

Abstract
This Article describes the development of a stable NiIV complex that mediates C(sp2)–H trifluoromethylation reactions. This reactivity is first demonstrated stoichiometrically and then successfully translated to a NiIV-catalyzed C–H trifluoromethylation of electron-rich arene and heteroarene substrates. Both experimental and computational mechanistic studies support a radical chain pathway involving NiIV, NiIII, and NiII intermediates.
Graphical Abstract
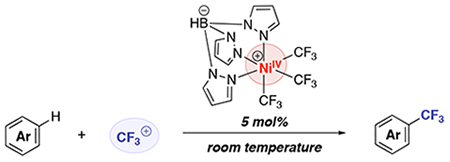
INTRODUCTION
Over the past three decades, nickel has emerged as an economical base-metal catalyst for a wide array of important chemical transformations.1 The vast majority of these Ni-catalyzed processes are proposed to proceed via Ni0/II, NiI/III or NiI/II/III catalytic cycles.2 In many cases, these mechanisms are supported by the isolation of organonickel intermediates and studies of their catalytic competence.3 While organonickel(I), (II), and (III) complexes are well-precedented, until 5 years ago there were very few examples of isolable Ni complexes in the +4 oxidation state.4 As a result, NiIV intermediates have rarely been proposed in catalysis. However, recent progress by our group and others has shown that organonickel(IV) complexes can be synthesized at room temperature using mild oxidants.4b–e Given the accessibility and unique reactivity of NiIV, we sought to investigate its relevance in catalysis.
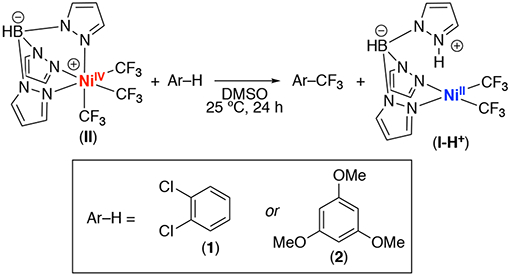
NiIV–CF3 complexes have been shown to exhibit superior stability when compared to other NiIV–alkyls.5 They are also easily prepared via the net two-electron oxidation of NiII precursors with CF3+ oxidants (for example, see A in Figure 1A).4,5 Additionally, a recent report by Nebra and coworkers demonstrated a NiIV–CF3 complex that effects the stoichiometric C–H trifluoromethylation of 1,2-dichlorobenzene under mild conditions (Figure 1B).6 Inspired by these results, we hypothesized that we could develop a NiIV-catalyzed method for the C–H trifluoromethylation of arenes by identifying the optimal combination of ligand, substrate, and CF3+ oxidant (Figure 1C).
Figure 1.
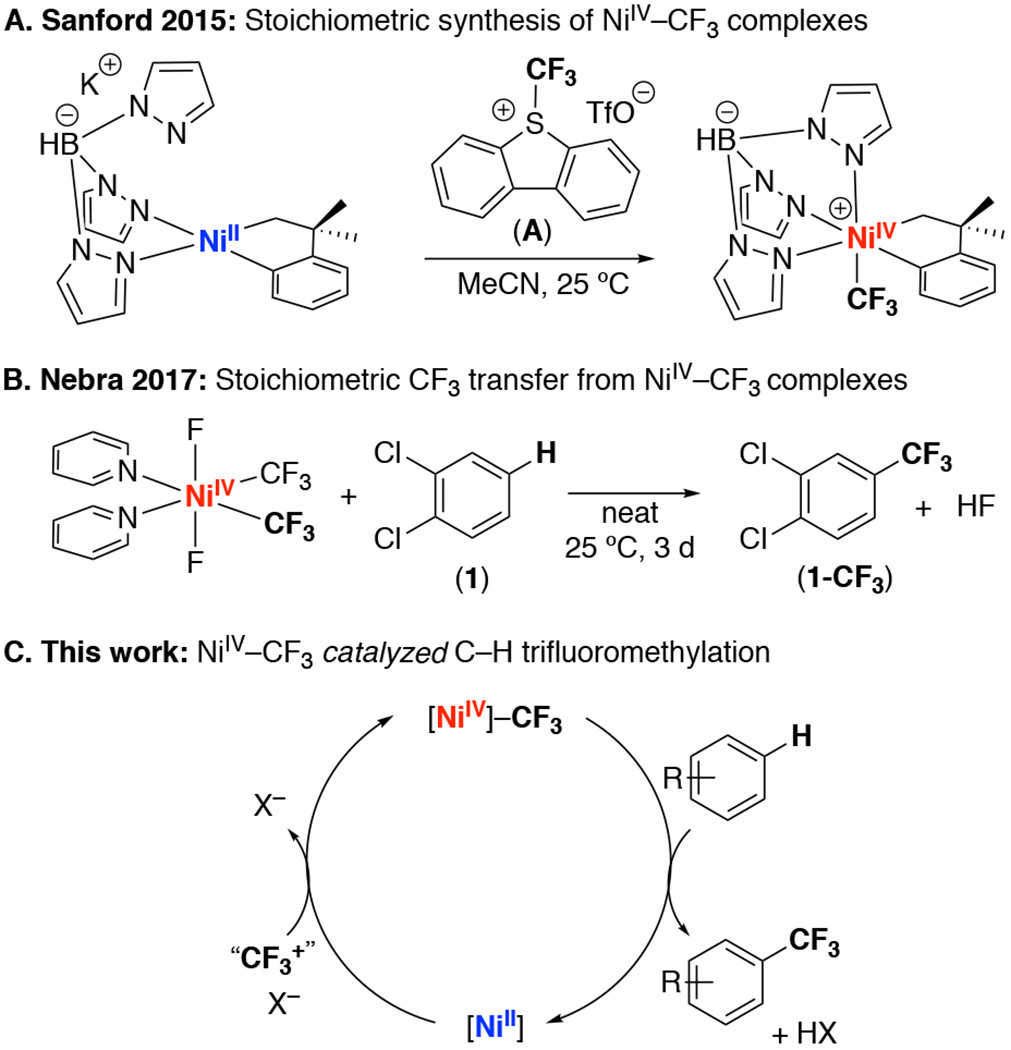
(A) Synthesis of NiIV–CF3 complexes. (B) Stoichiometric C(sp2)-H trifluoromethylation mediated by NiIV. (C) Overall cycle for NiIV-catalyzed trifluoromethylation.
We demonstrate herein the design and synthesis of a NiIV–CF3 complex that participates in a net transfer of CF3+ to electron-rich arenes. Furthermore, in the presence of an electrophilic trifluoromethylating reagent, this NiIV complex serves as an effective catalyst for the C–H trifluoromethylation of arene and heteroarene substrates. Mechanistic studies suggest that this reaction proceeds through a NiII/III/IV cycle involving CF3 radicals as key intermediates.
RESULTS AND DISCUSSION
Design and synthesis of NiIV–CF3 complex.
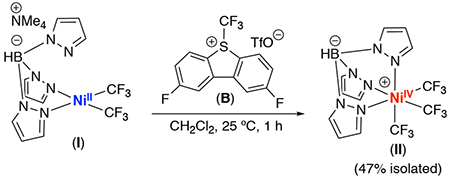
Our initial efforts focused on identifying a ligand scaffold that would enable both steps of the proposed catalytic cycle: (1) the net 2e− oxidation of a NiII precursor with a “CF3+” oxidant to afford a NiIV–CF3 complex and (2) the subsequent reaction of this NiIV–CF3 with arenes to afford C(sp2)–H trifluoromethylation products. Based on previous work from our group,4c,5a we hypothesized that the tris(pyrazolyl)borate (Tp) ligand would be effective in this context. Furthermore, based on Nebra’s results,6 we reasoned that the incorporation of multiple CF3 ligands would render the resulting complex reactive towards CF3 transfer reactions. With these considerations in mind, we initially targeted TpNiIV(CF3)3 (II) (eq. 1).
 |
(1) |
Complex II was synthesized via the oxidation of NiII complex I with 2,8-difluoro-5-(trifluoromethyl)-5H-dibenzo[b,d|thiophen-5-ium trifluoromethanesulfonate (B). The NiIV product was isolated in 47% yield as a yellow solid and was characterized by 1H, 19F, 13C, and 11B NMR spectroscopy. X-ray quality crystals were obtained by recrystallization from MeCN, and an ORTEP diagram is shown in Figure 2.
Figure 2.
