

Summary
The G2019S mutation in leucine-rich repeat kinase 2 (LRRK2) is a common cause of familial Parkinson’s disease (PD). This mutation results in dopaminergic neurodegeneration via dysregulated protein translation, although how alterations in protein synthesis contributes to neurodegeneration in human neurons is not known. Here we define the translational landscape in LRRK2-mutant dopaminergic neurons derived from human induced pluripotent stem cells (hiPSCs) via ribosome profiling. We found that mRNAs that have complex secondary structure in the 5’ untranslated region (UTR) are translated more efficiently in G2019S LRRK2 neurons. This leads to the enhanced translation of multiple genes involved in Ca2+ regulation and to increased Ca2+ influx and elevated intracellular Ca2+ levels, a major contributor to PD pathogenesis. This study reveals a link between dysregulated translation control and Ca2+ homeostasis in G2019S LRRK2 human dopamine neurons, which potentially contributes to the progressive and selective dopaminergic neurotoxicity in PD.
Keywords: Parkinson’s disease, LRRK2, ribosome profiling, translatome, calcium homeostasis, 5’UTR, RPS15, uS19
Graphical Abstract
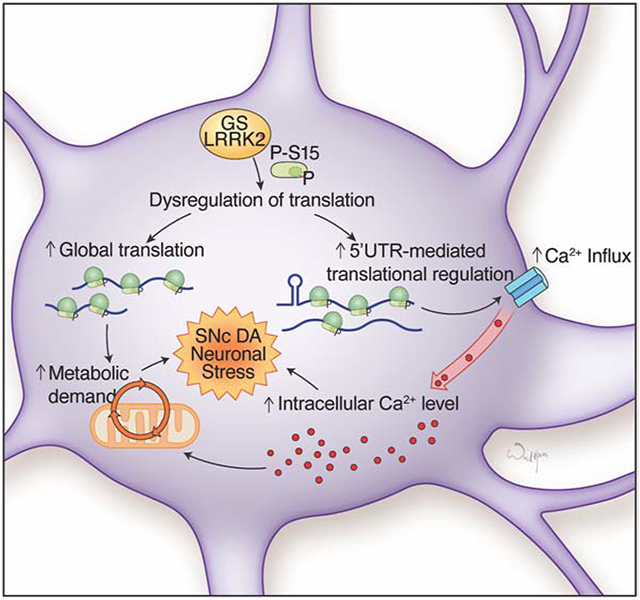
eTOC statement
Using ribosome profiling of human dopamine neurons, Dawson and colleagues show that protein synthesis is altered in Parkinson’s disease-linked G2019S LRRK2 neurons resulting in calcium dysregulation.
Introduction
PD is the second most prevalent neurodegenerative disorder, globally affecting approximately 2-3% of the population over 65 years of age (Poewe et al., 2017). Loss of dopamine (DA) neurons in the substantia nigra pars compacta (SNc) and subsequent reduction in striatal DA signaling are the pathological hallmarks of PD (Smith et al., 2006). PD is a multifactorial disorder with both environmental and genetic components, while familial PD consists of 5-10% of total PD cases.
Dominant mutations in the LRRK2 gene are the most common genetic cause of PD (Funayama et al., 2005; Paisan-Ruiz et al., 2004; Zimprich et al., 2004). LRRK2 mutations typically result in classical late-onset familial PD and these mu(tations are also observed in sporadic PD cases (Cookson, 2010). LRRK2 is a multi-domain enzyme with GTPase and kinase activities with yet unclear physiological functions, while most PD-associated mutations in LRRK2 were found within the enzymatic domains (Hernandez et al., 2016). The G2019S missense mutation residing in the kinase domain is the most frequent disease-causing mutation in LRRK2 (Martin et al., 2014a). Multiple lines of evidence support that the G2019S mutation enhances kinase activity of LRRK2 leading to neurotoxicity (Greggio et al., 2006; Luzon-Toro et al., 2007; MacLeod et al., 2006; Smith et al., 2006; West et al., 2005; West et al., 2007).
While diverse cellular pathways are associated with LRRK2 mutations, an emerging body of evidence shows that dysregulated protein synthesis downstream of LRRK2 kinase activity plays a role in PD pathogenesis. Eukaryotic initiation factor 4E (eIF4E)-mediated cap-dependent mRNA translation and the miRNA pathway were shown to be impaired in Drosophila models expressing LRRK2 mutants (Gehrke et al., 2010; Imai et al., 2008; Penney et al., 2016). Previously, we reported that G2019S LRRK2 increases global protein synthesis through phosphorylation of the ribosomal protein S15 (uS19), and suppression of global protein synthesis is protective against G2019S LRRK2 neurotoxicity using in vivo Drosophila models (Martin et al., 2014b). Although those findings from Drosophila support the roles of translational regulation in PD pathogenesis, the translational landscape of human DA neurons and the impact of G2019S LRRK2 on the human DA neuronal translatome remain to be answered.
We hypothesized that dysregulated translation in G2019S LRRK2 may bring about changes in the levels of a subset of proteins whose precise regulation is integral for the long-term survival of DA neurons. Therefore, we investigated the G2019S LRRK2 translatome via ribosome profiling in human DA neurons differentiated from PD patient-derived induced pluripotent stem cells (iPSCs). Our results revealed that G2019S LRRK2 causes an alteration in the translatome that changes expression levels of various genes involved in Ca2+ homeostasis in neurons, including voltage-gated Ca2+ channel subunits. This abnormality in gene expression regulation leads to an increase in Ca2+ influx leading to elevated intracellular Ca2+ concentration in DA neurons. We also found that translation of mRNAs with complex 5’UTR secondary structure is generally increased in G2019S LRRK2 human DA neurons, suggesting the molecular mechanisms for the translational abnormality.
Results
Generation and characterization of G2019S LRRK2 patient-derived iPSCs and DA neuronal differentiation
Human DA neurons were differentiated from patient-derived human iPSCs. First, fibroblasts from three unrelated G2019S LRRK2 PD patients and three healthy unrelated individuals without the LRRK2 mutation (wild-type) were reprogrammed into iPSCs (Figures 1A and S1A). All iPSC lines show pluripotent marker expression with normal karyotypes (Figures S1B and S1C). These six lines were differentiated into human DA neurons (Kriks et al., 2011). Post-differentiation marker analysis demonstrates that the cultures are enriched in DA neuronal population consisting of more than 70% of tyrosine hydroxylase (TH) positive neurons (Figures 1B and 1C). No difference was observed in the efficiency of neuronal differentiation between wild-type and G2019S LRRK2 iPSCs (Figures 1B and 1C). Basal electrophysiological characteristics, spontaneous and evoked firing patterns of DA neurons are also indistinguishable between wild-type and G2019S LRRK2 human DA neurons (Figures S2A and S2B).
Figure 1. Altered translatome in G2019S LRRK2 human dopamine neurons.

(A) A schematic for the generation of human DA neurons. (B and C) Assessing the efficiency of DA differentiation. n=6, independent measurements per line. Unpaired t-test, mean ± SEM. ns=no significance. (D) A schematic of ribosome profiling. (E and F) MA plots for RNA-seq and ribosome profiling, respectively. Genes with >2-fold changes with FDR<10% were highlighted. Read abundance: mapped reads in log10 scale. (G) Scatterplot comparing RNA-seq and ribosome profiling. Genes with FDR<10% were highlighted. All values are in log2, each data point represents a single transcript. See also Figures S1 to S3, Table S1 and S2.
Ribosome profiling of G2019S LRRK2 human DA neurons
We employed ribosome profiling to investigate the translatome in G2019S LRRK2 human DA neurons. In ribosome profiling, ribosome-protected mRNA fragments (RPFs or ribosome footprints) are captured and deep-sequenced, providing a quantitative measurement of translation and precise location of translating ribosomes at a genomic scale (Figure 1D) (Ingolia et al., 2009). Ribosome profiling libraries prepared from G2019S LRRK2 human DA neurons showed normal triplet periodicity and RPF length distribution (Figures S2C and S3A), and we could not find any noticeable changes in ribosomal disposition from a metagene-based global scoring approach (data not shown).
To identify differentially regulated genes, we analyzed the data with the DESeq2 R package (Love et al., 2014). Of note, we found that the gene expression profiles varied substantially between lines, even though they were cultured and differentiated on the same batch (Figures 1E and 1F). Since they were generated from unrelated individuals, their genetic backgrounds may have contributed to the variability. Nevertheless, using 10% false discovery rate (FDR) and a 2-fold cutoff, we found 84 differentially regulated genes from RNA-seq and 7 differentially regulated genes from ribosome profiling (Figures 1E and 1F, and Table S1). To estimate translation efficiency (TE) by subtracting the transcriptional contribution from the ribosome profiling readout, we used a multi-factor design of DESeq2 with a model:
Ribosome profiling = mRNA changes + translational changes
A comparison between the ribosome profiling expression changes and the RNA-seq expression changes shows that there are widespread translational effects in the translatome (Figure 1G). Analysis of the overall distribution of TE changes also demonstrated a global translational shift in G2019S LRRK2 neurons (Figures S3B and S3C). However, although we observed a broad alteration in TE distribution, DESeq2 did not report individual genes with significant TE changes (Figure 1G). The facts that translational effects are widespread but the fold changes are relatively mild, and the aforementioned genetic background variation between cell lines seem to be accountable for these results.
Gene expression profiles indicate elevated intracellular calcium levels
Gene Ontology (GO) and Ingenuity Pathway Analysis (IPA) with the TE values were used to identify affected cellular pathways (Table S3). GO analysis showed that the major cellular processes deregulated in G2019S LRRK2 neurons are involved in the regulation of synaptic activity and membrane potential (Figures S3D) (Bindea et al., 2009). Additional GO analysis with the human DA neuronal transcriptome as reference also showed that the pathways with highest fold enrichments are regulation of signaling receptor activity and glutamate receptor signaling pathway (Table S3) (Mi et al., 2019). Likewise, IPA analysis reported that various signaling pathways in neurons are affected; Ca2+ signaling is one of the top IPA pathways disturbed in human G2019S LRRK2 DA neurons, and the other top pathways contain voltage-gated Ca2+ channel genes as their constituents (Figures 2A and S3D). Based on the results, we hypothesized that deregulation of multiple channel and receptor genes may have affected the Ca2+ homeostasis in these neurons.
