China) at a final concentration of 2 μg/ml for 5–7 days. Subsequently, the LV-sgRNA lentivirus was seeded and the empty vector served as the control. The CHD5 overexpression cells, empty vector transfected cells, and untransfected cells were harvested 7–10 days later, which were defined as CHD5-OE, Ctrl-OE, and NC cells, respectively.
2.4. RNA Isolation and Quantitative Reverse Transcription PCR (RT-qPCR)
RNA was obtained by dissolving tissues and cell lines using TRIzol (Invitrogen, Carlsbad, CA, USA), then reverse transcribed into cDNA using a PrimeScript RT Reagent Kit with gDNA Eraser (TaKaRa, Japan), and the expression of CHD5 was detected by SYBR Premix Ex Taq II (TaKaRa, Japan). Primers sequences were as follows: CHD5: 5′-CGAAGGCTACAAGTATGAGCGG-3′ and 5′-GGTTGAGAGGAGGAAGCAGAAC-3′; MMP-9: 5′-CTGGAGACCTGAGAACCAATC-3′ and 5′-CAGAGATTTCGACTCTCCACG-3′; MMP-2: 5′-GATAACTCTGGACTTAGACCGC-3′ and 5′-CAGCCATAGAAGGTGTTCAGG-3′; and β-actin: 5′-CATGTACGTTGCTATCCAGGC-3′ and 5′-CTCCTTAATGTCACGCACGAT-3′. The relative expression of CHD5 was analyzed by the 2-ΔΔT method and normalized with β-actin.
2.5. Western Blot
Proteins were isolated from cell lines and tissues by the RIPA lysate (Beyotime), then separated by electrophoresis on 8–15% SDS-polyacrylamide gel and transferred to PVDF membrane (Bio-Rad). After blockage with 5% skim milk for 1 hour, the primary antibody (Cell Signaling Technology) was incubated at 4°C overnight and then probed with a secondary antibody (Beyotime). Signals were visualized by ECL Western Blotting Detection Reagents (Epizyme, Shanghai, China).
2.6. Cell Counting Kit-8 (CCK-8) Analysis
Cell proliferation was assessed by CCK-8 assay. 2 × 103cells were inoculated into 96-well plates and incubated for cell attachment. Ten microliters of the enhanced CCK-8 reagent (Beyotime) was added and incubated for 2 hours; then, the absorbance was measured at 450 nm with Infinite M200 (Tecan, Switzerland). Thereafter, measurements were taken every 24 hours until 96 hours.
2.7. Plate Colony Formation Assays
The 769-P and ACHN (500 cells per dish) cells were seeded onto 35 mm dishes. After two weeks of culture, colonies were stained with 0.1% crystal violet (Beyotime) and counted with a light microscope.
2.8. Flow Cytometry Analysis
To examine apoptotic cells, cells from each group were harvested and resuspended in 195 μl of binding solution. Then, 5 μl of Annexin V-FITC and 10 μl of propidium iodide staining solution were added and mixed gently. After incubation at room temperature in the dark for 15 minutes, the apoptotic cells were detected by flow cytometry (BD Biosciences, MA, USA).
For cell cycle analysis, cells were fixed with ice-cold 70% ethanol for 12 hours; then, propidium iodide staining solution and RNase A (Beyotime) were added and bathed for 30 minutes at 37°C. Red fluorescence was detected by flow cytometry (BD Biosciences), and DNA content was analyzed using ModFit LT software.
2.9. Wound Healing and Transwell Assays
Wound healing assay was carried out to assess migration ability. The 769-P and ACHN cells were seeded into 6-well plates and maintained until cell confluence reached 90–100%, scratched with 10 μl micropipette tips, and then cultured in serum-free medium. Images were captured with an inverted microscope at 0 and 24 hours after the scratch.
Transwell assay was performed in a 24-well transwell chamber (Corning, NY, USA) to evaluate cell invasion capacity. Matrigel (BD Biosciences) was diluted to 300 μg/ml to coat the chamber. Subsequently, the upper and low chambers were supplied with 100 μl of serum-free medium containing 105 cells and 600 μl medium with 10% FBS, respectively. Following 24 hours of incubation, the cells on the surface of the membrane were gently wiped with a cotton swab, and the cells invading into the membranes were stained with 0.1% crystal violet. Five fields were randomly observed with a microscope.
2.10. Xenograft Mouse Tumor Models
Eighteen 4-5-week-old BALB/c nude mice were obtained from the Experimental Animal Center of Southern Medical University. Mice were randomized into three groups (NC, Ctrl-OE, and CHD5-OE) with six mice per group. The ACHN cell suspensions were mixed with an equal volume of Matrigel (BD Biosciences), and then 5 × 106 cells were administered into the right axilla of mice. The length (L) and width (W) of the tumors were recorded once a week, and tumor volumes ((L × W2)/2) were calculated. On day 28, mice were sacrificed and tumors were resected. The experiments were approved by the Animal Research Ethics Committee of Southern Medical University.