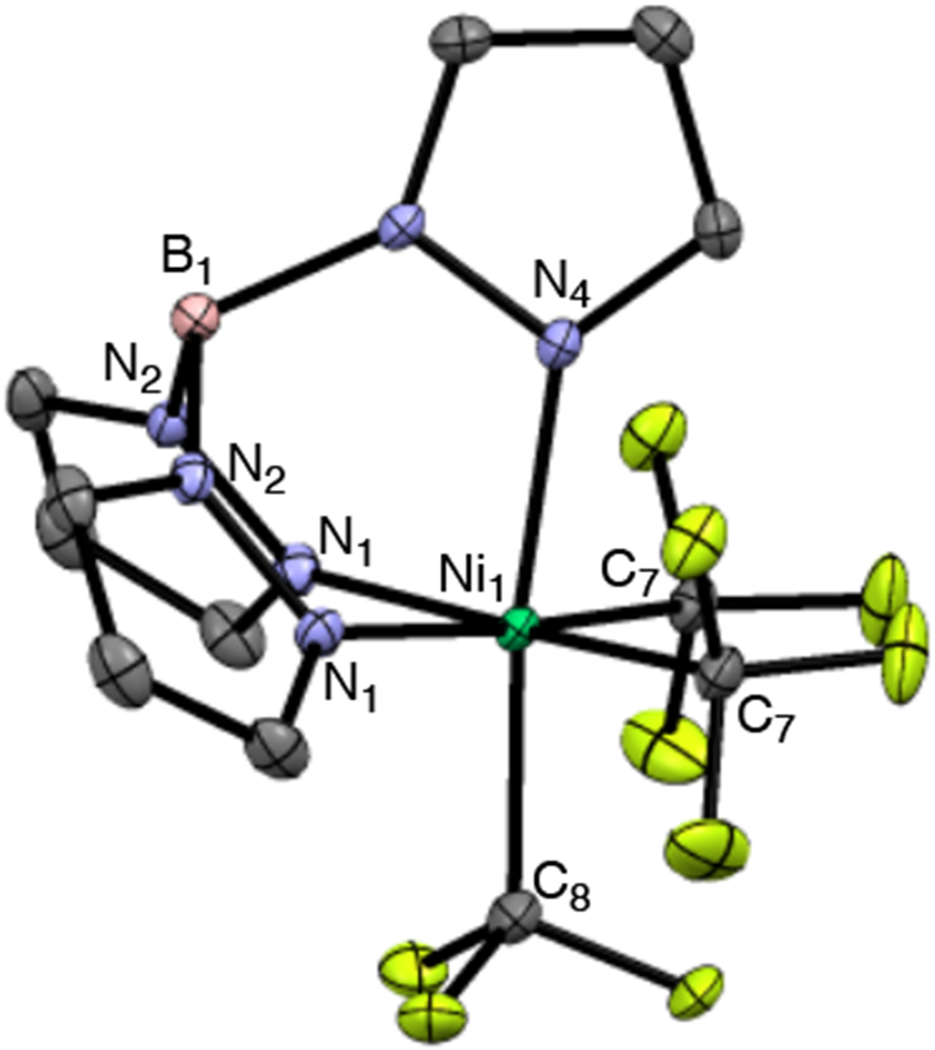
ORTEP diagram for complex II. Hydrogen atoms have been omitted for clarity, thermal ellipsoids drawn at 50% probability. Bond lengths (Å): Ni1–N1 = 2.013, Ni1–N4 = 1.974, Ni1–C7 = 2.011, Ni1–C8 = 1.983 and bond angles (deg): C7–Ni1–C7 = 99.81, C7–Ni1–C8 = 88.05.
In the solid state, complex II is extremely stable to ambient conditions, showing no signs of decomposition after more than a year of storage on the benchtop. However, under analogous conditions as a 0.01 M solution in MeCN, II begins to decompose within 72 h at room temperature. Over this time, the NMR resonances associated with complex II gradually disappear, along with concomitant formation of CF3H.
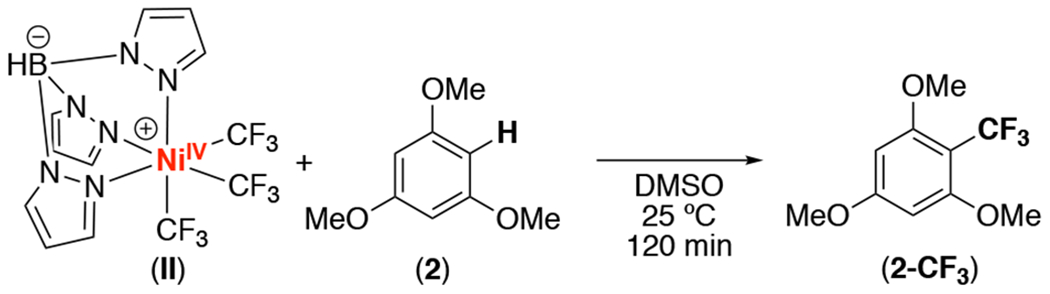
We next explored the reactivity of II in stoichiometric C(sp2)–H trifluoromethylation. Initial studies focused on 1,2-dichlorobenzene (1), the substrate originally employed by Nebra (Figure 1B).6 As shown in Table 1, entry 1, the reaction of II with 1 equiv of 1 afforded traces (<5%) of the C–H trifluoromethylation product 1-CF3 after 24 h at room temperature. This yield increased to 9% when 1 was used as the reaction solvent under otherwise analogous conditions (Table 1, entry 2). We next examined the more electron rich substrate 1,3,5-trimethoxybenzene (2). As shown in Table 1, entry 3, complex II reacted with 1 equiv of 2 to afford 2-CF3 in 47% yield as determined by 19F NMR spectroscopic analysis of the crude reaction mixture. The NiII product I-H+ was also formed in 56% yield.7 These yields increased to 72% and 74%, respectively, upon the use of 5 equiv of 1,3,5-trimethoxybenzene relative to II.
Table 1.
Reactions of II with Ar–H substrates.
Yields determined by 19F NMR spectroscopy using trifluorotoluene as an internal standard and are based on II. All reactions conducted using 1.0 equiv II.
1 : 1.2 ratio of 1-CF3 isomers.
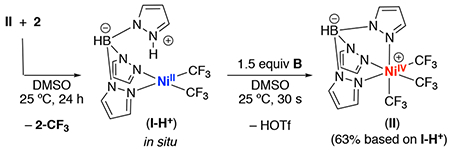
Finally, we evaluated the feasibility of regenerating NiIV complex II via the oxidation of I-H+ with B. As shown in eq. 2, the treatment of a solution of I-H+ (generated in situ from the reaction of II with 2) with 1.5 equiv of B afforded II in 63% yield after 30 s. Overall, thees studies demonstrate each individual step in the II-catalyzed trifluoromethylation of 2.
 |
(2) |
Complex II-catalyzed C–H trifluoromethylation.
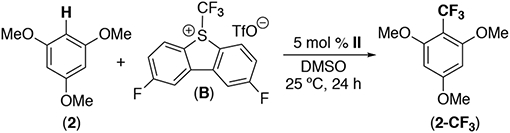
We next explored the use of II as a catalyst for the C-H trifluoromethylation of arenes using B as the “CF3+” source.8,9 Initial studies employed 5 mol % of II in combination with 1 equiv of 1,3,5-trimethoxybenzene and 1 equiv of B in DMSO. After 24 h at room temperature in the dark, this reaction afforded 37% yield of the C-H trifluoromethylation product 2-CF3 (Table 2, entry 1). Importantly, no reaction was observed in the absence of complex II under these conditions (Table 2, entry 2). The presence/absence of ambient light did not impact the yield, ruling out a photoredox-type pathway (Table 2, entries 1 and 3). Employing complex I as the catalyst resulted in slightly diminished yield (25%, entry 4). The use of A as an oxidant also afforded a lower yield (25%, entry 5). Employing an excess of 2 (2 or 5 equiv relative to B) led to an increase in the yield of product 2-CF3 (to 62% and 93%, respectively, entries 6 and 7). Optimization of the reaction time, concentration, temperature, and solvent revealed that the highest yield of 2-CF3 (93%) is obtained under the conditions in entry 7 (see SI for complete optimization details).
Table 2.