Figure 2. Altered translatome results in increased calcium signaling in G2019S LRRK2 neurons.

(A) IPA results with the TE values (log2 fold change>0.8 or <−0.8). (B) A heatmap with the significantly changed 84 mRNAs. Activity-dependent genes (5 genes) were denoted. mRNA expression: normalized mapped reads in log2. (C) Expression of activity-dependent genes compared to all genes. A group of induced activity-dependent transcripts were marked with an arrow. An activity-dependent gene list was from a previous study (Xiang et al., 2007). Kolmogorov–Smirnov test (p=0.002803). WT: wild-type, GS: G2019S. See also Figures S2 and S3, Tables S1 to S3.
Increased Ca2+ signaling in neurons is known to induce expression of activity-regulated genes, including the immediate early genes (Ebert and Greenberg, 2013). We noticed that several genes identified as differentially regulated in G2019S LRRK2 human DA neurons are well-known activity-dependent genes (RNA-seq: 5 genes out of 84 total differentially regulated genes, ribosome profiling: 1 out of 7 differentially regulated genes) (Table S1). Expression levels of 84 differentially regulated transcripts were visualized across individual cell lines, and the results showed that activity-regulated genes have higher expression in G2019S LRRK2 DA neurons in all three lines regardless of their varying genetic backgrounds (Figure 2B and Tables S1 and S2). Accordingly, we found that a group of known activity-regulated genes are induced in G2019S LRRK2 human DA neurons (Figure 2C) (Xiang et al., 2007). The findings collectively suggest a link between defective Ca2+ regulation and the G2019S LRRK2 mutation in human DA neurons.
Calcium currents and intracellular calcium levels are increased in G2019S LRRK2 human DA neurons
Fluorescent Ca2+ indicator Fluo-4 was used to measure intracellular Ca2+ concentrations in G2019 LRRK2 and control human DA neurons. The results revealed that G2019S LRRK2 DA neurons have overall higher intracellular Ca2+ levels than control DA neurons (Figure 3A). We previously reported that phosphorylation of ribosomal protein S15, a kinase substrate of LRRK2, is an important mediator for the hyperactive protein synthesis in G2019S LRRK2 neurons (Martin et al., 2014b). Therefore, we next assessed role of S15 phosphorylation in intracellular Ca2+ concentration. Adeno-associated virus (AAV)-mediated overexpression of T136A S15, a mutant that is not phosphorylatable by LRRK2, rescues the elevated intracellular Ca2+ levels (Figure 3A).
Figure 3. Intracellular calcium concentration is elevated in G2019S LRRK2 human dopamine neurons.

(A) Intracellular Ca2+ measurement by the Fluo-4. 6-8 independent measurements per line, cell lines with the same genotype were pooled for statistical analysis. n=165 (WT), 175 (WT + TA S15), 154 (GS LRRK2), 138 (GS LRRK2 + TA S15). One-way ANOVA with Bonferroni correction (F=180.7, ns (WT vs WT + TA S15), p<0.001 (WT vs GS LRRK2), p<0.001 (GS LRRK2 vs GS LRRK2 + TA S15)). (B) Whole-cell patch clamp recording to measure Ca2+ currents. (C) Quantification of Ca2+ peak currents. 10 independent measurements, pooled by genotype. Average Ca2+ peak current values: 218.11pA (WT), 199.77pA (WT + TA S15), 358.68pA (GS LRRK2), 242.75pA (GS LRRK2 + TA S15). One-way ANOVA with Bonferroni correction (F=21.96, ns (WT vs WT + TA S15), p<0.001 (WT vs GS LRRK2), p<0.001 (GS LRRK2 vs GS LRRK2 + TA S15)). Boxplot visualizes the median, the first and the third quartile along with the data distribution pattern. (D) Ca2+ peak current density measurement with L-VGCC antagonist isradipine. IC-I: control – isradipine, currents blocked by isradipine. (E) Quantification of Ca2+ peak current density. Total Ca2+ peak current was normalized to cell capacitance (pA/pF). 6-8 independent measurements per genotype, and three lines of each genotype were pooled. Mann-Whitney nonparametric test (p<0.05 (Total Ca2+), p<0.05 (L-VGCC)). n=22 (WT), 12 (WT + isradipine), 23 (GS LRRK2), 10 (GS LRRK2 + isradipine). (F) Fura-2 imaging with isradipine (1μM). 8-10 independent measurements per genotype, three different lines were pooled. One-way ANOVA with Bonferroni correction (F=140.5, p<0.05 (WT vs WT + isradipine), p<0.001 (WT vs GS LRRK2), p<0.001 (GS LRRK2 vs GS LRRK2 + isradipine)). Average Ca2+ concentrations: 104.21nM (WT), 112.33nM (WT + isradipine), 150.45nM (GS LRRK2), 112.25nM (GS LRRK2 + isradipine). n=166 (WT), 161 (WT + isradipine), 180 (GS LRRK2), 204 (GS LRRK2 + isradipine). Boxplot visualizes the median, the first and the third quartile along with the data distribution pattern. *p<0.05, ***p<0.001, ns=no significance. GS: G2019S, TA: T136A. See also Figure S2.
Multiple genes whose product could be responsible for activity-dependent Ca2+ influx, including voltage-gated Ca2+ channel (VGCC) and ionotropic glutamate receptors (GluRs), are induced in G2019S LRRK2 human DA neurons (Table S3) (Yap and Greenberg, 2018). Therefore, we performed whole cell Ca2+ current recordings on human DA neurons and found that G2019S LRRK2 neurons have increased Ca2+ currents in both the peak and sustained currents (Figures 3B and 3C). Overexpression of T136A S15 reduces the increased Ca2+ currents (Figures 3B and 3C).
Among VGCCs, L-type VGCCs (L-VGCCs) in SNc DA neurons are known to be engaged during autonomous pacemaking, thereby continuously allowing Ca2+ influx (Surmeier et al., 2012). L-VGCCs are also important for the induction of activity-dependent gene expression (Dolmetsch et al., 2001). Therefore, we hypothesized that suppression of L-VGCC activities by treating its antagonist isradipine may reduce the increased intracellular Ca2+ levels. Isradipine treatment restored the increased Ca2+ currents in G2019S LRRK2 human DA neurons similar to the levels of wild-type neurons (Figure 3D). Contribution of L-VGCC currents to the total Ca2+ influx, calculated by subtracting the isradipine-treated currents from the control currents, indicated that G2019S LRRK2 DA neurons have increased Ca2+ influx through L-VGCCs (Figures 3D and 3E). Intracellular Ca2+ measurements using the ratiometric Ca2+ indicator Fura-2 demonstrated that isradipine reduced the elevated intracellular Ca2+ level in G2019S LRRK2 human DA neurons (Figure 3F).
Correction of the G2019S LRRK2 mutation rescues elevated Ca2+ levels in human DA neurons
We examined the observed Ca2+ dysregulation using a pair of iPSC lines with the same genetic background (isogenic lines). A patient-derived G2019S LRRK2 iPSC line and its mutation-corrected derivative were obtained from the Gasser lab at University of TQbingen (Reinhardt et al., 2013). Isogenic pair lines do not show any noticeable differences in DA differentiation efficiency, and the differentiated DA neurons show indistinguishable morphology and electrophysiological characteristics (Figures S3E to S3G). Notably, we found that phosphorylation of S15 T136 residue is reduced in human DA neurons differentiated from the mutation-corrected line, which is consistent with our previous findings of increased S15 phosphorylation by G2019S LRRK2 in Drosophila brain (Figures S4A and S4B) (Martin et al., 2014b). We found that mutation-corrected human DA neurons have significantly lower cytosolic Ca2+ levels and Ca2+ influx that are similar to the levels of wild-type neurons (Figures 4A to 4C). Elevated Ca2+ currents mediated by L-VGCCs in G2019S LRRK2 DA neurons are also alleviated in the mutation-corrected neurons (Figures 4D and 4E). Isradipine treatment rescues the elevated Ca2+ levels in the G2019S LRRK2 human DA neurons (Figure 4F). The results from isogenic pair lines collectively support our findings that G2019S LRRK2 mutation is responsible for the observed Ca2+ dysregulation in human DA neurons.
Figure 4. Correction of the G2019S LRRK2 mutation rescues increased calcium concentration.

(A) Intracellular Ca2+ measurements of an isogenic pair hDA neurons by the Fluo-4. 6-8 independent measurements per line, same genotype were pooled for statistical analysis. n=60 for all samples. Student’s t-test (p<0.001). MC: mutation-corrected. (B) Whole-cell patch clamp Ca2+ currents recording. (C) Quantification of Ca2+ peak currents. 10 independent measurements per line, pooled by genotype. Average Ca2+ peak current values: 197.9 pA (MC), 303.0 pA (GS LRRK2). Student’s t-test (p=0.003). (D) Ca2+ peak current density measurement with isradipine (1μM). IC-I: control – isradipine, currents blocked by isradipine. (E) Quantification of Ca2+ peak current density. Total Ca2+ peak current was normalized to cell capacitance (pA/pF). 6-8 independent measurements per genotype, 3 lines of each genotype were pooled. Student’s t-test (p=0.002 for total Ca2+, p<0.001 for L-VGCCs), n=20 for all samples. (F) Fura-2 imaging with isradipine (1μM). 8-10 independent measurements per genotype and 3 different lines were pooled. One-way ANOVA with Bonferroni correction (F=31.17, ns (MC vs MC + isradipine), p<0.001 (MC vs GS LRRK2), p<0.001 (GS LRRK2 vs GS LRRK2 + isradipine)). Average Ca2+ concentrations: 109.0nM (MC), 102.7nM (MC + isradipine), 143.9nM (GS LRRK2), 115.1nM (GS LRRK2 + isradipine). n=88 (MC), 65 (MC + isradipine), 85 (GS LRRK2), 75 (GS LRRK2 + isradipine). Boxplot visualizes the median, the first and the third quartile along with the data distribution pattern. **p<0.01, ***p<0.001, ns=no significance. See also Figure S3.
