

Abstract
A universal and standardized sample preparation method becomes vital for denaturing top-down proteomics (dTDP) to advance the scale and accuracy of proteoform delineation in complex biological systems. It needs to have high protein recovery, minimum bias, good reproducibility, and compatibility with downstream mass spectrometry (MS) analysis. Here, we employed a lysis buffer containing sodium dodecyl sulfate for extracting proteoforms from cells and, for the first time, compared membrane ultrafiltration (MU), chloroform-methanol precipitation (CMP), and single-spot solid-phase sample preparation using magnetic beads (SP3) for proteoform cleanup for dTDP. The MU method outperformed CMP and SP3 methods, resulting in high and reproducible protein recovery from both Escherichia coli cell (59 ± 3%) and human HepG2 cell (86 ± 5%) samples without a significant bias. Single-shot capillary zone electrophoresis (CZE)-MS/MS analyses of the prepared E. coli and HepG2 cell samples using the MU method identified 821 and 516 proteoforms, respectively. Nearly 30 and 50% of the identified E. coli and HepG2 proteins are membrane proteins. CZE-MS/MS identified 94 histone proteoforms from the HepG2 sample with various post-translational modifications, including acetylation, methylation, and phosphorylation. Our results suggest that combining the SDS-based protein extraction and the MU-based protein cleanup could be a universal sample preparation method for dTDP. The MS raw data have been deposited to the ProteomeXchange Consortium with the data set identifier PXD018248.
Keywords: sample preparation, denaturing top-down proteomics, SDS, membrane ultrafiltration, chloroform-methanol precipitation, SP3, CZE-MS/MS, membrane proteins, histone, PTMs
Graphical Abstract
INTRODUCTION
Denaturing top-down proteomics (dTDP) aims to delineate proteoforms in cells with high throughput.1–3 It is becoming an important tool for gaining a better understanding of protein function in disease and development.3,4 For mass spectrometry (MS)-based dTDP, tremendous efforts have been made in boosting proteoform liquid-phase separation,5–15 improving MS instrumentation,8,16–18 and developing new bioinformatics tools for proteoform identifications (IDs) through database search,19–21 leading to thousands of proteoform IDs from a complex proteome. The Kelleher group integrated three-dimensional (3D) liquid-phase separations (isoelectric focusing (IEF), gel-eluted liquid fraction entrapment electrophoresis (GELFrEE), and reverse-phase liquid chromatography (RPLC)) and a 12 T FT-ICR mass spectrometer for large-scale dTDP of human cells, enabling over 3000 proteoform IDs.5 Anderson et al. showed that coupling 2D GELFrEE-RPLC separation to a 21 T FT-ICR mass spectrometer identified over 3000 proteoforms from human cancer cells.8 The Ge group combined 2D size exclusion chromatography (SEC)-RPLC separation and a Q-TOF mass spectrometer for dTDP, detecting 5000 different proteoforms from heart tissues.9 Our group coupled a 3D SEC-RPLC-capillary zone electrophoresis (CZE) separation to an Orbitrap mass spectrometer for dTDP and identified nearly 6000 proteoforms from Escherichia coli cells.11 The Wu group developed a 2D-RPLC system for high-capacity proteoform separation and identified 2778 proteoforms from HeLa cell lysates.12 The Paŝa-Tolić group developed a high-capacity RPLC system for proteoform separation via using an 80 cm-long RPLC column, enabling 1665 proteoform IDs from bacteria with an Orbitrap mass spectrometer.13 Recently, our group employed a 1.5 m-long capillary for CZE separation of proteoforms and coupling the CZE separation to an Orbitrap mass spectrometer enabled the identification and quantification of thousands of proteoforms from zebrafish brain samples using hundreds of nanograms of protein materials.14
The development of large-scale dTDP underlines the importance of a standardized and universal sample preparation method to achieve comprehensive extraction of proteins from biological samples with high recovery, good reproducibility, minimum bias, and the absence of MS incompatible salts, chaotropes, and detergents. Protein extraction using a cell lysis buffer containing chaotropic agents or detergents and protein sample cleanup before MS with ultrafiltration or precipitation have been suggested as efficient approaches for preparation of protein samples for MS.22 Sodium dodecyl sulfate (SDS) is an extremely efficient detergent for solubilizing and denaturing proteins, making it widely used in proteomics studies for protein extraction.23 However, higher than 0.01% (w/v) SDS can be detrimental to chromatography separation and suppress the ESI.24 Highly efficient depletion of SDS before MS analysis is critical. Multiple methods have been evaluated for SDS removal for bottom-up proteomics and/or dTDP, including membrane ultrafiltration,25 chloroform-methanol precipitation (CMP),26 and single-spot solid-phase sample preparation using magnetic beads (SP3).27,28
Membrane ultrafiltration (MU) has been widely used by the bottom-up proteomics community for the filter-aided sample preparation (FASP) method to remove SDS before enzymatic digestion of proteins.25 Basically, a protein sample in 1–5% (w/v) SDS solution is loaded onto a commercialized membrane filter unit with a 10–30 kDa molecular weight cutoff (MWCO), followed by washing with an 8 M urea solution to remove SDS, which is based on the fact that 8 M urea can destroy the hydrophobic interaction between SDS and proteins. MU has also been routinely deployed for buffer exchange for TDP sample preparation.22 CMP is a well-recognized method for removing SDS from proteins in the dTDP workflow, and the Kelleher group has utilized CMP for cleaning the protein samples after GELFrEE fractionation in their large-scale dTDP works.5,6,8 Briefly, a protein sample dissolved in an SDS solution is mixed with methanol, chloroform, and water. After centrifugation, three phases form and the proteins precipitate at the interphase. After removing the upper phase, more methanol is added and the purified protein pellet is obtained after centrifugation. SP3 has been suggested as an efficient sample preparation method for bottom-up proteomics and various detergents can be removed from proteins using the SP3 method.27,28 Recently, the Webb group evaluated the SP3 method for preparing intact protein samples for dTDP, demonstrating the great potential of the SP3 method as a universal sample preparation method for both bottom-up proteomics and dTDP.29 For SP3, a protein sample in an SDS buffer was mixed with magnetic beads and acetonitrile (ACN). Under a high concentration of ACN, proteins are adsorbed on the beads. Then, the beads are washed with organic solvents (i.e., ethanol and ACN) to clean up the proteins followed by on-bead digestion for bottom-up proteomics27,28 or recovering proteins from beads with cold 80% (v/v) formic acid for dTDP.29
In this work, for the first time, we compared MU with a 30 kDa MWCO membrane, CMP, and SP3 methods for cleaning up proteins extracted from E. coli cells using 1% (w/v) SDS for dTDP. The MU method showed the best results regarding the protein recovery and compatibility with the follow-up MS analysis. We further tested the MU method for human cells (HepG2). We analyzed the prepared E. coli and HepG2 samples using our CZE-MS/MS system. Our data demonstrated that coupling the SDS-based protein extraction with the MU-based sample cleanup could be a universal sample preparation method for dTDP with high protein recovery, no significant protein bias, good reproducibility, and great compatibility with follow-up MS analysis.
EXPERIMENTAL SECTION
Details of materials and reagents are listed in Supporting Information I.
Protein Extraction from E. coli and HepG2 Cells
E. coli (strain K-12 substrain MG1655) was cultured in the LB (Luria-Bertani) medium at 37 °C until OD600 reached 0.7. The E. coli cells were harvested by centrifugation at 4000 rpm for 10 min. The cell pellet was washed with PBS (phosphate-buffered saline) buffer three times to remove the leftover culture medium. After that, 400 μL of a lysis buffer containing 1% (w/v) SDS, 100 mM NH4HCO3, protease inhibitors, and phosphatase inhibitors (pH 8.0) was added into an Eppendorf tube containing the E. coli cells. The cells were pipetted up and down a couple of times and lysed by ultrasonication (Branson Sonifier 250, VWR Scientific, Batavia, IL) on ice for 10 min. After cell lysis, the cell lysates were then centrifuged at 14,000g for 5 min. After that, the protein concentration of the supernatant was measured with the BCA (bicinchoninic acid) assay. The supernatant was then aliquoted into 100 μg/tube (4 mg/mL protein concentration) and stored at −80 °C before use. The cultured HepG2 cells were kindly provided by Prof. David Lubman at the Department of Surgery Research of University of Michigan. After cell culture, the HepG2 cells were harvested through centrifugation at 100g for 5 min and were washed with the PBS buffer three times. The cell lysis protocol was the same as the E. coli cells described above. After the BCA assay for protein concentration measurement, the extracted proteins were aliquoted into 100 μg/tube (4 mg/mL protein concentration) and stored at −80 °C before use.
Protein Sample Cleanup with Various Methods before MS Analysis
SP3 Method.