Optimizing NiIV-catalyzed trifluoromethylation
 | ||
|---|---|---|
| entry | modification | yield (%)a |
| 1 | dark | 37 |
| 2 | no II | 0 |
| 3 | ambient light | 35 |
| 4 | I as catalyst | 25 |
| 5 | A used as oxidant | 25 |
| 6 | 2 equiv substrate | 62 |
| 7 | 5 equiv substrate | 93 |
Yields determined by 19F NMR spectroscopy using trifluorotoluene as an internal standard and are based on B as the limiting reagent. Standard conditions: 1.0 equiv 2, 1.0 equiv B, 5 mol% II in DMSO at room temperature for 24 h.
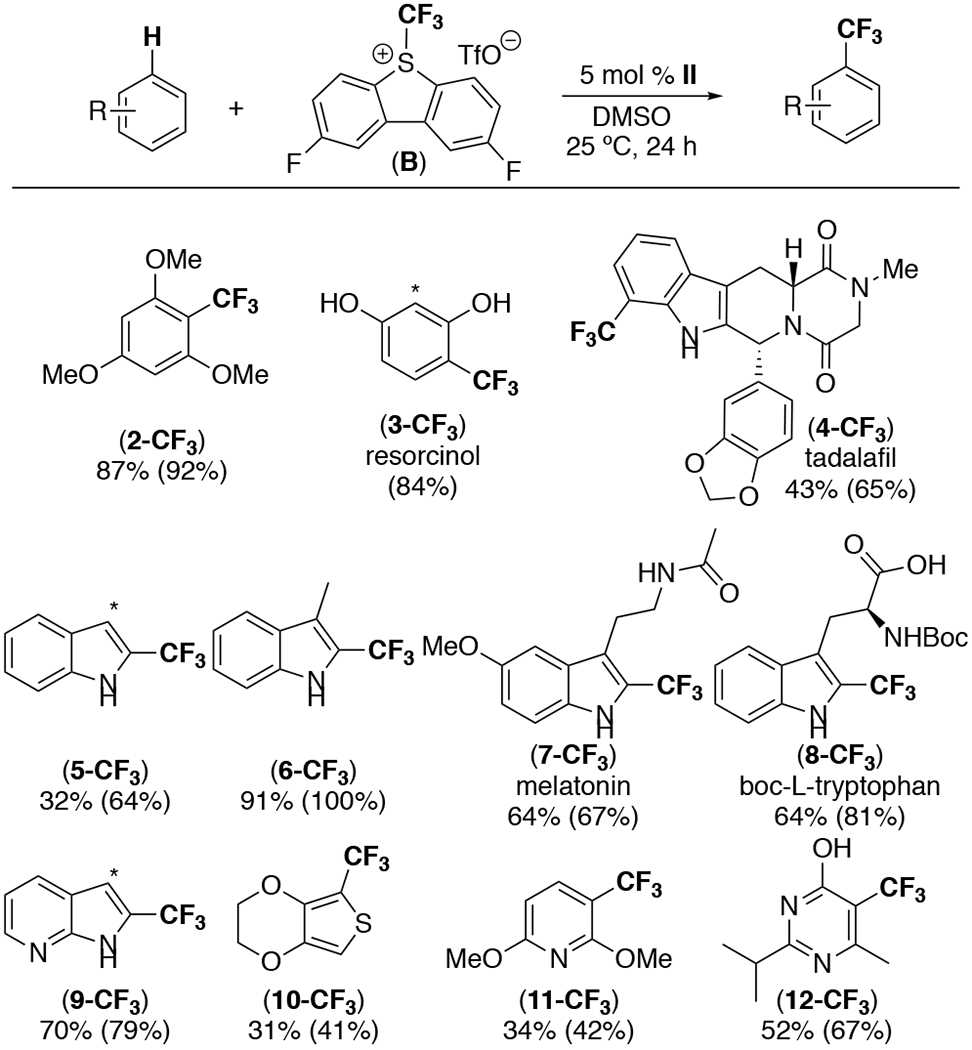
We next evaluated the scope of this II-catalyzed C–H trifluoromethylation. As shown in Scheme 1, this reaction proved applicable to a variety of electron-rich arene and heteroarene substrates (Scheme 1). In addition to trimethoxybenzene, electron rich pyridines as well as indole and thiophene derivatives showed good reactivity. A variety of biologically active molecules, including melatonin, Boc-L-tryptophan, resorcinol, and tadalafil, also served as effective substrates for this transformation. Notably, trifluoromethylation of tadalafil occurs at the 7-position of the indole backbone (rather than on the 1,3-dioxole-substituted ring), a site that has been functionalized in several other reported radical trifluoromethylation reactions.10 Overall, this transformation works best with highly electron-rich (hetero)arenes, while less electron rich substrates afforded moderate to low yields (see SI for details of other substrates examined). This relatively narrow substrate scope as well as the use of 5 equiv of (hetero)arene substrate relative to oxidant represent limitations of this method compared to state-of-the-art radical C–H trifluoromethylation protocols.11,12
Scheme 1.
Scope of NiIV-catalyzed trifluoromethylationa
a General conditions: 1.0 equiv B, 5.0 equiv arene, 5 mol % of II in DMSO for 24 h at 25 ºC. 19F NMR yields are in parentheses and were determined using trifluorotoluene as an internal standard. In cases where multiple isomers were formed, NMR yield is given as a combined yield. *Signifies the site of a minor isomer.
Mechanistic studies of CF3 transfer.
We next focused on interrogating the mechanism of the CF3 transfer step. We originally envisioned two possible pathways for this transformation (Scheme 2). The first (pathway i) involves a direct transfer of CF3+ from complex II to the organic substrate to form the cationic organic intermediate 13 along with the NiII product TpNiII(CF3)2− (I). Intermediate 13 could then undergo deprotonation to form the trifluoromethylated product 2-CF3 along with I-H+. An alternative pathway (ii in Scheme 2) would proceed via release of trifluoromethyl radical from NiIV and subsequent addition of CF3• to 2 to afford the organic intermediate 14 along with the NiIII complex TpNiIII(CF3)2 (III). Compound 14 could then undergo oxidation and deprotonation to generate 2-CF3 along with along with I-H+.
Scheme 2.
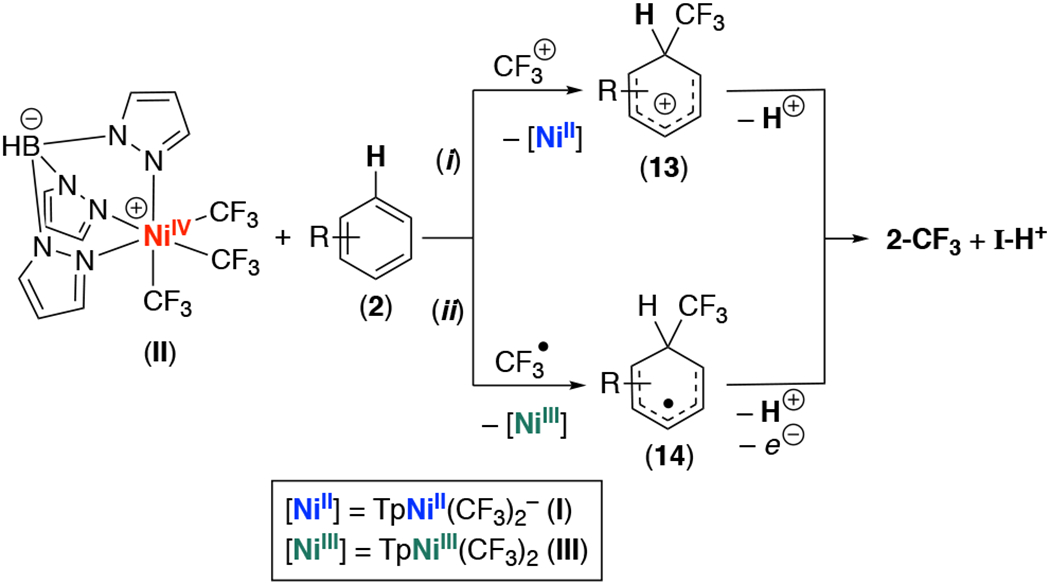
Two possible mechanisms for CF3 transfer from II
To gain further insights into this transformation, we first conducted a time study, monitoring the consumption of II and appearance of 2-CF3 via 19F NMR spectroscopy. As shown in Figure 3, an induction period of approximately 25 min is observed, followed by rapid consumption of starting material and concomitant appearance of 2-CF3. This reaction profile provides initial evidence against a direct transfer of CF3+ from II (Scheme 2, i), as no induction period would be expected for this pathway.