Ribosome profiling of isogenic wild-type and G2019S LRRK2 human DA neurons
We performed ribosome profiling with human DA neurons differentiated from the isogenic lines. The replicates are highly reproducible in both RNA-seq and ribosome profiling results, demonstrating that genetic background explains the differences in gene expression profiles in non-isogenic samples (Figures S4C and S4D). Metagene analyses do not show any noticeable differences between the two lines (Figures S4E and S5A). DESeq2 analysis pipeline reported 414 genes from RNA-seq, 515 genes from ribosome profiling, and 15 genes from TE as differentially regulated using the same 10% FDR and 2-fold cutoff (Figures 5A to 5C, Tables S1 and S2). Of note, activity-dependent genes are increased in G2019S LRRK2 neurons compared to mutation-corrected neurons (Figure 5D). In addition, we performed a combined DESeq2 analysis with both non-isogenic and isogenic datasets. The results show generally lower numbers of significantly affected genes, likely due to heterogeneous genetic backgrounds: 10 genes from RNA-seq, 2 genes from ribosome profiling, and 1 gene from TE analysis (Figure S5B, Tables S1 and S2). In accordance with the previous observations, 2 out of 10 differentially regulated mRNAs are activity-dependent genes (Table S1). G2019S LRRK2 neurons show generally increased expression of activity-dependent genes in the combined analysis as well (Figure S5C).
Figure 5. Dynamic nature of human dopamine neuronal translatome.

(A and B) MA plots for RNA-seq and ribosome profiling from an isogenic pair. Genes with FDR<10% were visualized. Read abundance: mapped reads in log10 scale. n=2 for each genotype. (C) Scatterplot comparing RNA-seq and ribosome profiling. Genes with a 2-fold cutoff and FDR<10% were visualized. All values are in log2, and each data point represents a single transcript. (D) Expression of activity-dependent genes compared to all genes from an isogenic pair. A group of induced activity-dependent transcripts were marked with an arrow. Kolmogorov–Smirnov test (p=0.03409). (E) TE change (GS LRRK2/control) comparison between non-isogenic and isogenic datasets. Dotted guidelines are TE=±0.5, Pearson’s correlation r=0.072. (F and G) TE up and TE down genes (TE>0.5 or <−0.5, respectively) from non-isogenic (F) or isogenic (G) datasets were selected and their TE distribution in the other dataset was plotted. Boxplot with violin plot visualizes the median, the first and the third quartile along with the data distribution pattern. *p<0.05, **p<0.01, ***p<0.001, ns=no significance. Wilcoxon signed-rank test ((F) p<0.001 for TE up, p=0.01129 for TE down (G) p=0.005743 for TE up, p<0.001 for TE down). See also Figures S4 and S5, and Tables S1 and S2.
We directly compared TE values of non-isogenic and isogenic datasets and observed no overall correlation (r=0.0726) (Figure 5E), reflecting the impact from genetic background diversity. However, when the genes with positive or negative TE values (log2FoldChange>0.5 or <−0.5) from the non-isogenic datasets were selected and their TE values from the isogenic datasets were plotted, they show significant shifts to the respective direction (Figure 5F). The shift was also observed when we selected and plotted in the opposite way (Figure 5G). Therefore, we hypothesized that there might be common translational effects underneath the seemingly prevalent baseline variability.
Secondary structure in 5’UTR correlates to translatome alteration
In order to identify systematic translational effects by G2019S LRRK2, we examined previously suggested modes of translational regulation. However, targets of let-7 family miRNAs or the mRNAs with 5’ terminal oligopyrimidine tract (TOP) do not show significant TE differences (Figures S5D and S5E) (Gehrke et al., 2010; Imai et al., 2008). Previous studies have suggested that deregulated translation caused by LRRK2 kinase activity may interfere the initiation step of translation (Imai et al., 2008; Martin et al., 2014b; Penney et al., 2016). Secondary structure in the 5’ UTR of the mRNA is an important regulatory feature for translational initiation (Hinnebusch et al., 2016), and more than 40% of the mRNAs in the human transcriptome have relatively complex (< −100kcal/mol) 5’UTR secondary structure. Therefore, predicted 5’UTR folding energy (FE) was compared between TE up and TE down (log2FoldChange>0.5 or <−0.5, respectively) genes from each dataset. The TE up genes in G2019S LRRK2 human DA neurons have significantly lower FE than randomly selected control genes, while the TE down genes have significantly higher FE compared to the control genes (Figures 6A and S5F). The trend remains unchanged when the 5’UTR FE is normalized by its length (Figures S5G to S5I). Conversely, when mRNAs with complex 5’UTR structure (< −350kcal/mol) are selected, G2019S LRRK2 human dopamine neurons show significantly higher TE than control genes, while mRNAs possessing simple 5’UTR secondary structure (> −15kcal/mol) show significantly lower TE than controls genes (Figure 6B). The results are consistent across non-isogenic lines, isogenic pair lines, and combined analyses.
Figure 6. Positive correlation between 5’UTR secondary structure and translation efficiency in G2019S LRRK2 human dopamine neurons.

(A and B) 5’UTR FE is from the UCSC genome database hg38: foldUtr5 field. (A) 5’UTR FE of TE up and TE down genes (TE>0.5 or <−0.5, respectively). (B) Genes with complex 5’UTR (estimated FE:<−350kcal/mol) or simple 5’UTR (> −15kcal/mol) were selected and TE differences were plotted. Boxplot overlaid with violin plot visualizes the median, the first and the third quartile along with the data distribution pattern. Wilcoxon signed-rank test (all comparisons are against all genes control) ((A): p<0.001 (TE up), p<0.05 (TE down), p<0.001 (TE up:isogenic), p=0.1718 (TE down:isogenic), (B): p<0.001 (Simple), p<0.001 (Complex), p=0.02483 (Simple:isogenic), p<0.001 (Complex:isogenic)). (C) 5’UTR stem-loop reporters. Inserted sequence length: 110nt. RNAfold from ViennaRNA for FE calculation (Lorenz et al., 2011). (D and E) 5’UTR stem-loop luciferase reporter assays with mouse primary neurons. (D) n=5; (E) n=5 of independent measurements, respectively. Each measurement consists of technical triplicates. One-way ANOVA with Bonferroni correction. (D) 0kcal/mol: F=6.355, ns (Control vs GS/DA LRRK2), p<0.001 (Control vs GS LRRK2), ns (GS/DA LRRK2 vs GS LRRK2), ns (GS LRRK2 vs GS LRRK2 + TA S15); −30kcal/mol: F=11.91, ns (Control vs GS/DA LRRK2), p<0.001 (Control vs GS LRRK2), p<0.01 (GS/DA LRRK2 vs GS LRRK2), p<0.001 (GS LRRK2 vs GS LRRK2 + TA S15). Two-way ANOVA to ensure the reporter effect (0kcal/mol vs −30kcal/mol) : F=5.688, p=0.0188. (E) 0kcal/mol: F=1.527, ns (Control vs WT S15), ns (Control vs TA S15), ns (Control vs TD S15); −30kcal/mol: F=6.731, ns (Control vs WT S15), ns (Control vs TA S15), p<0.001 (Control vs TD S15). (F) Stem-loop reporter assays with LRRK2 kinase inhibitors, LRRK2-IN-1 and CZC-25146 (0.1μM for both). n=4 for both, each measurement consists of technical replicates. Two-way ANOVA with Bonferroni post-test (0kcal/mol: inhibitor effect: F=8.17, p<0.01, GS LRRK2 effect: F=10.76, p<0.01, DMSO vs LRRK2-IN-1: ns (Control), p<0.01 (GS LRRK2), DMSO vs CZC-25146: ns (Control), ns (GS LRRK2); −30kcal/mol: inhibitor effect: F=29.08, p<0.001, GS LRRK2 effect: F=44.43, p<0.001, DMSO vs LRRK2-IN-1: ns (Control), p<0.001 (GS LRRK2), DMSO vs CZC-25146: ns (Control), p<0.001 (GS LRRK2)). Data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, ns=no significance. GS: G2019S, GS/DA: G2019S/D1994A, TA: T136A, TD: T136D. See also Figures S5 and S6.
To confirm our findings using a simple reporter system, we generated 5’UTR luciferase reporters. 5’UTR sequences with no predicted secondary structure or artificial hairpin structure were inserted into a firefly luciferase reporter vector (Figure 6C). The reporters with complex 5’UTR secondary structure is translated more efficiently in primary mouse neurons expressing G2019S LRRK2 (Figures 6D and S6A). The increased reporter expression by G2019S LRRK2 is suppressed by introducing kinase-dead D1994A mutation to G2019S LRRK2 or co-expressing of T136A S15 (Figures 6D and S6A) (Martin et al., 2014b). Overexpression of phosphorylation-mimetic T136D S15 recapitulates the effects of G2019S LRRK2 (Figures 6E and S6B). 5’UTR luciferase reporters were tested with potent LRRK2 kinase inhibitors, LRRK2-IN-1 and CZC-25146 (Deng et al., 2011; Ramsden et al., 2011). LRRK2 kinase inhibitors reduce the increased expression of the reporter (Figures 6F and S6C). These results collectively suggest that LRRK2 kinase activity is required for maximal translation efficiency for the transcripts with complex 5’UTR structure.
5’UTR secondary structure and translational regulation of voltage-gated calcium channels in G2019S LRRK2 neurons
We tested whether translation of VGCC or GluR genes, whose product mediates Ca2+ influx, are increased in G2019S LRRK2 neurons. Firstly, we plotted TE of the VGCC and GluR genes. Both VGCCs and GluRs show significantly increased translation in the non-isogenic and the combined datasets, while VGCCs have a more prominent shift (Figures 7A and S6D). In the isogenic lines, only VGCCs show a statistically significant increase (Figure 7B). Of note, in the non-isogenic datasets, VGCC alpha and gamma subunits are broadly increased while beta subunits are not. In the isogenic datasets, two beta subunits show the highest increase while the other subunits do not show a specific trend (Table S3). While it is unclear why different subsets of genes constituting the same channel complex are induced in the two conditions, we speculate that this observation may reflect that their expression levels are rapidly changing upon its cellular and physiological circumstances.
Figure 7. 5’UTR secondary structure and translation efficiency of L-type voltage-gated calcium channels.