The SP3 procedure was performed according to the literature with some modifications.27,28 The two types of carboxylate-modified paramagnetic beads (10, 100, and 500 μg) were added into 100 μg of E. coli protein extract followed by addition of acetonitrile (ACN), ensuring ACN concentration higher than 70% (v/v). The E. coli protein extract was incubated in the presence of magnetic beads and ACN for 18 min at room temperature and then was placed on a magnet for 2 min. The supernatant was taken out and dried, and the protein concentration was measured through the BCA assay. Ethanol (200 μL) was used to rinse the beads twice, and 200 μL of ACN was used to rinse the beads once. A 60 μL solution of 100 mM NH4HCO3 (pH 8) was then added into the bead solution, and the resulting solution was sonicated for 10 min. The solution was then placed on a hotplate at 95 °C for 15 min. The supernatant containing proteins was taken out and the protein concentration was measured with the BCA assay. The SP3 method was also applied on the HepG2 cell lysate with the same procedure.
CMP Method.
The CMP procedure was processed based on the literature.26 Briefly, 400 μL of methanol, 100 μL of chloroform, and 300 μL of water were added into 100 μg of E. coli cell lysate (1 μg/μL, 1% (w/v) SDS) successively. Every addition of the reagent was followed by a thorough vortex. The mixture was then centrifuged at 14,000g for 1 min. The solution separated into three layers after centrifugation. The top aqueous layer was carefully removed without disturbing the protein flake. Methanol (400 μL) was then added into the solution followed by a thorough vortex. The mixture was then centrifuged at 20,000g for 5 min. The supernatant was removed. The protein pellet was suspended in a 50 μL buffer containing 100 mM NH4HCO3 (pH 8) with or without 1% (w/v) SDS with gentle pipetting. We also vortexed and sonicated the sample solution gently for a short period of time to improve the protein recovery. After centrifugation, the protein solution was analyzed by the BCA assay to determine the protein concentration.
MU Method.
A 100 μL solution of 8 M urea in 100 mM NH4HCO3 (pH 8) was first added into 100 μg of E. coli cell lysate, producing a protein solution with an ~0.80 mg/mL protein concentration. The mixture was then loaded onto a membrane filtration unit (30 kDa MWCO membrane). The filtration unit was centrifuged at 14,000g to make sure that all the solution went through the membrane. The membrane was then washed with 100 μL of 8 M urea in 100 mM NH4HCO3 twice followed by membrane washing with 100 μL of 100 mM NH4HCO3 three times. After the washing, 50 μL of 100 mM NH4HCO3 was loaded onto the membrane followed by pipetting up and down a few times. The filtration unit was then vortexed for 5 min and flipped over followed by a quick spin-down to recover the proteins from the membrane. The protein concentration in the collected solution was measured through the BCA assay. The same procedure was utilized for the HepG2 cell lysate.
SDS-PAGE and CZE-MS/MS Analysis
The E. coli and HepG2 cell lysates before and after cleanup with the three methods were analyzed by SDS-PAGE according to the procedure in the literature.30
For CZE-MS/MS, a linear polyacrylamide (LPA)-coated capillary (50/360 μm i.d./o.d.) with one end etched by hydrofluoric acid was used for CZE separation.31–33 The commercialized electrokinetically pumped sheath flow CE-MS interface (EMASS II, CMP scientific, Brooklyn, NY) was used to couple CZE to MS.34,35 The automated CZE operations were implemented with an ECE-001 autosampler (CMP scientific). A Q-Exactive HF mass spectrometer (Thermo Fisher Scientific) was used for all CZE-MS/MS analyses. A data-dependent acquisition (DDA) method was employed. The details of SDS-PAGE and CZE-MS/MS analysis are described in Supporting Information I.
Data Analysis
The TopPIC (top-down mass spectrometry-based proteoform identification and characterization) software was applied for proteoform IDs via database search for all E. coli and HepG2 data.19 Briefly, the RAW files were converted into mzML files using the msconvert tool.36 The mzML files were then processed by the TopFD (Top-down mass spectrometry feature detection) tool for spectral deconvolution. The resulted msalign files were then processed by TopPIC (v1.3.1) for database searching. UniProt databases of E. coli (UP000000625) and Human (UP000005640) were used for search. For the database search, the maximum number of mass shifts was 1. All other parameters were kept as default. The target-decoy approach was employed to evaluate the false discovery rate (FDR) of proteoform spectrum match (PrSM) and proteoform IDs.37,38 The database search results were filtered with a 1% PrSM-level FDR and a 5% proteoform-level FDR. The proteoforms identified from E. coli and HepG2 cells are listed in Supporting Information II. The MS raw data have been deposited to the ProteomeXchange Consortium via the PRIDE39 partner repository with the data set identifier PXD018248.
The Retrieve/ID mapping tool from the UniProt was used for Gene Ontology (GO) analysis. Grand average of hydropathy (GRAVY) values of proteoforms were calculated through a GRAVY calculator (http://www.gravy-calculator.de/). Positive GRAVY values suggest hydrophobic, and negative values indicate hydrophilic. The transmembrane domains (TMDs) of identified membrane proteins were predicted using the TMHMM software (http://www.cbs.dtu.dk/services/TMHMM/).
RESULTS AND DISCUSSION
Comparison of MU, CMP, and SP3 Methods for Cleanup of Cell Lysates Containing SDS before MS
SDS has been widely used in proteomics studies to facilitate protein extraction from cells and protein solubilization. However, a trace amount of SDS could be detrimental to downstream processes such as enzymatic digestion in bottom-up proteomics, chromatographic separation, and MS detection.24,40 It is vital to remove SDS from cell lysates before top-down MS analysis. MU, CMP, and SP3 methods have been used in dTDP for removing detergents (e.g., SDS) from proteins.5,6,8,22,29 Here, for the first time, we compared the MU, CMP, and SP3 methods for preparation of E. coli and human (HepG2) cell lysates containing 1% (w/v) SDS for dTDP regarding protein recovery and protein bias. For each method, 100 μg of proteins dissolved in 1% (w/v) SDS was used as the starting material. The BCA assay and SDS-PAGE were used to evaluate the performance of the three methods. To make the sample preparation method compatible with follow-up dynamic pH junction-based CZE-MS/MS analysis,41 100 mM NH4HCO3 (pH 8.0) was used to redissolve the proteins after removing SDS with the three methods.
For the SP3 method, we first tested the loading capacity of magnetic beads by incubating 100 μg of E. coli proteins with three different amounts of magnetic beads, 10, 100, and 500 μg. The protein recovery based on the BCA assay was ~60% and had no obvious difference among the three different bead amounts, Figure 1A. We also measured the amount of proteins that were not bound to the magnetic beads at the first step with the BCA assay, Figure 1B. The unbound protein amount was ~5 μg, indicating that the magnetic beads captured proteins with high efficiency. Considering the recovered proteins (~60 μg) and unbound proteins (~ 5 μg), we noted that ~35% of the loaded proteins were lost somewhere during the SP3 process. We speculated that those proteins were still adsorbed on the magnetic beads and were not eluted by the 100 mM NH4HCO3 (pH 8.0) buffer. We further analyzed the proteins prepared by the SP3 method with the three different bead amounts using SDS-PAGE, Figure S1 in Supporting Information I. The three E. coli protein samples after the SP3 cleanup show no significant difference regarding the molecular weight (MW) distributions. The results indicate that 10 μg of magnetic beads is good enough to prepare 100 μg of proteins from a complex proteome, which agrees well with the data in the literature.27,28 We utilized 10 μg of beads for all the following SP3 experiments. We also noted that the SP3 method-based sample cleanup introduced an obvious bias against large proteins (higher than 50 kDa) compared to the sample before cleanup, Figure S1. The bias was also observed in the HepG2 human cell lysate processed by the SP3 method, Figure S2. We used 100 mM NH4HCO3 (pH 8.0) to extract the proteins from the beads in order to make the method compatible with follow-up CZE-MS/MS analysis, which might lead to relatively low efficiency of redissolving large proteins, because it has been suggested that a buffer containing detergents is essential for completely extracting proteins bound to beads in SP3.27–29
Figure 1.