2.11. Statistical Analysis
SPSS software version 20.0 (SPSS Inc., Chicago, IL, USA) was used to analyze the data, and the data were presented as mean ± standard error of the mean (SEM). t-test or Mann-Whitney U test were conducted to analyze differences between the two groups. One-way ANOVA was performed to compare the differences among multiple groups, and Kaplan-Meier survival analysis was used to compare patients' overall survival time. p < 0.05 was defined as statistically significant.
3. Results
3.1. CHD5 Is Downregulated and Correlated with Adverse Clinicopathological Characteristics in RCC Patients
To understand the role of CHD5 in RCC, we first measured the mRNA and protein levels in 24 paired tumor and adjacent tissues. The results showed that the expression of CHD5 was obviously downregulated in tumor tissues compared to matched adjacent tissues (Figure 1(a)). In advanced pathological stages (stages III and IV), CHD5 levels were lower than those in primary pathological stages (stages I and II) (Figure 1(b)). Similarly, downregulation of CHD5 was also observed in five human RCC cell lines compared to normal renal epithelial cells HK-2 and HEK293 (Figure 1(c)). Subsequently, the correlation between CHD5 expression levels and clinicopathological characteristics was assessed and the results showed that low CHD5 expression was closely related to advanced TNM stage, high Fuhrman grade, and lymph node metastasis; nevertheless, there was no relationship to patients' gender, age, and tumor size (Table 1). In addition, Kaplan-Meier survival analysis of tissue array immunohistochemical staining results showed that patients with low CHD5 expression had a shorter overall survival than patients with high CHD5 expression (Figures 1(d) and 1(e)). Taken together, these data indicate that CHD5 is involved in the tumorigenesis and has a significant correlation with the prognosis of patients with RCC.
Figure 1.
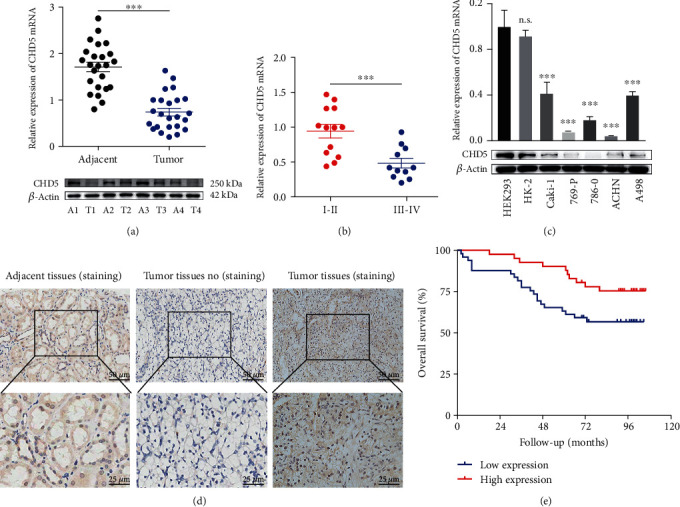
Expression of CHD5 in RCC tissues and cell lines. (a) CHD5 was downregulated in 24 RCC tumor tissues (T) compared to adjacent tissues (A). (b) CHD5 expression in TNM stage III-IV was lower than that in stage I-II. (c) CHD5 is downregulated in RCC cell lines Caki-1, 769-P, 786-0, ACHN, and A498, compared to HEK293 and HK-2. (d) Representative IHC staining with CHD5 in tumor and adjacent tissues. (e) Kaplan-Meier survival analysis assessed the correlation between CHD5 expression and overall survival. ∗∗∗p < 0.001. n.s.: no significance.
Table 1.
Correlation between CHD5 expression levels and clinicopathological features in RCC patients.
