

Abstract
The shapes of many eukaryotic cells depends on the actin cytoskeleton, and changes in actin assembly dynamics underlie many changes in cell shape. Ena/VASP-family actin polymerases, for example, modulate cell shape by accelerating actin filament assembly locally and slowing filament capping. When concentrated into discrete foci at the leading edge, VASP promotes filopodia assembly and forms part of a poorly understood molecular complex that remains associated with growing filopodia tips. Here we identify precursors of this filopodia tip complex in migrating B16F1 cells: small leading-edge clusters of the adaptor protein lamellipodin (Lpd) that subsequently recruit VASP and initiate filopodia formation. Dimerization, membrane association, and VASP binding are all required for lamellipodin to incorporate into filopodia tip complexes, and overexpression of monomeric, membrane-targeted lamellipodin mutants disrupts tip complex assembly. Once formed, tip complexes containing VASP and lamellipodin grow by fusing with each other, but their growth is limited by a size-dependent dynamic instability. Our results demonstrate that assembly and disassembly dynamics of filopodia tip complexes are determined, in part, by a network of multivalent interactions between Ena/VASP proteins, EVH1 ligands, and actin filaments.
INTRODUCTION
Filopodia are thin, finger-like protrusions of the plasma membrane that participate in fundamental cellular processes, including directed migration, substrate adhesion, and cell-cell communication (Mattila and Lappalainen, 2008; Blanchoin et al., 2014). Filopodia are defined by morphology rather than function or composition, and growing evidence suggests that different types of filopodia assemble via different mechanisms that employ different sets of actin regulators (Yang and Svitkina, 2011; Barzik et al., 2014; Young et al., 2015, 2018). One class of filopodia grows from dynamic, lamellipodial actin networks by a process of “convergent elongation.” In this mechanism, the growing barbed ends of several preexisting actin filaments converge to a point in the plasma membrane where they are held together by a self-assembling filopodial “tip complex” (Lewis and Bridgman, 1992; Svitkina et al., 2003; Mogilner and Rubinstein, 2005). The tip complex contains actin polymerases, such as Ena/VASP-family proteins that accelerate filament growth and inhibit filament capping (Breitsprecher et al., 2008; Hansen and Mullins, 2010) and are required for the formation of filopodia in several cell types (Kwiatkowski et al., 2007; Damiano-Guercio et al., 2020). The spatial constraint imposed on growing barbed ends of filaments associated with tip complexes causes them to become aligned and subsequently crosslinked by the protein fascin (Vignjevic et al., 2003).
Clustering of VASP tetramers is a key event in tip complex assembly and an important driver of convergent elongation, but the mechanisms underlying this clustering remain obscure. The Ena/VASP Homology-1 (EVH1) domain determines the subcellular localization of VASP by binding to proteins that contain an FPPPP (or less commonly LPPPP) motif, such as zyxin, vinculin, Abi1, RIAM, and lamellipodin (Brindle et al., 1996; Gertler et al., 1996; Reinhard et al., 1996; Niebuhr et al., 1997; Prehoda et al., 1999; Ball et al., 2000; Bear et al., 2000; Krause et al., 2004; Lafuente et al., 2004). Specifically, VASP molecules lacking an EVH1 domain fail to find the leading edge, and EVH1 ligand sequences artificially targeted to other cellular compartments, such as mitochondria, can deplete VASP from the leading edge (Bear et al., 2000). Although the EVH1 domain drives localization of VASP, it is unclear whether this domain is also responsible for its incorporation into filopodia tip complexes.
Evidence that EVH1 ligands could promote VASP clustering comes from in vitro studies of lamellipodin (Hansen and Mullins, 2015), a leading edge protein that contains six FPPPP motifs and forms membrane-associated dimers (Chang et al., 2013). In these studies, purified VASP formed dense coclusters with lamellipodin (Lpd) constructs on actin filaments. Preformed clusters of purified lamellipodin also recruited VASP and dramatically increased the processivity of its actin polymerase activity. In motile cells, lamellipodin helps recruit VASP tetramers to the leading edge via interaction of its Ras-binding and pleckstrin homology (RAPH) domains with membrane-associated small G-proteins and phospholipids, respectively (Bear et al., 2002; Krause et al., 2004; Hansen and Mullins, 2015). An attractive hypothesis is that VASP tetramers form clusters at the leading edge due to multivalent interactions between their four EVH1 domains and the twelve FPPPP motifs in a lamellipodin dimer. Because both VASP and lamellipodin also bind filamentous actin, clustering might be driven by mutual interactions between three critical components: VASP, lamellipodin, and actin.
An alternative set of candidates for driving cluster formation are SH3-containing proteins that interact with recognition motifs (based on pxxp sequences) in the proline-rich region of VASP. These interaction partners include IRSp53 (insulin receptor phosphotyrosine 53 kDa substrate), an I-BAR domain-containing protein that localizes to regions of membrane curvature and promotes formation of long filopodial protrusions (Yamagishi et al. 2004; Millard et al. 2005). IRSp53 is a particularly attractive candidate for initiating filopodia tip complexes (Welch and Mullins 2002; Ahmed et al. 2010), not only because it interacts with VASP, but also because its I-BAR domain tubulates membranes in vitro and localizes to membrane tubules in live cells (Disanza et al., 2013; Prévost et al., 2015). In vitro, purified IRSp53 can recruit VASP tetramers to lipid-coated beads, and a Cdc42-activated IRSp53-VASP complex has been proposed to trigger VASP clustering in cells (Lim et al., 2008; Disanza et al., 2013).
To determine whether EVH1 ligands or SH3 domains drive clustering of VASP and initiation of filopodial bundles, we performed high-resolution time-lapse microscopy of fluorescent actin regulators expressed from their endogenous gene loci in migrating B16F1 mouse melanoma cells. We chose B16F1 cells because they are an excellent model system for studying convergent elongation of leading-edge filopodia: during the initial spreading and polarization phase, VASP localizes along the leading-edge lamellipodial actin network of these cells, where it continually coalesces into discrete foci, each of which generates a filopodial actin bundle via convergent elongation (Rottner et al., 1999; Svitkina et al., 2003; Mejillano et al., 2004; Korobova and Svitkina, 2008). Our experiments revealed that nascent VASP clusters generally do not contain IRSp53, but rather arise from preexisting foci of lamellipodin, and that reducing the valency of binding between VASP and lamellipodin disrupts their ability to cocluster. Additionally, rapid capping of actin filament barbed ends induces immediate spatial separation of the VASP and lamellipodin components of the filopodia tip complex, indicating that free barbed ends play a key role in cluster stability. Surprisingly, our experiments also revealed that filopodia tip complexes undergo spontaneous size-dependent splitting, which appears to limit their maximum size. Together, our data indicate that filopodia tip complexes are held together by a dynamic network of multivalent interactions between VASP, lamellipodin, and actin barbed ends.
RESULTS
Leading-edge dynamics of VASP expressed at endogenous levels in B16F1 melanoma cells
To visualize VASP in live B16F1 cells, we used CRISPR/Cas9 to create a monoclonal cell line expressing VASP-eYFP from the endogenous gene locus (Figure 1A; Supplemental Figure 1A). Our knock-in strategy enabled us to study protein localization and dynamics without overexpression artifacts and to compare fluorescence intensities quantitatively between cells and across experiments. Similarly to previous studies (Reinhard et al., 1992; Gertler et al., 1996), we found that during the early stages of cell spreading and migration, VASP-eYFP localized to nascent focal adhesions (Figure 1A, dashed ellipse) as well as the leading edges of advancing lamellipodia, where it often concentrated into discrete foci (Figure 1A, arrowheads; Supplemental Figure 1B; Video 1). Compared with B16F1 cells transiently overexpressing GFP-VASP, our engineered cells had significantly fewer discrete VASP foci at the leading edge (Supplemental Figure 1C). We occasionally captured de novo formation of VASP foci in regions of otherwise uniform VASP density (Figure 1B), and consistent with previous studies (Svitkina et al., 2003), we observed that VASP clusters skate laterally along the leading edge, often fusing with each other to form larger clusters (Figure 1B). Stable VASP foci were associated with actin bundles that extended back into the lamellipodial network, and the birth of a VASP focus at the leading edge always heralded the appearance of an associated actin bundle (Figure 1C; Video 2). Whenever two VASP clusters fused, their associated actin bundles also zippered together—via a short-lived, lambda-shaped intermediate—to form a single, larger bundle (Figure 1C). Overall, the fluorescence intensity of leading-edge actin bundles correlates with the intensity of their associated VASP clusters, suggesting that VASP clusters play a role in creating and maintaining their structure (Figure 1D). Some VASP-associated actin bundles form filopodia that protrude several micrometers beyond the leading edge, while others barely dent the membrane surface. The shorter protrusions are often called “microspikes” to distinguish them from longer filopodia (Yang and Svitkina, 2011), but we observed occasional interconversion between these structures, and so, consistent with Svitkina et al. (2003), we refer to them collectively as “filopodial actin bundles.”
