

Abstract
Cellular actin arrays are often highly organized, with characteristic patterns critical to their in vivo functions, yet the mechanisms for establishing these higher order geometries remain poorly understood. In Saccharomyces cerevisiae, formin-polymerized actin cables are spatially organized and aligned along the mother–bud axis to facilitate polarized vesicle traffic. Here, we show that the bud neck–associated F-BAR protein Hof1, independent of its functions in regulating the formin Bnr1, binds to actin filaments and organizes actin cables in vivo. Hof1 bundles actin filaments and links them to septins in vitro. F-actin binding is mediated by the “linker” domain of Hof1, and its deletion leads to cable organization defects in vivo. Using superresolution imaging, we show that Hof1 and septins are patterned at the bud neck into evenly spaced axial pillars (∼200 nm apart), from which actin cables emerge and grow into the mother cell. These results suggest that Hof1, while bound to septins at the bud neck, not only regulates Bnr1 activity, but also binds to actin cables and aligns them along the mother–bud axis. More broadly, these findings provide a strong example of how an actin regulatory protein can be spatially patterned at the cell cortex to govern actin network geometry.
INTRODUCTION
Cells assemble a wide variety of actin-based structures that have distinct geometries, filamentous architectures, and mechanical properties tailored to their biological functions (Chhabra and Higgs, 2007; Blanchoin et al., 2014). Actin networks are polymerized by a suite of different actin nucleators and elongation factors, the mechanisms and functions of which are now coming into focus (Chesarone and Goode, 2010; Campellone and Welch, 2010). By comparison, it is far less clear how spatial organization of these actin arrays, including filament orientation and spacing, is established and maintained in cells. The polarized actin cable array of Saccharomyces cerevisiae provides a genetically amenable system for addressing this question. Cables are polymerized by formins, and run parallel to the mother–bud axis with their barbed ends oriented toward the bud tip. These linear actin arrays are essential for polarized cell growth and provide tracks for myosin V–based transport of secretory vesicles and other cargoes to the bud (Moseley and Goode, 2006). Cables are highly dynamic, extending toward the rear of the mother cell at a rate of ∼0.3–0.6 µm s−1 (or ∼100–200 subunits s−1), and are disassembled at similar rates (Yang and Pon, 2002; Okada et al., 2006; Yu et al., 2011).
Two formins, Bni1 and Bnr1, polymerize actin cables in S. cerevisiae and have genetically redundant yet distinct roles (Evangelista et al., 2002; Sagot et al., 2002; Pruyne et al., 2004). During polarized cell growth, Bni1 is localized to the bud tip, where it assembles cables that are released from the cortex into the bud compartment and grow to extend through the bud neck and into the mother cell compartment (Buttery et al., 2007). In contrast, the formin Bnr1 is recruited by septin networks found at the bud neck, where it polymerizes actin cables that specifically enter the mother compartment (Pruyne et al., 2004; Gao et al., 2010; Buttery et al., 2012). During most phases of polarized cell growth, the Bni1- and Bnr1-generated actin cables in S. cerevisiae are aligned parallel to the mother–bud axis and spatially distributed to extend into different regions of the mother cell. How this is achieved is not well understood.
We previously showed that the F-BAR domain protein Hof1, which localizes to the bud neck through an association with the septin ring (Vallen et al., 2000), is a direct inhibitor of Bnr1-mediated actin nucleation (Graziano et al., 2014; Garabedian et al., 2018). These Bnr1-inhibitory activities are mediated primarily by the F-BAR and SH3 domains of Hof1, and hof1∆ leads to defects in the organization of Bnr1-generated actin cables in vivo. Hof1 stably associates with the septin ring during polarized growth and cytokinesis (Oh et al., 2013), and is thought to perform its cellular functions from this location. Septins are conserved membrane-associated polymer networks, which in yeast recruit at least 50 other proteins (including Bnr1 and Hof1) to the bud neck to facilitate polarized growth and cytokinesis (Gladfelter et al., 2001; Bezanilla et al., 2015). Recent electron microscopy (EM) studies have demonstrated that the ultrastructure of the bud neck–associated yeast septin network changes dramatically over the cell cycle. Early in bud emergence septin networks consist primarily of axial filaments, which run parallel to the mother–bud axis. Then, just before cytokinesis the septin collar splits, and the networks consist primarily of circumferential filaments running perpendicular to the mother–bud axis (Bertin et al, 2012; Ong et al., 2014).
Our previous work demonstrated that bnr1∆ only partially suppresses hof1∆ defects in actin cable organization, raising the possibility that Hof1 has additional roles in actin regulation beyond regulating the activity of Bnr1 (Graziano et al., 2014; Garabedian et al., 2018). Here, we investigated this possibility by studying the effects of Hof1 on actin organization in a bnr1∆ background. Our results reveal a new and unanticipated role for Hof1 in directly binding to and spatially organizing actin cables. Even in the absence of Bnr1, loss of HOF1 leads to significant defects in the organization of cables polymerized by Bni1, which in turn disrupts polarized transport of secretory vesicles. Purified Hof1 binds to actin filaments using its previously uncharacterized linker domain (located between the F-BAR and SH3 domains). Hof1 also physically links actin filaments to septins in vitro. Using superresolution imaging, we show that septins, Hof1, and Bnr1 each form regularly spaced axial striations (or “pillars”) at the bud neck during polarized cell growth, with actin cables emerging from these sites. Thus, septins and the proteins they recruit (including Hof1) provide evenly spaced actin cable-organizing centers, which facilitate proper orientation and spatial distribution of cables into the mother cell.
RESULTS
A Bnr1-independent role for Hof1 in organizing actin cables in vivo
Our previous work on Hof1 demonstrated a role in directly regulating the actin nucleation activity of the formin Bnr1 (Graziano et al., 2014; Garabedian et al., 2018). Here, we investigated the possibility that Hof1 has additional, Bnr1-independent roles in regulating actin cable formation. As a first step, we compared wild-type, hof1∆, bnr1∆, and hof1∆bnr1∆ strains for differences in cell growth and actin cable organization. hof1∆ cells showed impaired growth at elevated temperatures in two different assays (Figure 1, A and B), consistent with previous reports (Lippincott and Li, 1998; Vallen et al., 2000; Oh et al., 2013), but bnr1∆ failed to suppress the cell growth defects of hof1∆. This observation suggests that Hof1 may have Bnr1-independent functions in promoting polarized cell growth.
FIGURE 1:

Hof1 has Bnr1-independent roles in spatially organizing actin cables. (A) Fivefold serial dilutions of indicated yeast strains grown on YEPD plates at 25, 30, 34, and 37°C for 2 d. The loss of BNR1 does not suppress the temperature sensitivity of hof1∆. (B) Growth rates of the same strains measured by optical density (OD600) at 35°C in liquid culture (YEPD) in a shaking microplate absorbance reader. Data were averaged from six independent trials. Error bars, SEM. (C) Structured illumination microscopy (SIM) of F-actin organization in fixed and phalloidin-stained wild-type, hof1∆, bnr1∆, and hof1∆bnr1∆ cells grown at 25°C. Scale bar, 5 µm. (D) Automated traces of actin cable organization from SIM images as in C with SOAX, showing actin cable segments (purple) and intersections (green). Scale bar, 5 µm. (E) Average number of actin cable segments per cell analyzed by SOAX, quantified for 20 cells per strain. (F) Average number of cable intersections per cell analyzed by SOAX, quantified for the same 20 cells per strain. (G) Coefficient of variation (CoV) of phalloidin staining in mother cells after 100 µM CK666 treatment averaged from 50 cells per strain from confocal images. (H) Fluorescence density measured from at least 20 individual actin cables in each strain from confocal images (WT, n = 29; hof1∆, n = 24; bnr1∆, n = 22; hof1∆bnr1∆, n = 26). Error bars (SD) in panels E and F. Statistical significance in all panels calculated by one-way ANOVA (n.s., no significance; p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001).
To investigate this idea further, we compared actin cable organization in wild-type, hof1∆, bnr1∆, and hof1∆bnr1∆ cells. Using superresolution structured illumination microscopy (SIM), not available to us at the time of our earlier Hof1 studies (Graziano et al., 2014), we were able to image actin cable networks at increased resolution, revealing finer detail (Figure 1C). This revealed clear defects in cable organization in both hof1∆ and hof1∆bnr1∆ strains. In addition, we used open-source software (SOAX; Xu et al., 2015) to perform an unbiased quantitative comparison of cable organization in the four strains (Figure 1D). As expected, hof1∆ cells showed a significant increase in the number of cable segments and cable intersections, indicating cable disorganization and entanglement (Figure 1, E and F). Importantly, bnr1∆ did not suppress these hof1∆ cable defects, suggesting a Bnr1-independent role for Hof1 in actin cable organization.
