

Abstract
Regulation of excitatory to inhibitory signaling balance is essential to nervous system health and is maintained by numerous enzyme systems that modulate the activity, localization, and abundance of synaptic proteins. SUMOylation is a key post-translational regulator of protein function in diverse cells, including neurons. There, its role in regulating synaptic transmission through pre- and postsynaptic effects has been shown primarily at glutamatergic central nervous system synapses, where the sole SUMO-conjugating enzyme Ubc9 is a critical player. However, whether Ubc9 functions globally at other synapses, including inhibitory synapses, has not been explored. Here, we investigated the role of UBC-9 and the SUMOylation pathway in controlling the balance of excitatory cholinergic and inhibitory GABAergic signaling required for muscle contraction in Caenorhabditis elegans. We found inhibition or overexpression of UBC-9 in neurons modestly increased muscle excitation. Similar and even stronger phenotypes were seen with UBC-9 overexpression specifically in GABAergic neurons, but not in cholinergic neurons. These effects correlated with accumulation of synaptic vesicle-associated proteins at GABAergic presynapses, where UBC-9 and the C. elegans SUMO ortholog SMO-1 localized, and with defects in GABA-dependent behaviors. Experiments involving expression of catalytically inactive UBC-9 [UBC-9(C93S)], as well as co-expression of UBC-9 and SMO-1, suggested wild type UBC-9 overexpressed alone may act via substrate sequestration in the absence of sufficient free SUMO, underscoring the importance of tightly regulated SUMO enzyme function. Similar effects on muscle excitation, GABAergic signaling, and synaptic vesicle localization occurred with overexpression of the SUMO activating enzyme subunit AOS-1. Together, these data support a model in which UBC-9 and the SUMOylation system act at presynaptic sites in inhibitory motor neurons to control synaptic signaling balance in C. elegans. Future studies will be important to define UBC-9 targets at this synapse, as well as mechanisms by which UBC-9 and the SUMO pathway are regulated.
Keywords: SUMO, E:I balance, inhibitory, synapse, presynaptic, Caenorhabditis elegans
Significance statement
Identifies a presynaptic role for the SUMO enzyme system in inhibitory neurons during neuromuscular signaling.
Shows SUMO E2 UBC-9 localizes to GABAergic motor neuron presynapses and affects vesicle localization and muscle excitation.
Aids understanding of conserved UBC-9 functions in E:I balance, which is lost in neurological diseases.
Introduction
Excitatory to inhibitory (E:I) signaling balance is critical for nervous system function, and many neurological diseases, including epilepsy, autism, and numerous neurodegenerative conditions, involve E:I imbalances.1,2 Hundreds of proteins reside at pre- and postsynaptic sites to control the amount and timing of signaling at specific synapses, contributing to regulation of this balance.3 A variety of post-translational modifications, including phosphorylation, ubiquitination, and palmitoylation tightly control the functions of synaptic proteins.4-8 Many of these modifications are activity-dependent and serve to modulate the localization, surface expression, activity, and/or complex formation of synaptic proteins to impact synaptic vesicle release and neurotransmitter signal reception.4,9–15 The extent of these modifications, as well as the specific enzymes and target proteins involved, remain incompletely understood.