FIGURE 1:

Endogenous VASP-eYFP localization and dynamics during B16F1 cell migration. (A) Representative image of monoclonal, endogenously expressed VASP-eYFP B16F1 cell line generated by CRISPR/Cas9. Yellow arrowheads: VASP clusters; yellow ellipse: focal adhesions. (B) Two examples of nascent VASP clusters being born from fluctuations in VASP intensity along the leading edge (Top) and fusion of existing VASP clusters (Bottom) by live cell microscopy of the VASP-eYFP B16F1 cell line. Yellow bars: coalescence of VASP-eYFP intensity; yellow arrowheads: VASP-eYFP clusters. (C) Coupled kinetics of VASP-eYFP (green) clustering and SNAP-actin bundle (magenta) formation and zippering. (D) Correlation between integrated VASP cluster intensity and underlying actin bundle intensity in 73 foci and 10 cells. (E) Fluorescent image of a representative VASP-eYFP B16F1 cell (Left) that was used to generate colored outlines of the leading edge position over time (Middle). Adaptive kymograph map (Right) remaps the leading-edge position to the y-axis and time (40 frames or 160 s) on the x-axis to visualize VASP-eYFP foci dynamics. Color map: blue (low fluorescence intensity) to red (high fluorescence intensity).
Movie S1.
Endogenous VASP-eYFP localization and dynamics at the leading edge. B16F1 cells expressing VASP-eYFP from the endogenous gene locus imaged by spinning disc confocal microscopy at 4-second time points.
Movie S2.
VASP-eYFP foci coalesce at the tips of filopodia actin bundles. B16F1 VASP-eYFP (green) cell line with overexpressed SNAP-actin (magenta) imaged by spinning disc confocal at 4-second time points.
Movie S3.
Lamellipodin localizes to dynamic VASP clusters and displays coupled dynamics at the leading edge. B16F1 VASP-eYFP (green) and lamellipodin-tdTomato (magenta) cell line imaged by spinning disc confocal at 4-second time points.
Movie S4.
IRSp53 displays punctate membrane localization but does not concentrate in VASP foci. B16F1 VASP-eYFP (green) and IRSp53-tdTomato (magenta) cell line imaged by spinning disc confocal at 4-second time points.
To track the creation and evolution of VASP-eYFP clusters more easily across the entire leading edge of spreading and crawling cells, we created automated image analysis tools using MATLAB. Briefly, we used VASP-eYFP fluorescence to find the cell edge in every frame of a time-lapse image sequence (Figure 1E, left panel), and then—based on membrane morphology and dynamics—identified leading-edge lamellipodia for further analysis (Figure 1E, middle panel). For each frame, we mapped VASP-eYFP intensity along the cell edge onto a vertical line to create a space–time plot, or kymograph (Figure 1E, right panel). Unlike kymographs that analyze a fixed region of space, these adaptive kymographs follow the advancing and retracting cell edge and simplify analysis of VASP cluster dynamics by 1) removing membrane movement; 2) reducing the number of spatial dimensions; and 3) mapping time onto space, enabling us to identify key events rapidly in the life cycles of VASP clusters across many cells. These key events include the birth of nascent clusters; lateral skating of mature clusters; and fusion of colliding clusters.
Lamellipodin is a stoichiometric component of leading-edge VASP foci
Until now, the earliest observable event in filopodial bundle formation has been the formation of leading-edge VASP clusters, so we aimed to identify additional molecules that might initiate and/or stabilize VASP clusters. Multivalent EVH1-binding proteins including Mig10, RIAM, and lamellipodin (collectively, the MRL family of proteins) associate with VASP in leading-edge lamellipodia, but previous studies have disagreed about the association of MRL proteins with VASP foci. Krause et al. (2004) initially observed lamellipodin at the tips of filopodial protrusions and colocalized with Mena and VASP foci at the leading edge, while Disanza et al. (2013) claimed that MRL proteins do not localize into discrete foci at filopodia initiation sites.
To settle the question of whether MRL-family proteins interact with VASP in leading edge clusters and form part of the filopodial tip complex, we created a double CRISPR knock-in B16F1 cell line expressing both VASP-eYFP and a fluorescent lamellipodin-tdTomato fusion protein from their endogenous loci (Figure 2A). Consistent with the original report of Krause et al. (2004), we found that Lpd-tdTomato localizes along leading edge lamellipodia and concentrates with VASP at the tips of filopodia. More significantly, all leading edge VASP foci also contain lamellipodin as a stoichiometric component (Figure 2A, arrowheads). That is, the ratio of time-averaged, integrated intensities of VASP and Lpd within leading edge foci was approximately constant (R2 = 0.79) across 58 stable foci observed in 10 cells over four separate experiments (Figure 2C, black circles). In contrast, the fluorescence intensities of Lpd and VASP measured along the entire leading edge were much less correlated (Supplemental Figure 2A). These results reveal that, even though lamellipodin and VASP are not present in a constant ratio across the entire leading edge, they aggregate with a fixed stoichiometry in stable VASP foci.
FIGURE 2:

Differential localization of the VASP binding partners, lamellipodin (Lpd) and IRSp53, at the leading edge. (A) Representative image of monoclonal, endogenously expressed VASP-eYFP and Lpd-tdTomato B16F1 cell line shows strong colocalization of the two proteins at VASP foci. Arrowheads: VASP-eYFP foci. (B) Representative image of monoclonal, endogenously expressed VASP-eYFP and IRSp53-tdTomato B16F1 cell line shows poor colocalization of the two proteins at VASP foci. Arrowheads: VASP-eYFP foci; yellow bar: filopodia shaft. (C) The time-averaged integrated intensities of VASP-eYFP and Lpd-tdTomato within leading edge foci are tightly correlated (black circles). A plot of average Lpd intensity versus VASP intensity fits a straight line (R2 = 0.79). In contrast, the integrated intensities of VASP-eYFP and IRSp53-tdTomato within leading edge foci are not correlated (red squares; R2 = 0.00014). (D) Zoomed plot of the VASP-eYFP and IRSp53-tdTomato dataset shows the lack of correlation in their intensities.
IRSp53 does not stably interact with VASP in leading edge foci
Disanza et al. (2013) proposed that IRSp53, a dimeric I-BAR domain-containing protein associated with the plasma membrane, binds VASP tetramers and induces them to form stable clusters. Other studies, however, suggest that the connection between VASP and IRSp53 may be less direct (Nakagawa et al. 2003; Sudhaharan et al. 2019). Therefore, to determine whether IRSp53 is present in the early stages of VASP cluster formation, we used CRISPR/Cas9 to create a double knock-in B16F1 cell line, expressing both IRSp53-tdTomato and VASP-eYFP from their endogenous loci (Figure 2B). We confirmed that the localization of our fluorescent IRSp53 derivative matched that of the endogenous protein using immunofluorescence (Supplemental Figure 3, A and B). In both live and fixed cells, IRSp53-tdTomato was concentrated in patches along the plasma membrane and often enriched in the shafts of mature filopodia (Figure 2B, arrowheads). Unlike lamellipodin, however, IRSp53-tdTomato was not a stoichiometric component of VASP foci, and the correlation between the time-averaged integrated intensities of VASP and IRSp53 in leading edge foci (R2 = 0.00014) was not significant (Figure 2, C and D; red squares). Finally, in addition to tagging IRSp53 at the endogenous gene locus, we also transiently overexpressed IRSp53 fused at its N-terminus to mRuby2. Overexpressing this fluorescent fusion protein induced formation of filopodial protrusions containing mRuby2-IRSp53, most of which localized along the shaft of the protrusions but rarely extended out to the tip (Supplemental Figure 3C, arrowhead). The majority of VASP-eYFP in these cells remained associated with lamellipodial actin networks, where it formed clusters that contained little or no detectable fluorescent IRSp53. Conversely, the longer protrusions associated with mRuby2-IRSp53 contained little detectable VASP-eYFP at their tips. Together, these localization data indicate that IRSp53 is not a stable or stoichiometric component of leading edge VASP foci.