We also pretreated cells with CK666 before fixation and imaging to remove cortical actin patches nucleated by Arp2/3 complex (Nolen et al., 2009; Burke et al., 2014). This provided a less obstructed view of the formin-generated actin cable networks (Supplemental Figure S1A), enabling us to perform coefficient of variation (CoV) analysis in the mother cell compartment. We measure the mean fluorescence of actin cable staining in the mother cell and divide this by the SD of the fluorescence. For wild-type cells, which have well-defined and brightly stained cables against a dark background, there is a higher SD, and thus a higher CoV. In contrast, for cells with disorganized actin cable networks, there is a lower SD, and consequently a lower CoV. This analysis revealed that wild-type and bnr1∆ cells have higher CoV values compared with hof1∆ and hof1∆bnr1∆ cells (Figure 1G). The more dispersed cables seen in hof1∆ and hof1∆bnr1∆ cells were specific to medium- and large-budded cell stages of polarized growth (Supplemental Figure S1B). On the other hand, we found no significant difference in average cable thickness (intensity of phalloidin staining) between wild-type and hof1∆ cells, or between bnr1∆ and hof1∆bnr1∆ cells (Figure 1H). This suggests that the defects in cable organization caused by hof1∆ are not due to altered cable thickness. Cables were thinner in bnr1∆ compared with wild-type cells, suggesting that when Bni1-polymerized cables reach the bud neck they may be stitched together with Bnr1-polymerized cables to produce the thicker cables extending into the mother compartment in wild-type cells.
To better understand how actin cables become disorganized in hof1∆ and hof1∆bnr1∆ cells, we performed live imaging on cables using an integrated Abp140-3GFP marker. We analyzed cable extension speeds, as well as angles of cable extension using custom software (Figure 2A; Graziano et al, 2014; Eskin et al., 2016). Although hof1∆ did not change cable extension speeds (Figure 2B), the angles of extension were significantly wider in hof1∆ and hof1∆bnr1∆ compared with wild-type and bnr1∆ cells (Figure 2C). From these results, we conclude that hof1∆ alters the angles of cables extending into the mother cell without changing the extension rate. The altered cable growth angles may contribute to the increased cable disorganization (cable segment numbers and crossovers), described above, in these mutants. Note that bnr1∆ did lead to an increase in cable extension rate, as previously reported (Chesarone-Cataldo et al., 2011), which may be due to the remaining formin (Bni1) having increased access to a limited pool of profilin-bound actin monomers (Suarez et al., 2015).
FIGURE 2:
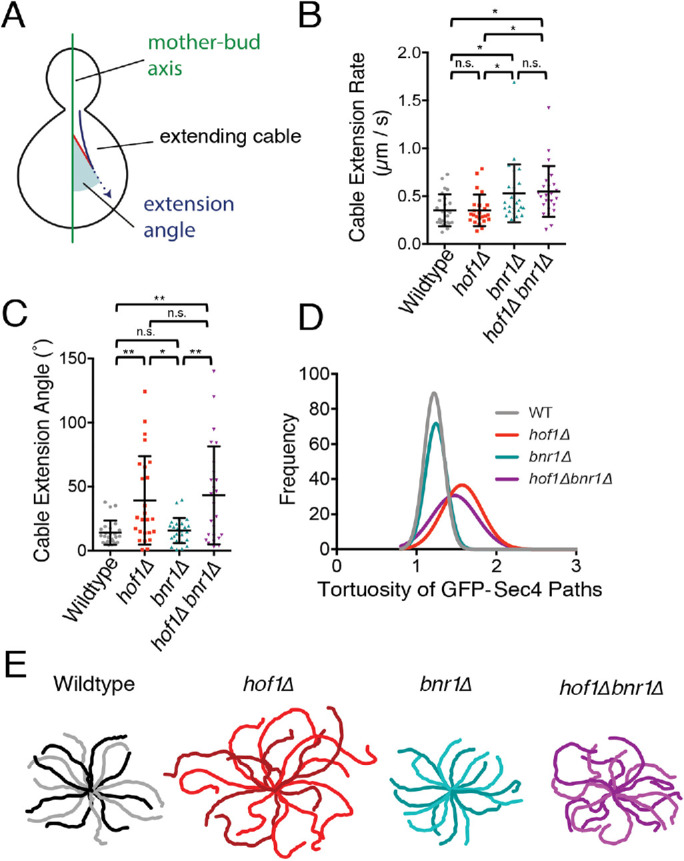
Live imaging of actin cable extension angles and secretory vesicle movements reveals defects in hof1∆ and hof1∆ bnr1∆ cells. Diagram of aspects of cable dynamics analyzed. Extending cables (blue arrow) and extension angles (shaded in red) corresponding to angles of cable extension relative to the mother–bud axis. (B) Extension rates of Abp140-3GFP marked actin cables measured in custom software written in MATLAB (n = 25 cables per strain). Error bars, SD. Statistical significance calculated by one-way ANOVA (n.s., no significance; p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001). (C) Extension angles of same actin cables as in B measured in custom MATLAB software (n = 25 cables per strain). Error bars, SD. Statistical significance calculated by one-way ANOVA (n.s., no significance; p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001). (D) Frequency distributions of the tortuosity (ratio of length to distance) of GFP-Sec4 paths observed in each indicated strain (n = 150 vesicles per strain). (E) Representative traces of GFP-Sec4 paths observed in the indicated strains. In each case, the outer tips of each trace represent the beginning of the vesicle path and the center of the traces represents the bud neck.
Because cable organization defects are predicted to alter polarized secretory traffic, we also used live imaging to compare secretory vesicle paths (marked with GFP-Sec4) in wild-type, hof1∆, bnr1∆, and hof1∆bnr1∆ cells. Vesicle paths were traced and the tortuosity (ratio of path length to distance traveled) was calculated. Vesicle traffic was more circuitous in hof1∆ and hof1∆bnr1∆ cells compared with wild-type and bnr1∆ cells (Figure 2D; example traces in Figure 2E). Thus, independent of its regulatory effects on Bnr1, Hof1 is required to maintain proper spatial organization and function of Bni1-polymerized actin cables.
Hof1 binds to and bundles actin filaments in vitro
Our observations above indicate that Hof1 regulates the organization of actin cables even in the absence of Bnr1. However, Hof1 has no appreciable biochemical effects on the activity of the remaining formin (Bni1) (Graziano et al., 2014; Garabedian et al., 2018). Given that Hof1 is anchored to septins at the bud neck in vivo, and must exert its influence on actin cable organization from this position, we considered whether Hof1 might bind to actin filaments. Indeed, F-actin binding has been reported for the human F-BAR protein syndapin/pacsin (Kostan et al., 2014). To test this possibility, we used total internal reflection fluorescence (TIRF) microscopy to monitor the interactions of fluorescently labeled full-length Hof1 with labeled actin filaments, and observed Hof1 binding to and bundling filaments (Figure 3A). Bundling also was observed with the same concentration of unlabeled Hof1 (Figure 3, B and C). Thus, Hof1 directly binds and bundles actin filaments, possibly stemming from Hof1 dimerization (Moravcevic et al., 2015; Garabedian et al., 2018).
FIGURE 3:

Purified Hof1 bundles F-actin. (A) Representative image of TIRF microscopy assay in which 2 µM actin (10% Oregon-green labeled) was allowed to polymerize and then 100 nM SNAP549-Hof1-FL was flowed in. SNAP-labeled Hof1 bundles actin filaments. Scale bar, 25 µm. (B) Representative images of TIRF microscopy assays in which 2 µM actin (10% Oregon-green labeled) was polymerized and then buffer or 100 nM Hof1-FL was flowed into the TIRF chamber. Scale bar, 20 µm. (C) Diagram of Hof1 constructs tested in TIRF assays as in B and CoV measurements for 100 nM (green) and/or 1 µM (blue) of each. Data averaged from three FOVs in each of two independent experiments. Error bars, SEM. Statistical significance compared with control (gray bar) calculated by one-way ANOVA (n.s., no significance; p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001). (D) Representative EM images of negatively stained actin filaments. Top, actin filaments alone (no Hof1). Bottom, actin filaments and 500 nM Hof1-FL. Scale bars, 100 nm. (E) Top, higher magnification view of EM as in D with Hof1-FL (colored in blue) decorating actin filaments. Bottom, original uncolored EM image. (F) Class averages of Hof1 particles imaged by EM. Left, class average of free Hof1 particles (not bound to actin filaments). Right, class average of particles bound to actin filaments. Both class averages have a crescent-shaped structure as expected for F-BAR proteins. Scale bar, 20 nm. (G) Model for Hof1 dimer binding to and bundling actin filaments (created with Biorender.com).