Figure 3.
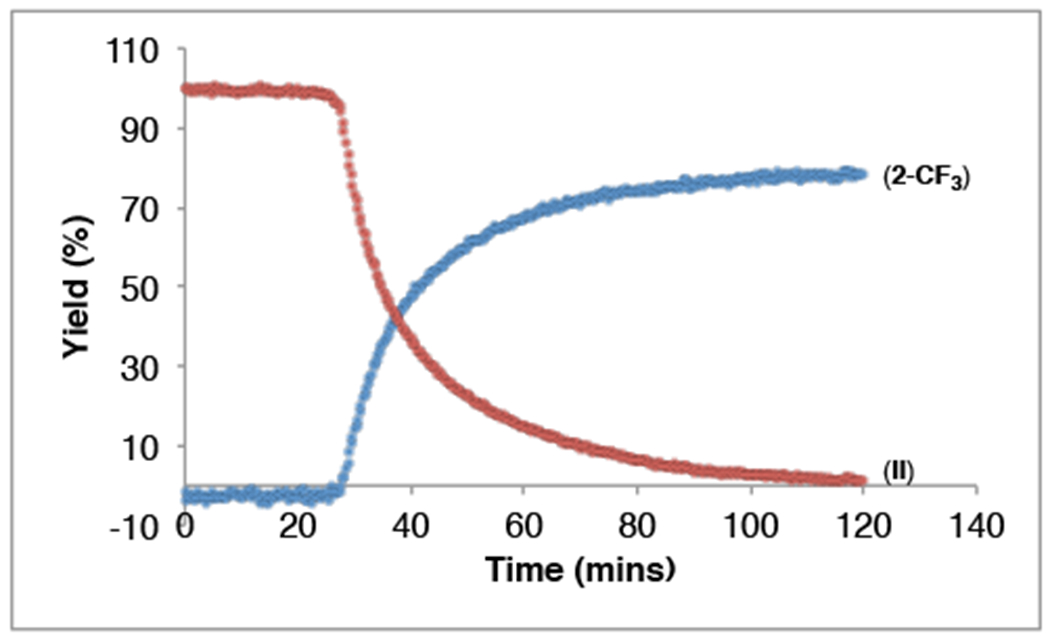
Reaction profile for the trifluoromethylation of trimethoxybenzene with complex II. Reaction conducted with 1.0 equiv II and 5.0 equiv 2.
Reaction profiles like that in Figure 3 are characteristic of radical chain processes.13 To trap putative radical intermediates, we next carried out the reaction between II and 2 in the presence of TEMPO. As shown in eq. 3, the addition of 1 equiv of TEMPO inhibited productive C–H trifluoromethylation, decreasing the yield of 2-CF3 from 72% to just 4% under otherwise analogous conditions. Furthermore, TEMPO–CF3 (15, 8%) was detected by 19F NMR spectroscopy.14
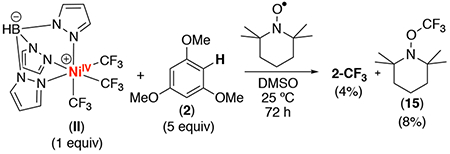 |
(3) |
The diminished yield of 2-CF3 and observation of 15 in the presence of TEMPO both implicate the intermediacy of CF3• in this transformation. Based on the mechanism in Scheme 2, ii, this could be accompanied by the formation of the NiIII intermediate III, a species that we have independently synthesized and characterized by EPR spectroscopy in a 3:1 mixture of PrCN:MeCN.15 To test for the formation of III, we conducted the reaction between II and 2 and removed aliquots at several time points before, during, and after the induction period (0.5, 35, 45, and 85 min). The samples were then diluted in PrCN, and EPR spectra were acquired for each time point. As shown in Figure S45, signals consistent with a Tp-ligated NiIII intermediate are observed; however, the EPR data for the major species present in these samples do not match those for III. This suggests that the major NiIII species formed under these conditions is not complex III. Furthermore, as shown in eq. 3, the addition of 1 equiv of III to the reaction between II and 2 inhibited the formation of 2-CF3, with none of the product observed after 45 min.16 Overall, these data suggest that complex III is not the major NiIII intermediate formed under these conditions.
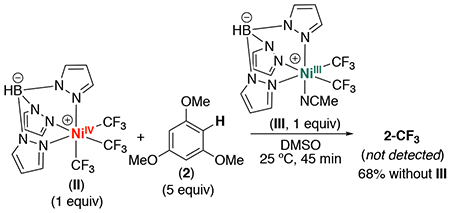 |
(4) |
Based on these studies, we propose a radical chain mechanism for the II-mediated C–H trifluoromethylation of 2. As shown in Scheme 3, i, the radical chain initiates via slow NiIV–CF3 bond homolysis. This liberates CF3•, which then enters the chain propagation sequence (Scheme 3, ii). Propagation is proposed to involve: (1) reaction of CF3• with 2 to afford intermediate 14; (2) oxidation and deprotonation of 14 by NiIV complex II to generate a protonated NiIII intermediate IV; and (3) NiIII–CF3 homolysis from IV to release I-H+ and regenerate CF3•. Importantly, the 17-electron NiIII complex IV is expected to be more reactive towards Ni–CF3 bond homolysis than the neutral, 18-electron NiIV starting material II.17 This should render chain propagation faster than initiation, resulting in a reaction profile like that in Figure 3.
Scheme 3.
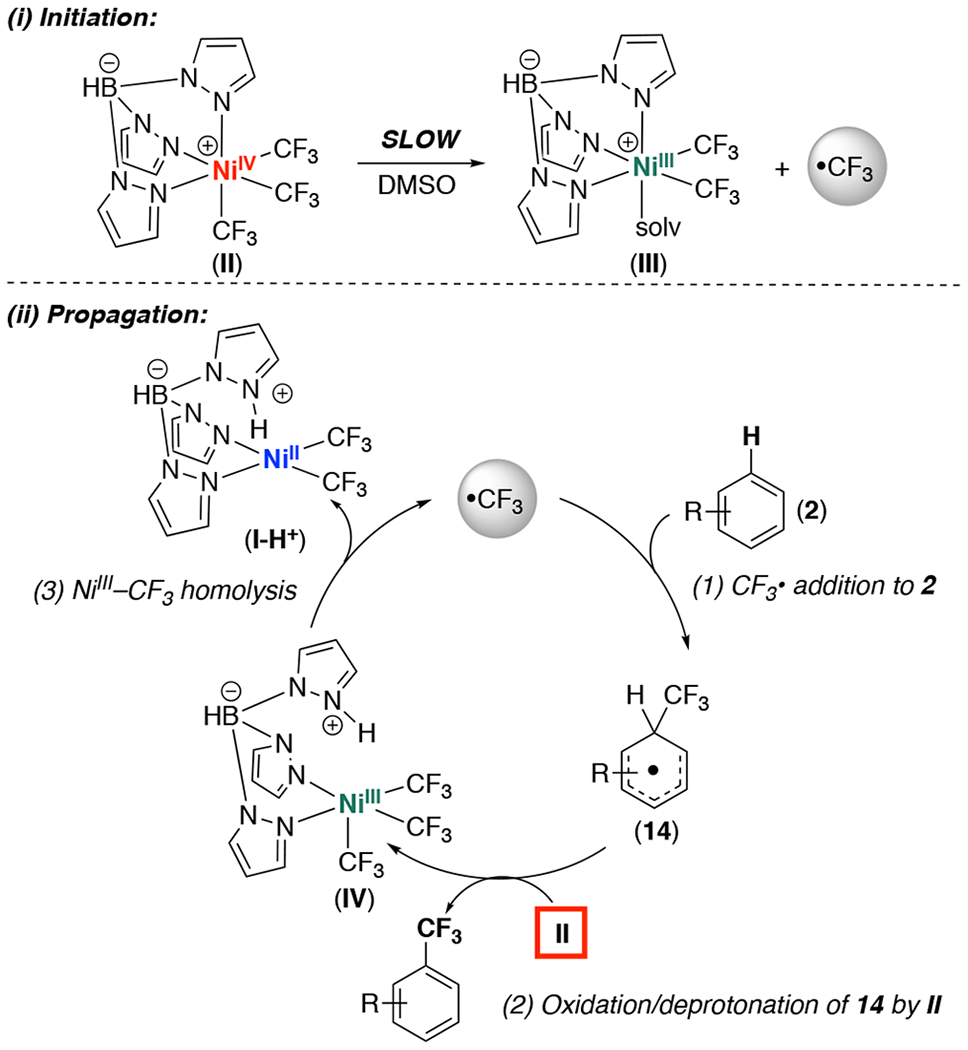
Proposed radical chain pathway for II-mediated C–H trifluoromethylation
If the mechanism in Scheme 3 is operative, then the induction period should be eliminated if the protonated (tris)trifluoromethyl NiIII complex IV is generated independently. Based on the cyclic voltammogram of 4 (Figure S52), Cp2Co (E1/2 = −1.33 V vs SCE) should effect the single electron reduction of II (E1/2 = –0.395 V vs SCE). In the presence of 1 equiv of TsOH•H2O, this is expected to afford IV, thus chemically initiating the reaction (Scheme 4).18
Scheme 4.