Dynamics of Lpd and IRSp53 within leading edge VASP foci
We next compared the time-dependent changes in the amounts of VASP and its interaction partners, Lpd and IRSp53, within filopodia tip complexes. For this analysis, we computed the cross-correlation between fluorescence intensities of VASP-eYFP and Lpd-tdTomato (or IRSp53-tdTomato) within individual VASP clusters over a time window of 160 s. Cross-correlation of VASP and Lpd intensities in leading edge foci revealed strongly coupled temporal fluctuations. The peak correlation between the two molecules (r = 0.75) occurs at a time lag of 0 s, implying that (within the time resolution of our image sequences) fluctuations in the intensity of one molecule do not systematically lead or lag fluctuations in the intensity of other (Figure 3A). In addition, the adaptive kymographs revealed numerous discrete foci of lamellipodin that lack a corresponding accumulation of VASP-eYFP (Figure 3B, white arrows). These Lpd-only foci were generally smaller and more transient and exhibited less lateral (skating) movement than foci that contained both VASP and lamellipodin (Figure 3B; Supplemental Figure 2B). Further examination revealed that all nascent VASP foci that we could detect emerged from these small preexisting clusters of lamellipodin (Figure 3B, white arrowhead). We corroborated these observations by examining the time-lapse movies of the corresponding VASP/Lpd clusters, which clearly show that an increased local density of lamellipodin precedes the accumulation of VASP into a detectable focus (Figure 3C, arrowheads).
FIGURE 3:

Dynamics of VASP clustering with its leading-edge binding partners, Lpd and IRSp53. (A) Temporal cross-correlation analysis of fluctuations in VASP and Lpd intensity at leading edge foci. Gray lines are individual cross-correlation measurements and the solid black line, which is symmetrical about zero, is the average of individual traces from 58 different clusters from four experiments. Red dotted lines indicate an estimated 95% confidence interval of significance. Peak cross correlation occurs at time lag 0 s with a correlation coefficient of r = 0.75. (B) Cell outline of changing leading edge positions over 40 frames (Left). Adaptive kymograph map (Right) displaying VASP-eYFP and Lpd-tdTomato dynamics over time (40 frames; 160 s). (C) Frames from time-lapse microscopy of VASP clusters being born from small preexisting Lpd clusters (arrowheads) at the leading edge in double knock-in VASP-eYFP(green)/Lpd-tdTomato (magenta) B16F1 cells. (D) Temporal cross-correlation of VASP-eYFP and IRSp53-tdTomato fluorescence signal at leading edge foci. Gray lines are individual cross-correlation measurements and the solid black line (the average of 34 clusters from three experiments) shows a weak peak occurring at time lag 8 s with a correlation coefficient of 0.09. Red dotted lines indicate an estimated 95% confidence interval of significance. (E) Cell outline of changing leading edge positions over 40 frames (left). Adaptive kymograph map (right) displaying VASP-eYFP foci and IRSp53-tdTomato dynamics over time (40 frames; 160 s). Color map: blue (low fluorescence intensity) to red (high fluorescence intensity). (F) Frames from time-lapse microscopy of VASP clusters (arrowhead) forming with no detectable accumulation of IRSp53-tdTomato until after the VASP foci is formed (arrow).
We also looked for possible transient interactions between VASP and IRSp53 by asking whether fluctuations in the intensities of these two molecules are also temporally correlated. Maximum cross-correlation between VASP and IRSp53 occurred with a time delay of 8 s, which, surprisingly, suggests that VASP might weakly recruit IRSp53 to preexisting filopodial tip complexes, although the correlation coefficient (r = 0.093) was just below the cutoff for statistical significance set by bootstrap analysis (Zoubir and Iskander, 2004; Figure 3D). Importantly, this fluctuation analysis argues that IRSp53 accumulation at the leading edge is unlikely to drive formation of VASP clusters. Furthermore, comparison of adaptive kymographs displaying VASP-eYFP and IRSp53-tdTomato dynamics along the leading edge of spreading cells revealed no consistent spatial colocalization of the two proteins (Figure 3E). We also could not detect IRSp53 persistently concentrated in either nascent or stable VASP clusters at the leading edge (Figure 3F). In fact, consistent with our temporal analysis, we occasionally detected accumulation of IRSp53 into preexisting VASP foci (Figure 3F, arrow at 24 s), suggesting that the VASP molecules occasionally recruit IRSp53 to filopodia tip complexes rather than vise versa.
VASP/Lpd clusters undergo size-dependent splitting events
Consistent with previous studies of VASP clusters and leading edge microspikes (Oldenbourg et al., 2000; Svitkina et al., 2003), we observed that VASP/Lpd foci grow both by incorporating molecules along the leading edge and by fusing with other foci. Because these filopodia tip complexes remain dynamic and dispersed along the leading edge rather than accumulating into a single large blob, we hypothesized that, in addition to growth and fusion, tip complexes must also undergo shrinkage and/or fission.
To explore tip complex disassembly, we quantified intensity fluctuations of dynamic VASP/Lpd clusters over time. The simplest assumption—that growth occurs by both the accumulation of individual molecules and the fusion of existing clusters, while shrinkage occurs only through the loss of individual molecules—suggests that relative size fluctuations should decrease as clusters grow larger and the ratio of their surface area to their volume decreases. By plotting intensity fluctuations as a function of cluster size, however, we found instead that the fluctuations increase nonlinearly with cluster size, implying that VASP/Lpd clusters become more unstable as they grow (Figure 4A). Across all of our experiments, size fluctuations over a 160-s window increased as the 1.75th power of the average cluster size (1.75 ± 0.12; Figure 4A, inset: log–log plot). A strong inverse relationship between cluster size and stability was apparent, no matter how we analyzed the data. For example, using the same data set, we also plotted the distribution of VASP/Lpd intensities of each individual cluster versus its average size (Supplemental Figure 4A) and observed the same nonlinear increase in vertical scatter with cluster size.
FIGURE 4:

VASP/Lpd clusters exhibit size-dependent splitting behavior. (A) Fluctuations in VASP and Lpd were quantified by plotting the SD of fluorescence intensity of VASP (black diamond) and Lpd (red circle) foci versus their time-averaged VASP (or Lpd) intensity. Inset: log plot of SD vs. time-averaged VASP/Lpd intensity fits a line with slope = 1.75 ± 0.12. (B) Intensity trace of the fluorescence intensity of a single VASP/Lpd focus from C. Red arrows indicate simultaneous drops in both VASP and Lpd fluorescence intensity, which correlate to splitting events. (C) Image (left panel) of double knock-in VASP-eYFP(green) Lpd-tdTomato(magenta) B16F1 cell with ROI indicating the VASP/Lpd cluster that undergoes a splitting event in which VASP material splits from the parental cluster (right panels, yellow arrow). (D) Kymograph of a ROI bisecting both the main VASP/Lpd cluster and secondary VASP clump that is shed indicates that the shed VASP does not move relative to the lab frame of view (asterisks). Dotted yellow line outlines the leading edge.