To determine which domain(s) of Hof1 mediate the interaction with F-actin, we compared the bundling activities of Hof1 constructs tagged with either monomeric maltose binding protein (MBP) (full-length, FL; N-terminus, NT; F-BAR) or dimeric glutathione S-transferase (GST) for constructs lacking the dimeric F-BAR domain (coiled coil 2, CC2; C-terminus, CT; Linker; SH3; Figure 3C and Supplemental Figure S2A). Bundling was quantified by CoV analysis of the labeled actin in TIRF fields. In this case, higher CoV values indicate greater bundling of actin filaments, while lower CoV values indicate a more dispersed network. Hof1-CT or Hof1 linker subfragment (100 nM; lacking the SH3 domain) bundled F-actin nearly as well as full-length Hof1. The SH3 domain alone lacked significant bundling activity even at 1 µM. At 100 nM, Hof1-NT (consisting of the F-BAR and CC2 domains) showed no significant bundling activity compared with control reactions. However, 1 µM Hof1-NT could bundle F-actin. Interestingly, neither of the two domains comprising Hof1-NT, i.e., the F-BAR and CC2, alone were sufficient to bundle. Thus, both domains are required for the modest bundling activity of Hof1-NT. Overall, these data show that the linker domain of Hof1 contains the strongest F-actin bundling activity, and that additional activity resides in the N-terminal half of Hof1.
Hof1 bundling activity was also confirmed by EM (Figure 3D). Hof1 particles could be seen decorating the actin filament bundles in a somewhat regular pattern, with roughly 20–30 nm spacing between particles (Figure 3, D and E, and Supplemental Figure S2B). Additional free Hof1 particles were dispersed around the actin bundles on the grids, allowing us to compare the shapes of the “bound” and “unbound” particles. Class averages showed that bound and unbound particles had a similar appearance to each other (Figure 3F), which was consistent with the ∼20-nm elongated structure of Hof1 dimers (Moravcevic et al., 2015; Garabedian et al., 2018). Based on these observations, and our understanding of Hof1 domain structure and function, we propose a working model for Hof1 dimers cross-linking actin filaments into bundles (Figure 3G). In this model, the two linker domains of a Hof1 dimer contact two different filaments, and the N-terminal half of Hof1 (F-BAR and CC2) is near the filaments, possibly contributing to binding and bundling.
The linker region of Hof1 is required for proper actin cable organization in vivo
Our biochemical observations above led us to ask whether the linker region of Hof1 contributes to actin cable organization in vivo. Therefore, we used CRISPR-mediated genome editing (Anand et al., 2017) to generate an internal deletion of the linker domain (hof1∆linker; ∆351-574), and then examined cable organization using SIM (Figure 4A). Actin cables in hof1∆linker cells were disorganized compared with wild-type cells, and reminiscent of hof1∆ cells, as indicated by their elevated levels of cable segments and intersections (Figure 4, B and C) and lower CoV values (Figure 4D). Similar results were observed in hof1∆linker bnr1∆ cells. Importantly, because these cells do not express Bnr1, the cable defects caused by hof1∆linker cannot be due to a loss of Hof1 inhibitory effects on Bnr1. The cable organization defects in hof1∆linker cells were not as severe as those in hof1∆ cells, suggesting that the N-terminal half of Hof1 makes contributions to cable organization, consistent with our biochemical observations (Figure 3). We also generated a hof1∆linker strain with a C-terminal GFP tag, and verified that it localizes to the bud neck and is expressed in cells at similar levels to Hof1-GFP (Supplemental Figure S3A and S3B). Thus, the actin cable phenotypes in hof1∆linker cells do not arise from lower expression levels, or a failure to localize to the bud neck. In the majority of Hof1∆linker-GFP cells, fluorescence was seen primarily at the bud neck, but additional signal at the bud tip was observed in a fraction of small budded Hof1∆linker-GFP cells. Finally, we note that Hof1∆linker-GFP signal at the bud neck is about twofold higher than Hof1-GFP throughout bud growth (Supplemental Figure S3C). The reason for this is unclear, but could stem from the linker region being a target of signaling pathways that help control Hof1 localization. Overall, these data demonstrate that the Hof1 linker domain, which mediates F-actin bundling in vitro, makes an important contribution to actin cable organization in vivo. Importantly, our data do not rule out the possibility that additional molecular interactions of the linker region, beyond F-actin binding, could contribute to actin cable organization.
FIGURE 4:

Internal deletion of the Hof1 linker domain disrupts actin cable organization in vivo. (A) Representative SIM images of wild-type, hof1∆, bnr1∆, hof1∆bnr1∆, hof1∆linker, and hof1∆linker bnr1∆ strains. Scale bar, 5 µm. (B) Average number of cable segments per cell analyzed by SOAX, quantified for 25 cells per strain. (C) Average number of cable intersections per cell analyzed by SOAX, quantified for same 25 cells per strain. (D) CoV measurements of phalloidin staining in mother cells after 100 µM CK666 treatment averaged from 50 cells per strain from confocal images. Error bars (SD) in all panels. Statistical significance in all panels calculated by one-way ANOVA (n.s., no significance; p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001).
Hof1 directly links actin and septin filaments in vitro
Given that Hof1 is recruited to the septin collar at the bud neck (Oh et al., 2013), we tested whether Hof1 might be capable of physically linking actin and septin filaments in vitro. Septin filaments (comprised of Cdc3, Cdc10-SNAP, Cdc11, and Cdc12) were purified, assembled, and visualized by TIRF microscopy in the presence of differentially labeled actin filaments (Figure 5A). In the absence of Hof1, there was minimal overlap between the septin and actin polymers; however, the addition of Hof1 led to their extensive overlap and coalignment (Figure 5B), and bundling of the F-actin over time (Supplemental Video S1). These results demonstrate that Hof1 can physically link actin and septin filaments in vitro, likely mediated by its separate Sfilaments in TIRF assays (Figure 5A), which was confirmed by EM (Supplemental Figure S4). This septin bundling effect is consistent with previous studies reporting that Hof1 overexpression leads to abnormally thick and disorganized septin structures at the bud neck (Lippincott and Li, 1998; Oh et al., 2013).
FIGURE 5:

Hof1 physically links septins and actin filaments in vitro. (A) Representative images from TIRF microscopy assays showing Hof1-FL physically links actin and septin filaments in vitro. All reactions contain 1 µM actin (10% Oregon-green labeled) and 50 nM labeled SNAP549-septin rods. Images show representative fields of view 200 s after initiation of actin and septin assembly in control reactions without Hof1 (top row) or reactions with 50 nM Hof1-FL (bottom row). Scale bar, 10 µm. (B) Pearson’s colocalization analysis of actin and septin filaments from TIRF microscopy assays in A in the presence and absence of Hof1-FL. Data averaged from four FOVs in each of three independent experiments. Error bars, SD. Statistical significance calculated by unpaired t test (****, p ≤ 0.0001). (C) Representative SIM images of actin organization in fixed and phalloidin-stained wild-type and temperature-sensitive septin mutants, cdc10-4, at 22°C and 34°C. Scale bar, 5 µm. (D) Average number of cable segments per cell analyzed by SOAX, quantified for 20 cells per strain. (E) Average number of cable intersections per cell analyzed by SOAX, quantified for the same 20 cells per strain. (F) CoV measurements of phalloidin staining in mother cells after 100 µM CK666 treatment averaged from 50 cells per strain. Error bars (SD) in all panels. Statistical significance in panels D–F calculated by one-way ANOVA (n.s., no significance; p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001).
Movie S1.
TIRF movies of septin-actin interactions with and without Hof1. Representative TIRF movies showing 1μM actin (10% Oregon green labeled) and 50 nM SNAP-547 labeled septin filaments in the absence (control) and presence of 50 nM Hof1-FL.
Given that Hof1 is anchored to septins at the bud neck in vivo, our results predict that disrupting septin organization should impair actin cable organization similar to hof1∆. To test this prediction, we examined actin cable morphology in a cdc10-4 strain (Weems et al., 2014) before and after shifting the mutant strain to the nonpermissive temperature (34°C). At permissive temperature (22°C), cable morphology was similar in mutant and wild-type cells (Figure 5C). However, shifting cdc10-4 cells to the nonpermissive temperature for 15 min led to highly disorganized cables similar to hof1∆ (Figure 5, D–F). These observations suggest that septins form the crucial scaffold at the bud neck, which in turn recruits Hof1 (and potentially other proteins) that bind and spatially organize actin cables.
Septins and Hof1 form evenly spaced “pillars” at the bud neck during polarized cell growth
Standard diffraction-limited light microscopy has previously been used to localize septins, and show that they form a broad band (or collar) that remains at the bud neck throughout polarized cell growth, then splits into two rings at cytokinesis (Marquardt et al., 2019). To gain additional insights into septin and Hof1 organization at the neck, we reexamined their localization patterns using superresolution SIM imaging. Throughout bud growth, we observed that tagged septins (Cdc3-yomApple, Cdc10-GFP, Cdc11-GFP, and Shs1-GFP) were organized into thick axial stripes, or “pillars,” which run parallel to the mother–bud axis (Figure 6, A and B) and are regularly spaced ∼200 nm apart (Figure 6C). Further, Hof1-Envy and Bnr1-Envy both colocalized with septins in pillars (Figure 6, D–G), and we observed alignment of actin cables with the pillars in fixed cells (Figure 6, H–J). In a few rare instances, when the bud was sheared off of the mother cell, we were able to observe septin and actin organization from the top down, revealing that actin cables emanate from the septin pillars (Figure 6, K and L). Taken together, these observations suggest that septins along with Hof1 and Bnr1 form evenly spaced bands at the neck, from which actin cables emerge as they enter the mother cell.