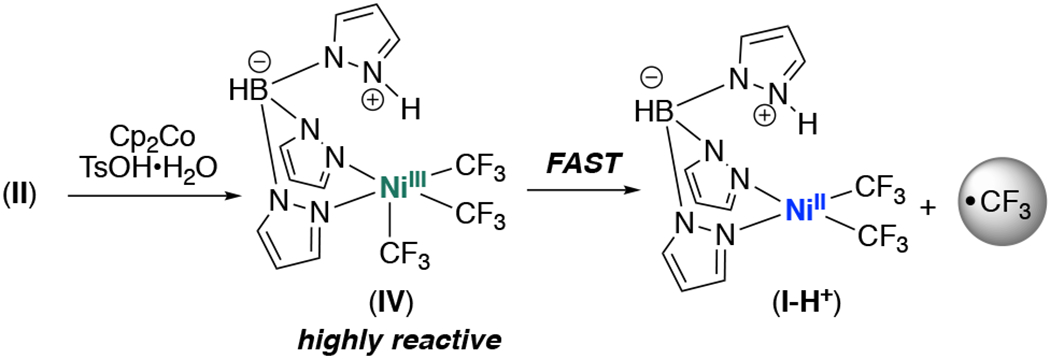
Reduction of II as a route to CF3•
As predicted, the addition of 0.1 equiv of Cp2Co and 0, 0.1, or 1.0 equiv of TsOH•H2O to the reaction between II and 2 under otherwise identical conditions eliminated the induction period and resulted in the steady formation of the trifluoromethylated product 2-CF3 (Figure 4).19 These experiments also show that the yield and rate of this reaction increases with increasing equiv of TsOH. This observation is consistent with the proposed intermediacy of the protonated NiIII complex IV.
Figure 4.
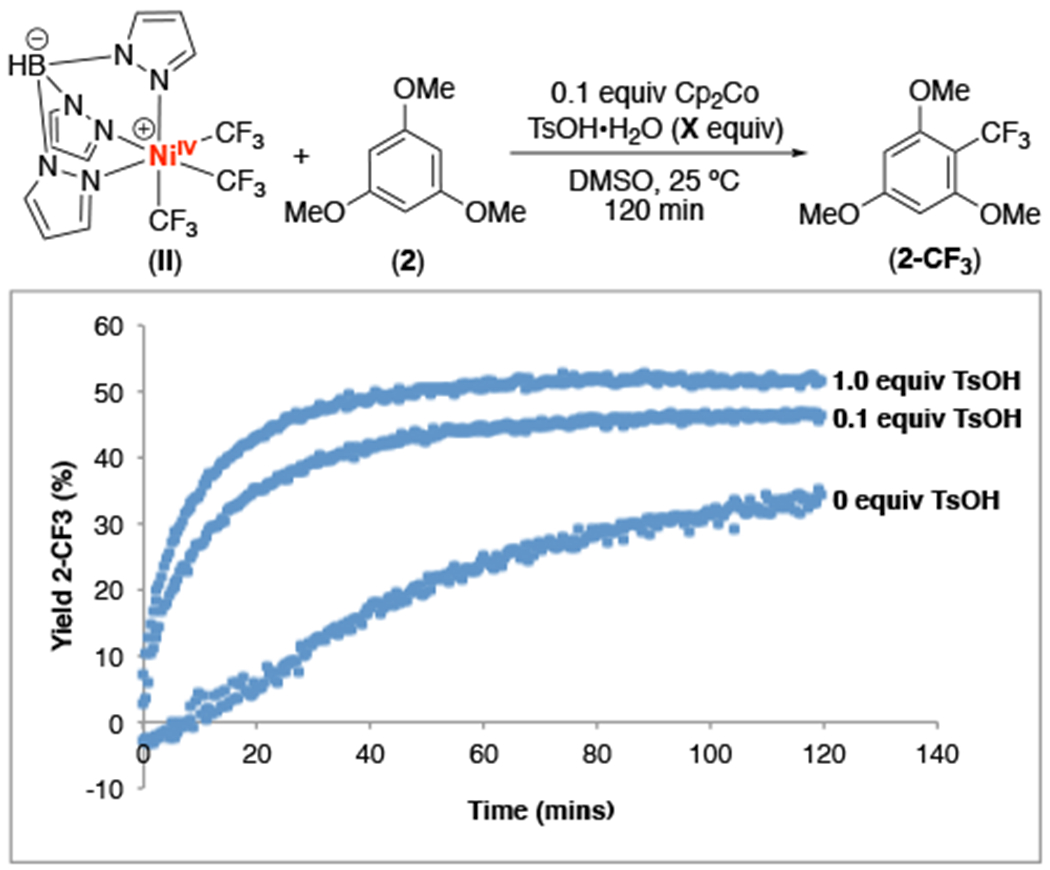
Reaction profile for the trifluoromethylation of trimethoxybenzene with complex II in the presence of Cp2Co and TsOH. Reaction conducted with 1.0 equiv II, 5.0 equiv 2, 0.1 equiv Cp2Co, and varied equiv of TsOH•H2O. Cp2Co was added to II and 2 immediately before the addition of TsOH•H2O.
It was recently reported that the generation of NiIV complexes through the net 2-electron oxidation of NiII precursors can proceed via two sequential one-electron oxidations involving the formation of transient NiIII intermediates.20 If complex II were generated from I under acidic conditions via this type of pathway, NiIII species IV would be a likely intermediate (Scheme 5). We explored this possibility by examining the time course of 2-CF3 formation, using a combination of 1 equiv of NiII complex I, 1 equiv of B, and 1 equiv TsOH•H2O to mediate the C–H trifluoromethylation of 2. As shown in Figure 5, using I/B/TsOH, the reaction showed no induction period, and 2-CF3 was formed in 70% yield within 1 h at room temperature.
Scheme 5.
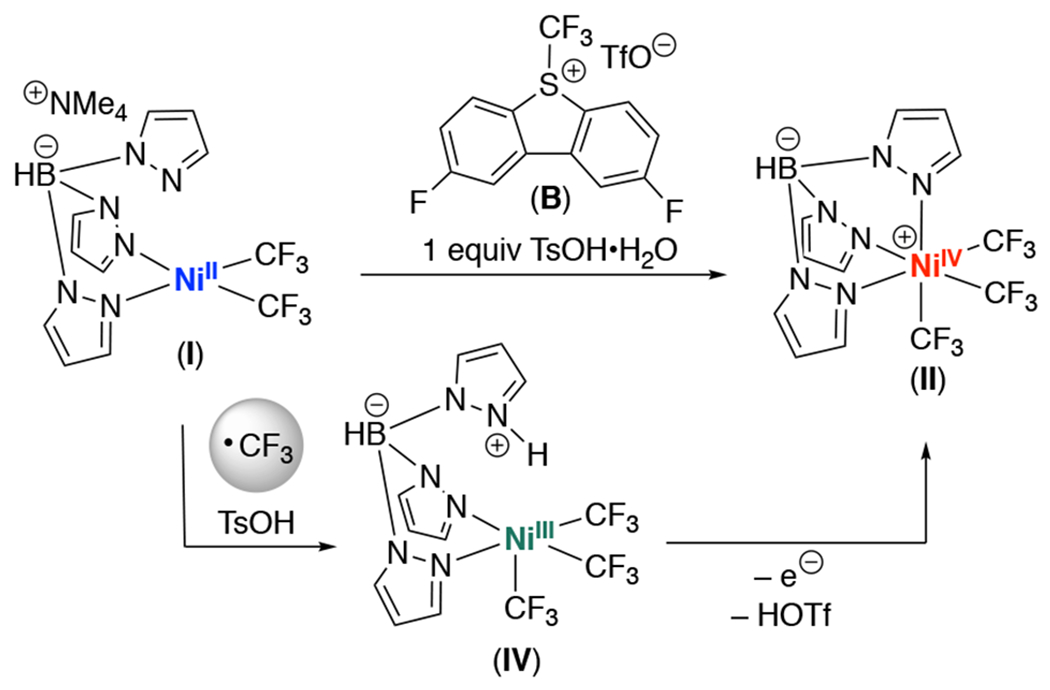
Proposed sequential single electron oxidation step to convert I to II in the presence of TsOH
Figure 5.