| Variables | Number (%) | mRNAa (mean ± SEM) | p value | Proteinb (mean ± SEM) | p value |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 17 (70.8) | 0.474 ± 0.061 | 0.573 | 0.629 ± 0.117 | 0.508 |
| Female | 7 (29.2) | 0.409 ± 0.094 | 0.494 ± 0.125 | ||
| Age | |||||
| <60 | 14 (58.3) | 0.532 ± 0.068 | 0.071 | 0.722 ± 0.135 | 0.101 |
| ≥60 | 10 (41.7) | 0.348 ± 0.064 | 0.404 ± 0.079 | ||
| Tumor size | |||||
| <7 | 14 (58.3) | 0.545 ± 0.072 | 0.069 | 0.738 ± 0.139 | 0.061 |
| ≥7 | 10 (41.7) | 0.330 ± 0.047 | 0.382 ± 0.050 | ||
| TNM stage | |||||
| I-II | 13 (54.2) | 0.630 ± 0.054 | <0.001 | 0.797 ± 0.138 | 0.008 |
| III-IV | 11 (45.8) | 0.249 ± 0.029 | 0.344 ± 0.049 | ||
| Fuhrman grade | |||||
| I-II | 12 (50.0) | 0.603 ± 0.074 | 0.004 | 0.811 ± 0.153 | 0.057 |
| III-IV | 12 (50.0) | 0.308 ± 0.035 | 0.368 ± 0.038 | ||
| Lymph node metastasis | |||||
| Yes | 6 (25.0) | 0.203 ± 0.014 | 0.002 | 0.307 ± 0.046 | 0.028 |
| No | 18 (75.0) | 0.539 ± 0.054 | 0.684 ± 0.111 |
aRelative expression of CHD5 ratio by RT-qPCR. bGrayscale ratio by western blot.
3.2. CHD5 Overexpressed in the ACHN and 769-P Cells by the CRISPR/dCas9 SAM System
The CRISPR/dCas9 SAM (synergistic activation mediator) system is a method for endogenously increasing the expression of target genes that was first proposed and established by Konermann and colleagues [23]. In brief, the dCas9-VP64 fusion protein (without endonuclease activity) binds to the target gene promoter region with the guidance of sgRNA to recruit transcription activation complex MS2-P65-HSF1, thereby increasing the expression of the target gene [23]. In the present study, we first designed three pairs of sgRNAs targeting the CHD5 gene on the CRISPR Design (http://crispr.mit.edu) and cloned them into the LV-sgRNA-MS2-P65-HSF1-Neo vector. Subsequently, the dCas9-VP64 fusion protein and sgRNA-MS2-P65-HSF1 sequence expression frame were introduced into the ACHN and 769-P cells by SAM double-vector lentivirus, respectively. RT-qPCR and western blot were performed to verify the transfected efficiency. The results indicated that the sgRNA3 sequence could significantly increase the expression of CHD5 in the ACHN and 769-P cells (Figure 2(a)).
Figure 2.
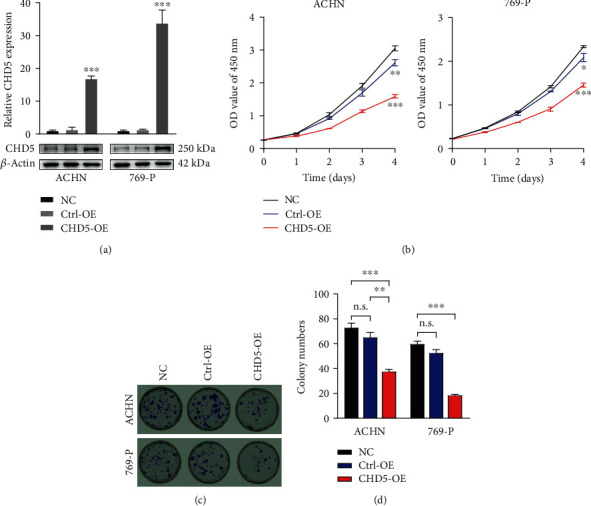
CHD5 inhibits cell proliferation in vitro. (a) CHD5 expression in the ACHN and 769-P cells transfected with lentivirus in comparison to the NC and Ctrl-OE groups. (b) CCK-8 assay examined the proliferation in the ACHN and 769-P cells. (c, d) Colony formation assays in the ACHN and 769-P cells. ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05. n.s.: no significance.
3.3. CHD5 Overexpression Inhibits RCC Cell Proliferation In Vitro
To illustrate the biological role of CHD5 in RCC, the CCK-8 and clone formation assays were performed to determine cell proliferation in vitro. CCK-8 assay demonstrated that the proliferation rate of the ACHN and 769-P cells overexpressing CHD5 was significantly lower than that of the Ctrl-OE and NC groups (Figure 2(b)), and clone formation assays also verified the inhibitory effect of CHD5 on proliferation in vitro (Figures 2(c) and 2(d)).
3.4. CHD5 Prompts Cell Cycle G1 Phase Arrest and Induces Apoptosis
We used flow cytometry to evaluate the cell cycle and apoptosis, as these factors are closely related to cell proliferation. After overexpression of CHD5 in the ACHN and 769-P cells, the cell ratio in G1 phase increased notably, while that in the S and G2 phases decreased compared with the those in the Ctrl-OE and NC groups (Figures 3(a) and 3(b)). Apoptosis detection results showed that the number of apoptotic cells also increased significantly in both the ACHN and 769-P cells overexpressing CHD5 (Figures 3(c) and 3(d)). The results indicate that CHD5 induces cell cycle G1 phase arrest and apoptosis to inhibit proliferation.
Figure 3.