FIGURE 6:

Septins and Hof1 form regularly spaced pillars at the bud neck and align with actin cables. (A) Top, representative SIM images of live yeast cells, grown in an asynchronous culture, showing the organization of septins (Cdc3-yomApple) at different stages of bud growth. Scale bar, 5 µm. Bottom, higher magnification views of axial septin structures, from the same images as above (coded by letters). Scale bar, 1 µm. (B) Representative SIM images of different GFP-tagged septins (Cdc10, Cdc11, and Shs1), showing similar axial pillars. Scale bar, 1 µm. (C) The average distance between axial pillars for Cdc3-yomApple, Cdc10-GFP, Cdc11-GFP, and Shs1-GFP is ∼200 nm. Statistical significance was calculated by one-way ANOVA (n.s., no significance; p > 0.05). (D) Representative SIM image showing Hof1-Envy colocalization with Cdc3-yomApple in live yeast cell. Scale bar, 1 µm. (E) Representative SIM image showing Bnr1-Envy colocalization with Cdc3-yomApple in live yeast cell. Scale bar, 1 µm. (F) Line scan analysis of merged image in D showing overlapping signals from Hof1-Envy and Cdc3-yomApple. (G) Line scan analysis of merged image in E showing overlapping signals from Bnr1-Envy and Cdc3-yomApple. (H) Representative SIM image of fixed and phalloidin-stained yeast cell expressing Cdc3-yomApple. Scale bar, 5 µm. (I) Zoom-in images denoted by box in H. Scale bar, 1 µm. Phalloidin-stained filaments appear aligned with axial septin pillars. (J) Line scan analysis of merged image in I showing overlapping signals from Bnr1-Envy and Cdc3-yomApple. (K) SIM image of fixed yeast cells expressing Abp140-3GFP and Cdc3-yomApple. Scale bar, 5 µm. (L) Higher magnification (zoom-in) view of yellow boxed area in K. Scale bar, 2 µm. Abp140-3GFP marked cables align with septin axial pillars in a cell where the bud has been sheared (yellow arrowheads).
DISCUSSION
We initiated this study by asking whether Hof1 might have Bnr1-independent functions in actin cable regulation, and discovered a new function for Hof1 in directly binding and organizing actin cables at the bud neck, and thereby facilitating polarized secretion. Consistent with our in vivo observations, we found that purified Hof1 bundles actin filaments in vitro, and physically links actin and septin filaments. Our results suggest that in S. cerevisiae, Hof1 serves as a bridge between septins and actin, whereas in some other species, e.g., Drosophila, septins bind directly to F-actin (Mavrakis et al., 2014). Our superresolution imaging reveals that Hof1, septins, and Bnr1 are organized into evenly spaced axial pillars at the bud neck (running parallel to the mother–bud axis), which coalign with emerging actin cables. Acute disruption of septin organization using a temperature-sensitive septin mutant, cdc10-4, disrupted cable organization similar to hof1∆. Together, these observations indicate that Hof1 and septins form a molecular scaffold at the bud neck that interacts with actin cables and helps orient and distribute them in the mother cell to facilitate polarized intracellular transport.
Our work assigns a new molecular function to the C-terminal “linker” domain of Hof1 in binding and organizing actin filaments. Until now, no specific functional roles have been assigned to this region of Hof1, and in general this region of F-BAR proteins has been poorly understood. Interestingly, a recent study in Schizosaccharomyces pombe showed that the equivalent region of the F-BAR protein Cdc15 is critical in vivo for cytokinesis (Mangione et al., 2019). This raises the intriguing possibility that this in vivo function of Cdc15 stems from molecular interactions of its linker region related to those we describe here for Hof1. In Hof1, the N-terminal F-BAR domain forms a dimer that binds to both the FH2 domain of Bnr1 and lipids (Moravcevic et al., 2015; Garabedian et al., 2018). The adjacent coiled-coil (CC2) domain in Hof1 interacts with septins and is required for Hof1 localization (Oh et al., 2013). The SH3 domain of Hof1 interacts with the proline-rich FH1 domain of Bnr1 to promote formin inhibition (Graziano et al., 2014; Garabedian et al., 2018), and later during cytokinesis binds to a separate set of Hof1 ligands (Vallen et al., 2000; Blondel et al., 2005; Nishihama et al., 2009; Oh et al., 2013, 2017; Wang et al., 2018). Taken together, these observations show that Hof1 is a multifunctional actin regulator, with its F-BAR and SH3 domains primarily regulating formin (Bnr1)-mediated actin nucleation, and its linker domain binding and organizing actin cables. Both of these functions contribute to building a proper actin cable network in cells.
How then does Hof1 coordinate its two distinct actin regulatory functions in vivo? During polarized cell growth, cables generated by Bni1 at the bud tip are released from the cortex and extend into the mother compartment, where it has been suggested that they may be incorporated (or stitched) into cables polymerized by Bnr1 (Buttery et al., 2007; Yu et al., 2011). We propose that Bni1-generated cables are transiently captured at bud neck by the Hof1-septin scaffold, through direct interactions with the Hof1 linker domain, and that these interactions serve to orient and spatially distribute cables within the mother cell (Figure 7). The regular spacing of the septin-Hof1 pillars lends itself well to evenly dispersing cables to different regions of the mother cells, for more efficient polarized vesicle traffic. This cable organization function may even be coupled to Hof1’s role as an inhibitor of Bnr1-mediated actin nucleation. Hof1 and Bnr1 are both recruited to the bud neck by septins, via independent mechanisms (Gao et al., 2010; Buttery et al., 2012; Graziano et al., 2014), and here we show that they colocalize to the axial septin pillars. Hof1 and Bnr1 also strongly interact to form stable complexes consisting of an elongated Hof1 dimer bound to two well-separated Bnr1 dimers positioned at either end of the complex (Garabedian et al., 2018). Based on these observations, we hypothesize that Hof1 may recruit Bni1-generated actin cables assembled in the bud and align them with Bnr1 dimers at the bud neck. In doing so, this would bring Bni1- and Bnr1-generated cables together as they enter the mother cell (Figure 7). It is also possible that capture of Bni1-generated cables at the neck by Hof1 could trigger the release of Bnr1 inhibition by Hof1, promoting Bnr1-mediated actin polymerization to thicken and reinforce cables as they enter the mother cell.
FIGURE 7:

Model for Hof1-septin–mediated actin cable organization. Working model showing actin cables polymerized in the bud compartment by Bni1 being captured and organized at the bud neck by a Hof1 and septin scaffold in wild-type cells. However, in hof1∆ cells this mechanism is disrupted, resulting in aberrant extension angles for cables entering the mother cell, and an entangled actin cable network.
Our data also have important implications for septin architecture in S. cerevisiae. Diffraction-limited light microscopy studies have described septins as forming a collar at the bud neck throughout polarized cell growth, where the collar later splits at cytokinesis into two rings (Marquardt et al., 2019). Our superresolution (SIM) imaging suggests that during polarized cell growth the collar is more elaborately organized into axial pillars that are evenly spaced ∼200 nm apart. We observed this pattern using integrated tags on four separate septin proteins and two different septin-associated proteins (Hof1 and Bnr1). Further, a similar pattern was observed using Airyscan superresolution microscopy (Supplemental Figure S5 and Supplemental Video 2), suggesting that the arrangement is not an artifact of the SIM method. It is worth noting that possibly related axial pillars were reported years ago in mating yeast cells (Longtine et al., 1998), leaving it unclear why mating and mitotically dividing cells had these differences in septin organization. Our results raise the possibility that septin organization is actually similar in both conditions, but that the distances separating the septin pillars in mating cells may be slightly larger, accounting for their detection by earlier studies using standard light microscopy.
Movie S2.
Three-dimensional projection of yeast septin scaffold. A rotating three-dimensional projection of yeast septins (Cdc10-GFP) at the bud neck from live cells imaged by Airyscan microscopy.