(A and B) Density plots comparing TE distributions of VGCCs (26 genes) and GluRs (18 genes) in non-isogenic and isogenic datasets, respectively. Wilcoxon signed-rank test (VGCC: non-isogenic (p<0.001), isogenic (p=0.03409), GluR: non-isogenic (0.0151), isogenic (p=0.0623)). (C) VGCC isoforms and 5’UTR FE from the hg38 reference genome. No. isoforms: number of reported transcript isoforms. Min. 5’UTR FE, Max. 5’UTR FE: minimum and maximum 5’UTR FE for the gene extracted from the hg38 foldUtr5 table. (D) L-VGCC 5’UTR luciferase reporters. 5’UTR sequences: CACNA1C (5’UTR length: 313nt, hg19: uc009zdu.1) or CACNA1D (5’UTR length: 118nt, hg19: uc003dgu.5). (E and F) 5’UTR luciferase reporter assays with mouse primary cortical neurons. Each experiment consists of technical triplicates. (E) n=3 (CaV1.2), 4 (CaV1.3), (F) n =3 (CaV1.2), 4 (CaV1.3) of independent measurements. One-way ANOVA with Bonferroni correction. (E) CaV1.2: F=5.026, ns (Control vs GS/DA LRRK2), p<0.05 (Control vs GS LRRK2), p<0.01 (GS LRRK2 vs GS LRRK2 + TA S15); CaV1.3: F=7.663, ns (Control vs GS/DA LRRK2), ns (Control vs GS LRRK2), ns (GS LRRK2 vs GS LRRK2 + TA S15). (F) CaV1.2: F=7.293, ns (Control vs WT S15), ns (Control vs TA S15), p<0.001 (Control vs TD S15); CaV1.3: F=4.274, p<0.01 (Control vs WT S15), ns (Control vs TA S15), ns (Control vs TD S15). * p<0.05, ** p<0.01, *** p<0.001, ns=no significance. Data are mean ± SEM. GS: G2019S, GS/DA: G2019S/D1994A, TA: T136A, TD: T136D. See also Figures S6 and S7.
To investigate the roles of 5’UTR in VGCC translation, we attempted to correlate their 5’UTR FE and TE values. Unexpectedly, we found that many of them have multiple isoforms with varying 5’UTR secondary structure (Figure 7C). Since this estimation process assumes that the expression is only coming from the principal transcript, correlation between TE and 5’UTR FE values does not fully reflect the actual effects when multiple isoforms present. Therefore, we designed 5’UTR luciferase reporters containing VGCC 5’UTR sequences. Based on the effectiveness of isradipine treatment, we designed two 5’UTR reporters from two L-VGCCs, one with CaV1.2 5’UTR the other one is CaV1.3 5’UTR (Figure 7D). The reporter assay showed that complex secondary structure existing in CaV1.2 5’UTR induces translation with G2019S LRRK2 expression through T136 phosphorylation of S15 while simple 5’UTR from CaV1.3 coding gene does not (Figures 7E and 7F, S7A and S7B). The results suggest that 5’UTR sequences of VGCC genes may contribute to the increased translation, while the exact isoform expression profile in DA neurons is required to fully address the translation regulatory mechanisms.
Discussion
Dysregulated calcium homeostasis and neuronal stress in G2019S LRRK2 DA neurons
Dysregulation of Ca2+ homeostasis and subsequent mitochondrial dysfunction have been ascribed as one of the major culprits in PD pathogenesis. Elevated Ca2+ increases cellular metabolic demands for cytosolic Ca2+ clearance, resulting in elevated mitochondrial respiration and cellular oxidant production (Camandola and Mattson, 2017; Nicholls, 2008; Surmeier et al., 2017). In addition, mitochondria are major reservoirs for intracellular Ca2+, making Ca2+ dysregulation critical to mitochondrial health (Perier and Vila, 2012). In line with the previous studies linking Ca2+ concentration to metabolic demands, transcript levels of ribosomal components and mitochondria respiratory chain complex genes are significantly increased in human G2019S LRRK2 DA neurons, reflecting elevated cellular metabolism (Figure S7C) (Dominy and Puigserver, 2013; Reinecke et al., 2009). Collectively, we speculate that dysregulation of Ca2+ homeostasis and altered translation together lead to the increased cellular metabolic needs and mitochondrial burden.
One question regarding the dopaminergic neurotoxicity is whether this Ca2+ dysregulation caused by G2019S LRRK2 only exists in DA neurons, or DA neurons are more susceptible to Ca2+ dysregulation. To address this question, we differentiated the isogenic iPSC lines into human cortical neurons and measured their cytosolic Ca2+ levels (Xu et al., 2016). We found that cortical neurons also have increased cytosolic Ca2+ concentrations (Figure S7D). The result suggests that G2019S LRRK2 could potentially elevate Ca2+ levels in other neuronal types as well, and DA neurons might be more vulnerable to the constitutive increase of intracellular Ca2+ levels. Previous studies speculating that DA neurons are more susceptible to Ca2+ dysregulation due to their extreme axonal arborization, continuous dendritic Ca2+ influx, and insufficient Ca2+ buffer proteins, and the fact that non-DA neurons are also affected in PD especially in late stages seem to be in line with this interpretation (Pacelli et al., 2015).
Proteostasis, calcium homeostasis and molecular pathogenesis in LRRK2 PD
There were previous reports implicating defective Ca2+ regulation in LRRK2 mutants, although the molecular mechanisms linking the function of LRRK2 to Ca2+ homeostasis remained unclear. It was reported that neurons expressing mutant LRRK2 have impaired Ca2+ clearance after KCl-induced neuronal depolarization (Cherra et al., 2013; Schwab and Ebert, 2015), or show ERK activation (Reinhardt et al., 2013). A recent study also reported that human neurons differentiated from G2019S LRRK2 iPSCs have altered ER Ca2+ regulation (Korecka et al., 2019). Our findings provide a plausible mechanism for the observed defects in Ca2+ homeostasis.
It is noteworthy that Ca2+ homeostasis may be a link connecting protein synthesis and protein degradation, the two axes in the proteostatic pathways dysregulated in LRRK2 mutants. LRRK2 mutations have been linked to impaired macroautophagy and endolysosomal pathways (Abeliovich and Gitler, 2016; Cookson, 2016; Dehay et al., 2013; Orenstein et al., 2013), functions that are regulated by cytosolic Ca2+ (Hoyer-Hansen et al., 2007). Of note, a previous study suggesting the function of LRRK2 in autophagic regulation through lysosomal Ca2+ signaling pathways (Gomez-Suaga et al., 2012) supports this hypothesis, although the detailed mechanisms need to be elucidated. Future studies aiming to investigate this molecular crosstalk between proteostasis and Ca2+ homeostasis will provide comprehensive insights on the neurodegenerative mechanisms in G2019S LRRK2 PD.
Voltage-gated calcium channels and Ca2+ dysregulation in G2019S LRRK2 neurons
In spite of the effectiveness of isradipine to reduce cytosolic Ca2+, it is yet unclear whether the L-type channels are specifically targeted by LRRK2 and their expression levels are increased by G2019S LRRK2. The observations that L-VGCC-mediated Ca2+ currents are increased and L-VGCC 5’UTR could increase reporter expression suggest that L-VGCC levels might be higher in G2019S LRRK2 neurons. However, ribosome profiling results reported only mild or insignificant TE changes of CACNA1C expression in G2019S LRRK2 human DA neurons (63.47% increase in non-isogenic, 8.25% decrease in isogenic) (Table S3). Since translation of many genes involved in Ca2+ homeostasis are broadly affected in G2019S LRRK2 human DA neurons, our interpretation is that deregulation of multiple genes including VGCC subunits collectively results in Ca2+ dysregulation. The next step would be testing the roles of other types of VGCC in the DA neuronal Ca2+ homeostasis and identifying pharmacologic and/or genetic methods which can effectively restore Ca2+ homeostasis in G2019S LRRK2 human DA neurons.
Expression of voltage-gated calcium channels and their 5’UTR isoforms
The GRCh38 genome assembly reports that there are multiple isoforms of VGCC genes with varying 5’UTR secondary structure. For example, CACNA1C has 12 different isoforms with the minimum (most complex) 5’UTR FE of −85 kcal/mol and the maximum (simplest) 5’UTR FE of −3.4 kcal/mol. As an extreme case, CACNB4 reportedly has 50 different isoforms and their 5’UTR FE spans from −277.2 kcal/mol to 0kcal/mol (Figure 7C and Table S3). The actual number of isoforms expressing in the DA neuron and the regulation for isoform selection have not yet been thoroughly addressed. Since the existence of multiple 5’UTR variants suggests that 5’UTR could be used to regulate expression of VGCCs, future studies focusing on the 5’UTR-mediated translational regulation in DA neuron are warranted.
It was previously suggested that 5’UTR secondary structure is involved in the gene expression control under neuronal activity (Dalal et al., 2017). In this regard, there is a possibility that the elevated Ca2+ resulting from increased expression of Ca2+ channels could further change the translational landscape of G2019S LRRK2 neurons. Thus, comprehensive understanding on the neuronal activity and 5’UTR-mediated translational regulation will deepen our knowledge on the G2019S LRRK2 PD pathology.
5’UTR-mediated translational effects of G2019S LRRK2
Previous studies have suggested that pathogenic LRRK2 mutants may cause abnormal translation initiation through multiple potential mechanisms (Imai et al., 2008; Martin et al., 2014b; Penney et al., 2016). The 5’UTR is known to possess regulatory roles in translation initiation and in particular, complex secondary structure in the 5’UTR is thought to serve as a negative modulator of translation initiation by hindering ribosomal scanning (Hinnebusch et al., 2016). Therefore, our findings on the structured 5’UTR preference indicates the significance of translation initiation in LRRK2-mediated translational abnormality.