This size-dependent instability imposes an effective limit on the maximum size of filopodia tip complexes, but the physical nature of these size-dependent intensity fluctuations was not clear until we returned to the time-lapse movies and followed the life histories of individual tip complexes. In the largest VASP/Lpd clusters, we identified abrupt and simultaneous drops in both VASP and lamellipodin intensity (Figure 4B, red arrows), which turn out to reflect dramatic splitting events in which large amounts of VASP and Lpd are simultaneously shed from the cluster (Figure 4C, yellow arrowhead; Video 5). In the early stages of tip complex splitting before cluster shrinkage occurs, both VASP and Lpd begin to take on an elongated shape (Figure 4C, yellow ellipse at 20 s). As splitting progresses, however, the inward-moving cluster begins to lose lamellipodin, eventually becoming a diffuse mass of VASP alone (Figure 4C, 32 s). The net result is that the lump of VASP is shed into the cytoplasm, while the lamellipodin is lost by redistribution across the plasma membrane, likely reflecting direct binding of the RAPH domain to phospholipids. Unlike the fusion of tip complexes, which takes place within the leading edge, fission occurs orthogonal to the leading edge, with a chunk of VASP and Lpd moving inward toward the cell body while the rest remains at the leading edge. The portion of the original VASP/Lpd cluster that remains associated with the leading edge exhibits the same dynamics and VASP/Lpd ratio as before the fission event, emphasizing the robustness of the mechanism that maintains the stoichiometry between VASP and lamellipodin in the clusters (Figure 4, A–C; see Supplemental Figure 4A for more examples). Additional, static kymograph analysis of splitting events revealed that the detached piece of VASP does not treadmill backward with the retrograde flow of actin, but remains stationary in the laboratory frame of reference as the leading edge moves forward (Figure 4D). In some examples, VASP shed from a leading edge focus adopted the elongated shape of a nascent focal adhesion and remained in the same spot for the duration of the movie (Figure 4C, arrowhead at 32 s; Figure 4D). In total, these data reveal that VASP/Lpd clusters exhibit a form of dynamic instability, becoming more unstable as they grow in size.
Movie S5.
Size-dependent splitting of VASP from VASP/lamellipodin clusters at the leading edge. B16F1 VASP-eYFP (green) and lamellipodin (magenta) cell line imaged by spinning disc confocal at 4-second time points. White arrow (left) indicates the parental cluster within the leading edge membrane; white arrow (right) indicates the VASP that splits away towards the cytoplasm.
Actin filaments with free barbed ends are required to form and maintain VASP/Lpd clusters
In vitro, lamellipodin drives clustering of VASP tetramers on actin filaments (Hansen and Mullins, 2015), and in cells the clustering of VASP and lamellipodin occurs only on the outer edge of lamellipodial actin networks, in the dynamic space between the plasma membrane and growing actin filaments. We therefore examined whether actin barbed ends might also contribute to cluster formation and/or stability. To test the requirement for barbed ends in VASP/Lpd clustering, we treated B16F1 cells expressing VASP-eYFP and Lpd-tdTomato with cytochalasin D (CD), a small molecule that rapidly caps free barbed ends. Within 100 s of drug treatment, VASP/Lpd clusters fell apart (Figure 5A, Video 6). Cluster dissolution was dramatic and proceeded in two phases. First, all of the leading edge-associated VASP, including the VASP molecules concentrated in filopodial tip complexes, translocated inward toward the cell body, leaving residual lamellipodin-only clusters behind at the leading edge (Figure 5, A and B). Next, the remaining lamellipodin clusters slowly dissolved as the constituent molecules redistributed laterally across the membrane (Figure 5A, 200 s). We quantified the relative change in lamellipodin clustering immediately following VASP dissociation and 100 s later by calculating the standard deviation (SD) of its fluorescence intensity along the leading edge normalized to the mean (Figure 5, C and D). Clustering increases the SD, while dissolution of clusters into a more uniform distribution causes the SD to fall. The disappearance of lamellipodin clusters suggests that their stability requires VASP and/or actin barbed ends near the membrane.
FIGURE 5:

Free barbed ends of actin filaments are required for VASP/Lpd cluster stability. (A) Representative example of a B16F1 cell expressing VASP-eYFP (green) and Lpd-tdTomato (magenta) before and after acute treatment with 500 nM cytochalasin D. (B) Kymograph of a slice taken perpendicular to the leading edge shows change in membrane localization of VASP (green) and Lpd (magenta) upon addition of cytochalasin D. (C) Schematic of quantification of changes in clustering. The SD along a defined leading edge before (0 s) and after drug treatment (100 s, 200 s) was calculated and normalized to the mean fluorescence intensity. (D) Quantification of Lpd clustering at foci following acute treatment with cytochalasin D or DMSO (vehicle) shows that free barbed ends are required for robust clustering of VASP and Lpd. Clustering is represented by SD along the leading edge ROI before and after treatment. Symbols are averages from three biological replicates, each with >8 cells. Paired t test of unnormalized data: p = 0.02.
Movie S6.
Capping barbed ends disrupts clustering of VASP and lamellipodin. B16F1 VASP-eYFP (green) and lamellipodin-tdTomato (magenta) cell line were treated with 500 nM Cytochalasin D at a 30-second time point and imaged by spinning disc confocal at 4-second time points.
Lamellipodin requires membrane-targeting and multiple EVH1-binding sequences to incorporate into VASP clusters
Although lamellipodin is a stoichiometric component of filopodia tip complexes and marks sites where they will form, our cross-correlation data do not reveal whether VASP/Lpd interactions are required to hold the filopodia tip complex together. To investigate the molecular determinants of VASP/Lpd cluster formation, we expressed various lamellipodin mutants and chimeras in our double knock-in B16F1 cell line (Figure 6A).
FIGURE 6:

Lpd incorporation into dynamic VASP foci depends on its number of VASP binding sites. (A) Schematic of the domain architecture of wild-type and various Lpd mutants that were overexpressed in B16F1 cells already endogenously expressing VASP-eYFP and Lpd-tdTomato. Leucine zipper (LZ) was used to induce dimerization of the Lpd C-term orthogonally; Lyn11 sequence was used to replace the N-term and orthogonally target C-term Lpd to the plasma membrane; VASP-binding mutant contains F/LPPPP6→ AAPPP6. (B–I) Representative images from three-color microscopy of endogenous VASP-eYFP (top panel) and Lpd-tdTomato (middle panel) and various overexpressed Lpd mutants (bottom panel). All Lpd mutants were overexpressed using a CMV promoter to drive expression of the N-terminal SNAP-Lpd (labeled with Janelia Fluor 646 SNAP ligand) fusion protein. Cartoon schematic below describes the effect that overexpression of each SNAP-Lpd mutant (red) has on endogenous VASP(orange)/Lpd(blue) clustering. Yellow arrowhead: example in which dimerized Lyn11 C-term Lpd mutant is able to incorporate into VASP/Lpd clusters. Adaptive kymographs display leading edge VASP/Lpd clusters in (F) wild-type double knock-in cells and (G) double knock-in cells with overexpressed monomeric Lyn11 C-term, which has a dominant negative effect.
We began by separately overexpressing N- and C-terminal truncation mutants of lamellipodin and asking 1) whether they were competent to incorporate into endogenous VASP/Lpd clusters and 2) whether they affected formation of these endogenous clusters (Figure 6B). The N-terminal region of lamellipodin, comprising the RAPH domain, drives dimerization of the protein and mediates membrane association via interaction with small G-proteins and acidic phospholipids, but lacks domains that bind VASP. When expressed alone, the RAPH domain localized to the plasma membrane but failed to concentrate at the leading edge, did not incorporate into endogenous VASP/Lpd clusters, and had no effect on cluster formation (“N-term Lpd”; Figure 6C). This result suggests that lamellipodin does not enter tip complexes via interaction with small G-proteins or acidic phospholipids. We next tested our C-terminal lamellipodin construct, which contains an actin-binding region and six VASP EVH1-binding motifs. Without the RAPH domain, this C-terminal construct failed to strongly localize to the plasma membrane and had no effect on VASP/Lpd cluster formation (“C-term Lpd”; Figure 6D).
To test whether the inactivity of our C-terminal lamellipodin construct was due to its inability to bind the plasma membrane, we fused C-term Lpd to a membrane-targeting sequence from Lyn11 (“Lyn11 C-term Lpd”; Figure 6E). When overexpressed, this monomeric, membrane-associated lamellipodin mutant dramatically disrupted the incorporation of VASP into filopodia tip complexes, resulting in fewer VASP-containing clusters but leaving many small, lamellipodin-only clusters (Figure 6G vs. Figure 6F control). This construct failed to incorporate into any of the endogenous VASP/lamellipodin clusters, suggesting that it acts by sequestering endogenous VASP tetramers away from the clusters rather than by replacing full-length lamellipodin within the clusters. To test this idea, we mutated all six VASP binding sites (FPPPP6→ AAPPP6) in Lyn11 C-term Lpd, to create a monomeric membrane-associated version of lamellipodin incapable of binding VASP. Consistent with a VASP sequestering effect, this mutant construct lost the ability to disrupt endogenous filopodia tip complexes (“VASP-binding mutant”; Figure 6H).