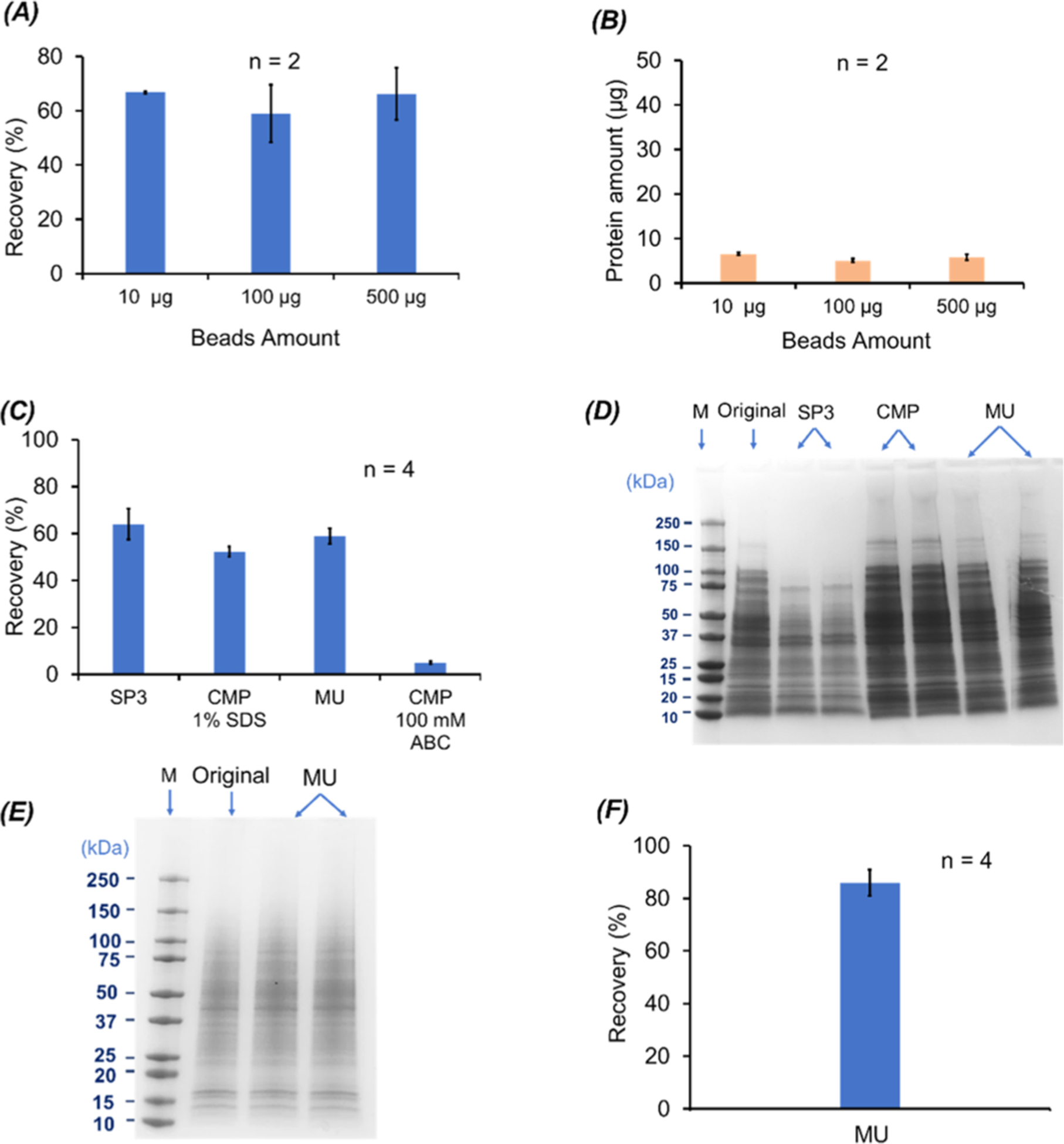
BCA and SDS-PAGE results on the (A–D) E. coli cell proteins and (E, F) HepG2 cell proteins when different SDS removal methods were applied. (A) Protein recovery (%) of the SP3 method for 100 μg of E. coli proteins when different amounts of magnetic beads were used (n = 2). (B) Amounts of unbound proteins to magnetic beads as a function of the magnetic bead amount (n = 2). (C) Protein recovery (%) of the SP3, CMP, and MU methods. The protein pellets from the CMP method were dissolved in 100 mM NH4HCO3 (ABC is short for ammonium bicarbonate) (pH 8) with or without 1% (w/v) SDS (n = 4). (D) SDS-PAGE data of the recovered E. coli proteins using the SP3, CMP, and MU methods (n = 2) as well as the E. coli cell lysate in 1% (w/v) SDS before sample cleanup (original). For the CMP method, the protein pellet dissolved in 100 mM NH4HCO3 (pH 8) with 1% (w/v) SDS was used for the analysis. For each sample, an aliquot of 10 μg of proteins was loaded for SDS-PAGE. (E) SDS-PAGE data of the HepG2 cell protein samples before (original) and after sample cleanup with the MU method (n = 2). For each sample, an aliquot of 6 μg of proteins was loaded for SDS-PAGE. (F) Protein recovery data of the HepG2 cell samples after the MU method-based sample cleanup (n = 4). The error bars in the figures represent the standard deviations of protein recovery or protein amount.
We then employed the MU, CMP, and SP3 methods for preparing aliquots of the E. coli cell lysate dissolved in 1% (w/v) SDS. Each aliquot contained 100 μg of proteins, and four aliquots were prepared by each method. The MU and SP3 methods generated much higher protein recovery than the CMP method (~60% vs 5%) with good reproducibility (RSD < 12%) when a solution containing 100 mM NH4HCO3 (pH 8.0) was used to redissolve the protein pellet from CMP, Figure 1C. We noted that the protein pellet from CMP was hard to be dissolved in the NH4HCO3 buffer, which resulted in a low protein recovery. We further tried to use a buffer containing 1% (w/v) SDS and 100 mM NH4HCO3 (pH 8.0) to redissolve the protein pellet and obtained a 50% protein recovery with high precision (RSD, 4%). We then analyzed the E. coli cell lysates before and after cleanup using the three methods by SDS-PAGE, Figure 1D. For the CMP method, we used the protein sample redissolved in the 1% (w/v) SDS solution for SDS-PAGE. Two batches of prepared samples with the three methods were analyzed. The MU and CMP method show comparable protein MW distributions, which are similar to the original E. coli sample without cleanup. As we discussed before, the SP3 method had trouble recovering large proteins with the 100 mM NH4HCO3 (pH 8.0) buffer. All the three methods show good reproducibility regarding the SDS-PAGE data. Based on the discussed protein recovery, protein bias, and compatibility with the CZE-MS/MS analysis of the three sample cleanup methods, the MU method outperformed the CMP and SP3 methods. We further employed the MU method for preparation of the HepG2 cell lysate in 1% (w/v) SDS. The SDS-PAGE and BCA assay data clearly show that the MU method can achieve reproducible preparation of the human cell lysate with high protein recovery and precision (86 ± 5%), Figure 1E,F. All the results demonstrate that the MU method could be a universal method for sample preparation in dTDP of complex proteomes. We obtained a higher protein recovery for the human cell lysate than the E. coli cell lysate (86% vs 60%) using the MU method, presumably due to the fact that E. coli proteins tend to be smaller than human proteins in the length range of 1–250 amino acids based on the data in Swiss-Prot database, Figure S3, resulting in a higher chance for protein to flow through the membrane (30 kDa MWCO) for the E. coli sample.
We also noted that for the MU method, when the centrifugal force is too high (i.e., 16,800g), the protein recovery can be reduced drastically compared to the typical centrifugal force (14,000g) used in the procedure (33% vs 86%), possibly due to membrane clogging by proteins or impurities in the extraction solution. We suggest a pre-centrifugation operation for protein samples to remove any precipitate before the MU procedure, which will ensure the straightforward MU operations and good protein recovery.
Coupling SDS-Based Protein Extraction and MU-Based Sample Cleanup to CZE-MS/MS for dTDP
We further coupled the SDS-based protein extraction and the MU-based protein sample cleanup to our dynamic pH junction-based CZE-MS/MS for dTDP of E. coli cells. Approximately 500 nL of the E. coli protein solution in 100 mM NH4HCO3 (pH 8.0) after cleanup was injected into the CZE capillary for analysis. The injected protein amount was roughly 400 ng. The BGE of CZE was 20% (v/v) acetic acid. We performed CZE-MS/MS analysis of two batches of the E. coli sample prepared by the MU method. Figure 2A shows the base peak electropherograms of the two E. coli samples, and Figure 2B shows the numbers of identified proteins, proteoforms, and PrSMs. The whole workflow shows good reproducibility regarding the CZE separation profile, base peak intensity (Table S1), and identifications. Single-shot CZE-MS/MS identified 832 ± 65 proteoforms (n = 2) with a 5% proteoform-level FDR. When we used a 1% proteoform-level FDR, 821 ± 67 (n = 2) proteoforms corresponding to 219 ± 21 proteins were identified in a single CZE-MS/MS run, Figure 2B. On average, ~20 fragment ions were matched to each identified proteoform, Figure 2C, suggesting the high confidence of the proteoform identifications. We noted that mass of identified proteoforms ranged from 1 to 25 kDa and over 70% of the identified proteoforms had a mass smaller than 6 kDa. We also analyzed the GO information of the identified proteins, Figure 2D, and ~30% of the proteins were membrane proteins. We finally analyzed the hydrophobicity of the identified proteoforms and compared it with our previous work in which 8 M urea was used for protein extraction from E. coli cells.41 As shown in Figure 2E, the E. coli proteoforms identified in this work show higher hydrophobicity than the ones identified in our previous work, most likely due to the fact that SDS has stronger solubility for hydrophobic proteins than 8 M urea. We also noted that compared to the protein samples extracted with 8 M urea,41 the samples from the 1% (w/v) SDS extraction required a higher acetic acid concentration in the BGE of CZE (20% vs 5% (v/v) acetic acid) to achieve reproducible CZE separations, which might be due to the higher hydrophobicity of proteoforms from the 1% (w/v) SDS extraction. We need to point out that when a high amount of acetic acid (i.e., 20%) is used as the BGE for CZE separation, the sample dissolved in the NH4HCO3 buffer in a sample vial could be acidified by the BGE during the sample injection process, which will influence the dynamic pH junction sample stacking obviously. When the sample volume is small (i.e., <5 μL), the issue becomes severe. Immersing the sample injection end of the capillary in a 100 mM NH4HCO3 buffer for seconds before moving it into the sample vial for sample injection can eliminate the issue based on our experience.