EM studies have shown that yeast septin ultrastructure changes at different stages of the cell cycle. Rodal and coworkers first examined septin ultrastructure by platinum-replica EM on spheroplasted yeast cells from an asynchronous population. They observed two types of septin structures (abundant highly ordered “gauzes” and more rare loosely associated rings), and proposed that these types of septin networks might be assembled at different stages of the cell cycle (Rodal et al., 2005). Subsequently, Ong and colleagues elegantly adapted and extended this approach using correlative light and electron microscopy (CLEM) on synchronized yeast cells. They powerfully demonstrated that septin ultrastructure indeed changes over the cell cycle, switching from networks composed of primarily axial filaments during early bud emergence to networks composed of primarily circumferential filaments at cytokinesis (Ong et al., 2014). Using a complementary approach, Bertin and coworkers performed electron tomography on freeze-substituted large-budded yeast cells (in and around the time of cell division), and observed circumferential filaments interconnected by short axial filaments (Bertin et al., 2012). Notably, none of these studies examined septin organization at the stages between bud emergence and cytokinesis, i.e., during polarized cell growth. Some aspects of our SIM imaging on live yeast cells agree with the EM studies. Early in the cell cycle, we predominantly observe axial filaments, and later in the cycle we observe the collar splitting into two rings. However, during polarized cell growth, we observe that septins are organized into broad axial pillars, evenly spaced ∼200 nm apart. This observation suggests that the axial filaments seen earlier in the EM studies during bud emergence may subsequently become organized into thick pillars during polarized growth. Our results also suggest that some of the higher organization observed in living cells by SIM may be altered in EM studies due to cell fixation and/or sample preparation. In light of these findings, it may be useful for future studies to examine septin organization at different stages of the yeast cell cycle using superresolution correlative light and electron microscopy (SR-CLEM).
In conclusion, our work has revealed an unanticipated role for Hof1 in directly binding actin filaments and linking them to septin networks, which is critical for orienting and spatially organizing actin cables as they enter the mother cell. Additionally, our findings provide new insights into septin organization at the bud neck, and suggest that septin organization into pillars concentrates actin-binding proteins into the same regularly spaced pillars, creating actin cable organization centers. These findings also have broad implications for understanding how cells establish and maintain the orientation of their actin networks, suggesting that one mechanism is to pattern an actin-binding protein at the cell cortex. In this specific case, an actin-binding protein (Hof1) is patterned at the cell cortex through its molecular interaction with septins. Indeed, a similar scheme is used in cytokinesis, where anillin physically links actin filaments to septins to help organize the contractile ring (Oegema et al., 2000; Kinoshita et al., 2002; Tasto et al., 2003). Thus, while one critical factor in establishing actin network geometry is the local recruitment and activation of actin nucleators, another key determinant is the patterning of network attachment points at the cell cortex.
MATERIALS AND METHODS
Plasmids and yeast strains
Standard methods were followed for S. cerevisiae work (Sambrook et al., 1989; Guthrie and Fink, 1991). Low copy centromeric plasmids for expression of GFP-Sec4 and genomic integration of ABP140-3GFP::LEU2 were gifts and have been described (Calero et al., 2003; Buttery et al., 2007). Plasmids used to express MBP-Hof1 NT (1–340) and MBP-F-BAR (1–275) in Escherichia coli were gifts from E. Bi (University of Pennsylvania, Philadelphia, PA) and have also been described (Oh et al., 2013). Plasmids used for expression of MBP-Hof1-FL-6xHis (1–669), GST-Hof1-CT (350–669), GST-Linker (350–575), and GST-SH3 (576–669) have been previously described (Graziano et al., 2014; Garabedian et al., 2018). The GST-Hof1-CC2 plasmid (pBG2197) was generated by PCR amplification of the Hof1 CC2 (forward, 5′-ATCGCTGGATCCCATAAGACTTCCAAAGGTGACATG-3′; reverse, 5′-TCAGTAGCGGCCGCCTAGACTTCTGGAGATGGCAATGG-3′) and cloned in-frame with GST into a pET-GST vector using the BamHI and NotI sites. Plasmids used to express and purify yeast septins were gifts from T. Gronemeyer (Ulm University, Ulm, Germany; Renz et al., 2013). The hof1∆linker (BGY4210), hof1∆linker bnr1∆::KANMX6 (BGY4247), and hof1∆linker-GFP::HIS3 strains (BGY4211) were generated by CRISPR-based gene editing as described (Anand et al., 2017). Complementary 20-nt DNA oligos (Integrated DNA Technologies) homologous to nucleotides 1400–1419 in the linker domain were generated and included 3′ overhangs for a BplI cut site. These were duplexed and cloned into the BplI restriction site in pJH2972 (Anand et al., 2017) to be used as the gRNA for Cas9-mediated gene editing. This plasmid was then cotransformed with 80 nt oligos homologous to regions flanking the linker domain (5′-TCCAGAAGTCACCATGGCTACACAATTTAGAAATTCCACAGATGAGAGAGGTGTGGTCAGGGATAGAGGTATTACTGTTA-3′). Transformants were grown on synthetic media lacking uracil to select for the Cas9 plasmid. Internal deletion of the linker domain was confirmed by PCR and sequencing. The Cas9 plasmid was then removed by growing the hof1∆linker strains on 5-FOA. ABP140-3xGFP::LEU2 was integrated into yeast strains after cutting pB1994 with NdeI and standard yeast transformation. Cdc10-GFP::HIS3, Cdc11-GFP::HIS3, and Shs1-GFP::HIS3 strains were obtained from the yeast GFP collection (Huh et al., 2003; ThermoFisher Scientific). The cdc10-4::KANMX septin mutant strain (CBY06421) have been previously described (Li et al., 2011; Weems et al., 2014). Strains expressing Cdc3-yomApple::HIS3 (BGY4123) were generated by integrating a C-terminal tag using conventional methods (Buttery et al., 2007; Lee et al., 2013; Slubowski et al., 2015). The Cdc3-yomApple::HIS3 Hof1-Envy::HIS5 strain (BGY4130) and the Cdc3-yomApple::HIS3 Bnr1-Envy::HIS5 strain (BGY4148) were generated by cross. Yeast strains in this study are in the Research Genetics background (Mata, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0 or Matα, ura3Δ0, leu2Δ0, his3Δ1, lys2Δ0).
Fixed cell imaging and analysis
Yeast cells were grown to midlog phase (OD600 0.4–0.8) in Yeast Extract-Peptone-Dextrose (YEPD) media at 25°C, fixed in 3.7% formaldehyde for 45 min at room temperature (RT), and washed three times with 1× PBS (phosphate-buffered saline). Cells were stained overnight with Alexa Fluor 488 phalloidin (Life Technologies) and then washed twice with 1× PBS. For experiments in which actin patches were removed, cells were treated with 100 μM CK666 for 10–15 min at 25°C before formaldehyde fixation. For comparing actin cable organization in cdc10-4 and wild-type cells at elevated temperature, cells were grown to midlog phase at 22°C and then shifted to 34°C for 15 min, fixed, and then fixed and stained for F-actin as above. Fixed cells were imaged in 1× PBS at RT by SIM on a Ti-2 SIM-E inverted microscope with a 100× oil objective (NA 1.49), and Hamamatsu Orca Flash 4.0 camera controlled by NIS-Elements software. In general, 100–200 ms exposure times at 50% laser power were used to image Alexa Fluor 488 phalloidin-stained actin. Images were captured at 0.1-µm steps. A violet-to-red diffraction grating was applied at three angles and five phases of illumination, resulting in 15 raw images for SIM. Raw images were then reconstructed with the default stack reconstruction settings within NIS-Elements software. Using SIM reconstructions, individual cells were cropped, background was subtracted from maximum intensity images in ImageJ, and actin cables were analyzed using an open-source program for biopolymer networks, SOAX (Xu et al., 2015). For all SOAX analyses, default settings were used, with two exceptions, to optimize detection of cables: R-threshold value was set to 0.005 and k-stretch factor was set to 2.0. The number of actin cable segments and intersections was automatically generated by the software. For CoV analysis and measurements of cable thickness, imaging was performed by confocal microscopy, allowing for a larger number of cells to be analyzed. Confocal imaging was performed on an i-E upright microscope with a CSU-W1 spinning disk head, 100× oil objective (NA 1.4), and an Ixon 897 Ultra-CCD camera controlled by NIS-Elements software. In general, 100–200 ms exposure times at 80–100% laser power were used to image Alexa Fluor 488 phalloidin-stained actin. The CoV measurements were made by first tracing the outline of the mother cell compartment in ImageJ, and then measuring the mean fluorescence of actin cable staining and the SD. The CoV is a ratio of the SD over the mean. Wild-type cells typically have well-defined, brightly stained cables against a dark cellular background, yielding a high SD, and thus a higher CoV. Mutant cells with more disorganized and dispersed actin cable networks give rise to lower stand deviation values and, consequently lower CoVs. Cable thickness was measured in single Z-slices in ImageJ by drawing a line thick enough to encompass an entire cable along the length of a phalloidin-stained cable and measuring the fluorescence density (signal per unit area) as a metric for cable thickness.