Finally, we asked what feature of the RAPH domain confers the ability of full-length lamellipodin to incorporate into filopodia tip complexes with VASP. The fact that N-terminal lamellipodin truncations containing the RAPH domain fail to disrupt or incorporate into VASP/Lpd clusters suggested to us that phospholipid and G-protein binding were not involved. However, this leaves open the possibility that dimerization mediated by lamellipodin’s RAPH domain is important for coclustering by increasing the valency of interactions with VASP. We therefore tested whether artificially inducing dimerization of the Lyn11 C-term Lpd mutant suffices to revert its dominant negative effect on VASP/Lpd clustering and restore wildtype cluster dynamics. For this experiment, we created a dimeric variant of Lyn11 C-term Lpd (“dimerized Lyn11 C-term Lpd”; Figure 6I) by introducing a leucine zipper motif into the N-terminal region. Strikingly, this dimeric membrane-associated C-terminal truncation mutant did not disrupt filopodia tip complexes like its monomeric counterpart, but was instead concentrated in tip complexes along with wild-type lamellipodin. The effects of dimerization and mutation of EVH1 ligand sequences on the ability of lamellipodin constructs to disrupt or participate in cluster formation argue that VASP/Lpd clusters are held together by multivalent interactions between EVH1 domains of VASP and the FPPPP motifs of lamellipodin and that the valency of these interactions is critical to the stability of the cluster.
DISCUSSION
The size, shape, and stability of actin-filled pseudopods depend on the dynamic localization of actin regulators at the plasma membrane (Weiner et al., 2007; Schmeiser and Winkler, 2015; Fritz-Laylin et al., 2017). These regulators include VASP and its relatives Mena and Evl, which are weakly processive polymerases that promote rapid growth of actin filaments near the membrane and help construct sheetlike lamellipodia (Lacayo et al., 2007) and finger-like filopodia (Han et al., 2002; Svitkina et al., 2003; Kwiatkowski et al. 2007). To form filopodia, VASP tetramers must be collected into dense clusters, called filopodia tip complexes. By monitoring the leading-edge dynamics of VASP and its binding partners, we discovered a precursor of the filopodia tip complex: a molecular cluster containing the VASP binding partner lamellipodin (Figure 7). We also discovered that filopodia tip complexes experience a size-dependent form of dynamic instability.
FIGURE 7:
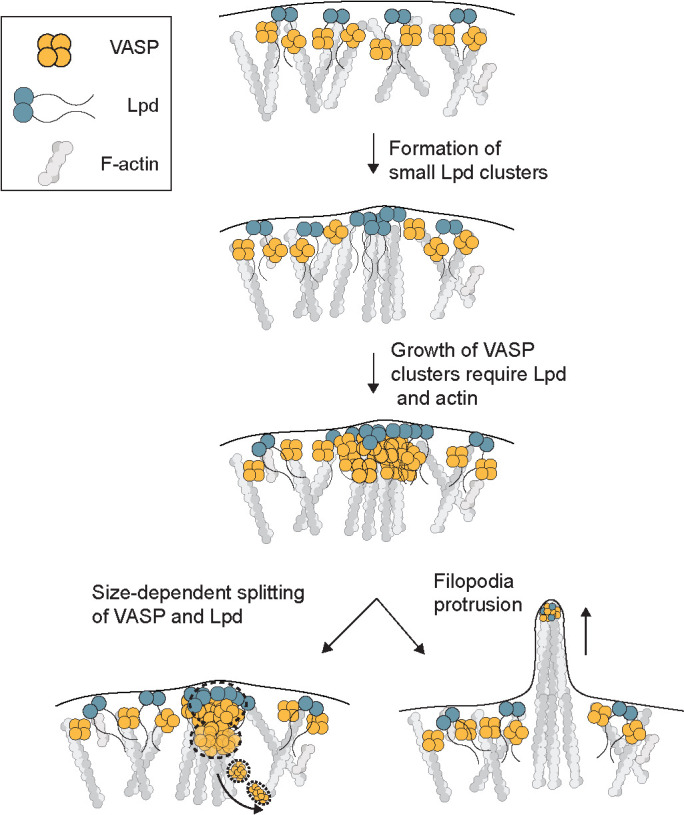
Model. Coalescence of VASP into leading edge clusters arises from small preexisting lamellipodin clusters. The interaction between VASP and Lpd at clusters is mediated through multivalent interactions between EVH1 domains (VASP) and short FPPPP proline motifs (Lpd) that require membrane tethering and dimerization of Lpd. The assembly of VASP clusters involve Lpd and free barbed ends of actin filaments at the leading edge, which can protrude into long filopodia or disassemble through size-dependent splitting of VASP into the cytoplasm.
The role of I-BAR proteins in filopodia assembly
Similarly to previous studies, we find that overexpressing the membrane-associated protein, IRSp53, strongly induces formation of filopodia (Krugmann et al., 2001; Yamagishi et al., 2004; Lim et al., 2008). Both overexpressed and endogenous IRSp53 localize along the shaft of filopodial protrusions, consistent with the preferential binding of its I-BAR (inverse-Bin-Amphiphysin-Rvs) domain to regions of negative membrane curvature (Yamagishi et al., 2004; Saarikangas et al., 2009). In vitro, the I-BAR domain from IRSp53 can tubulate PI(4,5)P2-containing vesicles (Mattila et al., 2007; Prévost et al., 2015), and when overexpressed in cells, it can even drive formation of tubular membrane protrusions that lack filamentous actin altogether (Yang et al., 2009). Although these results illustrate how easily membrane protrusion can be uncoupled from actin filament assembly, IRSp53 has also been shown to interact, via its SH3 domain, with various actin regulators, including VASP, Mena, N-WASP, mDia1, and Eps8 (Krugmann et al., 2001; Ahmed et al., 2010). Based on its interactions with curved membranes, actin regulators, and signaling molecules such as Cdc42, we and others (Welch and Mullins, 2002; Ahmed et al., 2010) suggested that IRSp53 might trigger filopodium formation by coupling actin assembly to membrane bending. Disanza et al. (2013) further proposed that IRSp53 actually drives formation of filopodial actin bundles by coclustering with VASP molecules. This attractive idea is generally consistent with the localization of IRSp53 to filopodial protrusions, but the strong preference of IRSp53 for curved membranes means it can localize to filopodia and lamellipodia even in Ena/VASP-deficient MVD7 cells (Nakagawa et al. 2003). We posed a more stringent test of IRSp53’s involvement in VASP clustering by asking whether IRSp53 associates with nascent VASP foci, or forms foci that subsequently recruit VASP. Using fluorescent proteins expressed from the endogenous loci, we found that the coalescence of VASP molecules into foci at the leading edge did not correlate with the presence of IRSp53. Moreover, we observed that VASP foci do not consistently contain detectable amounts of IRSp53 (Figures 2, 3). These results fit with recent superresolution light microscopy studies that found IRSp53 predominantly associated with the lateral edges of filopodia and not consistently enriched at the tip with VASP (Sudhaharan et al., 2019). Our data support the idea that IRSp53 induces protrusion of long filopodia by promoting and stabilizing membrane curvature rather than by clustering VASP tetramers.
The role of EVH1 ligands in VASP localization and clustering
VASP is recruited to membranes by the interaction of its Ena/VASP Homology 1 (EVH1) domain with proteins that contain FPPPP motifs. This was most clearly demonstrated by Bear et al. (2002), who found that VASP can be displaced from leading edge membranes by removing the EVH1 domain or ectopically expressing FPPPP-containing proteins on intracellular membranes. In addition to lamellipodin, several other proteins with EVH1-binding motifs are found on leading edge membranes. These include Mig10 and RIAM, which, together with lamellipodin, define the MRL protein family (Coló et al., 2012).