Figure 2.
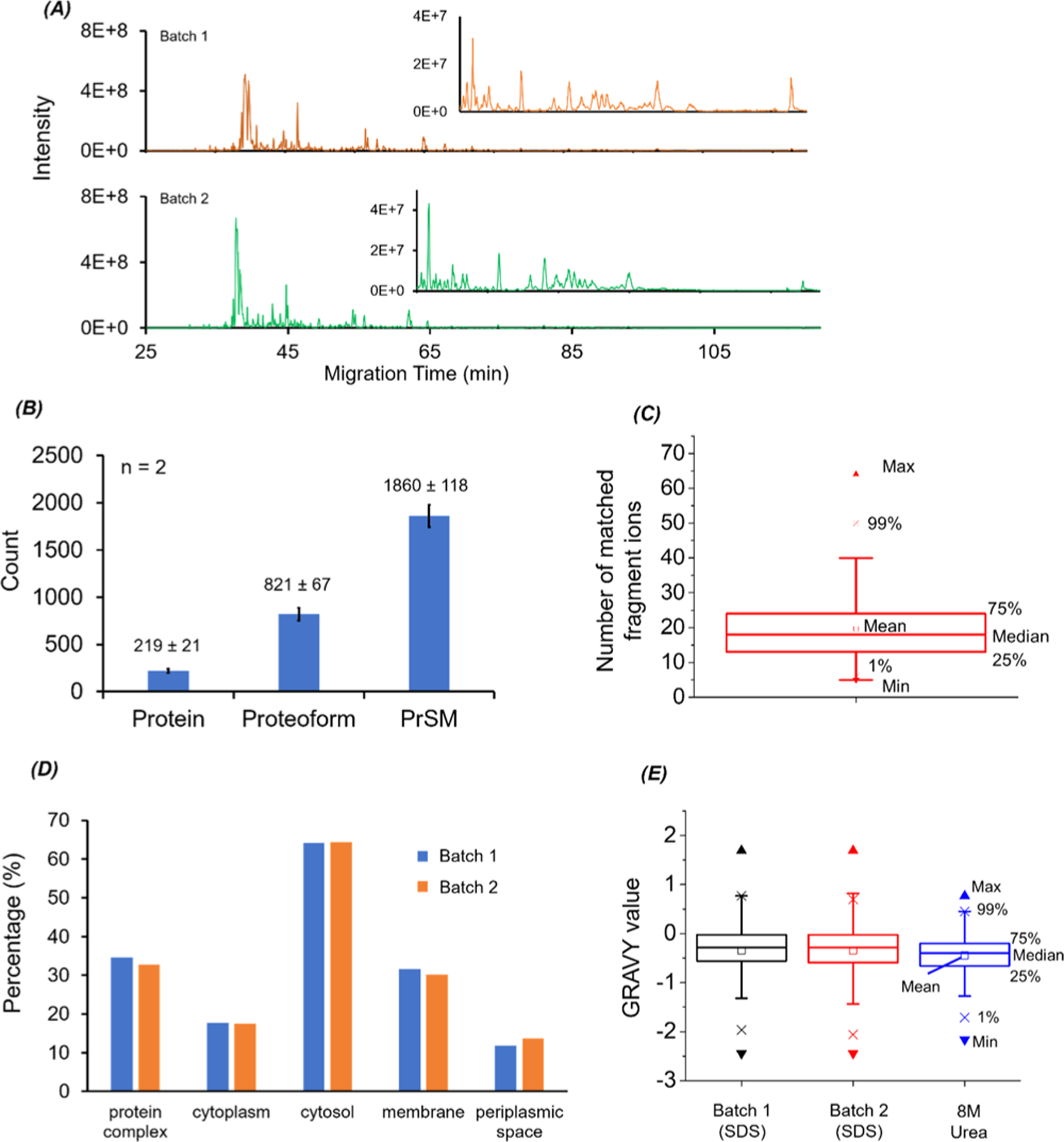
CZE-MS/MS data of E. coli samples prepared with the MU method. (A) Base peak electropherograms of two batches of prepared E. coli protein samples after CZE-MS/MS analysis. (B) Numbers of proteins, proteoforms, and PrSM identifications from the two CZE-MS/MS runs. The error bars represent the standard deviations of the number of identifications. (C) Box chart of the number of matched fragment ions of identified E. coli proteoforms. (D) Gene Ontology cellular component analysis of identified E. coli proteins from the two CZE-MS/MS analyses. (E) Box charts of GRAVY values of the identified proteoforms from the two CZE-MS/MS analyses in this work (SDS-batch 1 and SDS-batch 2) and from our previous work in ref 41 (8 M urea).
We also analyzed the HepG2 cell proteins prepared by the MU method using our dynamic pH junction-based CZE-MS/MS. The same CZE and MS conditions as those for the E. coli samples were used here except that we employed 40% (v/v) acetic acid as the BGE of CZE due to the much higher complexity of the human cell line sample compared to the E. coli sample. The CZE-MS/MS identified 534 proteoforms and 248 proteins in a single run with a 5% proteoform-level FDR. When a 1% proteoform-level FDR was used, 516 proteoforms corresponding to 241 proteins were identified. Figure 3A shows the base peak electropherogram of the CZE-MS/MS run. The mass of identified proteoforms ranged from ~1 kDa to roughly 24 kDa, Figure 3B. Over 200 proteoforms had a mass higher than 10 kDa. Out of the 248 identified proteins, 125 proteins are membrane proteins, 112 proteins are located in the nucleus, and 22 proteins belong to chromatin according to the information from the UniProt Knowledgebase (https://www.uniprot.org/). Sequences and fragmentation patterns of two transmembrane proteins (6.8 kDa mitochondrial proteolipid and cytochrome c oxidase subunit 6A1, mitochondrial) are shown in Figures 3C,D. The two membrane proteins were identified with high confidence, and the TMDs were cleaved reasonably well in the gas phase by HCD. Figure 3E,F shows the mass spectrum and fragmentation pattern of one proteoform of C1QBP (complement component 1 Q subcomponent-binding protein, mitochondrial) having a mass of 23767.7 Da. The proteoform had clear signals in the mass spectrum and was identified by MS/MS through the database search with 18 matched fragment ions and a 2.53 × 10−11 E value. An N-terminal truncation was determined for the proteoform.
Figure 3.
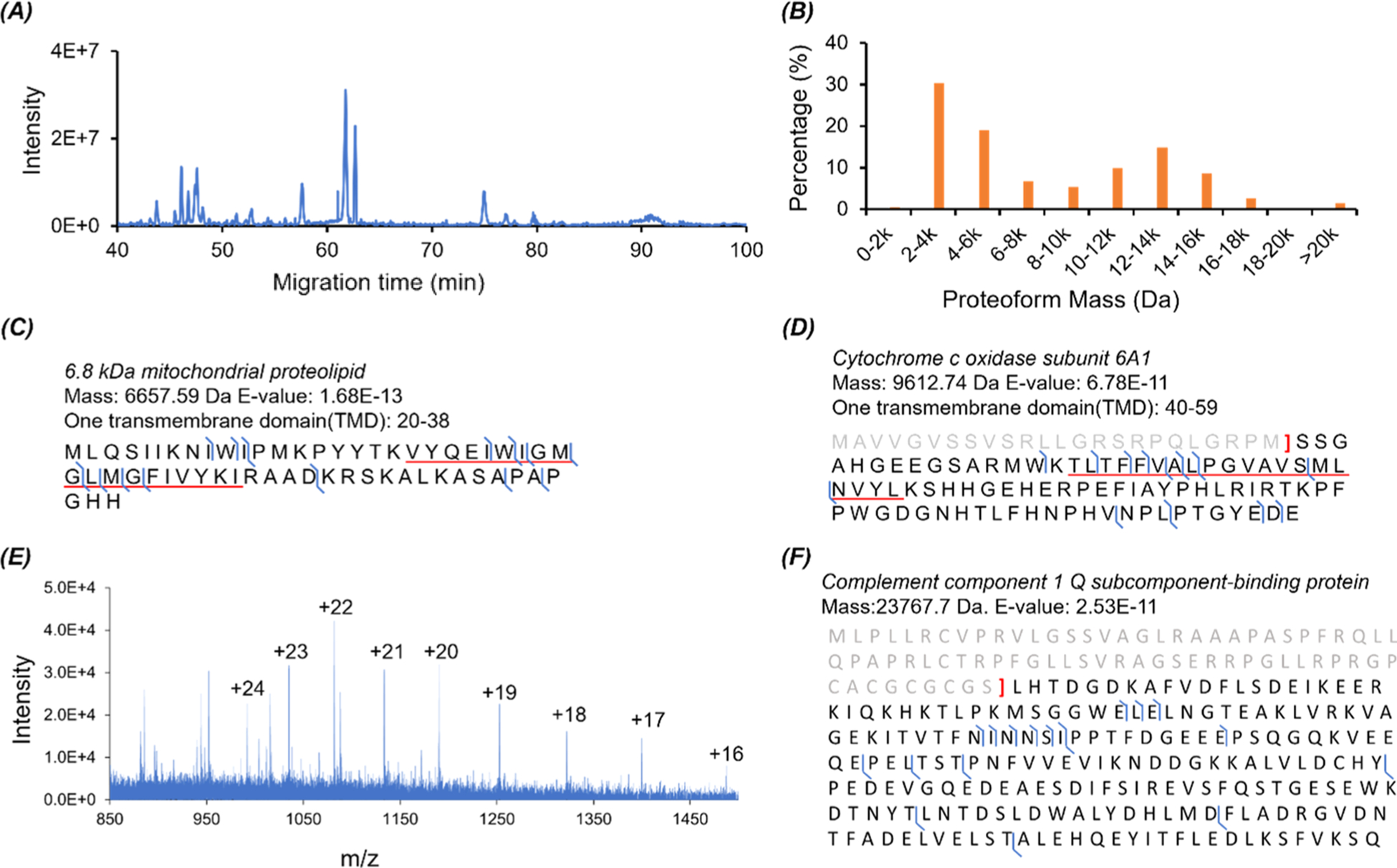
CZE-MS/MS data of the HepG2 cell protein sample prepared with the MU method. (A) Base peak electropherogram of the protein sample after CZE-MS/MS analysis. (B) Mass distribution of the identified proteoforms from the HepG2 protein sample. (C, D) Sequences and fragmentation patterns of two transmembrane proteins with one TMD. The regions corresponding to TMDs are underlined. (E) Mass spectrum of the identified proteoform of C1QBP (Complement component 1 Q subcomponent-binding protein, mitochondrial) with a mass of 23767.7 Da. (F) Sequence and fragmentation pattern of the C1QBP proteoform in (E).