Live cell imaging and analysis
For measuring in vivo actin cable extension rates, yeast cells expressing Abp140-3xGFP from the endogenous locus were grown to midlog phase (OD600 0.4–0.8) in synthetic media at 25°C. Live cells in media were mounted on slides with coverslips and immediately imaged at RT on an i-E upright confocal microscope (Nikon Instruments) with a CSU-W1 spinning disk head (Yokogawa), 100× oil objective (NA 1.4; Nikon Instruments), and an Ixon 897 Ultra-CCD camera (Andor Technology) controlled by NIS-Elements software (Nikon Instruments). An exposure time of 80 ms at 100% laser power was used. Actin cables were analyzed in a single optical plane, capturing images for 2 min. Individual cells were cropped using ImageJ. Actin cable extension speeds and angles were measured using custom software written in MATLAB (MathWorks; Eskin et al., 2016). For each cell, a movie is opened in the software and the user defines the cell boundaries and mother–bud axis. Then the user selects the starting point and ending points for the actin cable. The program calculates the velocity of the cable and the angle of its extension relative to the mother–bud axis. For imaging secretory vesicle traffic, wild-type and mutant yeast strains were transformed with a CEN plasmid expressing GFP-Sec4 (Calero et al., 2003). Cells were grown to midlog phase (OD600 0.4–0.8) in synthetic selective media at 25°C, then mounted on a microscope slide and imaged at RT on an inverted Ti200 microscope equipped with an Intensilight excitation source (Nikon Instruments), 100× objective (NA 1.30; Nikon Instruments), 1.5× magnifier, and iXon EMCCD camera. Exposure times of 100–200 ms at 20–50% intensity were used to image cells for 1–2 min. Focus was maintained with the Perfect Focus System (Nikon Instruments). Movies were subsequently analyzed in ImageJ. Secretory vesicle traffic was monitored within the mother cells of each strain by manually tracking the positions over time for 5–10 GFP-Sec4 puncta in each of 15 or more cells. Tortuosity measurements were made by dividing the length of the path (from the initial point of movement to the bud neck) by the distance between the point of origin and the bud neck. For imaging fluorescently tagged yeast septins, Hof1-Envy, and Bnr1-Envy cells were grown in synthetic media as described above. Live cells in media were mounted on slides with coverslips and immediately imaged by SIM on a Ti-2 SIM-E inverted microscope with a 100× oil objective (NA 1.49), and Hamamatsu Orca Flash 4.0 camera controlled by NIS-Elements software. Cdc3-yomApple strains were imaged at 200–300 ms exposure times while GFP and Envy tagged proteins were imaged at 100–00 ms exposures. Images were captured at 0.1-µm steps at 50% laser power in each case. A violet-to-red diffraction grating was applied at three angles and five phases of illumination, resulting in 15 raw images for SIM. Raw images were then reconstructed with the default stack reconstruction settings within NIS-Elements software. Distances between septin pillars were measured by drawing a line between individual pillars in ImageJ. Airyscan images of Hof1-GFP, Hof1∆linker-GFP, and fluorescently tagged yeast septins (Cdc3-yomApple and Cdc10-GFP) were acquired on a Zeiss AxioObserver LSM 880 equipped with an “Airyscan” superresolution module and gallenium arsenide phosphide detectors (GaAsP) using a Plan-Apochromat 63×/1.40 oil DIC M27 objective. GFP-tagged proteins were excited using an argon laser (488) and mApple-tagged proteins were excited using a DPSS 561-10 laser, each with a 0.5% laser power, pixel dwell time of 1.05 s, and z-step size of 0.175 µm. Image acquisition and Airyscan processing were done using Zen 2 (black) software. An analysis of Hof1-GFP and Hof1∆linker-GFP expression levels was performed using ImageJ. Sum intensity projections were background subtracted and total GFP signal in a single cell was measured by calculating the integrated density (product of the area and the mean fluorescence value) per cell. To compare signal intensities at the neck, the mean GFP signal at the neck of each cell was measured from a 1.1-µm-wide line drawn across the neck.
Protein purification
Rabbit muscle actin was purified as has been described (Spudich and Watt, 1971) from acetone powder made from frozen ground skeletal muscle of young rabbits (PelFreez). Lyophilized acetone powder stored at −80°C was mechanically sheered in a coffee grinder, resuspended in G-buffer (5 mM Tris-HCl, pH 7.5, 0.5 mM dithiothreitol [DTT], 0.2 mM ATP, and 0.1 mM CaCl2). The actin was then cleared by centrifugation for 20 min at 28,403 × g. Actin was polymerized by the addition of 2 mM MgCl2 and 50 mM NaCl and incubated overnight at 4°C. F-Actin was pelleted by centrifugation for 2.5 h at 113,613 × g. The F-actin pellet was solubilized by Dounce homogenization and dialyzed against G-buffer for 60 h at 4°C to depolymerize the actin. The G-actin was then precleared at 264,499 × g and gel filtered on an S200 (16/60) column (GE Healthcare) equilibrated in G-buffer. Peak actin-containing fractions were stored at 4°C and used within a 2-wk period. To label actin with Oregon green (OG) dye on cysteine 374, an F-actin pellet was dounced and dialyzed against G-buffer lacking DTT. The G-actin was polymerized by adding an equal volume of 2× labeling buffer (50 mM imidazole, pH 7.5, 200 mM KCl, 0.3 mM ATP, and 4 mM MgCl2). After a 5-min incubation at 25°C, the actin was mixed with a fivefold molar excess of OG-488 iodoacetamide (Invitrogen), resuspended in anhydrous dimethylformamide, and then incubated in the dark for 15 h at 4°C. The labeled F-actin was pelleted as above, and the pellet was rinsed with G-buffer, depolymerized by Dounce homogenization, and dialyzed against G-buffer for 60 h at 4°C. Labeled, monomeric actin was then applied to an S200 (16/60) gel filtration column as above. Peak fractions were dialyzed for 15 h against G-buffer with 50% glycerol and stored at −20°C.
MBP-Hof1 constructs (F-BAR and NT) were expressed in BL21(DE3) E. coli cells with the pRARE plasmid (MilliporeSigma). Bacteria were grown to late log phase (OD600 0.7–0.9) in terrific broth supplemented with ampicillin and chloramphenicol to maintain selection of the expression plasmid and the pRARE plasmid, respectively. Expression was induced with 0.8 mM Isopropyl β-d-1-thiogalactopyranoside (IPTG) overnight at 18°C, and then cells were pelleted and stored at −80°C. Cell pellets were thawed, resuspended in lysis buffer (20 mM Tris-HCl, pH 8.0, and 300 mM NaCl) supplemented with a protease inhibitor cocktail (1 mM phenylmethylsulfonyl fluoride and 0.5 μM each of pepstatin A, antipain, leupeptin, aprotinin, and chymostatin), and lysed by treatment with 1 mg/ml lysozyme and sonication. Lysates were cleared by centrifugation at 20,064 × g for 20 min in an F21S-8 × 50y rotor (Thermo Fisher Scientific), and the supernatant (20–30 ml) was mixed with 0.5 ml of amylose resin (New England Biolabs) and rotated at 4°C for 1 h. The resin was then washed five times with 5 ml of the lysis buffer. Hof1 constructs were eluted with lysis buffer plus 20 mM maltose and 1 mM DTT. Finally, eluted protein was exchanged into HEK buffer (20 mM HEPES, pH 7.5, 1 mM EDTA, and 50 mM KCl) supplemented with 10% glycerol (vol/vol) and 1 mM DTT using a PD10 desalting column (GE Life Sciences) and collected in 0.3-ml fractions. Peak fractions were aliquoted and snap frozen in liquid N2 and stored at −80°C.
MBP-Hof1-FL-6His and MBP-Hof1-FL-SNAP-6His were expressed and purified as above except for an initial nickel affinity purification. Cleared lysates were mixed with 0.5 ml Ni-NTA agarose resin (Qiagen, Valencia, CA) and rotated at 4°C for 1 h. The beads were then washed five times with 5 ml of the lysis buffer containing 50 mM imidazole. MBP-Hof1-FL-6His was eluted with lysis buffer with 300 mM imidazole in 0.5-ml fractions. Eluted proteins were diluted fourfold with the lysis buffer lacking imidazole and then further purified with amylose resin as described above. To label MBP-Hof1-FL-SNAP-6His, peak fractions were mixed with a fivefold molar excess of BG-549 SNAP dye overnight at 4°C. The protein was then exchanged into HEK buffer supplemented with 10% glycerol and 1 mM DTT on a PD10 desalting column as described above. For EM experiments, all proteins were purified as above except the final desalting buffer was HEK (20 mM HEPES, pH 7.5, 1 mM EDTA, and 50 mM KCl) supplemented with 150 mM NaCl and 1 mM DTT.
GST-Hof1-CT-6His, GST-Hof1-Linker-6His, and GST-Hof1-SH3-6His were expressed and purified as above using nickel affinity followed by glutathione resin. Peak fractions from Ni-NTA purification were pooled, diluted fourfold with buffer lacking imidazole, and mixed with glutathione agarose beads (Thermo Fisher Scientific) and rotated at 4°C for 1 h. The resin was then washed five times with 5 ml of lysis buffer, and protein was eluted using lysis buffer plus 30 mM glutathione and 1 mM DTT. The eluted protein was exchanged into HEK buffer with 10% glycerol (vol/vol) and 1 mM DTT on a PD10 desalting column (GE Life Sciences), aliquoted, snap frozen in liquid N2, and stored at −80°C. For GST-Hof1-CC2, a single GST purification step was used, omitting the nickel step.