When we compared the dynamics of endogenous VASP and lamellipodin during filopodia initiation, we found that VASP clusters form on pre-existing foci of lamellipodin. The small lamellipodin puncta form independent of VASP, but “ripen” and grow much larger when they begin to accumulate VASP. This observation reveals that small lamellipodin puncta are precursors to larger VASP/lamellipodin clusters that concentrate at the tips of nascent filopodia bundles and raises the question of how small lamellipodin puncta form in the first place. Two possibilities are that 1) lamellipodin can independently form small homooligomers, which is supported by a structural study that identified two dimerization motifs at its N-terminal RAPH domain (Chang et al. 2013), or 2) lamellipodin foci mark the positions of larger complexes that contain other, as yet unidentified proteins. We observed that lamellipodin-only clusters exhibit significantly less lateral mobility than clusters that contain VASP, and they only rarely fuse (Supplemental Figure 2b). Lateral “skating” of VASP-containing clusters occurs when the associated filopodial bundle is tilted away from normal to the leading edge (Katoh et al., 1999). The fact that lamellipodin-only clusters do not “skate” in the same way suggests that 1) small Lpd clusters are not as strongly coupled to the growing actin filaments beneath them (purified Lpd has weak affinity for actin filaments in vitro; Kd ∼ 200 nM; Hansen and Mullins, 2015) and 2) that lamellipodin by itself is not sufficient to locally rearrange lamellipodial actin filaments into parallel bundles. VASP appears to be required for both coupling to and rearranging the underlying filaments. This explanation also fits with the effects of perturbations such as the addition of cytochalasin D and the overexpression of Lyn11-C-term Lpd, which drive VASP out of the clusters, leaving behind small Lpd-only clusters that cannot skate or fuse. In other words, the dynamics of the Lpd-only clusters produced by these perturbations mirror the dynamics of endogenous Lpd-only clusters found in wild-type cells.
Our results provide compelling evidence that interactions between VASP, lamellipodin, and actin barbed ends help hold filopodia tip complexes together. We found, for example, that monomeric C-terminal Lpd mutants disrupt formation of VASP clusters, a result that could be rescued by simply dimerizing the mutant. We also identified the key features of Lpd that promote VASP clustering: membrane association, dimerization, and EVH1-binding. Surprisingly, the N-terminal Ras-association (RA) pleckstrin homology (PH) signaling domain of Lpd appears largely dispensable for VASP clustering, as we could functionally replace it with a Lyn11 membrane tether and a leucine zipper dimerization domain. Based on our results, we propose that the primary function of the RAPH domain is membrane localization and dimerization, rather than clustering per se. This proposal agrees with several studies demonstrating that the PH domain Lpd recruits the protein to the plasma membrane by binding PI(3,4)P2 (Bae et al., 2010). Our proposal also fits with a recent structural study of lamellipodin’s N-terminal region which suggests that its dimeric RAPH domain has lower affinity for Ras GTPases than individual RA domains found in other proteins, and may make minor contributions to clustering on membranes (Chang et al., 2013).
While multiple lines of evidence indicate that FPPPP-EVH1 interactions are responsible for VASP localization to the leading edge, Dimchev et al. reported that knocking out lamellipodin by CRISPR/Cas9 in B16F1 cells does not significantly change VASP recruitment to lamellipodia (Bear et al., 2002; Dimchev et al., 2019). This result suggests the existence of redundancy or compensation in the expression of FPPPP-containing proteins. For example, Dimchev et al. (2019) also reported that leading-edge localization of a member of the WAVE Regulatory Complex, Abi1, which binds EVH1 domains through an alternate proline motif was increased upon Lpd knockout and may allow it cocluster with VASP in the absence of lamellipodin (Chen et al., 2014; Dimchev et al., 2019). In addition, lamellipodin is just one member of the MRL family of proteins, which all interact with VASP through FPPPP proline motifs and could potentially replace Lpd’s VASP-specific functions at the leading edge.
Disassembly of VASP/Lpd clusters
VASP/Lpd clusters continuously undergo assembly and disassembly in order to maintain a dynamic distribution along the leading edge. Growth of clusters through gradual accumulation of monomers or abrupt fusion events must be counterbalanced by a disassembly mechanism to prevent the total coalescence of VASP into a few large, static foci at the leading edge. By analyzing VASP cluster size fluctuations, we discovered a disassembly mechanism whereby VASP/Lpd clusters undergo size-dependent splitting events. During splitting, a blob of VASP detaches from a cluster and moves into the cytoplasm, while a stoichiometric amount of Lpd redistributes across the plasma membrane. The larger the VASP/Lpd cluster grows, the more unstable it becomes, and the instability grows as the approximate square of the cluster size. The fact that these splitting events occur perpendicular to the plasma membrane while fusion events occur parallel to the membrane indicates that splitting is not simply the reverse of fusion, but a separate process with a different underlying molecular mechanism. In addition to maintaining dynamic VASP clusters, splitting might help relocalize VASP tetramers to other parts of the cell. For example, in some cases where nascent focal adhesions are proximal to the leading edge, we observed blobs of VASP splitting from the plasma membrane focus and almost immediately becoming immobilized in the elongated shape of a focal adhesion. As several focal adhesion proteins (such as vinculin and zyxin) directly bind VASP through the FPPPP-EVH1 interaction module, it is possible that fission of large VASP/Lpd clusters helps deposit VASP directly onto nascent focal adhesions.
MATERIALS AND METHODS
Constructs and reagents
The following primary antibodies and dyes were used for staining: polyclonal rabbit antibody against IRSp53 (4 μg/ml, Atlas Prestige antibodies, HPA023310), monoclonal rabbit antibody against VASP (1:100 dilution, Cell Signaling, #3132), Alexa-647 phalloidin (Thermo Fisher #A22287). Various Lpd constructs were derived from an original plasmid encoding human Lpd1-1250 provided by Matthias Krause (King’s College, London) and subsequently cloned into a pCMV-SNAP vector.
Cell culture
B16F1 (ATCC-CRL-6323) mouse melanoma cells were cultured in DMEM with 4.5 g/mL glucose and supplemented with 10% fetal bovine serum (FBS, Life Technologies Certified, US) and penicillin/streptomycin. Cells were maintained at 37°C and 5% CO2 and split every 2–3 d. Lipid-based transient transfection was performed using Lipofectamine 3000. For the six-well plate format, 7.5 μl Lipofectamine 3000 was combined with 1 μg DNA and 5 μl P3000 reagent. The transfection mixture was incubated with 50–70% confluent cells for 8-16 h and then replaced with fresh media. Cells were assayed or harvested 48 h later.
Cell line generation
The VASP locus was tagged with eYFP at the C terminus using CRISPR/Cas9-mediated genome engineering in B16F1 cells. The oligonucleotide guide sequences used to cut the targeted genomic sites are listed in Table 1 above. Cas9 and single-guide RNA were expressed using pX330 as previously described (Ran et al. 2013). Both pX330 and the eYFP-neomycin donor plasmid were a gift of Kara McKinley (University of California, San Francisco). pX330 and the donor plasmid were cotransfected into B16F1 cells at 1.25 μg each and selected after 72 h with 0.8 mg/ml G418 for 2 wk. Cells that survived antibiotic selection were then analyzed for positive fluorescence (compared with control wild-type B16F1 cells) by fluorescence-activated cell sorting (FACS). Fluorescence-positive cells were then individually seeded into 96-well plates and expanded to create stable monoclonal cell lines. Correct fluorescent protein knock-in was verified by PCR/sequencing and fluorescence microscopy. Lpd was sequentially tagged with tdTomato-puromycin at the C terminus using the same strategy as above. eYFP-VASP and tdTomato-Lpd (or tdTomato-IRSp53) double knock-in cell lines were selected using 2 ng/μl puromycin for 5 d and subsequently sorted for monoclonal colonies by FACS.
TABLE 1:
Knock-in guide sequences.
| Cell line generated | Parental cell line | Guide # | Guide sequence | PAM |
|---|---|---|---|---|
| B16F1_VASP-eYFP | B16F1 | 1 | CCACTTGGAAGATTCCACGT | GGG |
| 2 | CCCACTTGGAAGATTCCACG | TGG | ||
| B16F1_VASP-eYFP_Lpd-TdTomato | B16F1_VASP-eYFP | 1 | GGCTATGCAACATTGCGAAG | AGG |
| 2 | CACTACCTGAATAACATATC | AGG | ||
| B16F1_VASP-eYFP_IRSp53-TdTomato | B16F1_VASP-eYFP | 1 | TTCAGATGGACGTTGGCGAA | GGG |
| 2 | GGCCGATGTGAGGCGAGGCT | AGG |
Sample preparation for microscopy
Glass chambers of varying sizes were cleaned by plasma cleaning for 5 min, coated with 50 μg/ml mouse laminin (Sigma #L2020) diluted in phosphate-buffered saline (PBS), and incubated overnight at 4°C. On the day of imaging, laminin-coated glass was rinsed with PBS and replaced with imaging medium (Ham’s/F12 medium + 10% FBS + 50 mM HEPES). B16F1 cells were trypsinized and plated on laminin-coated glass and began to spread within 30 min. Cells transfected with SNAP-fusion proteins were labeled with 50 nM Janelia Fluor SNAP646-ligand diluted in imaging media for 1 h upon plating on laminin-coated glass. Janelia Fluor SNAP ligands were a generous gift of Luke Lavis (Janelia Farms). Prior to imaging, the cells were washed with PBS and replaced with fresh imaging media.