The CZE-MS/MS data further indicate that the sample preparation procedure (SDS-based protein extraction and MU-based sample cleanup) is efficient for extraction and preparation of proteins including membrane proteins from bacterial and human cells. The sample preparation procedure should be also compatible with widely used RPLC-MS/MS, although we only used CZE-MS/MS in this work.
Proteoforms with Post-translational Modifications (PTMs)
We also performed another CZE-MS/MS run of the prepared E. coli sample from the MU method under very clean CZE and MS conditions to pursue a higher number of proteoform identifications, leading to an identification of 1336 proteoforms corresponding to 301 proteins with a 1% proteoform-level FDR. Various protein modifications were detected, including but not limited to N-terminal methionine removal, N-terminal truncation, N-terminal acetylation, and disulfide bond, Figure 4A. Two truncated proteoforms of 50S ribosomal protein L7/L12 at the N-terminus with or without lysine methylation are shown in Figures 4B,C. The fragmentation patterns show extensive backbone cleavages of the two proteoforms. We also observed that the abundance of the methylated proteoform was ~50% of the non-methylated proteoform according to the mass spectrum in Figure 4D. The methylation at Lys-82 detected in our work agrees well with the data in the literature.42 We identified 15 proteoforms with one or two disulfide bonds, and those proteoforms are listed in Supporting Information II. Sequences and fragmentation patterns of two proteoforms with one and two disulfide bonds are shown in Figure 4E,F. Interestingly, for Figure 4E, the location of the disulfide bond was previously reported as the zinc ion binding position.43 For the 50S ribosomal protein L31, the literature data suggested that C16 was responsible for zinc ion binding, but our data show that C16, C18, C37, and C40 form two disulfide bonds, Figure 4F. The disulfide bonds might form endogenously or develop after cell lysis due to the loss of zinc ions during sample preparation.
Figure 4.
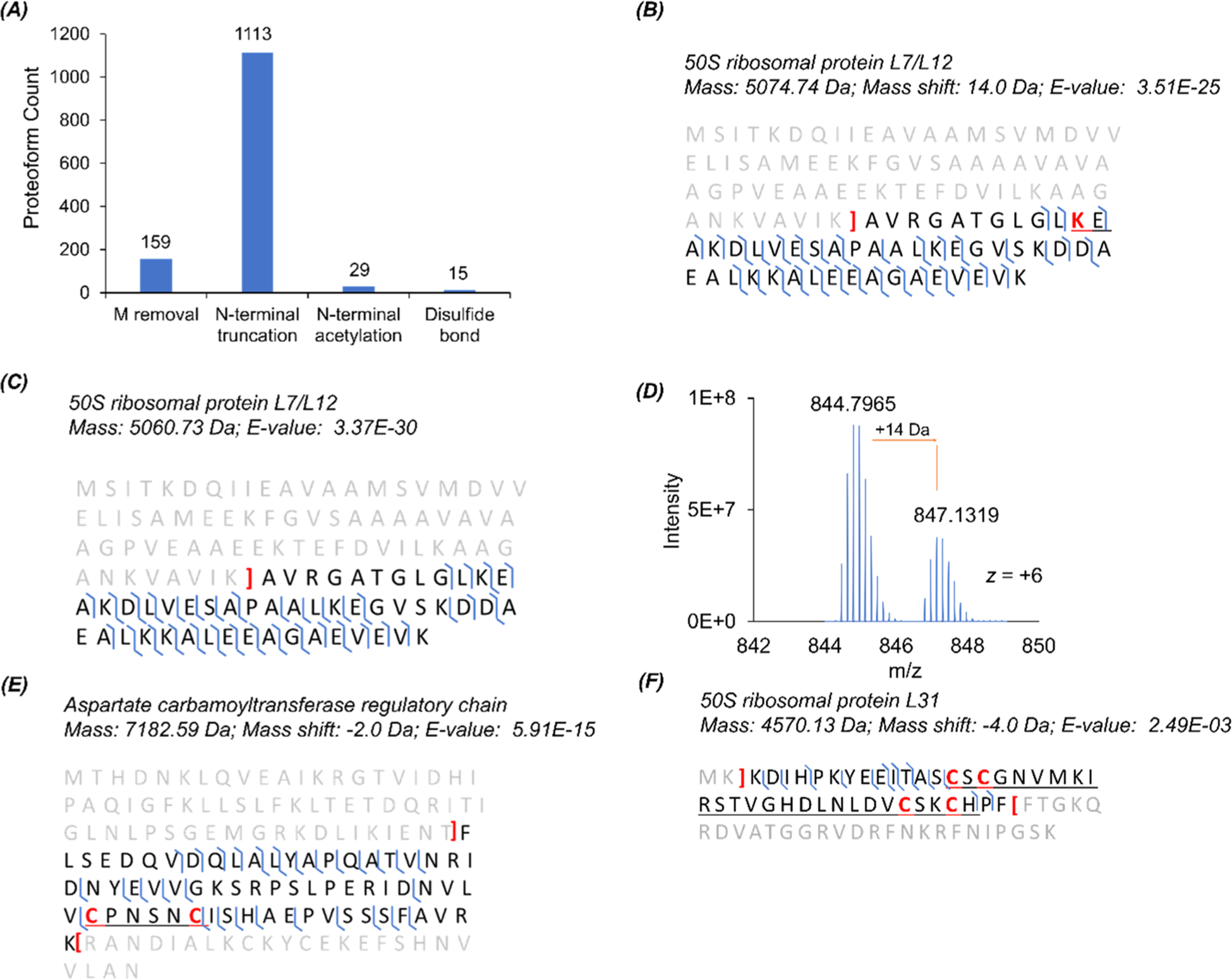
CZE-MS/MS data of the E. coli sample regarding PTMs. (A) Distribution of some modifications on the identified proteoforms. (B) Sequence and fragmentation pattern of the 50S ribosomal protein L7/L12 proteoform with one methylation at the marked lysine residue and N-terminal truncation. (C) Sequence and fragmentation pattern of the 50S ribosomal protein L7/L12 proteoform with only N-terminal truncation. (D) Mass spectrum of one charge state (+6) of the 50S ribosomal protein L7/L12 proteoforms in (B) and (C). (E) Sequence and fragmentation pattern of one proteoform of the aspartate carbamoyltransferase regulatory chain with one disulfide bond between the two marked cysteine residues and truncations at the termini. (F) Sequence and fragmentation pattern of one proteoform of 50S ribosomal protein L31 with two disulfide bonds among the four marked cysteine residues, removal of two amino acid residues at the N-terminus, and truncation at the C-terminus.