Protein SUMOylation is a post-translational modification, conserved from yeast to humans, that has emerged as a key regulator of neuronal and synaptic biology.8,16–18 During SUMOylation, proteins are modified by the covalent attachment of small ubiquitin-like modifier (SUMO) polypeptides. These approximately 100-amino acid globular SUMO molecules (SUMO-1, -2/3, and -4 in humans) are similar in size and shape to ubiquitin and are likewise added to lysine residues in target proteins through the activity of a multi-step enzymatic cascade. For SUMO, this involves activation of mature SUMO molecules by the E1 enzyme complex Aos1-Uba2 /SAE1-SAE2 followed by SUMO transfer to the E2 SUMO-conjugating enzyme Ubc9.16,19 Ubc9 alone, or in combination with any of a number of E3 ligases, mediates the attachment of SUMO to the target protein.19 Removal of SUMO peptides from substrates, as well as activation of immature SUMO peptides, occurs via the activity of SUMO isopeptidases, also known as ubiquitin-like-specific proteases (Ulp) or sentrin-specific proteases (SENPs).19,20
Originally identified for its role in regulating transcriptional activity and other nuclear functions, SUMOylation is now known to modulate the activity and localization of diverse cytosolic proteins, including many involved in neuronal and synapse physiology.8,17 Several known SUMOylated neuronal proteins, including huntingtin, amyloid precursor protein, ataxins, tau, alpha-synuclein, and BACE1, are linked to neurodegenerative diseases.18,21,22 SUMO-1 has also been detected in synaptosomes from mouse models of Alzheimer’s Disease,23 and global SUMOylation levels are reported to be differentially affected by varying levels of oxidative stress, a component of age- and infection-related neurodegeneration.24 Thus, the SUMOylation pathway has been proposed as a therapeutic target in these diseases. Biochemical studies of synaptic fractions from rat hippocampi indicate the presence of multiple SUMOylated synaptic proteins, although the identities of many of these remain unknown.25 Synaptic proteins shown to be regulated by SUMOylation, primarily at mammalian glutamatergic synapses, include postsynaptic scaffold proteins CASK and GISP,11,26 along with a number of presynaptic regulators of vesicle release.9,10,27,28 Data from other studies indicate certain K+ and Na+ channels also are regulated either directly or indirectly via SUMOylation to promote general neuronal excitability, as is the AMPA receptor regulator Arc.16,29 Together these data demonstrate the importance of protein SUMOylation in controlling synaptic function at central synapses.
Because it is the sole E2 SUMO conjugating enzyme in any organism, Ubc9 represents a critical component of the SUMO pathway. Ubc9 activity itself is regulated by SUMOylation, as well as by phosphorylation and acetylation.30–33 Ubc9 typically localizes to sites where SUMO is found,18 and organisms lacking Ubc9 are not viable, suggesting the importance of SUMOylation in development.34–36 Multiple studies also have implicated Ubc9 in synaptic function and plasticity. Work in Drosophila uncovered a genetic requirement for Ubc9 in long term memory formation where it appears to act with histone deacetylase 4 (HDAC4).37 Studies in rat hippocampal neurons demonstrated the ability of Ubc9 to interact with GluR6 kainate receptor subunits, which are SUMOylated following kainate application to promote postsynaptic receptor internalization and decreased kainate-evoked currents.38 Activity-dependent pre- and post-synaptic localization of Ubc9, as well as other SUMOylation and deSUMOylation machinery, at glutamatergic central synapses was confirmed by several other studies.11,13–15,25 These reports underscore the importance of Ubc9 and SUMOylation to synaptic function. However, questions as to whether Ubc9’s role is conserved across phylogeny and at diverse synapse types, including at inhibitory or peripheral synapses, remain. Just one study to date has described a role for the SUMO system at GABAergic synapses, where it regulates the postsynaptic GABAA receptor scaffold protein gephyrin,39 and a single paper identified SUMOylation of a muscarinic acetylcholine receptor in heterologous cell culture.40 Moreover, only a few papers have explored any neuronal SUMO or Ubc9 functions in non-mammalian organisms.37,41–45