A recent study reported that multiple S15 mutations are linked to chronic lymphocytic leukemia, and the disease-relevant mutants, mostly located at its C-terminus, show varying levels of translational defects (Bretones et al., 2018). Several studies on the ribosomal structure provide us a clue to understand the potential role of the C-terminus of S15 (positions 131-145), which contains the T136 residue phosphorylated by LRRK2. The C-terminus of S15 is unstructured and found only in eukaryotes, and studies on the ribosomal structure revealed that this C-terminal tail projects into the decoding sites of the ribosome (Figures S7E and S7F) (Anger et al., 2013; Khatter et al., 2015). The S15 C-terminus was shown to interact with the A site codon of mRNA on the 48S pre-initiation complex (PIC), and the A- and P-sites tRNAs, ribosomal RNA helices, and the mRNA on the 80S ribosome (Bhaskar et al., 2020; Khairulina et al., 2010; Sharifulin et al., 2015). A cryo-EM study focusing on the C-terminal tail of S15 demonstrated that T136 forms a hydrogen bond with the P-site tRNA (N28) on the 80S ribosome (Bhaskar et al., 2020). The C-terminus of S15 has also been suggested to play a role in the transition of ribosomal conformation which completes the scanning process and results in a stable 48S PIC formation (Lomakin and Steitz, 2013). Our findings warrant future studies on the structural properties of phospho-S15 ribosome, which will expand our knowledge on the intricate regulatory mechanisms in mRNA translation.
Limitations of the Study
In this study, a total of 8 lines of iPSCs – 6 unrelated lines from different individuals and one pair of isogenic lines with a mutation correction – were used. We recognize limitations associated with the number and configuration of iPSC lines used. Testing additional iPSC lines including mutation-corrected, paired lines can strengthen the conclusions of the study.
We found that annotations for 5’UTR and isoforms in DA neurons are not complete yet. Since we report 5’UTR-related correlations in this study, we recognize potential limitations ascribable to incomplete annotations in the current reference genome.
STAR Methods:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources (including genetic models) and reagents should be directed to and will be fulfilled by the Lead Contact, Valina Dawson (vdawson@jhmi.edu).
Materials Availability
Four plasmids generated in this study for 5’UTR luciferase assays have been deposited to Addgene (pJWK_VD_01: #158102, pJWK_VD_02: #158103, pJWK_VD_03: #158104, pJWK_VD_04: #158105). The iPSC lines generated in this study will be made available on request, but it will require a completed Materials Transfer Agreement.
Data and Code Availability
The high-throughput sequencing data generated in this study have been deposited in the NCBI GEO database under accession number GSE90469. Codes for ribosome profiling analysis are available on Nicholas Ingolia’s Github (https://github.com/ingolia-lab/RiboSeq).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal protocols are in accordance with the regulations of the Johns Hopkins University Animal Care and Use Committee.
Generation and characterization of human iPSC lines
Deidentified human fibroblasts were obtained and expanded in DMEM with 10% fetal bovine serum (FBS, Life Technologies). Human iPSCs were generated using CytoTune®-iPS Sendai Reprogramming Kit (Life Technologies) following manufacturer’s instruction; fibroblasts were plated at a density of 2.5-5×104 cells/cm2 on gelatin-coated plate and transduced with Sendai viruses expressing Oct4, Klf4, Sox2, and c-Myc at MOI of 3 and/or 6. At the day 6 of transduction, cells were dissociated with accutase and re-plated on multiple plates with mouse embryonic fibroblasts (MEFs) growing at various densities. Cells were fed with human embryonic stem cell (hESC) media (Gibco) from the next day of re-plating and replaced the medium daily until they formed hESC-like colonies approximately for 2-3 weeks. Mycoplasma contamination was tested and controlled regularly. Individual colonies were isolated and clonally expanded to establish iPSC lines. For the LRRK2 isogenic lines, the generation and characterization of the iPSC lines were previously published (Reinhardt et al., 2013).
Differentiation of human iPSCs to human DA neurons
Human iPSCs were cultured using standard protocol on inactivated mouse embryonic fibroblast. DA neuron differentiation was done as previously described (Kriks et al., 2011). Briefly, iPSCs were cultured on Matrigel (Corning)-coated plate at a density of 4×104 cells/cm2 in SRM media containing growth factors and small molecules including FGF8a (100ng/mL, R&D Systems), SHH C25II (100ng/mL, R&D Systems), LDN193189 (100nM, Stemgent), SB431542 (10μM, R&D Systems), CHIR99021 (3μM, Stemgent), and purmorphamine (2μM, Sigma) for the first five days. For the next six days, cells were maintained in Neurobasal medium (Gibco) containing B-27 supplement minus vitamin A (Gibco), N-2 supplement (Gibco) along with LDN193189 and CHIR99021. In the final stage, colonies were made into single cell suspension and seeded at density of 4×106 cells/cm2 on poly-ornithine and laminin coated plate in neurobasal media containing B27 minus vitamin A, BDNF (20ng/mL, Peprotech), GDNF (20ng/mL, Peprotech), TGFβ (1ng/mL, R&D Systems), ascorbic acid (0.2mM, Sigma), dibutyryl-cAMP (0.5mM, Sigma) and DAPT (10μM, Stemgent) until maturation. DA neurons were cultured for >60 differentiation days before measurements.
Differentiation of human iPSCs to human cortical neurons
For cortical neurons, the detailed procedure was previously published (Xu et al., 2016). Briefly, First, iPSCs were detached and grown as embryoid body (EB) suspensions. On the day 2, dual SMAD inhibition with noggin and SB431542 was initiated for 4 days until day 7 to allow the robust induction of rosette formation. Day 7, the EBs were plated on Matrigel- or laminin-precoated plates. Day 9, rosettes began to form with profound cellular expansion in a defined central area of each colony (rosette type neural stem cells, RONA). On the day 17, RONAs were manually isolated and grown as neuro spheres in suspension for 1 day. Day 18, the neurospheres were disassociated into single cells and plated on laminin/ poly-D-lysine–coated plates or coverslips where they formed neural precursor cell (NPC) clusters. For neuronal differentiation, retinoic acid (2 mM, Sigma), SHH (50 ng/ml, R&D Systems), purmorphamine (2 mM, Sigma), or the combination of retinoic acid, SHH, and purmorphamine was supplemented in neural differentiation medium containing Neurobasal/B27 (NB/B27, Invitrogen), brain-derived neurotrophic factor (BDNF; 20 ng/ml, R&D Systems), glial cell line–derived neurotrophic factor (GDNF; 20 ng/ml, R&D Systems), ascorbic acid (0.2 mM, Sigma), dibutyryl adenosine 3′,5′-monophosphate (0.5 mM, Sigma) at the indicated time points after neurospheres were dissociated into single cells. The neurons were cultured for >60 differentiation days before measurements.
Mouse primary cortical neuron culture
Dissipated primary cortical neurons were prepared from mouse E15 developing brain (CD1, Charles River). Developing cortices were dissected in the dissecting medium (Dulbecco's Modified Eagle Medium (DMEM) with 20% horse serum, 0.5mM GlutaMax, 6μM glucose, Gibco), digested with TrypLE (Gibco), and plated at a concentration of 3 × 106 cells for a plate. Culture plates were pre-coated with 15μg/mL poly-L-ornithine. Cultures were maintained under Neurobasal (Gibco) medium with a serum-free supplement B-27 (Gibco) and 0.5mM GlutaMax (Gibco).
METHOD DETAILS
Immunocytochemistry of human iPSCs and DA neurons
Cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature, then permeabilized with 0.03% Triton X-100 for 15 min. The cells were washed, and blocked for 1 hour with 10% goat serum in PBS. The blocked cells were subsequently incubated with primary antibody for overnight at 4°C. On the following day, the cells were incubated with secondary antibody for 1 hour at room temperature in a light controlled condition. After 3× wash with PBS buffer, the cells were mounted on cover slides with mounting media containing DAPI (ThermoScientific). All images were taken for analysis with Zeiss AxioObserver Z1 or LSM710 (Carl Zeiss) confocal laser scanning microscope under 20× or 40× oil objectives. For pluripotent stem cell markers, 2-3 random fields were captured for analysis. For DAPI, TH and TUJ1 counting, 6 random fields were captured and total 600-800 cells were counted in each condition for analysis. Blinding was not performed with immunocytochemistry experiments. The following primary antibodies were used for immunocytochemistry: α-TUJ1 (1:2000, Covance MMS-435P), α-TH (1:1000, EMD Millipore AB152), α-OCT4, α-NANOG, α-SOX2, α-SSEA4 (1:200 for all, Cell Signaling, StemLight™ Pluripotency Antibody Kit #9656).
T136A S15 overexpression and isradipine treatment in human DA neurons
Human cDNA expressing T136A S15 allele was cloned into pAM/CBA-pl-WPRE-bGH vector, and AAV virus (AAV2 serotype) was generated (Vector Biolabs). 2μl of 1×1013 GC/mL virus was used for one well of human DA neurons (approx. 4×106 cells, differentiation day>60), and incubated for 5 days with minimum culture medium change (half-change, once every 3 days) before measurements. Cells were exposed to isradipine (1μM) for 2 hours in prior to measurements.
Ribosome profiling library generation
Ribosome footprinting and RNA-seq libraries were prepared by following a previously described protocol with several modifications made for neurons (Ingolia et al., 2012). Dopamine neurons: 100μg/mL cycloheximide was applied to the cultures for 15 minutes, then neurons were lysed on ice in lysis buffer (10mM Tris pH 7.5, 150mM NaCl, 5mM MgCl2, 0.5mM DTT, 100μg/mL cycloheximide, EDTA-free protease inhibitor (Roche), 1% NP-40, 20U/mL SuperAse-In (Ambion)). The lysates were incubated in ice for 15 minutes, and centrifuged for 10 minutes at 20,000×g. Total RNA concentration of lysate was measured by Qubit RNA BR Assay (Life Technologies), and the same amount of RNA was used across samples. The supernatant was split into two tubes for ribosome footprinting and RNA-seq library generation.
Ribosome footprinting: The lysates were treated with 15μL of RNase I (Ambion) in 600μL total reaction volume for 45 min at room temperature, and the reaction was stopped by adding 30μL of SuperAse-In (Ambion). Sucrose cushion was performed with 1.7g sucrose in 3.9mL polysome buffer (10mM Tris pH 7.5, 150mM NaCl, 5mM MgCl2, 0.5mM DTT, 100μg/mL cycloheximide, 20U/mL SuperAse-In), 4 hours at 70,000rpm. The pellet was resuspended with 700μL QIAzol (QIAGEN) reagent, incubated for 5 minutes at room temperature, 140μL chloroform was added, vortexed for 15 seconds, and incubated again for 2 minutes at room temperature. The sample was centrifuged for 15 minutes at 12,000×g, the 350μL supernatant was mixed with 525μL 100% EtOH. The mixture was loaded on an RNeasy Mini column (QIAGEN), and the RNA was extracted. 26~34nt ribosome footprints were size-selected by Urea-PAGE, gel extraction and RNA purification. Ribo-Zero Gold Kit (Illumina) was used for rRNA removal after the size selection. The rRNA depleted ribosome footprints were dephosphorylated by T4 polynucleotide kinase treatment, then Universal miRNA Cloning Linker (NEB) was added to the 3’ ends. Reverse transcription reaction was performed with NI-NI-9 primers as previously described (Ingolia et al., 2012). NI-NI-9: 5’-(Phos)-AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC-(SpC18)-CACTCA-(SpC18)-TTCAGACGTGTGCTCTTCCGATCTATTGATGG TGCCTACAG-3’ (Phos:5’ phosphorylation, SpC18: a hexa-ethyleneglycol spacer). The cDNA was circularized by CircLigase I or CircLigase II (Epicentre) reaction, and subjected to the PCR for final library generation.