We identified proteoforms with various PTMs in the HepG2 data, including but not limited to N-terminal acetylation (205), phosphorylation (11), and disulfide bonds (8), Figure 5A. The proteoforms with phosphorylation and disulfide bonds are listed in Supporting Information II. We identified one proteoform of programmed cell death protein 5 with N-terminal acetylation and one serine phosphorylation (Figure 5B), one proteoform of 60S acidic ribosomal protein P2 with two serine phosphorylations (Figure 5C), and one proteoform of small ubiquitin-related modifier 1 with both acetylation and phosphorylation at the N-terminal serine residue (Figure 5D). The PTM information of these three proteoforms matches well with the UniProt Knowledgebase (https://www.uniprot.org/). We noted that the three serine residues marked in red in the underlined region in Figure 5C could be phosphorylated according to the UniProt Knowledgebase, and our data show that only two of them are actually phosphorylated in the proteoform. We also identified one proteoform of 60S ribosomal protein L32 with one disulfide bond, Figure S4, which is not reported in the literature according to the UniProt Knowledgebase. Prothymosin alpha (PTMA) is a histone-binding protein, and it can regulate gene transcription.44 Prothymosin alpha has eight phosphorylation sites according to the UniProt Knowledgebase. Our data revealed one phosphorylation site (mass shift of 79.97 Da) in the underlined region (S85 or T87) in Figure S5A, which is not reported previously. We also compared the relative abundance of the identified phosphorylated proteoform of PTMA and the corresponding unphosphorylated proteoform based on the extracted base peak electropherogram, Figure S5B. The unphosphorylated proteoform had ~5-times higher abundance than the phosphorylated one. Additionally, CZE separated the phosphorylated and unphosphorylated proteoforms very well with an 8 min difference in migration time and the phosphorylated one migrated obviously slower than the unphosphorylated one in CZE due to the charge reduction from the phosphorylation, which agrees well with the previous reports.45–48 The migration time shift between unphosphorylated and phosphorylated proteoforms provides additional evidence for the phosphorylation PTM. Figure S5C,D shows mass spectra of the unphosphorylated and phosphorylated proteoforms, indicating a difference between them regarding charge distribution. We speculate that the phosphorylation could influence the ESI of prothymosin alpha to some extent.
Figure 5.
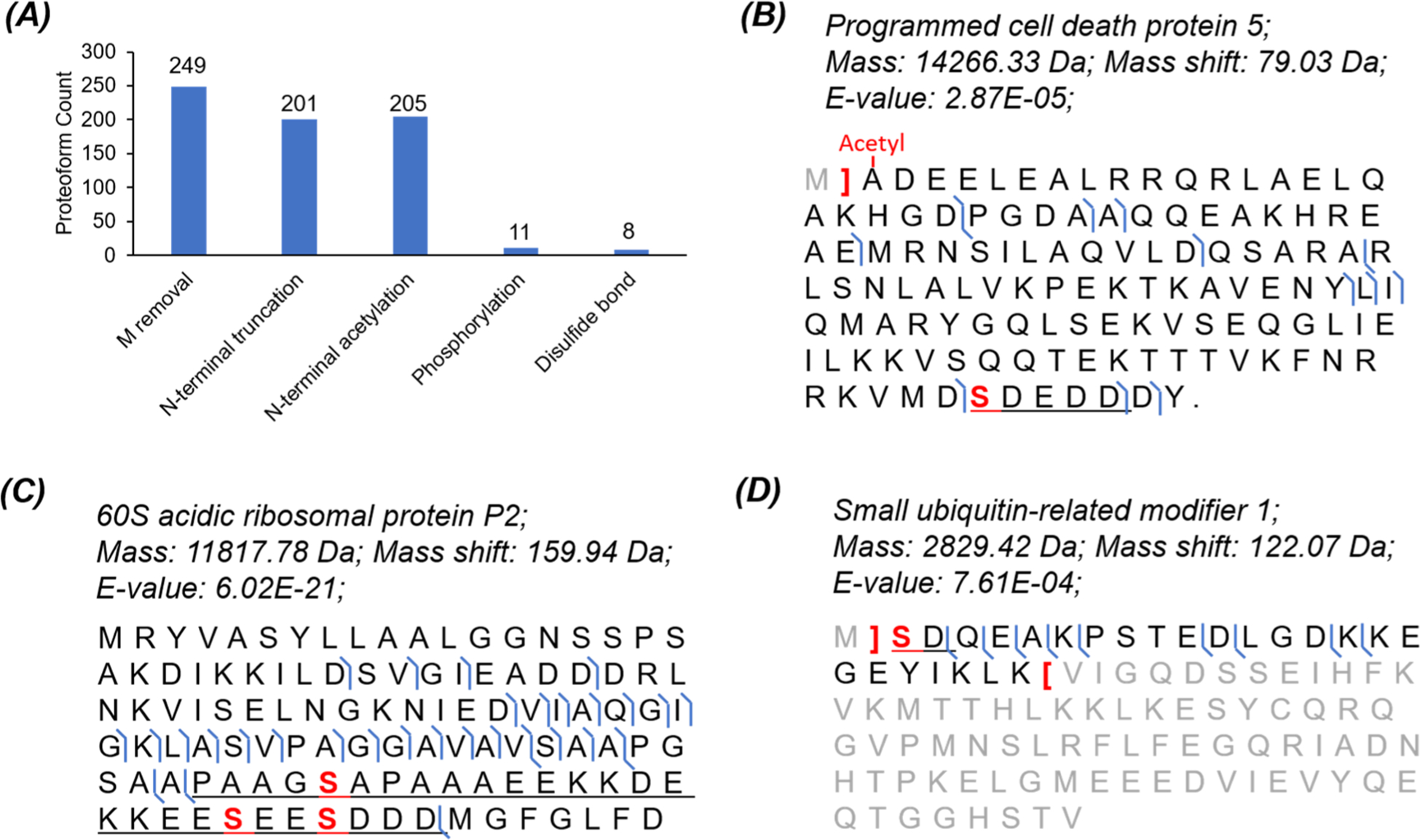
CZE-MS/MS data of the HepG2 sample regarding PTMs. (A) Distribution of some modifications on the identified proteoforms. Sequences and fragmentation patterns of some proteoforms (B) with one phosphorylation site and N-terminal acetylation, (C) with two phosphorylation sites, and (D) with phosphorylation and acetylation on the N-terminal serine residue.
Histone PTMs are extremely important for regulating gene expression, and dTDP is an invaluable approach for delineating the histone code in a proteoform-specific manner.49–52 In this work, we identified 94 histone proteoforms from the HepG2 sample in a single CZE-MS/MS run without any histone purification. The histone proteoforms are listed in Supporting Information II. The 94 histone proteoforms covered the five major histone variants, H1 (11), H2A (39), H2B (36), H3 (1), and H4 (7), Figure 6A. We observed various PTMs on the histone proteoforms, including acetylation, methylation, and phosphorylation, Figure 6B–F. Sequences and fragmentation patterns of two histone H4 proteoforms are shown in Figure 6B,C. We observed both N-terminal acetylation and a 28 Da mass shift, most likely corresponding to two methylations within the underlined region in the two proteoforms. Due to the limited backbone cleavage coverages for the two proteoforms, it is difficult to localize the methylation PTM. Interestingly, there are no literature reports about methylation or dimethylation PTM in the two regions of histone H4 underlined in Figure 6B,C according to the UniProt Knowledgebase. We also identified one histone H4 proteoform with a 337 Da mass shift, Figure 6D. The mass shift corresponds to a region with four lysine residues (K6, K9, K13, and K17). According to the UniProt Knowledgebase, these four lysine residues could have acetylation (+42 Da), propionylation (+56 Da), crotonylation (+68 Da), butyrylation (+70 Da), succinylation (+100 Da), and glutarylation (+114 Da). We speculate that the 337 Da mass shift is most likely produced by a combination of these various PTMs. The data further suggest the importance of improving the backbone cleavage coverage for comprehensive characterization of proteoforms.
Figure 6.