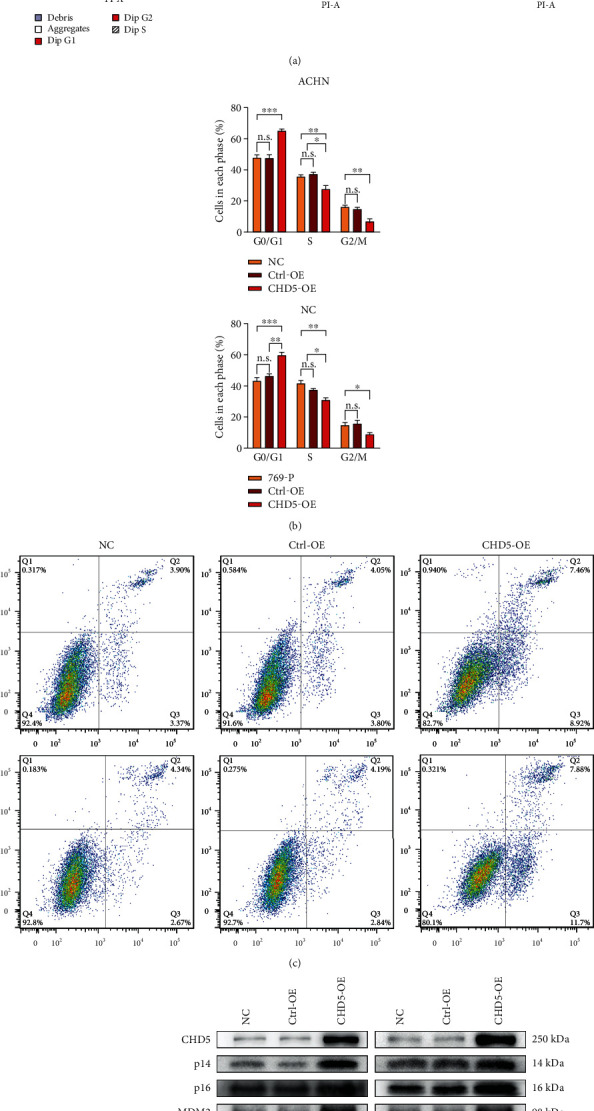
Effect of CHD5 on cell cycle and apoptosis. (a, b) Distribution of cell cycle in different phases analyzed by flow cytometry. (c, d) Percentage of apoptotic cells detected by flow cytometry. (e) The influence of CHD5 on the p14ARF/p53 and p16INK4a/RB pathways by western blot. ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05. n.s.: no significance.
3.5. CHD5 Activates the p16INK4a/p53 and p14ARF/RB Pathways
The INK4a/ARF locus encoded the tumor suppressor proteins, p16INK4a and p14ARF (p19ARF in mice), prompting the activities of RB and p53 to regulate cell growth [24]. Studies have shown that the p53 and RB pathways are inactivated in a variety of tumors, leading to cell cycle disorder and disruption of apoptosis [25]. A previous study has shown that CHD5 positively regulated p19ARF/p53-mediated pathways in mouse models and loss of CHD5 function is prone to malignant transformation by impairing the p19ARF/p53 pathway [5]. However, it is unclear whether CHD5 has similar regulatory effects on INK4a/ARF in different species and cells. Therefore, we predicted that CHD5 mediates the involvement of INK4a/ARF in the regulation of biological behavior in RCC. We examined the key proteins of the p16INK4a/RB- and p14ARF/p53-mediated pathways using western blot. As shown in Figure 3(e), the expression of p14ARF, MDM2, p53, p21, Bax, caspase-9, p16INK4a, and RB increased, while the expression of p-RB, cyclin D1, and CDK4 decreased. This suggests that CHD5 regulates cell cycle and apoptosis by activating the p14ARF/p53 and p16INK4a/RB pathways.
3.6. CHD5 Suppresses Migration and Invasion
As shown in Table 1, CHD5 low expression is related to lymph node metastasis. Therefore, we next investigated the effect of CHD5 on the migration and invasion in RCC cell lines. Wound healing and transwell assays demonstrated that overexpression of CHD5 significantly suppressed the migration and invasion of the ACHN and 769-P cells (Figures 4(a) and 4(b)). We further explored the potential mechanism of CHD5 inhibition of cell motility. Western blot and RT-qPCR were performed to detect previously reported MMPs (MMP-2 and MMP-9), which were closely correlated with the progression of RCC [26]. The results showed that MMP-9 significantly decreased in cells overexpressing CHD5 (Figures 4(c) and 4(d)). Therefore, we conclude that CHD5 may inhibit RCC metastasis, at least in part, through the regulation of MMP-9 expression.
Figure 4.