RNA-seq: Total RNA was purified by a combination of QIAzol and RNeasy Mini as described. Ribo-Zero Gold Kit was used for rRNA removal. RNA-seq library was generated from the total RNA by ScriptSeq v2 Library Preparation Kit (Epicentre).
Ribosome profiling data processing
Illumina HiSeq 2000/4000 were used for deep sequencing of the libraries. Sequencing results were processed by following a previously published pipeline (Ingolia et al., 2012). FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) was used for the initial processing of the reads.
Ribosome footprinting libraries: Only adapter-containing reads were clipped. The first nucleotide of the reads was trimmed. rRNA-mapped reads were discarded before genomic alignment.
RNA-seq libraries: Adapter-containing reads were clipped, rRNA-mapped reads were discarded.
The processed reads were mapped to the UCSC genome database (human: hg38) by Tophat (2.1.1) with Bowtie2 (2.3.4.1) (Langmead and Salzberg, 2012; Trapnell et al., 2009). Maximum 1 mismatch was allowed for the alignments.
Sequencing read counts
| Sample | Type | Mapped Reads |
|---|---|---|
| SC1014 | Ribo | 30,020,158 |
| mRNA | 67,674,841 | |
| SC1015 | Ribo | 59,114,954 |
| mRNA | 49,057,592 | |
| SC1055 | Ribo | 29,525,048 |
| mRNA | 78,870,616 | |
| SC1007 | Ribo | 21,384,758 |
| mRNA | 54,542,831 | |
| SC1034 | Ribo | 32,715,717 |
| mRNA | 43,651,560 | |
| SC1041 | Ribo | 27,376,583 |
| mRNA | 51,138,107 | |
| MC 1 (isogenic) | Ribo | 11,308,226 |
| mRNA | 47,089,189 | |
| MC 2 (isogenic) | Ribo | 11,459,344 |
| mRNA | 56,183,246 | |
| GS LRRK2 1 (isogenic) | Ribo | 19,286,120 |
| mRNA | 43,499,529 | |
| GS LRRK2 2 (isogenic) | Ribo | 15,900,455 |
| mRNA | 47,894,005 |
SC1014, SC1015, SC1055 lines are non-PD control. SC1007, SC1034, SC1041 lines are G2019S LRRK2 heterozygotes (GGC -> AGC). Ribo: ribosome profiling, mRNA: RNA-Seq.
5’UTR and IRES reporter assays
5’UTR stem-loop reporters: Additional 101 nucleotides were inserted into the 5’UTR region of the pGL4.53(luc2/PGK) vector (Promega). Estimated folding energy of the anticipated 5’UTR sequences were calculated by the Vienna RNA Package RNAfold (2.1.1) (Lorenz et al., 2011).
0kcal/mol:
AAGATCACAACTATACCTAAGCATCACCTCTACAACTACGATCACCACTATAAAGATCACAACTATACCTAAGCATCACCTCTACAACTACGATCACCACT
30kcal/mol:
AAGATCACAACTATACCTAAGCATCACCTCTACAACTACGATCACCACTATAAGGTGGAATTTGTAACGTAGAGGCTTGTATTGAGTACTAGTACGATCTA 5’UTR CACNA1C/1D reporters: 5’UTR sequence of CACNA1C/1D genes were retrieved from UCSC genome database (hg19, CACNA1C: uc009zdu.1, CACNA1D: uc003dgu.5). and cloned into the 5’UTR region of the pGL4.53(luc2/PGK) (Promega) vector.
C-terminal myc-tagged LRRK2 vectors and N-terminal V5-tagged S15 vectors were used as previously described (Martin et al., 2014b). Control plasmids are the same backbone without insert. The vectors were transfected into mouse cortical neurons at DIV 5 by Lipofectamine 2000 (Invitrogen) reagent. Culture medium was replaced (half-change) every 24 hours to minimize any potential effects from the growth condition including starvation. Luciferase activity was measured at DIV 7 by Luciferase Assay System (Promega) (for the 5’UTR reporters) with Glomax 20/20 Luminometer (Promega). The lysates were subjected to the total RNA purification with DNase treatment for the transcript level measurement. For the 5’UTR reporters, luciferase mRNAs were spiked-in for qPCR input normalization.
Immunoblotting
For P-S15 western blot, neurons were lysed in polysome buffer (10mM Tris pH 7.5, 150mM NaCl, 5mM MgCl2, 0.5mM DTT, 100μg/mL cycloheximide, EDTA-free protease inhibitor (Roche), 1% NP-40, 20U/mL SuperAse-In (Ambion)). Lysates were incubated in ice for 10 min, then spun down for 10 min × 12,000g at 4°C. Supernantant was loaded onto sucrose cushion with 1.7g sucrose in 3.9mL polysome buffer (10mM Tris pH 7.5, 150mM NaCl, 5mM MgCl2, 0.5mM DTT, 100μg/mL cycloheximide, 20U/mL SuperAse-In), 4 hours at 70,000rpm. Pellets were recovered using 2x Laemmli sample buffer, and proteins were precipitated from supernantant using methanol precipitation (4× volume methanol followed by 1× volume chloroform). The following antibodies were used for western blots: α-LRRK2 (1:1000, Cell Signaling #13046), α-S6 (1:1000, Cell Signaling #2217), α-S15 and α-P-S15 (in house, previously published (Martin et al., 2014b)).
Intracellular Ca2+ imaging of human DA neurons
Membrane permeable acetoxymethylester (AM) form of Fluo-4 (Thermo Fisher Scientific) or Fura-2 (Kd 0.14M; Invitrogen) was used to monitor intracellular Ca2+ levels. Blinding was not performed with Ca2+ imaging experiments. Human DA neurons (at differentiation day 60-80) were loaded with Fluo-4-AM or Fura-2-AM for 30min at 2μM final concentration. Neurons were washed three times with HEPES-buffered ACSF (NaCl 125mM, KCl 5mM, HEPES 10mM, MgCl2 1mM, CaCl2 2mM, glucose 25mM, pH 7.4, osmolality 310 mosM), and incubated at RT in the dark for additional 20–25 minutes to allow for complete dye de-esterification. Cells were placed in a 30-32°C heated adaptor and images was taken by inverted epifluorescence microscope (Nikon TE300) with a 40×/1.35NA oil-immersion objective. The imaging chamber was superfused with HEPES-buffered ACSF at a flow rate of 1mL/min. For Fluo-4-AM, live imaging was conducted with excitation wavelength of 485 nm and emission wavelength of 525nm. For Fura-2-AM, cells were illuminated at two excitation wavelengths (340 and 380nm) by Polychrome V (TILL Photonics) and emission was captured with a cooled CCD camera (Hamamatsu ImagEM) connected to a computer running Slidebook imaging software (Intelligent Imaging Innovations). Ratiometric images (F340/F380) were taken every 120ms with exposure time of 20ms. Regions of interests (ROIs) in the soma were selected for further analysis. ROI F340/F380 ratios were converted to Ca2+ concentrations, using a standard curve generated under the exact acquisition conditions (Calcium Calibration Buffer Kits, Invitrogen).
Whole-cell patch clamp Ca2+ current recordings of human DA neurons
Spontaneous and evoked action potentials (APs): HEKA EPC10 amplifier (HEKA Elektronik) was used. Human DA neurons were visualized under a 40× water immersion objective by fluorescence and DIC optics (Carl Zeiss), and the chamber was constantly perfused at a rate of 1-2mL/min at 32°C with bath solution (NaCl 137mM, KCl 5mM, CaCl2 2mM, MgCl2 1mM, HEPES 10mM, glucose 10mM). Blinding was not performed with Ca2+ imaging experiments. pH of bath solution was adjusted to 7.4 with NaOH, and osmolarity was at 300–310mosM. Patch pipettes (2–5 MΩ) were pulled from borosilicate glass (BF-150, Sutter Instruments) using a Flaming-Brown micropipette puller (P-1000, Sutter Instruments) and filled with pipette solution (K-gluconate 126mM, KCl 8mM, HEPES 20mM, EGTA 0.2mM, NaCl 2mM, MgATP 3mM, Na3GTP 0.5mM, adjusted to pH 7.3 with KOH, adjusted to 290-300mosM with sucrose). Resting membrane potential was recorded in current clamp mode at 0pA immediately after establishing a whole-cell configuration. Series resistance (Rseries) and input resistance (Rin) were calculated from a 5mV pulse and monitored throughout the experiment. Unstable recordings (>10% fluctuation of Rseries value) during the course of experiments were rejected from further analysis. Series of hyperpolarizing and depolarizing step currents were injected to measure intrinsic properties and to elicit APs. Presence of a sag in the membrane potential and the AP firing pattern were checked in current-clamp immediately after rupturing the membrane.