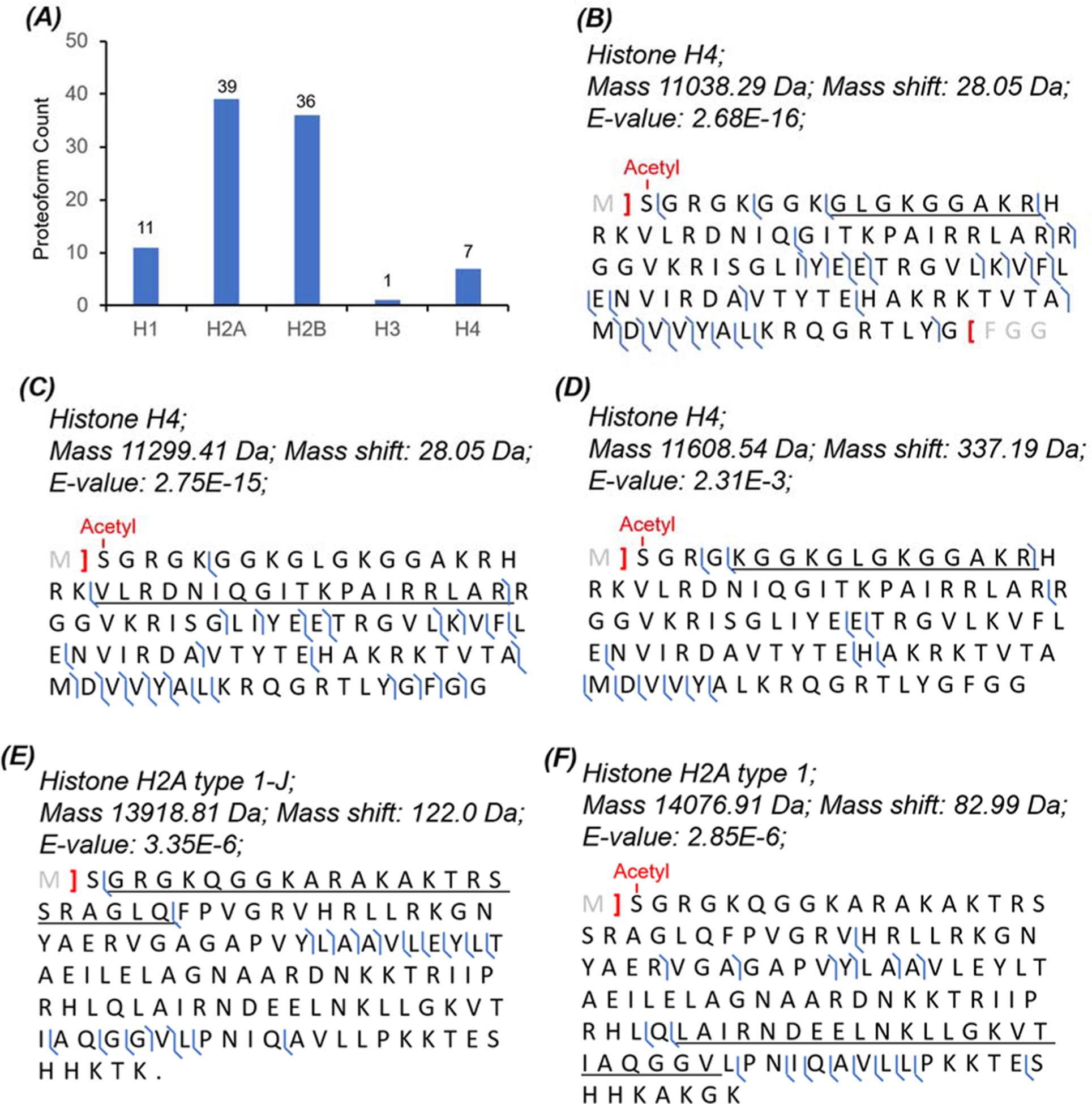
CZE-MS/MS data of the HepG2 sample regarding histone proteoforms. (A) Distribution of the identified histone proteoforms as a function of major histone variants. Sequences and fragmentation patterns of three H4 proteoforms with (B) a 28 Da mass shift, (C) a 28 Da mass shift, and (D) a 337 Da mass shift. Sequences and fragmentation patterns of (E) histone H2A type 1-J proteoform with a 122 Da mass shift and (F) histone H2A type 1 proteoform with an 83 Da mass shift.
We identified one proteoform of histone H2A type 1-J with a 122 Da mass shift in the underlined region, Figure 6E. We speculate that the mass shift corresponds to an acetylation (+42 Da) and a phosphorylation (+80 Da). It has been reported that the K6 and K10 residues could be acetylated.53 However, no literature information about the phosphorylation at T17, S19, or S20 in the mass shift corresponding region according to the UniProt Knowledgebase. We also identified one histone H2A type 1 proteoform with an 83 Da mass shift in the underlined region, Figure 6F. The K96 and K100 in the mass shift-corresponding region could be acetylated based on the previous reports53,54 and the information from PhosphoSitePlus v6.5.8 (https://www.phosphosite.org/). Two lysine acetylation modifications produce an 84 Da mass shift, which is 1 Da heavier than the observed mass shift. The 1 Da difference could be due to a misassignment of the monoisotopic peak of the protein, which resulted in a 1 Da error of the proteoform’s monoisotopic mass. Therefore, the observed 83 Da mass shift is most likely due to the acetylation at both K96 and K100.
CONCLUSIONS
We performed comprehensive comparisons of the MU, CMP, and SP3 methods for cleanup of proteome samples in a lysis buffer containing SDS regarding protein recovery, protein bias, and compatibility with follow-up MS analysis. Our data indicate that the SDS-based protein extraction and the MU-based protein cleanup could be a universal sample preparation procedure for dTDP of complex proteome samples. The procedure produced a reproducible sample preparation with high protein recovery for both E. coli and human cell line samples. Single-shot CZE-MS/MS analysis of the prepared E. coli and HepG2 cell proteome samples (400 ng proteins consumed) identified up to 1336 proteoforms (301 proteins) and 516 proteoforms (241 proteins) with a 1% proteome-level FDR, respectively. Single-shot CZE-MS/MS analysis of the HepG2 cell sample identified 125 membrane proteins and 94 histone proteoforms. The sample preparation procedure including the SDS-based protein extraction and the MU-based protein cleanup should be also compatible with the widely used RPLC-MS/MS approach, although we only used CZE-MS/MS in this work.
We need to point out that when the sample complexity and protein hydrophobicity increase, the BGE composition of CZE needs to be adjusted to ensure good solubility of proteins during CZE separation. We are working on optimizations of CZE-MS conditions for characterization of proteome samples with high hydrophobicity.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Heedeok Hong’s group at the Department of Chemistry of Michigan State University and Prof. David M. Lubman’s group at the University of Michigan for kindly providing the E. coli cells and HepG2 human cells for this project. We thank the support from the National Science Foundation (CAREER Award, grant DBI1846913) and the National Institutes of Health (grant R01GM125991).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00226.
Supporting Information I: supplemental experimental section; Table S1: intensity and migration time information of eight high-intensity peaks from the CZE-MS/MS analyses of two batches of E. coli samples prepared with the MU method; Figure S1: SDS-PAGE data of E. coli proteins processed by the SP3 method with three different amounts of beads; Figure S2: SDS-PAGE data of HepG2 proteins processed by the SP3 method; Figure S3: cumulative distribution of the length of E. coli proteins and human proteins; Figure S4: sequence and fragmentation pattern of one proteoform of 60S ribosomal protein L32 with one disulfide bond; Figure S5: protein phosphorylation data of prothymosin alpha; and supplementary references (PDF)
Supporting Information II: information of the identified proteoforms from the E. coli and HepG2 samples prepared with the MU method (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Smith LM; Kelleher NL; Consortium for Top Down, Proteomics. Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Toby TK; Fornelli L; Kelleher NL Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem 2016, 9, 499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Smith LM; Kelleher NL Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ntai I; Fornelli L; DeHart CJ; Hutton JE; Doubleday PF; LeDuc RD; van Nispen AJ; Fellers RT; Whiteley G; Boja ES; Rodriguez H; Kelleher NL Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 4140–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tran JC; Zamdborg L; Ahlf DR; Lee JE; Catherman AD; Durbin KR; Tipton JD; Vellaichamy A; Kellie JF; Li M; Wu C; Sweet SM; Early BP; Siuti N; LeDuc RD; Compton PD; Thomas PM; Kelleher NL Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Catherman AD; Durbin KR; Ahlf DR; Early BP; Fellers RT; Tran JC; Thomas PM; Kelleher NL Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Mol. Cell. Proteomics 2013, 12, 3465–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Paša-Tolić L; Jensen PK; Anderson GA; Lipton MS; Peden KK; Martinović S; Tolić N; Bruce JE; Smith RD High Throughput Proteome-Wide Precision Measurements of Protein Expression Using Mass Spectrometry. J. Am. Chem. Soc 1999, 121, 7949–7950. [Google Scholar]
- (8).Anderson LC; DeHart CJ; Kaiser NK; Fellers RT; Smith DF; Greer JB; LeDuc RD; Blakney GT; Thomas PM; Kelleher NL; Hendrickson CL Identification and Characterization of Human Proteoforms by Top-Down LC-21 Tesla FT-ICR Mass Spectrometry. J. Proteome Res 2017, 16, 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cai W; Tucholski T; Chen B; Alpert AJ; McIlwain S; Kohmoto T; Jin S; Ge Y Top-Down Proteomics of Large Proteins up to 223 kDa Enabled by Serial Size Exclusion Chromatography Strategy. Anal. Chem 2017, 89, 5467–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liang Y; Jin Y; Wu Z; Tucholski T; Brown KA; Zhang L; Zhang Y; Ge Y Bridged Hybrid Monolithic Column Coupled to High-Resolution Mass Spectrometry for Top-Down Proteomics. Anal. Chem 2019, 91, 1743–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McCool EN; Lubeckyj RA; Shen X; Chen D; Kou Q; Liu X; Sun L Deep Top-Down Proteomics Using Capillary Zone Electrophoresis-Tandem Mass Spectrometry: Identification of 5700 Proteoforms from the Escherichia coli Proteome. Anal. Chem 2018, 90, 5529–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yu D; Wang Z; Cupp-Sutton KA; Liu X; Wu S Deep Intact Proteoform Characterization in Human Cell Lysate Using High-pH and Low-pH Reversed-Phase Liquid Chromatography. J. Am. Soc. Mass Spectrom 2019, 30, 2502–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ansong C; Wu S; Meng D; Liu X; Brewer HM; Deatherage Kaiser BL; Nakayasu ES; Cort JR; Pevzner P; Smith RD; Heffron F; Adkins JN; Pasa-Tolic L Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 10153–10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lubeckyj RA; Basharat AR; Shen X; Liu X; Sun L Large-Scale Qualitative and Quantitative Top-Down Proteomics Using Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry with Nanograms of Proteome Samples. J. Am. Soc. Mass Spectrom 2019, 30, 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Liu Z; Wang R; Liu J; Sun R; Wang F Global Quantification of Intact Proteins via Chemical Isotope Labeling and Mass Spectrometry. J. Proteome Res 2019, 18, 2185–2194. [DOI] [PubMed] [Google Scholar]