Microscopy
Cells were imaged by confocal spinning disk microscopy using a 60× Nikon Plan Apo TIRF (NA 1.49) objective on a Nikon Eclipse microscope fitted with an Andor iXon emCCD camera and controlled by Micromanager 1.4. Cells were maintained at 37°C and 5% CO2 during imaging using an Okolab stage top incubator and controller. Image processing was performed using ImageJ/Fiji and custom Matlab code. For experiments in which the barbed ends of actin filaments were blocked using cytochalasin D (Sigma #C8273), B16F1 cells were first plated on laminin-coated glass as described above. To obtain a baseline before drug addition, we imaged cells for 30 s and then added 500 nM cytochalasin D (or DMSO) directly on the microscope stage for acute perturbation. The drug was prepared at 2× concentration in imaging media before addition to cells to achieve a final concentration of 500 nM.
Image analysis
To track individual VASP-containing clusters, we developed custom Matlab code to identify and track fluorescent foci through entire time-lapse image sequences. All time-lapse sequences analyzed in this work comprised 40 images of each fluorophore, acquired individually or in pairs at 15 acquisitions/minute. Image acquisition parameters were identical across all experiments. For each frame, we generated an ROI centered on the fluorescent focus and used it to calculate a background-subtracted integrated intensity. To analyze size fluctuations of individual foci over time, we computed the time-averaged intensity and SD for each series. For cross-correlation analysis, we simultaneously imaged fluorescent derivatives of both VASP and either lamellipodin or IRSp53. We computed integrated fluorescence intensities for each time point from the two sets of images to create a pair of intensity versus time traces. We then subtracted the mean intensity along with the linear and quadratic trends from each data and used the Matlab xcorr() function to compute a cross-correlation for each pair of traces. We then averaged cross-correlations of multiple foci (>30) from multiple cells (>10) over multiple experiments (>3). We used a naive bootstrap analysis to set the level of statistical significance for each cross-correlation (Zoubir and Iskander 2004). Briefly, we randomly swapped the data from the two fluorescent channels and calculated the cross-correlations of these unrelated data sets. We calculated average cross-correlations from more than 30 such random permutations for each pair of fluorescent molecules and then calculated the SD at each time lag position. We marked a minimum criterion for significance on the plots at twice the value of this SD, corresponding to a false positive rate of 0.05.
To generate adaptive kymographs to follow fluorescent protein dynamics along a specified leading edge, we developed a Matlab function that takes in two color time-lapse data sets, uses the marker fluorescence of a designated fluorescence channel to identify the cell edge, and, with user input, creates a dynamic region of interest around the cell edge. Tracking and plotting parameters include the following: gaussian blurring used to smoothen images; threshold to find the cell edge; absolute maximum intensity of channel 1 for plot display; absolute maximum intensity of channel 2 for plot display. The moving leading edge is aligned frame by frame using autocorrelation. Using this region of interest, the function creates a kymograph for the original channel (VASP) and a second, independent channel (i.e., lamellipodin or IRSp53). Matlab code is available at the Mullins lab Github repository: https://github.com/mullinslabUCSF/edge-kymograph.
Clustering quantification
VASP clustering at the leading edge was quantified by measuring the background-subtracted fluorescence intensity over an eight-point linewidth ROI traced along the leading edge in ImageJ/Fiji. The clustering metric was calculated as the SD along the leading edge ROI normalized to the average fluorescence intensity (SD/mean). For paired samples (such as in the cytochalasin D treatments), the drug-treated condition was normalized to the control condition.
Supplementary Material
Acknowledgments
We thank Kara McKinley, Scott Hansen, Luke Lavis, Samuel Lord, and members of the Mullins lab for reagents, experimental advice, and feedback on the manuscript. We thank Ron Vale, Sophie Dumont, Geeta Narlikar, Dan Fletcher, and Orion Weiner for helpful conversations. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (R35-GM118119 to R.D.M.), by the Howard Hughes Medical Institute Investigator program (R.D.M.), and by a predoctoral fellowship from the American Heart Association (17PRE33630125 to K.W.C.).
Abbreviations used:
- CD
cytochalasin D
- EVH1
Ena/VASP homology-1
- Lpd
lamellipodin
- RA
Ras-association
- PH
pleckstrin homology
- VASP
vasodilator-stimulated phosphoprotein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-04-0270) on June 24, 2020.
REFERENCES
- Ahmed S, Goh WI, Bu W. (2010). I-BAR domains, IRSp53 and filopodium formation. Semin Cell Dev Biol , 350–356. [DOI] [PubMed] [Google Scholar]
- Bae YH, Ding Z, Das T, Wells A, Gertler F, Roy P. (2010). Profilin1 regulates PI(3,4)P2 and lamellipodin accumulation at the leading edge thus influencing motility of MDA-MB-231 cells. Proc Natl Acad Sci USA , 21547–21552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LJ, Kühne R, Hoffmann B, Häfner A, Schmieder P, Volkmer-Engert R, Hof M, Wahl M, Schneider-Mergener J, Walter U, et al. (2000). Dual epitope recognition by the VASP EVH1 domain modulates polyproline ligand specificity and binding affinity. EMBO J , 4903–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzik M, McClain LM, Gupton SL, Gertler FB. (2014). Ena/VASP regulates mDia2-initiated filopodial length, dynamics, and function. Mol Biol Cell , 2604–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Loureiro JJ, Libova I, Fässler R, Wehland J, Gertler FB. (2000). Negative regulation of fibroblast motility by Ena/VASP proteins. Cell , 717–728. [DOI] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, et al. (2002). Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell , 509–521. [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. (2014). Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev , 235–263. [DOI] [PubMed] [Google Scholar]
- Breitsprecher D, Kiesewetter AK, Linkner J, Urbanke C, Resch GP, Small JV, Faix J. (2008). Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. EMBO J , 2943–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindle NP, Holt MR, Davies JE, Price CJ, Critchley DR. (1996). The focal-adhesion vasodilator-stimulated phosphoprotein (VASP) binds to the proline-rich domain in vinculin. Biochem J (Pt 3), 753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y-C, Zhang H, Brennan ML, Wu J. (2013). Crystal structure of Lamellipodin implicates diverse functions in actin polymerization and Ras signaling. Protein Cell , 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Squarr AJ, Stephan R, Chen B, Higgins TE, Barry DJ, Martin MC, Rosen MK, Bogdan S, Way M. (2014). Ena/VASP proteins cooperate with the WAVE complex to regulate the actin cytoskeleton. Dev Cell , 569–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coló GP, Lafuente EM, Teixidó J. (2012). The MRL proteins: adapting cell adhesion, migration and growth. Eur J Cell Biol , 861–868. [DOI] [PubMed] [Google Scholar]
- Damiano-Guercio J, Kurzawa L, Mueller J, Dimchev G, Schaks M, Nemethova M, Pokrant T, Brühmann S, Linkner J, Blanchoin L, et al. (2020). Loss of Ena/VASP interferes with lamellipodium architecture, motility and integrin-dependent adhesion. eLife . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimchev G, Amiri B, Humphries AC, Schaks M, Dimchev V, Stradal TEB, Faix J, Krause M, Way M, Falcke M, et al. (2019). Lamellipodin tunes cell migration by stabilizing protrusions and promoting adhesion formation. J Cell Sci . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disanza A, Bisi S, Winterhoff M, Milanesi F, Ushakov DS, Kast D, Marighetti P, Romet-Lemonne G, Müller H-M, Nickel W, et al. (2013). CDC42 switches IRSp53 from inhibition of actin growth to elongation by clustering of VASP. EMBO J , 2735–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz-Laylin LK, Riel-Mehan M, Chen B-C, Lord SJ, Goddard TD, Ferrin TE, Nicholson-Dykstra SM, Higgs H, Johnson GT, Betzig E, et al. (2017). Actin-based protrusions of migrating neutrophils are intrinsically lamellar and facilitate direction changes. eLife . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. (1996). Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell , 227–239. [DOI] [PubMed] [Google Scholar]