Yeast septins were purified as previously described (Renz et al., 2013). Two bicistronic plasmids encoding yeast septins (Flag-Cdc3/Cdc10-SNAP and 6His-Cdc12/Cdc11-S-Tag) were expressed in BL21(DE3) E. coli cells. Cells were grown to late log phase (OD600 of 0.7–0.9) in terrific broth supplemented with ampicillin and chloramphenicol to maintain selection of both expression plasmids. Expression was induced with 0.4 mM IPTG overnight at 18°C, and then cells were pelleted and stored at −80˚°C. The pellet was then thawed and resuspended in septin resuspension buffer (50 mM Tris-HCl, pH 8, 300 mM NaCl, 2 mM MgCl2, 15 mM imidazole, 12% glycerol [vol/vol]) supplemented with 0.1% Tween 20, 40 μM GDP, 5 mM β-mercaptoethanol, and the same protease inhibitor cocktail as described above. Cells were then lysed with 1 mg/ml lysozyme and sonication. Lysates were cleared by centrifugation as above. The cleared lysate was then loaded onto a 1 ml HisTrap column (GE Healthcare) equilibrated with IMAC buffer A (300 mM NaCl, 2 mM MgCl2, 15 mM imidazole, and 5 mM DTT) and eluted with a three-step elution: 10 column volumes of each of 3%, 25%, and 100% of IMAC Buffer B (50 mM Tris-HCl, pH 8, 500 mM NaCl, 2 mM MgCl2, 500 mM imidazole, 5 mM DTT, and 12% glycerol [vol/vol]). The septins eluted at 25% IMAC Buffer B. Eluted septins were pooled and buffered in BisTris 7.5 using a PD10 desalting column. This was then applied to 5 ml HiTrapQ column (GE Healthcare) for anion exchange chromatography with a linear elution gradient (7 column volumes). Septins eluted at 600 mM NaCl. The eluted protein was exchanged and concentrated in storage buffer (50 mM Tris-HCl, pH 8, 300 mM NaCl, 10% glycerol [vol/vol]) and stored at −80°C.
TIRF microscopy and analysis
For TIRF microscopy experiments, 24 × 60-mm coverslips (Thermo Fisher Scientific) were cleaned by sonication in dish detergent for 60 min and rinsed thoroughly in sterile water, followed by sonication in 1 M HCl for 20 min and 1 M KOH for 20 min and then sonication in ethanol for 60 min. Coverslips were then washed extensively with sterile water, dried in an N2 stream, and coated with 200 μl of 80% ethanol, pH 2.0, 2 mg/ml methoxy-poly(ethylene glycol)-silane, and 2 μg/ml biotin-poly(ethylene glycol)-silane (Laysan Bio). Coverslips were incubated at 70°C for 1–3 d before use. Flow cells were assembled just before imaging by rinsing coverslips extensively with sterile water, attaching coverslips to a plastic flow chamber (Ibidi) using double-sided tape 2.5 cm × 2 mm × 120 μm (Grace Bio-Labs), and sealing both ends with epoxy resin (Devcon).
Just before each experiment, flow cells were incubated for 30 s with HBSA (20 mM HEPES, pH 7.5, 1 mM EDTA, 50 mM KCl, and 1% bovine serum albumin) and then washed with TIRF buffer (10 mM imidazole, 50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 0.2 mM ATP, 10 mM DTT, 15 mM glucose, 20 μg/ml glucose oxidase, and 0.5% methylcellulose [4000 cP], pH 7.4). Indicated proteins were mixed in TIRF buffer and then flowed into the chamber. For actin bundling, 2 µM actin (10% OG-labeled) was polymerized in the TIRF chamber for 15–20 min and then three volumes of TIRF buffer was flowed through to wash away free G-actin. Indicated concentrations of Hof1 polypeptides were then flowed in during image acquisition. For assays with actin and septins, 1 µM actin (10% OG-labeled) and 50 nM septins (SNAP549) were mixed together in TIRF buffer with or without 50 nM MBP-Hof1-FL-6His and flowed into the TIRF chamber. Images were acquired in ambient temperature using an inverted Ti200 TIRF microscope (Nikon Instruments) equipped with 100-mW solid-state lasers (Agilent Technologies), a CFI Apo 60× TIRF objective (NA 1.49; Nikon Instruments), and an iXon EMCCD camera (Andor Technology). Focus was maintained with the Perfect Focus System (Nikon Instruments).
Image analysis was performed in ImageJ. TIRF images were background subtracted using a rolling ball radius of 5 pixels. To quantify the effects of Hof1 polypeptides on actin bundling, the mean and SD of the actin fluorescence intensity were measured and used to calculate the CoV (ratio of the SD over the mean). Fields of view (FOVs) with low actin bundling have many single filaments over the entire view and a low SD. FOVs with high actin bundling have bright actin bundles against a dark background resulting in a higher SD and therefore CoV. To quantify the effect of Hof1 on septin and actin bundling, the colocalization of actin with septin filaments was analyzed using the Coloc 2 ImageJ plugin to calculate the Pearson’s R correlation coefficient for all FOVs.
EM
To examine actin bundling, 500 nM MBP-Hof1-FL-6His was mixed with 1 µM preformed F-actin filaments. Then, 3 µl of protein complexes were applied to the carbon-coated glow-discharged copper grids. Samples were blotted and negatively stained with 1.0% uranyl acetate two times for 30 s each. To examine Hof1–septin interactions, 500 nM MBP-Hof1-FL-6His was mixed with 50 nM of septin rods and applied to glow-discharged grids and negatively stained as above. Air-dried grids were imaged on a JEOL 2100 transmission electron microscope equipped with a Gatan Ultrascan 1000XP CCD camera and operated at 200 kV. The data were collected under low-dose conditions at a ∼1.5 μm defocus and 40,000 magnification resulting in 2.5 A pixel size. Class averages of Hof1 particles on actin bundles and on the carbon substrate shown in Figure 3 were collected in EMAN2 (Tang et al., 2007). A total of 50 particles for each set were cut from the micrographs and aligned against one another. The total sums of bound Hof1 and free Hof1 have comparable size and shape.
Statistical analysis
For comparisons of two categories, an unpaired, two-tailed t test was used. To compare more than two categories, one-way ANOVA was used. In all cases, n.s. indicates no significance and p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
Supplementary Material
Acknowledgments
We are grateful to Alan Tartakoff for hosting M.V.G. in his lab, to Andrey Moiseenko for help with EM data collection, and to Erfei Bi for advice throughout this project. We are also grateful to Alison Wirshing, Julian Eskin, M. Angeles Juanes, Thomas Rands, and Luther Pollard for comments on the manuscript. This work was supported by a National Institutes of Health R35 award (GM-134895) to B.L.G., and the Brandeis National Science Foundation Materials Research Science and Engineering Center (DMR-1420382). O.S. was supported by a grant from the Russian Foundation for Basic Research (Grant no.19-34-90178) and by a Moscow State University (MSU) grant for Leading Scientific Schools “Depository of the Living Systems” in the frame of the MSU Development Program, and A.V. was supported by the Council on grants of the President of the Russian Federation (Grant no. MK-2614.2018.4. Electron microscopy). We also thank the User Facility Center for Electron Microscopy in Life Sciences at Moscow State University. The JEOL2100 electron microscope used in this study was supported by the Ministry of Science and Higher Education of the Russian Federation (RFMEFI61919×0014).
Abbreviations used:
- F-BAR
Fer/Cip4 homology–Bin amphiphysin Rvs
- FH
formin homology
- GST
glutathione S-transferase
- MBP
maltose binding protein
- OG
Oregon Green
- SH3
Src homology 3
- SIM
structured illumination microscopy
- TIRF
total internal reflection fluorescence.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-12-0693) on June 24, 2020.