- (16).Riley NM; Westphall MS; Coon JJ Activated Ion-Electron Transfer Dissociation Enables Comprehensive Top-Down Protein Fragmentation. J. Proteome Res 2017, 16, 2653–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shaw JB; Li W; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J. American Chem. Soc 2013, 135, 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Shaw JB; Malhan N; Vasil’ev YV; Lopez NI; Makarov A; Beckman JS; Voinov VG Sequencing Grade Tandem Mass Spectrometry for Top-Down Proteomics Using Hybrid Electron Capture Dissociation Methods in a Benchtop Orbitrap Mass Spectrometer. Anal. Chem 2018, 90, 10819–10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kou Q; Xun L; Liu X TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics 2016, 32, 3495–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cai W; Guner H; Gregorich ZR; Chen AJ; Ayaz-Guner S; Peng Y; Valeja SG; Liu X; Ge Y MASH Suite Pro: A Comprehensive Software Tool for Top-Down Proteomics. Mol. Cell. Proteomics 2016, 15, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sun RX; Luo L; Wu L; Wang RM; Zeng WF; Chi H; Liu C; He SM pTop 1.0: A High-Accuracy and High-Efficiency Search Engine for Intact Protein Identification. Anal. Chem 2016, 88, 3082–3090. [DOI] [PubMed] [Google Scholar]
- (22).Donnelly DP; Rawlins CM; DeHart CJ; Fornelli L; Schachner LF; Lin Z; Lippens JL; Aluri KC; Sarin R; Chen B; Lantz C; Jung W; Johnson KR; Koller A; Wolff JJ; Campuzano IDG; Auclair JR; Ivanov AR; Whitelegge JP; Paša-Tolić L; Chamot-Rooke J; Danis PO; Smith LM; Tsybin YO; Loo JA; Ge Y; Kelleher NL; Agar JN Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Speers AE; Wu CC Proteomics of integral membrane proteins - theory and application. Chem. Rev 2007, 107, 3687–3714. [DOI] [PubMed] [Google Scholar]
- (24).Botelho D; Wall MJ; Vieira DB; Fitzsimmons S; Liu F; Doucette A Top-down and bottom-up proteomics of SDS-containing solutions following mass-based separation. J. Proteome Res 2010, 9, 2863–2870. [DOI] [PubMed] [Google Scholar]
- (25).Wiśniewski JR; Zougman A; Nagaraj N; Mann M Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- (26).Wessel D; Flügge UI A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem 1984, 138, 141–143. [DOI] [PubMed] [Google Scholar]
- (27).Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol 2014, 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hughes CS; Moggridge S; Müller T; Sorensen PH; Morin GB; Krijgsveld J Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc 2019, 14, 68–85. [DOI] [PubMed] [Google Scholar]
- (29).Dagley LF; Infusini G; Larsen RH; Sandow JJ; Webb AI Universal Solid-Phase Protein Preparation (USP3) for Bottom-up and Top-down Proteomics. J. Proteome Res 2019, 18, 2915–2924. [DOI] [PubMed] [Google Scholar]
- (30).Blancher C; Jones A SDS-PAGE and Western Blotting Techniques. Methods Mol. Med 2001, 57, 145–162. [DOI] [PubMed] [Google Scholar]
- (31).McCool EN; Lubeckyj R; Shen X; Kou Q; Liu X; Sun L Large-scale Top-down Proteomics Using Capillary Zone Electrophoresis Tandem Mass Spectrometry. J. Visualized Exp 2018, 140, No. e58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhu G; Sun L; Dovichi NJ Thermally-initiated free radical polymerization for reproducible production of stable linear polyacrylamide coated capillaries, and their application to proteomic analysis using capillary zone electrophoresis-mass spectrometry. Talanta 2016, 146, 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sun L; Zhu G; Zhao Y; Yan X; Mou S; Dovichi NJ Ultrasensitive and fast bottom-up analysis of femtogram amounts of complex proteome digests. Angew. Chem 2013, 52, 13661–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sun L; Zhu G; Zhang Z; Mou S; Dovichi NJ Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. J. Proteome Res 2015, 14, 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wojcik R; Dada OO; Sadilek M; Dovichi NJ Simplified capillary electrophoresis nanospray sheath-flow interface for high efficiency and sensitive peptide analysis. Rapid Commun. Mass Spectrom 2010, 24, 2554–2560. [DOI] [PubMed] [Google Scholar]
- (36).Kessner D; Chambers M; Burke R; Agus D; Mallick P ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Keller A; Nesvizhskii AI; Kolker E; Aebersold R Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem 2002, 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- (38).Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- (39).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Pérez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaíno JA The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Arribas J; Castaño JG Kinetic studies of the differential effect of detergents on the peptidase activities of the multicatalytic proteinase from rat liver. J. Biol. Chem 1990, 265, 13969–13973. [PubMed] [Google Scholar]
- (41).Lubeckyj RA; McCool EN; Shen X; Kou Q; Liu X; Sun L Single-Shot Top-Down Proteomics with Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry for Identification of Nearly 600 Escherichia coli Proteoforms. Anal. Chem 2017, 89, 12059–12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chang CN; Chang FN Methylation of the ribosomal proteins in Escherichia coli. Nature and stoichiometry of the methylated amino acids in 50S ribosomal proteins. Biochemistry 1975, 14, 468–477. [DOI] [PubMed] [Google Scholar]
- (43).Monaco HL; Crawford JL; Lipscomb WN Three-dimensional structures of aspartate carbamoyltransferase from Escherichia coli and of its complex with cytidine triphosphate. Proc. Natl. Acad. Sci. U. S. A 1978, 75, 5276–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Karetsou Z; Kretsovali A; Murphy C; Tsolas O; Papamarcaki T Prothymosin alpha interacts with the CREB-binding protein and potentiates transcription. EMBO Rep 2002, 3, 361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Chen D; Lubeckyj RA; Yang Z; McCool EN; Shen X; Wang Q; Xu T; Sun L Predicting Electrophoretic Mobility of Proteoforms for Large-Scale Top-Down Proteomics. Anal. Chem 2020, 92, 3503–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wojcik R; Vannatta M; Dovichi NJ Automated enzyme-based diagonal capillary electrophoresis: application to phosphopeptide characterization. Anal. Chem 2010, 82, 1564–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kim J; Zand R; Lubman DM Electrophoretic mobility for peptides with post-translational modifications in capillary electrophoresis. Electrophoresis 2003, 24, 782–793. [DOI] [PubMed] [Google Scholar]
- (48).Faserl K; Sarg B; Gruber P; Lindner HH Investigating capillary electrophoresis-mass spectrometry for the analysis of common post-translational modifications. Electrophoresis 2018, 39, 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zheng Y; Huang X; Kelleher NL Epiproteomics: quantitative analysis of histone marks and codes by mass spectrometry. Curr. Opin. Chem. Biol 2016, 33, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Janssen KA; Sidoli S; Garcia BA Recent Achievements in Characterizing the Histone Code and Approaches to Integrating Epigenomics and Systems Biology. Methods in enzymol 2017, 586, 359–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wang T; Holt MV; Young NL Early butyrate induced acetylation of histone H4 is proteoform specific and linked to methylation state. Epigenetics 2018, 13, 519–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Gargano AFG; Shaw JB; Zhou M; Wilkins CS; Fillmore TL; Moore RJ; Somsen GW; Paša-Tolić L Increasing the Separation Capacity of Intact Histone Proteoforms Chromatography Coupling Online Weak Cation Exchange-HILIC to Reversed Phase LC UVPD-HRMS. J. Proteome Res 2018, 17, 3791–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wu Q; Cheng Z; Zhu J; Xu W; Peng X; Chen C; Li W; Wang F; Cao L; Yi X; Wu Z; Li J; Fan P Suberoylanilide hydroxamic acid treatment reveals crosstalks among proteome, ubiquitylome and acetylome in non-small cell lung cancer A549 cell line. Sci. Rep 2015, 5, 9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhao S; Xu W; Jiang W; Yu W; Lin Y; Zhang T; Yao J; Zhou L; Zeng Y; Li H; Li Y; Shi J; An W; Hancock SM; He F; Qin L; Chin J; Yang P; Chen X; Lei Q; Xiong Y; Guan KL Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.