- Han Y-H, Chung CY, Wessels D, Stephens S, Titus MA, Soll DR, Firtel RA. (2002). Requirement of a vasodilator-stimulated phosphoprotein family member for cell adhesion, the formation of filopodia, and chemotaxis in dictyostelium. J Biol Chem , 49877–49887. [DOI] [PubMed] [Google Scholar]
- Hansen SD, Mullins RD. (2010). VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J Cell Biol , 571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SD, Mullins RD. (2015). Lamellipodin promotes actin assembly by clustering Ena/VASP proteins and tethering them to actin filaments. eLife . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Hammar K, Smith PJ, Oldenbourg R. (1999). Birefringence imaging directly reveals architectural dynamics of filamentous actin in living growth cones. Mol Biol Cell , 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Svitkina T. (2008). Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol Biol Cell , 1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Leslie JD, Stewart M, Lafuente EM, Valderrama F, Jagannathan R, Strasser GA, Rubinson DA, Liu H, Way M, et al. (2004). Lamellipodin, an Ena/VASP ligand, is implicated in the regulation of lamellipodial dynamics. Dev Cell , 571–583. [DOI] [PubMed] [Google Scholar]
- Krugmann S, Jordens I, Gevaert K, Driessens M, Vandekerckhove J, Hall A. (2001). Cdc42 induces filopodia by promoting the formation of an IRSp53:Mena complex. Curr Biol , 1645–1655. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski AV, Rubinson DA, Dent EW, Edward van Veen J, Leslie JD, Zhang J, Mebane LM, Philippar U, Pinheiro EM, Burds AA, et al. (2007). Ena/VASP is required for neuritogenesis in the developing cortex. Neuron , 441–455. [DOI] [PubMed] [Google Scholar]
- Lacayo CI, Pincus Z, VanDuijn MM, Wilson CA, Fletcher DA, Gertler FB, Mogilner A, Theriot JA. (2007). Emergence of large-scale cell morphology and movement from local actin filament growth dynamics. PLoS Biol , e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafuente EM, van Puijenbroek AAFL, Krause M, Carman CV, Freeman GJ, Berezovskaya A, Constantine E, Springer TA, Gertler FB, Boussiotis VA. (2004). RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev Cell , 585–595. [DOI] [PubMed] [Google Scholar]
- Lewis AK, Bridgman PC. (1992). Nerve growth cone lamellipodia contain two populations of actin filaments that differ in organization and polarity. J Cell Biol , 1219–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KB, Bu W, Goh WI, Koh E, Ong SH, Pawson T, Sudhaharan T, Ahmed S. (2008). The Cdc42 effector IRSp53 generates filopodia by coupling membrane protrusion with actin dynamics. J Biol Chem , 20454–20472. [DOI] [PubMed] [Google Scholar]
- Mattila PK, Lappalainen P. (2008). Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol , 446–454. [DOI] [PubMed] [Google Scholar]
- Mattila PK, Pykäläinen A, Saarikangas J, Paavilainen VO, Vihinen H, Jokitalo E, Lappalainen P. (2007). Missing-in-metastasis and IRSp53 deform PI(4,5)P2-rich membranes by an inverse BAR domain-like mechanism. J Cell Biol , 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejillano MR, Kojima S, Applewhite DA, Gertler FB, Svitkina TM, Borisy GG. (2004). Lamellipodial versus filopodial mode of the actin nanomachinery: pivotal role of the filament barbed end. Cell , 363–373. [DOI] [PubMed] [Google Scholar]
- Millard TH, Bompard G, Heung MY, Dafforn TR, Scott DJ, Machesky LM, Fütterer K. (2005). Structural basis of filopodia formation induced by the IRSp53/MIM homology domain of human IRSp53. EMBO J , 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilner A, Rubinstein B. (2005). The physics of filopodial protrusion. Biophys J , 782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Miki H, Nozumi M, Takenawa T, Miyamoto S, Wehland J, Small JV. (2003). IRSp53 is colocalised with WAVE2 at the tips of protruding lamellipodia and filopodia independently of Mena. J Cell Sci , 2577–2583. [DOI] [PubMed] [Google Scholar]
- Niebuhr K, Ebel F, Frank R, Reinhard M, Domann E, Carl UD, Walter U, Gertler FB, Wehland J, Chakraborty T. (1997). A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASP family. EMBO J , 5433–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenbourg R, Katoh K, Danuser G. (2000). Mechanism of lateral movement of filopodia and radial actin bundles across neuronal growth cones. Biophys J , 1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehoda KE, Lee DJ, Lim WA. (1999). Structure of the enabled/vasp homology 1 domain–peptide complex. Cell , 471–480. [DOI] [PubMed] [Google Scholar]
- Prévost C, Zhao H, Manzi J, Lemichez E, Lappalainen P, Callan-Jones A, Bassereau P. (2015). IRSp53 senses negative membrane curvature and phase separates along membrane tubules. Nat Commun , 8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc , 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M, Halbrügge M, Scheer U, Wiegand C, Jockusch BM, Walter U. (1992). The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J , 2063–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M, Rüdiger M, Jockusch BM, Walter U. (1996). VASP interaction with vinculin: a recurring theme of interactions with proline-rich motifs. FEBS Lett , 103–107. [DOI] [PubMed] [Google Scholar]
- Rottner K, Behrendt B, Small JV, Wehland J. (1999). VASP dynamics during lamellipodia protrusion. Nat Cell Biol , 321–322. [DOI] [PubMed] [Google Scholar]
- Saarikangas J, Zhao H, Pykäläinen A, Laurinmäki P, Mattila PK, Kinnunen PKJ, Butcher SJ, Lappalainen P. (2009). Molecular mechanisms of membrane deformation by I-BAR domain proteins. Curr Biol , 95–107. [DOI] [PubMed] [Google Scholar]
- Schmeiser C, Winkler C. (2015). The flatness of Lamellipodia explained by the interaction between actin dynamics and membrane deformation. J Theor Biol , 144–155. [DOI] [PubMed] [Google Scholar]
- Sudhaharan T, Hariharan S, Lim JSY, Liu JZ, Koon YL, Wright GD, Chiam KH, Ahmed S. (2019). Superresolution microscopy reveals distinct localisation of full length IRSp53 and its I-BAR domain protein within filopodia. Sci Rep , 2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG. (2003). Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol , 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignjevic D, Yarar D, Welch MD, Peloquin J, Svitkina T, Borisy GG. (2003). Formation of filopodia-like bundles in vitro from a dendritic network. J Cell Biol , 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner OD, Marganski WA, Wu LF, Altschuler SJ, Kirschner MW. (2007). An actin-based wave generator organizes cell motility. PLoS Biol , e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MD, Mullins RD. (2002). Cellular control of actin nucleation. Annu Rev Cell Dev Biol , 247–288. [DOI] [PubMed] [Google Scholar]
- Yamagishi A, Masuda M, Ohki T, Onishi H, Mochizuki N. (2004). A novel actin bundling/filopodium-forming domain conserved in insulin receptor tyrosine kinase substrate p53 and missing in metastasis protein. J Biol Chem , 14929–14936. [DOI] [PubMed] [Google Scholar]
- Yang C, Hoelzle M, Disanza A, Scita G, Svitkina T. (2009). Coordination of membrane and actin cytoskeleton dynamics during filopodia protrusion. PLoS One , e5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Svitkina T. (2011). Filopodia initiation: focus on the Arp2/3 complex and formins. Cell Adh Migr , 402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LE, Heimsath EG, Higgs HN. (2015). Cell type-dependent mechanisms for formin-mediated assembly of filopodia. Mol Biol Cell , 4646–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LE, Latario CJ, Higgs HN. (2018). Roles for Ena/VASP proteins in FMNL3-mediated filopodial assembly. J Cell Sci . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoubir AM, Iskander DR. (2004). Bootstrap Techniques for Signal Processing. Cambridge: Cambridge University Press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.