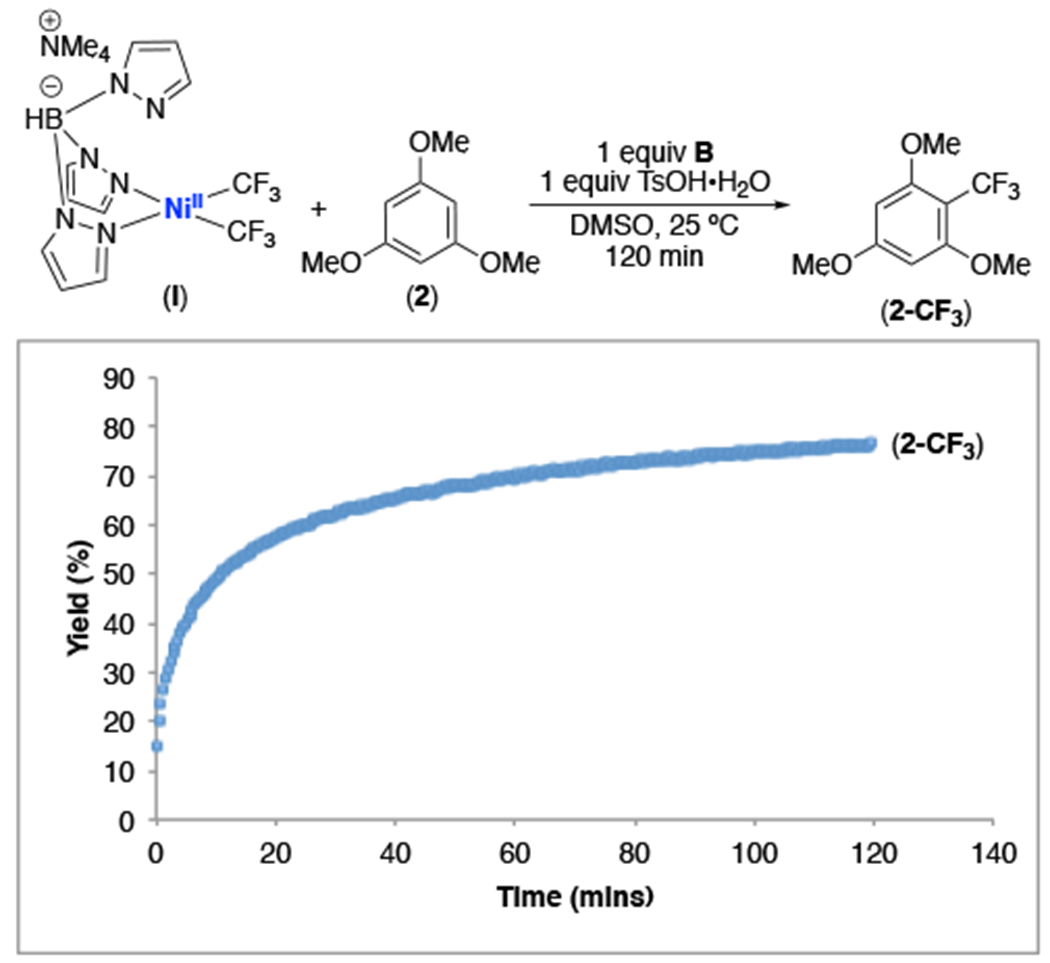
Reaction profile for the trifluoromethylation of trimethoxybenzene meditated by complex I in the presence of B and TsOH. Reaction conducted with 1.0 equiv I, 5.0 equiv 2, 1.0 equiv B, and 1.0 equiv TsOH•H2O. B was added to I and 2 immediately before the addition of TsOH•H2O.
The results in Figure 5 can be rationalized based on the formation of the protonated NiIII intermediate IV during oxidation of I. We propose that IV then rapidly expels CF3• to initiate the reaction and enter the propagative cycle. However, notably, our proposed mechanism also implicates the intermediacy of NiIV complex II, which serves as an oxidant during propagation. Consistent with this proposal, 19F NMR spectroscopic monitoring of the trifluoromethylation of 2 with I/B showed that the Ni largely rests as NiIV complex II during this transformation (Figure S48). This NiIV complex persists throughout the product-forming stage, decaying at a rate that is similar to the rate of 2-CF3 formation. These results are mirrored in the catalytic variant of this reaction (see SI for details).
DFT studies of CF3 transfer from complex II.
We next examined the feasibility of the proposed pathway using DFT calculations.21 Gaussian 09 was used at the B3LYP22 level of density functional theory (DFT) for geometry optimization (see SI for complete details). Figure 6 illustrates the energy profile for propagation obtained following the initiation step that forms CF3 radical.
Figure 6.
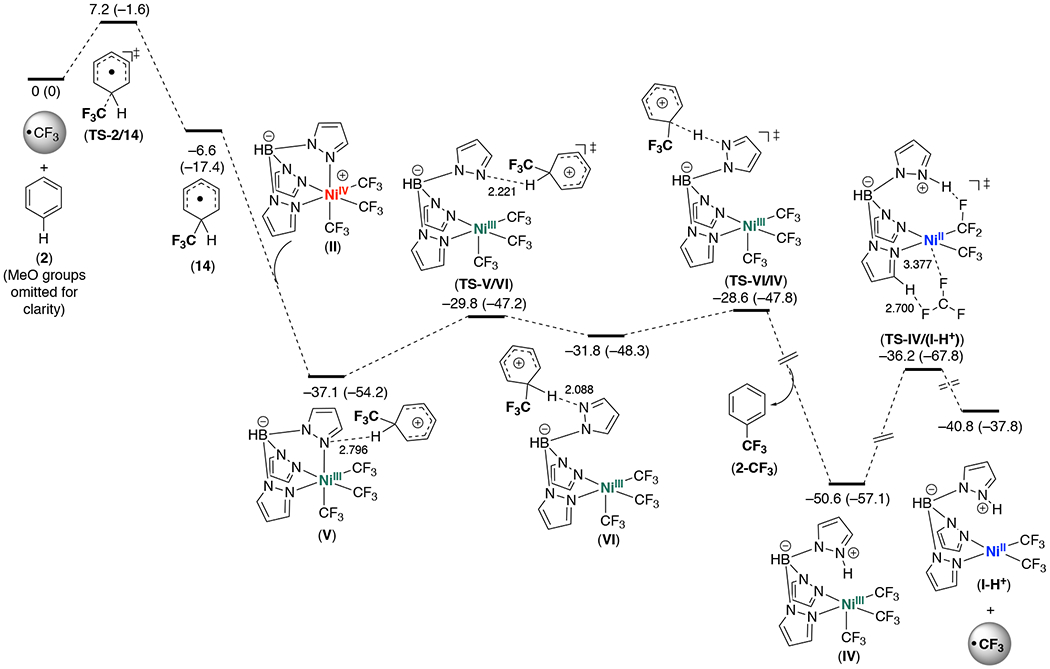
Energy profile for the propagation steps in the reaction of TpNiIV(CF3)3 (II) with 1,3,5-trimethoxybenzene (2). Methoxy groups are omitted for clarity. Details for additional species: adduct after TS-VI/IV (−48.8 (−67.3)), conformer after IV formed by pyrazole rotation (−46.9 (−55.0), and adduct after VII (−36.1 (−40.7)) are provided in Supporting Information. Distances for selected interatomic interactions are given in Å. Energies ΔG (ΔH) in kcal/mol are relative to (CF3• + 2).