Voltage clamp: External solution (TEA-MeSO3 140mM, HEPES 10mM, BaCl2 or CaCl2 10mM, pH was adjusted to 7.4 with CsOH, osmolarity was adjusted to 300-310mosM with glucose) and internal solution (pipet solution) (CsMeSO3 135mM, CsCl 5mM, MgCl2 1mM, MgATP 4mM, HEPES 5mM, EGTA 5mM, pH was adjusted to 7.3 with CsOH, and osmolarity was adjusted to 290-300mosM with glucose) were used. Currents were recorded by holding the cell at −90mV, before stepping to various potentials from −60mV to +50mV for 250ms in 10mV increments. Tetrodotoxin (1μM) was used to ensure blockade of voltage-gated sodium currents. Data were acquired by PatchMaster software (HEKA Elektronik), sampled at 10kHz, and filtered at 2.9kHz. Ca2+ currents were analyzed using Clampfit 10.5 software (Molecular devices). Ramp voltage clamp: Cultures were transferred to a recording chamber on a fixed-stage inverted microscope (Diaphot 200; Nikon). The recording chamber was perfused (2 ml/min, 25°C) with HEPES-based solution: 130mM NaCl, 1mM MgCl2, 10mM BaCl2, 10mM HEPES, 10mM dextrose, 10mM sucrose and 15mM CsCl at pH 7.4 and osmolarity of ~320mosM. Internal pipette solutions contained the following: 180mM N-methyl-D-glucamine, 40mM HEPES, 4mM MgCl2, 12mM phosphocreatine, 0.1mM leupeptin, 2mM Na2ATP, 0.5mM Na3GTP, 5mM BAPTA, pH 7.2–7.3 and osmolarity of ~290 mosM. Somatic whole-cell patch-clamp recordings were obtained with a MultiClamp 700A amplifier (Molecular Devices) connected to an Intel-based PC running pClamp10 (Molecular Devices). The signals were filtered at 1kHz and digitized at 10kHz with Digidata 1440A (Molecular Devices). Recording voltage was either fixed to +10mV, or current–voltage relationships were calculated with current responses to voltage ramps (0.7mV/ms) from −60 to 80 mV. Cell capacitance (measured using PClamp 10 automatic compensation) was used to calculate peak Ca2+ current densities (pA/pF). At the end of each experiment, Ca2+ current–voltage relationships were corrected for leaks currents measured in the presence of Cd2+ (200μM), a non-selective Ca2+ channels antagonist. L-VGCC currents were estimated by subtracting Ca2+ currents in the presence of isradipine (1 μM) from total Ca2+ currents. Peak Ca2+ current densities were measured from current–voltage relationships using Clampfit 10 (Axon Instruments) and IgorPro 6 software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance, type of the test used for individual testing, number of independent experiments are indicated in the figure legends. Non-parametric test is used and denoted when samples do not meet normality assumptions. Error bars indicate SEM without further clarification.
Ribosome profiling data analysis
Aligned reads were counted by custom scripts written by Nicholas Ingolia (McGlincy and Ingolia, 2017). Annotations and sequencing reads were handled using an R package GenomicFeatures (Lawrence et al., 2013). To avoid multiple counting on isoforms, transcript reference data were processed to have one unique annotation covering all isoforms (union of isoforms) per gene. Reads only in the CDS regions were counted. Transcripts with low read counts (< 64 reads) were discarded. An R package DESeq2 (1.22.2) was used for calculating normalized expression from either ribosome footprinting or RNA-seq data based on a negative binomial distribution and generalized linear model (Love et al., 2014). For the human neuron data, biological triplicates (non-isogenic) or replicates (isogenic) were handled by DESeq2. 5’UTR estimated folding energy table was extracted from the UCSC genome database (fold5UTR field: hg38). For the 5’UTR estimated folding energy comparison, a control group with similar group size was randomly selected for each comparison to avoid potential bias from sample size differences. Transcript coordinates were calculated by a custom R script and realigned based on the rounded half point of the ribosome footprint (5’ end + (footprint length/2)) or by fp-framing (https://github.com/ingolia-lab/RiboSeq).
Ingenuity pathway analysis and gene ontology
Ingenuity Pathway Analysis (QIAGEN) was used for the pathway analysis in G2019S LRRK2 human dopamine neurons. For gene ontology, a Cytoscape (3.7.1) plug-in ClueGo (2.5.4) and PANTHER (15.0) were used (Bindea et al., 2009; Mi et al., 2019). For PANTHER GO analysis, a total transcript list from non-isogenic datasets was used as reference genome.TE values calculated by DESeq2 were used for analysis, the same cutoff (TE>0.8 or <−0.8) was used for both analyses.
Human 80S ribosome structure
The original structure was from PDB 4V6X (Anger et al., 2013). The structure was visualized using UCSF Chimera (Pettersen et al., 2004).
Supplementary Material
Table S1. Related to Figures 1, 2, and 5. LRRK2 human DA neurons ribosome profiling DESeq2 analysis
Table S2. Related to Figures 1, 2, and 5. RNA-seq and ribosome profiling normalized counts by cell line
Table S3. Related to Figure 2. Gene Ontology and Ingenuity Pathway Analysis results, VGCC 5’UTR and TE table
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-LRRK2 (1:1000) | Cell Signaling | Cat#13046 RRID: AB_2798098 |
| Rabbit monoclonal anti-S6 (1:1000) | Cell Signaling | Cat#2217 RRID: AB_331355 |
| Rabbit polyclonal anti-S15 (1:1000) Rabbit polyclonal anti-phospho-S15 (1:1000) |
Dawson Lab Martin I. et al., 2014 |
N/A |
| Rabbit monoclonal anti-β-actin (HRP conjugated, 1:2000) | Cell Signaling | Cat#4970 RRID: AB_2223172 |
| Mouse monoclonal anti-TUJ1 (Mouse monoclonal, 1:2000) | Covance | Cat#MMS-435P RRID:AB_2313773 |
| Rabbit polyclonal anti-TH (1:1000) | EMD Millipore | Cat#AB152 RRID:AB_390204 |
| Rabbit monoclonal anti-OCT4 (1:200) | Cell Signaling | Cat#2840 RRID:AB_2167691 |
| Rabbit monoclonal anti-NANOG (1:200) | Cell Signaling | Cat#4903 RRID:AB_10559205 |
| Rabbit monoclonal anti-SOX2 (1:200) | Cell Signaling | Cat#3579 RRID:AB_2195767 |
| Mouse monoclonal anti-SSEA4 (1:200) | Cell Signaling | Cat#4755 RRID:AB_1264259 |
| Bacterial and Virus Strains | ||
| pAM/CBA-RPS15(T136A)-WPRE-bGH (AAV2) | Vector Biolabs | N/A |
| Biological Samples | ||
| Fibroblasts (control and PD-patient, deidentified) | Mayo Clinic | N/A |
| iPSCs (isogenic) L1-2Mut, L1-2GC | Gasser Lab Reinhardt et al. ,2013 | N/A |
| PD patient brain tissue | Mayo Clinic Brain Bank (Florida) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Sigma | Cat#C7698 |
| EDTA-free protease inhibitor | Roche | Cat#11836170001 |
| SuperAse-IN | Thermo Fisher | Cat#AM2694 |
| RNase I | Thermo Fisher | Cat#AM2294 |
| QIAzol | QIAZEN | Cat#79306 |
| Universal miRNA Cloning Linker | NEB | Cat#S1315S |
| CircLigase II | Epicentre | Cat#CL9021K |
| Matrigel | Corning | Cat#356231 |
| FGF-8a | R&D Systems | Cat#4745-F8-050 |
| SHH (C25II) | R&D Systems | Cat#464-SH-025 |
| LDN193189 | Stemgent | Cat#04-0074 |
| SB431542 | R&D Systems | Cat#1614/1 |
| CHIR99021 | Stemgent | Cat#04-0004 |
| Purmorphamine | Sigma | Cat#SML0868 |
| BDNF | R&D Systems | Cat#248-BD-025/CF |
| GDNF | R&D Systems | Cat#212-GD-010/CF |
| TGF-β3 | R&D Systems | Cat#243-B3-002/CF |
| Dibutyryl-cAMP | Sigma | Cat#D0627 |
| DAPT | Stemgent | Cat#04-0041 |
| Ascorbic Acid | Sigma | Cat#A4034 |
| Laminin | Sigma | Cat#L2020 |
| Y-27632 | Sigma | Cat#Y0503 |
| Accutase | Life Technologies | Cat#A1110501 |
| B-27 Supplement | Life Technologies | Cat#12587010 |
| Neurobasal Medium | Life Technologies | Cat#21103049 |
| Retinoic acid | Sigma | Cat#R2625 |
| SHH | R&D Systems | Cat#8908-SH |
| Purmorphamine | Sigma | Cat#540220 |
| Fluo-4-AM | Thermo Fisher | Cat#F14201 |
| Fura-2-AM | Thermo Fisher | Cat#F1201 |
| Isradipine | Sigma | Cat#I6658 |
| Tetrodotoxin | Sigma | Cat#T8024 |
| Critical Commercial Assays | ||
| Ribo-Zero Gold Kit | Illumina | Cat#MRZG126 |
| ScriptSeq v2 Library Preparation Kit | Illumina | Cat#SSV21106 |
| Luciferase Assay | Promega | Cat#E1500 |
| Dual-Glo ® Luciferase Assay | Promega | Cat#E2920 |
| CytoTune®-iPS Sendai Reprogramming Kit | Life Technologies | Cat#A13780 |
| Human Embryonic Stem Cell Media Kit | Life Technologies | Cat#A1412901 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GSE90469 |
| Human reference genome: hg19, hg38 (knownGene, foldUtr5) | UCSC Genome Browser | https://genome.ucsc.edu/ |
| Oligonucleotides | ||
| NI-NI-9 (reverse transcription primer) | Ingolia et al., 2012 | N/A |
| 5’UTR sequences: CACNA1C (human) (hg19) | UCSC Genome Browser | uc009zdu.1 (5’UTR) |
| 5’UTR sequences: CACNA1D (human) (hg19) | UCSC Genome Browser | uc009zdu.1 (5’UTR) |
| Recombinant DNA | ||
| pGL4.53(luc2/PGK) | Promega | Cat#E5011 |
| pcDNA3.1/LRRK2 G2019S-myc-His | Martin et al., 2014b | N/A |
| pcDNA3.1/LRRK2 G2019S/D1994A-myc-His | Martin et al., 2014b | N/A |
| pcDNA3.1/nV5-DEST-RPS15 WT | Martin et al., 2014b | N/A |
| pcDNA3.1/nV5-DEST-RPS15 T136A | Martin et al., 2014b | N/A |
| pcDNA3.1/nV5-DEST-RPS15 T136D | Martin et al., 2014b | N/A |
| Software and Algorithms | ||
| FASTX-Toolkit | Hannon Lab | http://hannonlab.cshl.edu/fastx_toolkit/ |
| Bowtie2 (2.3.4.1) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Tophat (2.1.1) | Trapnell et al., 2009 | https://ccb.jhu.edu/software/tophat/index.shtml |
| ClueGO (2.5.4) | Bindea et al., 2009 | http://apps.cytoscape.org/apps/cluego |
| PANTHER (v14.0) | Mi et al., 2019 | http://www.pantherdb.org |
| Ingenuity Pathway Analysis | QIAGEN | N/A |
| RNAfold (2.1.1) | Lorenz et al., 2011 | http://rna.tbi.univie.ac.at |
| GenomicAlignments | Anders et al., 2015 | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html |
| GenomicFeatures | Anders et al., 2015 | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html |
| DESeq2 (1.22.2) | Anders and Huber, 2010 | http://bioconductor.org/packages/release/bioc/html/DESeq.html |
| fp-framing | Nicholas Ingolia | https://github.com/ingolia-lab/RiboSeq |
| PClamp 10 | Molecular Devices | N/A |
| Clampfit 10 | Molecular Devices | N/A |
Highlights.