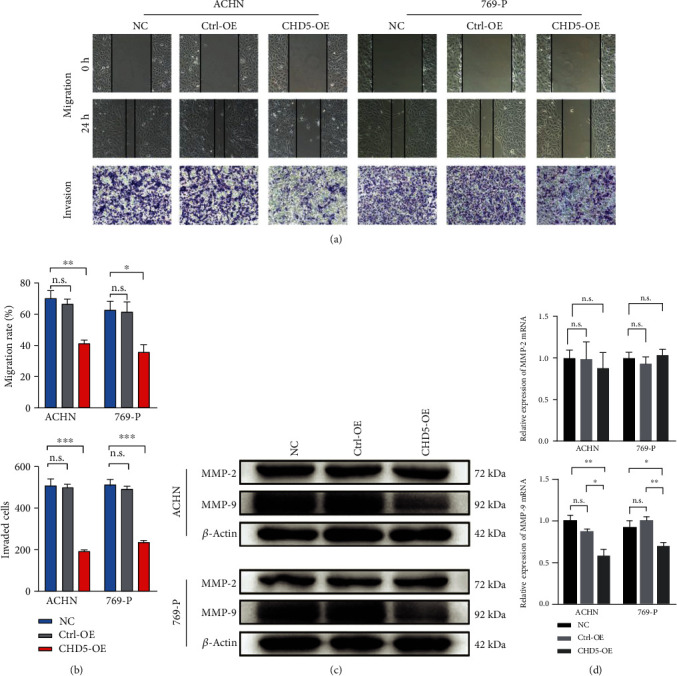
CHD5 suppresses migration and invasion of the ACHN and 769-P cells. (a, b) Migration and invasion ability were examined by wound healing and transwell assays. (c, d) MPP-2/9 expression levels were detected by western blot and RT-qPCR. ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05. n.s.: no significance.
3.7. CHD5 Inhibits Tumor Growth In Vivo
To further investigate the effect of CHD5 on tumorigenesis in vivo. ACHN cells with stable expression of CHD5 (Figure 5(a)) were injected subcutaneously into BALB/c nude mice. Four weeks later, mice were sacrificed and tumors were resected (Figure 5(b)). The tumor mass was weighed, and the tumor volume was calculated. It was found that the average tumor weight (Figure 5(c)) and average tumor volume (Figure 5(d)) of the CHD5-OE group were significantly lower than those of the Ctrl-OE and NC groups. Therefore, overexpression of CHD5 significantly inhibits tumorigenesis in vivo.
Figure 5.
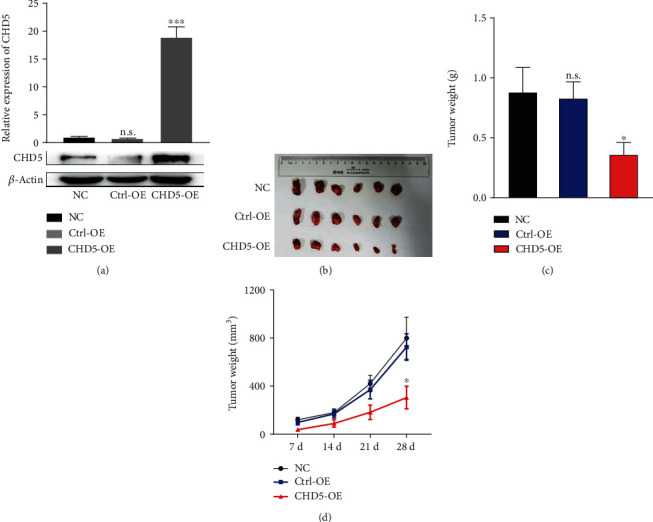
CHD5 inhibits tumor growth in vivo. (a) The efficiency of CHD5 overexpressing in ACHN verified by WB and RT-qPCR. (b) Tumors were resected after 4 weeks of ACHN cell injection. (c) Tumor weight was measured after 4 weeks. (d) Tumor size was recorded every week. ∗∗∗p < 0.001 and ∗p < 0.05. n.s.: no significance.
4. Discussion
RCC is the malignant tumor with the highest mortality rate in the urogenital system. Over the past decade, there has been some progress in the treatment of RCC, such as the advent of targeted agents and immune checkpoint inhibitors [27]; however, the clinical outcomes of patients with RCC are still unsatisfactory [28, 29]. Hence, it is necessary to identify new biomarkers that provide valuable information for clinicians to choose appropriate treatment options and predict prognosis of patients.