The addition of CF3• to 2 to afford 14 is downhill by −6.6 kcal/mol (with ΔG† = 7.7 kcal/mol). The organic radical 14 exhibits a distorted tetrahedral geometry at the “C(H)(CF3)C2” center. As anticipated for p-delocalization, the C–Cortho bonds are ~0.1 Å longer than those between the other carbon atoms, and the SOMO exhibits p-character located on these carbon and adjacent oxygen atoms. Compound 14 can undergo a highly favorable [DG (ΔH) = −30.5 (−36.8) kcal/mol] electron transfer with NiIV complex II to afford the arenium/NiIII pair V in a barrierless process. Structure V exhibits a weak interaction between the organic fragment and a nitrogen atom coordinated to Ni (N⋯··H = 2.796 Å). Spin density for V is located primarily at Ni (0.82 e/Å3), and the SOMO is an antibonding orbital located in the Ni coordination sphere (Figure 7). This is consistent with electron transfer from 14 to nickel to give NiIII and an arenium fragment.
Figure 7.
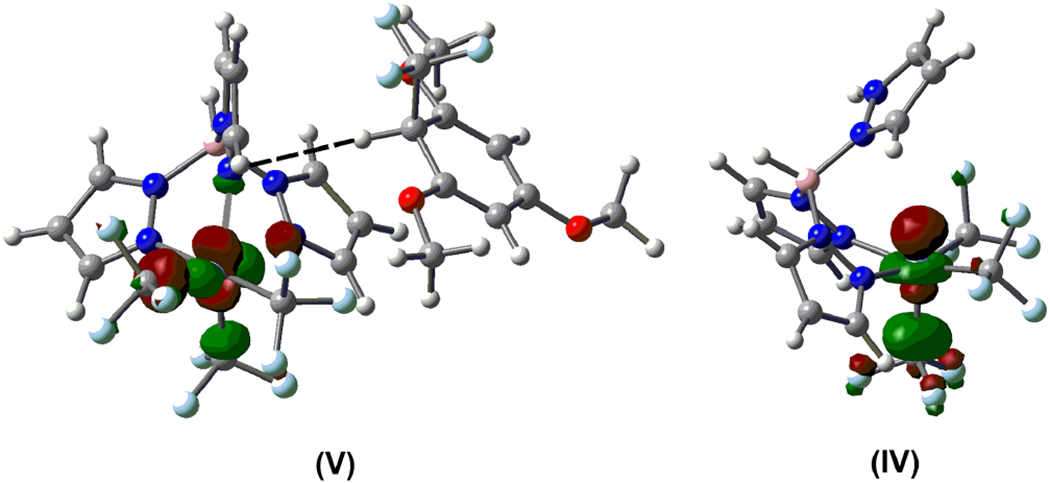
Nickel-centered SOMOs for V and IV, illustrating nickel-centered antibonding character in both structures.
The sequence V → IV was obtained by initially locating transition state TS-VI/IV, followed by potential energy scans and optimizations from this structure. Notably, barriers within this sequence are very low: ΔG† = 3.2 and 7.3 kcal/mol). Dissociation of the axial pyrazole occurs via transition state TS-V/VI to give VI, followed by proton transfer via transition state TS-VI/IV to afford a weak adduct that dissociates to form organic product 2-CF3 and square-pyramidal NiIII (IV). Consistent with the formulation of IV as a NiIII complex, spin-density is located primarily at Ni (Ni 0.68 e/Å3, Caxial 0.28 e/Å3) and a LUMO can be readily assigned as Ni–C s* (Figure 7). Computation for loss of CF3• from IV to give the diamagnetic NiII product I-H+ led to detection of transition state TS-IV/I-H+ for CF3• dissociation.
Overall, these calculations show that the proposed steps of the propagative sequence have low barriers (<14.4 kcal/mol for IV → I-H+) and are thus expected to be fast at 25 °C. We next turned our focus on the initiation step. A transition state for the formation of TpNiIII(CF3)2 and CF3• from II could not be detected computationally. Thus, in accord with the protocol presented by Hartwig and Hall,23 the Gibbs initiation barrier is estimated as ~ΔH, i.e. ΔG‡ ~ 17.9 kcal/mol. This barrier is significantly higher than that for IV ® I-H+ (14.4 kcal/mol), consistent with the proposed mechanism. In addition to experimental evidence for a higher barrier for initiation versus propagation, the initiation step computes as endergonic (ΔG = 3.4 kcal/mol) and only slightly favorable after solvation to form III ligated by DMSO (ΔG‡ = −0.5 kcal/mol).
These studies support the challenging release of CF3• from complex II. This is consistent with the induction period observed in both stoichiometric and catalytic reactions that start with NiIV complex II. Subsequent entrance into the proposed propagative regime through the addition of CF3• to trimethoxybenzene represents a transition into a much lower energy phase of the reaction profile. In summary, these computations provide additional support for the proposed radical chain mechanism involving an energy-intensive initiation event that then provides access to a more favorable propagative regime. The NiIV-catalyzed C–H trifluoromethylation reaction is expected to have analogous initiation and propagation as the stoichiometric variant. The reaction then turns over via the oxidation and deprotonation of I-H+ with the CF3+ reagent B.
SUMMARY AND CONCLUSIONS
In conclusion, this study represents the first example where NiIV intermediates are implicated spectroscopically in catalysis. Detailed experimental and computational studies of the trifluoromethylation of trimethoxybenzene mediated by complex II support a radical chain pathway in which this NiIV intermediate plays a role in both the initiation and propagation regimes. Future work will investigate the use of NiIV as a mild source of other carbon-centered radicals. Ultimately, we anticipate that the insights acquired in this study will inform the development of novel reactions involving NiIV intermediates.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (NSF) Grant CHE-1111563, the Australian Research Council, and the Australian National Computing Infrastructure. We also thank Rackham Graduate School and the NSF for graduate fellowships to E.A.M, the NSF for a REU summer fellowship for S.N.N., and the NSERC for a postdoctoral fellowship for E.C. We gratefully acknowledge Dr. Jeff Kampf for X-ray crystallographic analysis of complex II, as well as funding from NSF Grant CHE-0840456 for X-ray instrumentation.
Footnotes
ASSOCIATED CONTENT
Experimental details, optimization tables, and complete characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- 1.(a) Tasker SZ; Standley EA; Jamison TF Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ananikov VP Nickel: The “spirited horse” of transition metal catalysis. ACS Catal. 2015, 5, 1964–1971. [Google Scholar]; (c) Keim W Nickel: an element with wide application in industrial homogeneous catalysis. Angew. Chem., Int. Ed 1990, 29, 235–244. [Google Scholar]; (d) Rosen BM; Quasdorf KW; Wilson DA; Zhang N; Resmerita A-M; Garg NK; Percec V Nickel-catalyzed cross-coupling involving carbon–oxygen bonds. Chem. Rev 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Montgomery J, in Organometallics in Synthesis: Fourth Manual, B. H. Lipschutz, Ed. (Wiley, Hoboken, NJ, 2013), pp. 319–428. [Google Scholar]; (b) Hu X Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective. Chem. Sci 2011, 2, 1867–1886. [Google Scholar]
- 3.For some examples, see:; (a) Tsou TT; Kochi JK Mechanism of oxidative addition. Reaction of nickel(0) complexes with aromatic halides. J. Am. Chem. Soc 1979, 101, 6319–6332. [Google Scholar]; (b) Laskowski CA; Bungum DJ; Baldwin SM; Del Ciello SA; Iluc VM; Hillhouse GL Synthesis and reactivity of two-coordinate Ni(I) alkyl and aryl complexes. J. Am. Chem. Soc 2013, 135, 18272–18275. [DOI] [PubMed] [Google Scholar]; (c) Grove DM; van Koten G; Zoet R Unique stable organometallic nickel(III) complexes: syntheses and molecular structure of Ni[C6H3(CH2NMe2)2-o,o’]2I2. J. Am. Chem. Soc 1983, 105, 1380–1381. [Google Scholar]
- 4.For select examples of NiIV complexes that have been isolated and studied, see:; (a) Klein H-F; Bickelhaupt A; Jung T; Cordier G Syntheses and properties of the first octahedral diorganonickel(IV) compounds. Organometallics, 1994, 13, 2557–2559. [Google Scholar]; (b) Dimitrov V; Linden A A pseudotetrahedral, high-oxidation-state organonickel compound: synthesis and structure of bromotris(1-norbornyl)nickel(IV). Angew. Chem., Int. Ed 2003, 42, 2631–2633. [DOI] [PubMed] [Google Scholar]; (c) Camasso NM; Sanford MS Design, synthesis, and carbon-heteroatom coupling reactions of organometallic nickel(IV) complexes. Science 2015, 347, 1218–1220. [DOI] [PubMed] [Google Scholar]; (d) Martinez GE; Ocampo C; Park YJ; Fout AR Accessing pincer bis(carbene) Ni(IV) complexes from Ni(II) via halogen and halogen surrogates. J. Am. Chem. Soc 2016, 138, 4290–4293. [DOI] [PubMed] [Google Scholar]; (e) Schultz JW; Fuchigami K; Zheng B; Rath NP; Mirica LM Isolated organometallic nickel(III) and nickel(IV) complexes relevant to carbon-carbon bond formation reactions J. Am. Chem. Soc 2016, 138, 12928–12934. [DOI] [PubMed] [Google Scholar]
- 5.(a) Bour J; Camasso NM; Sanford MS Oxidation of Ni(II) to Ni(IV) with aryl electrophiles enables Ni-mediated aryl–CF3 coupling. J. Am. Chem. Soc 2015, 137, 8034–8037. [DOI] [PubMed] [Google Scholar]; (b) Chong E; Kampf JW; Ariafard A; Canty AJ; Sanford MS Oxidatively induced C-H activation at high valent nickel. J. Am. Chem. Soc 2017, 139, 6058–6061. [DOI] [PubMed] [Google Scholar]; (c) Zhang C-P; Wang H; Klein A; Biewer C; Stirnat K; Yamaguchi Y; Xu L; Gomez-Benitez V; Vicic DA A five-coordinate nickel(II) fluoroalkyl complex as a precursor to a spectroscopically detectable Ni(III) species. J. Am. Chem. Soc 2013, 135, 8141–8144. [DOI] [PubMed] [Google Scholar]
- 6.D’Accriscio F; Borja P; Saffon-Merceron N; Fustier-Boutignon M; Mézailles N; Nebra N C–H bond trifluoromethylation of arenes enabled by a robust, high-valent NiIV complex. Angew. Chem., Int. Ed 2017, 56, 12898–12902. [DOI] [PubMed] [Google Scholar]
- 7.The identity of NiII product I-H+ was confirmed by comparing the 19F NMR shift observed for the NiII product of this reaction to the signal generated upon treatment of NiII complex I with TsOH in DMSO. See SI for details.