G2019S LRRK2 induces translatome alterations in human dopamine neurons
5’UTR secondary structure mediates translational regulation by G2019S LRRK2
Ca2+ homeostasis is disrupted due to aberrant protein synthesis
Intracellular Ca2+ levels are increased in G2019S LRRK2 human dopamine neurons
Acknowledgements:
This work was supported by grants from the NIH P50 NS38377 and the JPB Foundation. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Program M-2014. J.W.K. was supported by Korea Foundation of Advanced Studies. I.M. was supported by NIH/NIA grant K01-01AG050718. L.A-A was supported by a La Caixa Foundation grant. We thank Rachel Green and Mollie Meffert for discussion; Hyesoo Kim for iPSC generation; Joo Heon Shin for deep sequencing; Thomas Gasser and Hans Schöler for isogenic iPSC lines. I-H. Wu created Figure 1D and Graphical Abstract.
Footnotes
Declaration of Interests:
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Abeliovich A, and Gitler AD (2016). Defects in trafficking bridge Parkinson's disease pathology and genetics. Nature 539, 207–216. [DOI] [PubMed] [Google Scholar]
- Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, and Beckmann R (2013). Structures of the human and Drosophila 80S ribosome. Nature 497, 80–85. [DOI] [PubMed] [Google Scholar]
- Bhaskar V, Graff-Meyer A, Schenk AD, Cavadini S, von Loeffelholz O, Natchiar SK, Artus-Revel CG, Hotz HR, Bretones G, Klaholz BP, et al. (2020). Dynamics of uS19 C-Terminal Tail during the Translation Elongation Cycle in Human Ribosomes. Cell Rep 31, 107473. [DOI] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, and Galon J (2009). ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretones G, Álvarez MG, Arango JR, Rodríguez D, Nadeu F, Prado MA, Valdés-Mas R, Puente DA, Paulo JA, Delgado J, et al. (2018). Altered patterns of global protein synthesis and translational fidelity in RPS15-mutated chronic lymphocytic leukemia. Blood 132, 2375–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camandola S, and Mattson MP (2017). Brain metabolism in health, aging, and neurodegeneration. EMBO J 36, 1474–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ 3rd, Steer E, Gusdon AM, Kiselyov K, and Chu CT (2013). Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 182, 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR (2010). The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat Rev Neurosci 11, 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR (2016). Cellular functions of LRRK2 implicate vesicular trafficking pathways in Parkinson's disease. Biochem Soc Trans 44, 1603–1610. [DOI] [PubMed] [Google Scholar]
- Dalal JS, Yang C, Sapkota D, Lake AM, O'Brien DR, and Dougherty JD (2017). Quantitative Nucleotide Level Analysis of Regulation of Translation in Response to Depolarization of Cultured Neural Cells. Front Mol Neurosci 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, and Bezard E (2013). Lysosomal impairment in Parkinson's disease. Mov Disord 28, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR, et al. (2011). Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat Chem Biol 7, 203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, and Greenberg ME (2001). Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333–339. [DOI] [PubMed] [Google Scholar]
- Dominy JE, and Puigserver P (2013). Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, and Greenberg ME (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, Tsuji S, and Obata F (2005). An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol 57, 918–921. [DOI] [PubMed] [Google Scholar]
- Gehrke S, Imai Y, Sokol N, and Lu B (2010). Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 466, 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Suaga P, Luzon-Toro B, Churamani D, Zhang L, Bloor-Young D, Patel S, Woodman PG, Churchill GC, and Hilfiker S (2012). Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet 21, 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, et al. (2006). Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 23, 329–341. [DOI] [PubMed] [Google Scholar]
- Hernandez DG, Reed X, and Singleton AB (2016). Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 139 Suppl 1, 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG, Ivanov IP, and Sonenberg N (2016). Translational control by 5'-untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. (2007). Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 25, 193–205. [DOI] [PubMed] [Google Scholar]
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, and Lu B (2008). Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J 27, 2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairulina J, Graifer D, Bulygin K, Ven'yaminova A, Frolova L, and Karpova G (2010). Eukaryote-specific motif of ribosomal protein S15 neighbors A site codon during elongation and termination of translation. Biochimie 92, 820–825. [DOI] [PubMed] [Google Scholar]
- Khatter H, Myasnikov AG, Natchiar SK, and Klaholz BP (2015). Structure of the human 80S ribosome. Nature 520, 640–645. [DOI] [PubMed] [Google Scholar]
- Korecka JA, Talbot S, Osborn TM, de Leeuw SM, Levy SA, Ferrari EJ, Moskites A, Atkinson E, Jodelka FM, Hinrich AJ, et al. (2019). Neurite Collapse and Altered ER Ca. Stem Cell Reports 12, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 480, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin IB, and Steitz TA (2013). The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 500, 307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol Biol 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzon-Toro B, Rubio de la Torre E, Delgado A, Perez-Tur J, and Hilfiker S (2007). Mechanistic insight into the dominant mode of the Parkinson's disease-associated G2019S LRRK2 mutation. Hum Mol Genet 16, 2031–2039. [DOI] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, and Abeliovich A (2006). The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593. [DOI] [PubMed] [Google Scholar]
- Martin I, Kim JW, Dawson VL, and Dawson TM (2014a). LRRK2 pathobiology in Parkinson's disease. J Neurochem 131, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, Kim JW, Lee BD, Kang HC, Xu JC, Jia H, Stankowski J, Kim MS, Zhong J, Kumar M, et al. (2014b). Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson's disease. Cell 157, 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, and Ingolia NT (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, and Thomas PD (2019). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 14, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG (2008). Oxidative stress and energy crises in neuronal dysfunction. Ann N Y Acad Sci 1147, 53–60. [DOI] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacelli C, Giguere N, Bourque MJ, Levesque M, Slack RS, and Trudeau LE (2015). Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr Biol 25, 2349–2360. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44, 595–600. [DOI] [PubMed] [Google Scholar]
- Penney J, Tsurudome K, Liao EH, Kauwe G, Gray L, Yanagiya A, M RC, Sonenberg N, and Haghighi AP (2016). LRRK2 regulates retrograde synaptic compensation at the Drosophila neuromuscular junction. Nat Commun 7, 12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier C, and Vila M (2012). Mitochondrial biology and Parkinson's disease. Cold Spring Harb Perspect Med 2, a009332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, and Lang AE (2017). Parkinson disease. Nat Rev Dis Primers 3, 17013. [DOI] [PubMed] [Google Scholar]
- Ramsden N, Perrin J, Ren Z, Lee BD, Zinn N, Dawson VL, Tam D, Bova M, Lang M, Drewes G, et al. (2011). Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson's disease-related toxicity in human neurons. ACS Chem Biol 6, 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke F, Smeitink JA, and van der Westhuizen FH (2009). OXPHOS gene expression and control in mitochondrial disorders. Biochim Biophys Acta 1792, 1113–1121. [DOI] [PubMed] [Google Scholar]
- Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, Hoing S, Hargus G, Heck SA, Dhingra A, et al. (2013). Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367. [DOI] [PubMed] [Google Scholar]
- Schwab AJ, and Ebert AD (2015). Neurite Aggregation and Calcium Dysfunction in iPSC-Derived Sensory Neurons with Parkinson's Disease-Related LRRK2 G2019S Mutation. Stem Cell Reports 5, 1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifulin DE, Grosheva AS, Bartuli YS, Malygin AA, Meschaninova MI, Ven'yaminova AG, Stahl J, Graifer DM, and Karpova GG (2015). Molecular contacts of ribose-phosphate backbone of mRNA with human ribosome. Biochim Biophys Acta 1849, 930–939. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, and Ross CA (2006). Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9, 1231–1233. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez J, and Schumacker PT (2012). Physiological phenotype and vulnerability in Parkinson's disease. Cold Spring Harb Perspect Med 2, a009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Schumacker PT, Guzman JD, Ilijic E, Yang B, and Zampese E (2017). Calcium and Parkinson's disease. Biochem Biophys Res Commun 483, 1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, and Dawson TM (2005). Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 102, 16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, et al. (2007). Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 16, 223–232. [DOI] [PubMed] [Google Scholar]
- Xiang G, Pan L, Xing W, Zhang L, Huang L, Yu J, Zhang R, Wu J, Cheng J, and Zhou Y (2007). Identification of activity-dependent gene expression profiles reveals specific subsets of genes induced by different routes of Ca(2+) entry in cultured rat cortical neurons. J Cell Physiol 212, 126–136. [DOI] [PubMed] [Google Scholar]
- Xu JC, Fan J, Wang X, Eacker SM, Kam TI, Chen L, Yin X, Zhu J, Chi Z, Jiang H, et al. (2016). Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci Transl Med 8, 333ra348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap EL, and Greenberg ME (2018). Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100, 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figures 1, 2, and 5. LRRK2 human DA neurons ribosome profiling DESeq2 analysis
Table S2. Related to Figures 1, 2, and 5. RNA-seq and ribosome profiling normalized counts by cell line
Table S3. Related to Figure 2. Gene Ontology and Ingenuity Pathway Analysis results, VGCC 5’UTR and TE table
Data Availability Statement
The high-throughput sequencing data generated in this study have been deposited in the NCBI GEO database under accession number GSE90469. Codes for ribosome profiling analysis are available on Nicholas Ingolia’s Github (https://github.com/ingolia-lab/RiboSeq).