CHD5 is the fifth member of the chromatin remodeling family and is involved in regulating the structure and transcription of chromatin [21]. CHD5 binds to unmodified N-terminus of histone 3 for tumor suppression [30]. Increasing evidence shows that CHD5 plays a critical role in human cancer initiation and progression [21]. Studies on neuroblastoma have found that high CHD5 expression is closely related to favorable clinical and biologic characteristics, while low expression or deletion is related to adverse characteristics such as MYCN amplification and poor prognosis. Interestingly, induction chemotherapy could restore the expression of CHD5 in tumor tissues of half of high-risk neuroblastoma patients and present good response to chemotherapy and radiotherapy [7, 31]. The study by Du et al. revealed that CHD5 was downregulated in gallbladder cancer, and low expression of CHD5 was associated with poor clinical and pathological characteristics; and the patients with low expression of CHD5 had shorter overall survival and disease-free survival [32]. Similar results were observed in other tumors [19, 20, 33–35]. Herein, we reported the prognostic value of CHD5 expression in RCC patients. In brief, CHD5 is significantly downregulated in RCC tissues and cell lines, and downregulation of CHD5 was closely related to advanced TNM stage, high Fuhrman grade, and lymph node metastasis, as well as poor overall survival. Taken together, CHD5 was involved in the initiation and progression of RCC, and a decrease in CHD5 expression was associated with poor clinical prognosis.
Unlimited proliferation of tumor cells often involves cell cycle disorder and apoptotic response inactivation [36, 37]. The INK4a/ARF locus encoded two gene products, p16INK4a and p14ARF (p19ARF in mice), which function by promoting the activity of RB and p53 transcription factors to regulate the cell cycle and apoptosis [24]. The disruption of p14ARF/p53 and p16INK4a/RB is involved in tumorigenesis of multiple tumors [38]. Moreover, the p16INK4a/p14ARF axis has been previously reported to be involved in the development of RCC [39]. In addition, Bagchi et al. found that CHD5 could positively regulate INK4a/ARF in mice and CHD5 deficiency is prone to malignant transformation by impairing the p19ARF/p53 pathway [5]. Nevertheless, it is unclear whether it has a similar effect on p14ARF in human RCC cells. In our present study, we revealed that CHD5 overexpression increased the expression level of p14ARF and p16INK4a and then activated the p53 and RB pathways, resulting in a decrease in cyclin D1 and CDK4 expression and an increase in Bax and caspase-9 expression, thereby prompting G1 phase arrest and inducing apoptosis. These results suggested that a potential mechanism underlying CHD5 inhibition of RCC proliferation might be dependent on the activation of the p53 and RB signaling pathways by the INK4a/ARF locus.
Distant metastasis is the leading cause of death in RCC patients. Degradation of the extracellular matrix (ECM) by matrix metalloproteinase (MMP) is a key process in tumor metastasis and progression. Studies have shown that MMPs are frequently upregulated in RCC, including MMP-2/7/9/14/17 [40, 41]. In particular, MMP-2/9 is strongly correlated with poor survival in clear cell RCC, as MMP-2/9 is also involved in angiogenesis, which has a crucial role in the progression of highly vascularized malignancies, such as RCC [26]. Therefore, MMP-2/9 expression is higher in patients with metastatic RCC, compared with localized RCC [42]. In addition, a study has shown that the introduction of wild-type p53 into p53 mutant human soft tissue sarcoma cells could reduce the expression of MMP-9 mRNA and protein and decrease the proteolytic activity of MMP-9 [43]. Our data showed that low expression of CHD5 in RCC was closely associated with advanced TNM stage and lymph node metastasis. Moreover, CHD5 overexpression in the ACHN and 769-P cells suppressed migration and invasion, upregulated p53, and decreased the expression of MMP-9. This suggested that CHD5 might be involved in RCC metastasis through downregulation of MMP-9 expression mediated by p53. Certainly, this needs to be confirmed by a series of follow-up studies.
5. Conclusions
Our study indicates that CHD5 is downregulated in RCC and is closely associated with adverse clinicopathological features and poor outcomes. CHD5 inhibits the tumorigenesis by activating the p14ARF/p53 and p16INK4a/RB pathways. These data suggest that CHD5 could be a diagnostic biomarker and potential therapeutic target for RCC.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (No. 81771710), the Guangzhou Science and Technology Plan Project (Nos. 20170701006 and 201610010174), and the Guangdong Natural Science Foundation (No. 2017A030310100).
Contributor Information
Sheng Huang, Email: sheng_uro@163.com.
Qitao Yan, Email: yanqitao1979@126.com.
Rui Zhao, Email: zhaorui1981@126.com.
Chunxiao Liu, Email: liuchx888@163.com.