- 8.(a) Umemoto T; Zhang B; Zhu T; Zhou X; Zhang P; Hu S; Li Y Powerful, thermally stable, one-pot-preparable, and recyclable electrophilic trifluoromethylating agents: 2,8-difluoro- and 2,3,7,8-tetrafluoro-S-(trifluoromethyl)bibenzothiophenium salts. J. Org. Chem 2017, 82, 7708–7719. [DOI] [PubMed] [Google Scholar]; (b) Egami H; Ito Y; Ide T; Masuda S; Hamashima Y Simple photo-induced trifluoromethylation of aromatic rings. Synthesis 2018, 50, 2948–2953. [Google Scholar]
- 9.Gao X; Geng Y; Han S; Liang A; Li J; Zou D; Wu Y; Wu Y Nickel-catalyzed direct C–H trifluoromethylation of free anilines with Togni’s reagent. Org. Lett 2018, 20, 3732–3735. [DOI] [PubMed] [Google Scholar]
- 10.(a) Rey-Rodriguez R; Retailleau P; Bonnet P; Gillaizeau I Iron-catalyzed trifluoromethylation of enamide. Chem. Eur. J 2015, 21, 3572–3575. [DOI] [PubMed] [Google Scholar]; (b) Jacquet J; Blanchard S; Derat E; Desage-El Murr M; Fensterbank L Redox-ligand sustains controlled generation of CF3 radicals by well-defined copper complex. Chem. Sci 2016, 7, 2030–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chang B; Shao H; Yan P; Qiu W; Weng Z; Yuan R Quinone-mediated trifluoromethylation of arenes and heteroarenes with visible light. ACS Sustainable Chem. Eng 2017, 5, 334–341. [Google Scholar]
- 11.Nagib DA; MacMillan DWC Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji Y; Brueckl T; Baxter RD; Fujiwara Y; Seiple IB; Su S; Blackmond DG; Baran PS Innate C-H trifluoromethylation of heterocycles. Proc. Nat. Acad. Sci 2011, 108, 14411–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helfferich FG Chain Reactions In Comprehensive Chemical Kinetics. Elsevier Science B.V.: Amsterdarm, The Netherlands: 2001; Vol. 38, pp 263–297. [Google Scholar]
- 14.(a) Seo S; Taylor JB; Greaney MF Silver-catalysed trifluoromethylation of arenes at room temperature. Chem. Commun 2013, 49, 6385–6387. [DOI] [PubMed] [Google Scholar]; (b) Chen S; Feng D-F; Li D-Y; Liu P-N Radical cyanotrifluoromethylation of isocyanides: step-econmoical access to CF3-containing nitriles, amines, and imines. Org. Lett 2018, 20, 5418–5422. [DOI] [PubMed] [Google Scholar]; (c) Liang A; Han S; Liu Z; Wang L; Zou D; Wu Y; Wu Y Regioselective synthesis of N-heteroaromatic trifluoromethoxy compounds by direct O–CF3 bond formation. Chem. Eur. J 2016, 22, 5102–5106. [DOI] [PubMed] [Google Scholar]
- 15.Bour JB; Camasso NM; Meucci EA; Kampf JW; Canty AJ; Sanford MS Carbon-carbon bond-forming reductive elimination from isolated nickel(III) complexes. J. Am. Chem. Soc 2016, 138, 16105–16111. [DOI] [PubMed] [Google Scholar]
- 16.We investigated the possible role of the MeCN ligand on III by adding and removing (via vacuum) CH2Cl2 from III several times. The resulting complex was also observed to severely inhibit the trifluoromethylation of 2 by II. The trifluoromethylation of 2 catalyzed by II performs similarly in MeCN and DMSO. These data suggest that the MeCN on III does not inhibit the reaction.
- 17.(a) Tilset M One-electron oxidation of cyclopentadienylchromium carbonyl hydrides: thermodynamics of oxidative activation of metal-hydrogen bonds toward homolytic and heterolytic cleavage. J. Am. Chem. Soc 1992, 114, 2740–2741. [Google Scholar]; (b) Skagestad V; Tilset M Thermodynamics of heterolytic and homolytic metal-hydrogen bond cleavage reactions of 18-electron and 17-electron group 6 hydridotris(pyrazolyl)borate metal hydrides. J. Am. Chem. Soc 1993, 115, 5077–5083. [Google Scholar]; (c) Tilset M; Hamon J-R; Hamon P Relative M–X bond dissociation energies in 16-, 17- and 18-electron organotransition-metal complexes (X = halide, H). Chem. Commun 1998, 765–766. [Google Scholar]
- 18.Connelly NG; Geiger WE Chemical redox agents for organometallic chemistry. Chem. Rev 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- 19.Attempts to utilize Cp2Co and T sOH to initiate the catalytic reaction did not result in higher yields, see SI for details.
- 20.Bour JR; Ferguson DM; McClain EJ; Kampf JW; Sanford MS Connecting organometallic Ni(III) and Ni(IV): Reactions of carbon-centered radicals with high-valent organonickel complexes. J. Am. Chem. Soc 2019, 141, 8914–8920. [DOI] [PubMed] [Google Scholar]
- 21.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; and Fox DJ Gaussian 09, revision A.02; Gaussian, Inc.; Wallingford CT, 2009. [Google Scholar]
- 22.Lee CT; Yang WT; Parr RG Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]; (b) Miehlich B; Savin A; Stoll H; Preuss H Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett 1989, 157, 200–206. [Google Scholar]; (c) Becke AD Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- 23.(a) Hartwig JF; Cook KS; Hapke M; Incarvito CD; Fan Y; Webster CE; Hall MB Rhodium boryl complexes in the catalytic, terminal functionalization of alkanes. J. Am. Chem. Soc 2005, 127, 2538–2552. [DOI] [PubMed] [Google Scholar]; (b) Wei CS; Jimenez-Hoyos CA; Videa MF; Hartwig JF; Hall MB Origins of the selectivity for borylation of primary over secondary C–H bonds catalyzed by Cp*-Rhodium complexes. J. Am. Chem. Soc 2010, 132, 3078–3091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.