REFERENCES
- Anand R, Memisoglu G, Haber J. (2017). Cas9-mediated gene editing in Saccharomyces cerevisiae. Protoc Exch. 10.1038/protex.2017.021a [DOI] [Google Scholar]
- Bertin A, McMurray MA, Pierson J, Thai L, McDonald KL, Zehr EA, Garcia G, 3rd, Peters P, Thorner J, Nogales E. (2012). Three-dimensional ultrastructure of the septin filament network in Saccharomyces cerevisiae. Mol Biol Cell , 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla M, Gladfelter AS, Kovar DR, Lee WL. (2015). Cytoskeletal dynamics: a view from the membrane. J Cell Biol , 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. (2014). Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev , 235–263. [DOI] [PubMed] [Google Scholar]
- Blondel M, Bach S, Bamps S, Dobbelaere J, Wiget P, Longaretti C, Barral Y, Meijer L, Peter M. (2005). Degradation of Hof1 by SCF(Grr1) is important for actomyosin contraction during cytokinesis in yeast. EMBO J , 1440–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TA, Christensen JR, Barone E, Suarez C, Sirotkin V, Kovar DR. (2014). Homeostatic actin cytoskeleton networks are regulated by assembly factor competition for monomers. Curr Biol , 579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttery SM, Kono K, Stokasimov E, Pellman D. (2012). Regulation of the formin Bnr1 by septins anda MARK/Par1-family septin-associated kinase. Mol Biol Cell , 4041–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttery SM, Yoshida S, Pellman D. (2007). Yeast formins Bni1 and Bnr1 utilize different modes of cortical interaction during the assembly of actin cables. Mol Biol Cell , 1826–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero M, Chen CZ, Zhu W, Winand N, Havas KA, Gilbert PM, Burd CG, Collins RN. (2003). Dual prenylation is required for Rab protein localization and function. Mol Biol Cell , 1852–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campellone KG, Welch MD. (2010). A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol , 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra ES, Higgs HN. (2007). The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol , 1110–1121. [DOI] [PubMed] [Google Scholar]
- Chesarone MA, DuPage AG, Goode BL. (2010). Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol , 62–74. [DOI] [PubMed] [Google Scholar]
- Chesarone-Cataldo M, Guerin C, Yu JH, Wedlich-Soldner R, Blanchoin L, Goode BL. (2011). The myosin passenger protein Smy1 controls actin cable structure and dynamics by acting as a formin damper. Dev Cell , 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskin JA, Rankova A, Johnston AB, Alioto SL, Goode BL. (2016). Common formin-regulating sequences in Smy1 and Bud14 are required for the control of actin cable assembly in vivo. Mol Biol Cell , 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista M, Pruyne D, Amberg DC, Boone C, Bretscher A. (2002). Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat Cell Biol , 260–269. [DOI] [PubMed] [Google Scholar]
- Gao L, Liu W, Bretscher A. (2010). The yeast formin Bnr1p has two localization regions that show spatially and temporally distinct association with septin structures. Mol Biol Cell , 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garabedian MV, Stanishneva-Konovalova T, Lou C, Rands TJ, Pollard LW, Sokolova OS, Goode BL. (2018). Integrated control of formin-mediated actin assembly by a stationary inhibitor and a mobile activator. J Cell Biol , 3512–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladfelter AS, Pringle JR, Lew DJ. (2001). The septin cortex at the yeast mother-bud neck. Curr Opin Microbiol , 681–689. [DOI] [PubMed] [Google Scholar]
- Graziano BR, Yu HY, Alioto SL, Eskin JA, Ydenberg CA, Waterman DP, Garabedian M, Goode BL. (2014). The F-BAR protein Hof1 tunes formin activity to sculpt actin cables during polarized growth. Mol Biol Cell , 1730–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. (1991). Guide to yeast genetics and molecular biology. Methods Enzymol , 1–863. [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. (2003). Global analysis of protein localization in budding yeast. Nature , 686–691. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Field CM, Coughlin ML, Straight AF, Mitchison TJ. (2002). Self- and actin-templated assembly of mammalian septins. Dev Cell , 791–802. [DOI] [PubMed] [Google Scholar]
- Kostan J, Salzer U, Orlova A, Toro I, Hodnik V, Senju Y, Zou J, Schreiner C, Steiner J, Merilainen J, et al (2014). Direct interaction of actin filaments with F-BAR protein pacsin2. EMBO Rep , 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lim WA, Thorn KS. (2013). Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS One , e67902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Vizeacoumar FJ, Bahr S, Li J, Warringer J, Vizeacoumar FS, Min R, Vandersluis B, Bellay J, Devit M, et al. (2011). Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat Biotechnol , 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott J, Li R. (1998). Dual function of Cyk2, a cdc15/PSTPIP family protein, in regulating actomyosin ring dynamics and septin distribution. J Cell Biol , 1947–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, Fares H, Pringle JR. (1998). Role of the yeast Gin4p protein kinase in septin assembly and the relationship between septin assembly and septin function. J Cell Biol , 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangione MC, Snider CE, Gould KL. (2019). The intrinsically disordered region of the cytokinetic F-BAR protein Cdc15 provides a unique essential function in maintenance of cytokinetic ring integrity. Mol Biol Cell , 2790–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt J, Chen X, Bi E. (2019). Architecture, remodeling, and functions of the septin cytoskeleton. Cytoskeleton , 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis M, Azou-Gros Y, Tsai FC, Alvarado J, Bertin A, Iv F, Kress A, Brasselet S, Koenderink GH, Lecuit T. (2014). Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nat Cell Biol , 322–334. [DOI] [PubMed] [Google Scholar]
- Moravcevic K, Alvarado D, Schmitz KR, Kenniston JA, Mendrola JM, Ferguson KM, Lemmon MA. (2015). Comparison of Saccharomyces cerevisiae F-BAR domain structures reveals a conserved inositol phosphate binding site. Structure , 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley JB, Goode BL. (2006). The yeast actin cytoskeleton: from cellular function to biochemical mechanism. Microbiol Mol Biol Rev , 605–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihama R, Schreiter JH, Onishi M, Vallen EA, Hanna J, Moravcevic K, Lippincott MF, Han H, Lemmon MA, Pringle JR, et al (2009). Role of Inn1 and its interactions with Hof1 and Cyk3 in promoting cleavage furrow and septum formation in S. cerevisiae. J Cell Biol , 995–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. (2009). Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature , 1031–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oegema K, Savoian MS, Mitchison TJ, Field CM. (2000). Functional analysis of a human homologue of the Drosophila actin binding protein anillin suggests a role in cytokinesis. J Cell Biol , 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Schreiter J, Nishihama R, Wloka C, Bi E. (2013). Targeting and functional mechanisms of the cytokinesis-related F-BAR protein Hof1 during the cell cycle. Mol Biol Cell , 1305–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Schreiter JH, Okada H, Wloka C, Okada S, Yan D, Duan X, Bi E. (2017). Hof1 and Chs4 Interact via F-BAR d and Sel1-like repeats to control extracellular matrix deposition during cytokinesis. Curr Biol , 2878–2886.e2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Ravi H, Smith EM, Goode BL. (2006). Aip1 and cofilin promote rapid turnover of yeast actin patches and cables: a coordinated mechanism for severing and capping filaments. Mol Biol Cell , 2855–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong K, Wloka C, Okada S, Svitkina T, Bi E. (2014). Architecture and dynamic remodelling of the septin cytoskeleton during the cell cycle. Nat. Commun , 5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne D, Gao L, Bi E, Bretscher A. (2004). Stable and dynamic axes of polarity use distinct formin isoforms in budding yeast. Mol Biol Cell , 4971–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz C, Johnsson N, Gronemeyer T. (2013). An efficient protocol for the purification and labeling of entire yeast septin rods from E. coli for quantitative in vitro experimentation. BMC Biotechnol , 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal AA, Kozubowski L, Goode BL, Drubin DG, Hartwig JH. (2005). Actin and septin ultrastructures at the budding yeast cell cortex. Mol Biol Cell , 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagot I, Klee SK, Pellman D. (2002). Yeast formins regulate cell polarity by controlling the assembly of actin cables. Nat Cell Biol , 42–50. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Slubowski CJ, Funk AD, Roesner JM, Paulissen SM, Huang LS. (2015). Plasmids for C-terminal tagging in Saccharomyces cerevisiae that contain improved GFP proteins, Envy and Ivy. Yeast (Chichester, England) , 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich JA, Watt S. (1971). The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem , 4866–4871. [PubMed] [Google Scholar]
- Suarez C, Carroll RT, Burke TA, Christensen JR, Bestul AJ, Sees JA, James ML, Sirotkin V, Kovar DR. (2015). Profilin regulates F-actin network homeostasis by favoring formin over Arp2/3 complex. Dev Cell , 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. (2007). EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol , 38–46. [DOI] [PubMed] [Google Scholar]
- Tasto JJ, Morrell JL, Gould KL. (2003). An anillin homologue, Mid2p, acts during fission yeast cytokinesis to organize the septin ring and promote cell separation. J Cell Biol , 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallen EA, Caviston J, Bi E. (2000). Roles of Hof1p, Bni1p, Bnr1p, and myo1p in cytokinesis in Saccharomyces cerevisiae. Mol Biol Cell , 593–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Nishihama R, Onishi M, Pringle JR. (2018). Role of the Hof1-Cyk3 interaction in cleavage-furrow ingression and primary-septum formation during yeast cytokinesis. Mol Biol Cell , 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weems AD, Johnson CR, Argueso JL, McMurray MA. (2014). Higher-order septin assembly is driven by GTP-promoted conformational changes: evidence from unbiased mutational analysis in Saccharomyces cerevisiae. Genetics , 711–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Vavylonis D, Tsai FC, Koenderink GH, Nie W, Yusuf E, Lee IJ, Wu JQ, Huang X. (2015). SOAX: a software for quantification of 3D biopolymer networks. Sci Rep , 9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HC, Pon LA. (2002). Actin cable dynamics in budding yeast. Proc Natl Acad Sci USA , 751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JH, Crevenna AH, Bettenbuhl M, Freisinger T, Wedlich-Soldner R. (2011). Cortical actin dynamics driven by formins and myosin V. J Cell Sci , 1533–1541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.