Data Availability
The data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
Authors' Contributions
Sheng Huang and Qitao Yan contributed to this work equally.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R. L., Torre L. A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a Cancer Journal for Clinicians. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Capitanio U., Bensalah K., Bex A., et al. Epidemiology of renal cell carcinoma. European Urology. 2019;75(1):74–84. doi: 10.1016/j.eururo.2018.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jonasch E., Futreal P. A., Davis I. J., et al. State of the science: an update on renal cell carcinoma. Molecular cancer research: MCR. 2012;10(7):859–880. doi: 10.1158/1541-7786.MCR-12-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagchi A., Mills A. A. The quest for the 1p36 tumor suppressor. Cancer Research. 2008;68(8):2551–2556. doi: 10.1158/0008-5472.CAN-07-2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bagchi A., Papazoglu C., Wu Y., et al. CHD5 is a tumor suppressor at human 1p36. Cell. 2007;128(3):459–475. doi: 10.1016/j.cell.2006.11.052. [DOI] [PubMed] [Google Scholar]
- 6.Fujita T., Igarashi J., Okawa E. R., et al. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. Journal of the National Cancer Institute. 2008;100(13):940–949. doi: 10.1093/jnci/djn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia I., Mayol G., Rodríguez E., et al. Expression of the neuron-specific protein CHD5 is an independent marker of outcome in neuroblastoma. Molecular Cancer. 2010;9(1):p. 277. doi: 10.1186/1476-4598-9-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X., Lau K. K., So L. K., Lam Y. W. CHD5 is down-regulated through promoter hypermethylation in gastric cancer. Journal of Biomedical Science. 2009;16(1):p. 95. doi: 10.1186/1423-0127-16-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mokarram P., Kumar K., Brim H., et al. Distinct high-profile methylated genes in colorectal cancer. PLoS One. 2009;4(9, article e7012) doi: 10.1371/journal.pone.0007012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fatemi M., Paul T. A., Brodeur G. M., Shokrani B., Brim H., Ashktorab H. Epigenetic silencing of CHD5, a novel tumor-suppressor gene, occurs in early colorectal cancer stages. Cancer. 2014;120(2):172–180. doi: 10.1002/cncr.28316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berger M. F., Lawrence M. S., Demichelis F., et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470(7333):214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J., Chen H., Fu S., Xu Z. M., Sun K. L., Fu W. N. The involvement of CHD5 hypermethylation in laryngeal squamous cell carcinoma. Oral Oncology. 2011;47(7):601–608. doi: 10.1016/j.oraloncology.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Zhao R., Yan Q., Lv J., et al. CHD5, a tumor suppressor that is epigenetically silenced in lung cancer. Lung Cancer. 2012;76(3):324–331. doi: 10.1016/j.lungcan.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 14.Baykara O., Tansarikaya M., Bulut P., Demirkaya A., Buyru N. CHD5 is a potential tumor suppressor in non small cell lung cancer (NSCLC) Gene. 2017;618:65–68. doi: 10.1016/j.gene.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 15.Zhao R., Wang N., Huang H., Ma W., Yan Q. CHD5 a tumour suppressor is epigenetically silenced in hepatocellular carcinoma. Liver international: official journal of the International Association for the Study of the Liver. 2014;34(6):e151–e160. doi: 10.1111/liv.12503. [DOI] [PubMed] [Google Scholar]
- 16.Gorringe K. L., Choong D. Y., Williams L. H., et al. Mutation and methylation analysis of the chromodomain-helicase-DNA binding 5 gene in ovarian cancer. Neoplasia. 2008;10(11):1253–IN32. doi: 10.1593/neo.08718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulero-Navarro S., Esteller M. Chromatin remodeling factor CHD5 is silenced by promoter CpG island hypermethylation in human cancer. Epigenetics. 2014;3(4):210–215. doi: 10.4161/epi.3.4.6610. [DOI] [PubMed] [Google Scholar]
- 18.Zhao R., Meng F., Wang N., Ma W., Yan Q. Silencing of CHD5 gene by promoter methylation in leukemia. PLoS One. 2014;9(1, article e85172) doi: 10.1371/journal.pone.0085172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall W. A., Petrova A. V., Colbert L. E., et al. Low CHD5 expression activates the DNA damage response and predicts poor outcome in patients undergoing adjuvant therapy for resected pancreatic cancer. Oncogene. 2014;33(47):5450–5456. doi: 10.1038/onc.2013.488. [DOI] [PubMed] [Google Scholar]
- 20.Wang L., He S., Yanyang T. U., et al. Downregulation of chromatin remodeling factor CHD5 is associated with a poor prognosis in human glioma. Journal of clinical neuroscience. 2013;20(7):958–963. doi: 10.1016/j.jocn.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 21.Kolla V., Zhuang T., Higashi M., Naraparaju K., Brodeur G. M. Role of CHD5 in human cancers: 10 years later. Cancer Research. 2014;74(3):652–658. doi: 10.1158/0008-5472.CAN-13-3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du Z., Li L., Huang X., et al. The epigenetic modifier CHD5 functions as a novel tumor suppressor for renal cell carcinoma and is predominantly inactivated by promoter CpG methylation. Oncotarget. 2016;7(16):21618–21630. doi: 10.18632/oncotarget.7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konermann S., Brigham M. D., Trevino A. E., et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherr C. J. The INK4a/ARF network in tumour suppression. Nature Reviews Molecular Cell Biology. 2001;2(10):731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 25.Polager S., Ginsberg D. p53 and E2f: partners in life and death. Nature Reviews Cancer. 2009;9(10):738–748. doi: 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- 26.Kallakury B. V., Karikehalli S., Haholu A., Sheehan C. E., Azumi N., Ross J. S. Increased expression of matrix metalloproteinases 2 and 9 and tissue inhibitors of metalloproteinases 1 and 2 correlate with poor prognostic variables in renal cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2001;7(10):3113–3119. [PubMed] [Google Scholar]
- 27.Barata P. C., Rini B. I. Treatment of renal cell carcinoma: current status and future directions. CA: a Cancer Journal for Clinicians. 2017;67(6):507–524. doi: 10.3322/caac.21411. [DOI] [PubMed] [Google Scholar]
- 28.Ljungberg B., Albiges L., Abu-Ghanem Y., et al. European Association of Urology Guidelines on Renal Cell Carcinoma: The 2019 Update. European Urology. 2019;75(5):799–810. doi: 10.1016/j.eururo.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Porta C., Cosmai L., Leibovich B. C., Powles T., Gallieni M., Bex A. The adjuvant treatment of kidney cancer: a multidisciplinary outlook. Nature Reviews Nephrology. 2019;15(7):423–433. doi: 10.1038/s41581-019-0131-x. [DOI] [PubMed] [Google Scholar]
- 30.Paul S., Kuo A., Schalch T., et al. Chd5 requires PHD-mediated histone 3 binding for tumor suppression. Cell Reports. 2013;3(1):92–102. doi: 10.1016/j.celrep.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koyama H., Zhuang T., Light J. E., et al. Mechanisms of CHD5 Inactivation in neuroblastomas. Clinical cancer research. 2012;18(6):1588–1597. doi: 10.1158/1078-0432.CCR-11-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du X., Wu T., Lu J., et al. Decreased expression of chromodomain helicase DNA-binding protein 5 is an unfavorable prognostic marker in patients with primary gallbladder carcinoma. Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 2013;15(3):198–204. doi: 10.1007/s12094-012-0903-2. [DOI] [PubMed] [Google Scholar]
- 33.Wu X., Zhu Z., Li W., et al. Chromodomain helicase DNA binding protein 5 plays a tumor suppressor role in human breast cancer. Breast cancer research : BCR. 2012;14(3):p. R73. doi: 10.1186/bcr3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma Z., Song J., Liu S., et al. Decreased expression of the CHD5 gene and its clinicopathological significance in breast cancer: correlation with aberrant DNA methylation. Oncology Letters. 2016;12(5):4021–4026. doi: 10.3892/ol.2016.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong R. R. Y., Chan L. K. Y., Tsang T. P. T., et al. CHD5 Associated with Poor Prognosis in Epithelial Ovarian Cancer. Gynecologic and Obstetric Investigation. 2011;72(3):203–207. doi: 10.1159/000323883. [DOI] [PubMed] [Google Scholar]
- 36.Aubrey B. J., Kelly G. L., Janic A., Herold M. J., Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death and Differentiation. 2018;25(1):104–113. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ingham M., Schwartz G. K. Cell-cycle therapeutics come of age. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology. 2017;35(25):2949–2959. doi: 10.1200/JCO.2016.69.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim W. Y., Sharpless N. E. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127(2):265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Esteller M., Corn P. G., Baylin S. B., Herman J. G. A gene hypermethylation profile of human cancer. Cancer Research. 2001;61(8):3225–3229. [PubMed] [Google Scholar]
- 40.Gobin E., Bagwell K., Wagner J., et al. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer. 2019;19(1):p. 581. doi: 10.1186/s12885-019-5768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y., Wang Y., Liu F. A 44-gene set constructed for predicting the prognosis of clear cell renal cell carcinoma. International Journal of Molecular Medicine. 2018;42(6):3105–3114. doi: 10.3892/ijmm.2018.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slaton J. W., Inoue K., Perrotte P., et al. Expression levels of genes that regulate metastasis and angiogenesis correlate with advanced pathological stage of renal cell carcinoma. The American Journal of Pathology. 2001;158(2):735–743. doi: 10.1016/S0002-9440(10)64016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J., Zhan M., Hannay J. A. F., et al. Wild-type p53 inhibits nuclear factor-kappaB-induced matrix metalloproteinase-9 promoter activation: implications for soft tissue sarcoma growth and metastasis. Molecular cancer research : MCR. 2006;4(11):803–810. doi: 10.1158/1541-7786.MCR-06-0201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included within the article.