The neuromuscular junction (NMJ) of Caenorhabditis elegans is an ideal system in which to investigate the broader regulation of synaptic signaling and E:I balance by the SUMO pathway. The body wall muscles of these microscopic roundworms contain both excitatory inputs from cholinergic motor neurons that promote muscle contraction and inhibitory inputs from GABAergic motor neurons that promote muscle relaxation by counteracting the excitatory signals.46 Tight regulation of the balance of excitatory and inhibitory transmission is required for proper muscle contraction and movement of the worms. Behavioral assays, including sensitivity to paralysis induced by the acetylcholinesterase inhibitor, aldicarb, can be used to measure muscle excitation as a readout for neuromuscular signaling balance.47 In the presence of aldicarb, populations of worms paralyze over time. Animals carrying mutations that increase cholinergic signaling or decrease GABAergic signaling show increased paralysis relative to wild type worms, whereas those with decreased cholinergic or increased GABAergic signaling exhibit decreased paralysis.47 Numerous genes now known to regulate E:I signaling balance across phylogeny were identified based on the aldicarb phenotypes of mutant worms.48–50 C. elegans contain homologs of the SUMO E1 (AOS-1 and UBA-2 subunits) and E2 (UBC-9) enzymes, as well as homologs of several E3 ligases and SENPs (called ULPs in C. elegans).34 A single C. elegans SUMO ortholog, SMO-1, exists and has structural and functional similarity to SUMO-1 and -2 in humans. The smo-1 gene is essential and expressed throughout development, and animals lacking smo-1 expression that survive due to maternal contributions are sterile with abnormal somatic gonads, vulvas, and germlines.51 Other studies have identified SUMOylated C. elegans proteins,52 and current estimates indicate as much as 15% to 20% of the worm proteome is SUMOylated at any given time, similar to estimates in human cells.53,54
In this study, we set out to assess the potential requirements for the SUMO pathway in controlling E:I balance at the C. elegans NMJ. Given the localization of Ubc9 to synapses in mammalian neurons and the general importance of presynaptic protein SUMOylation for neurotransmitter release at glutamatergic hippocampal synapses, we hypothesized that SUMO pathway enzymes are required in motor neurons to maintain appropriate neuroromuscular E:I signaling balance and muscle excitation. Here, we show that the SUMO-conjugating enzyme UBC-9 can act in GABAergic neurons to regulate neuromuscular function. Overexpressed UBC-9 localized to presynaptic sites in GABAergic motor neurons, caused accumulation of GABA-containing synaptic vesicles, and increased muscle contraction, changes that correlated with defects in a pharmacologic test of GABAergic signaling. Similar results were seen with overexpression of the SUMO E1 subunit AOS-1, supporting a role for the SUMO pathway beyond just UBC-9. Enhanced muscle contraction also was seen with inhibition of UBC-9 or AOS-1 expression, as well as with overexpression of a SUMO conjugation-deficient UBC-9, consistent with dominant negative substrate sequestration effects. Overexpression of SMO-1 also modestly increased muscle excitation in a manner non-additive with UBC-9 overexpression, suggesting UBC-9 and SMO-1 act in a common pathway to impact neuromuscular function and further underscoring the need for precise control of SUMO enzyme activity at this synapse. Based on these data, we propose that endogenous UBC-9 acts in GABAergic neurons, likely via a SUMO-dependent pathway, to promote appropriate levels GABA neurotransmitter release and prevent excess muscle contraction. Our results are consistent with those of prior studies that demonstrate the critical need for tight regulation of synaptic protein SUMOylation and SUMO enzyme function for proper synaptic signaling.
Materials and Methods
C. elegans strains
All strains were grown on nematode growth medium (NGM) agar plates spotted with OP50 Escherichia coli and maintained at 20°C according to standard protocols.55 Young adult hermaphrodite worms were used for all experiments. Table 1 lists the strains used in this study.
Table 1.
C. elegans strains used (*indicates additional times outcrossed to wild type).
| Strain number | Genotype |
|---|---|
| N2 (Bristol) | Wild type |
| NM467 | snb-1(md247) |
| RB1500 | ulp-1(ok1768) |
| FJ413 | ulp-3(tm1287) |
| FJ415 | ulp-1(tm1174) |
| JRK10 | ulp-5(tm3071)*2 |
| JRK18 | kjrEx4 (Punc-25::mcherry::snb-1; Pmyo-2::NLS::mcherry)]48 |
| JRK24 | ulp-1(ok1768)*3;ulp-3(tm1287) |
| JRK25 | ulp-1(ok1768)*3;ulp-5(tm3071) |
| JRK34 | uIs60 (Punc-119::yfp; Punc-119::sid-1); nre-1(hd120) lin-15(hd126) |
| JRK48 | juIs1*1 |
| JRK50 | kjrEx12 (Psnb-1::aos-1; Pmyo-2::NLS::gfp) |
| JRK64 | kjrEx20 (Pmyo-2::NLS::gfp) |
| JRK68 | nuIs152*2 |
| JRK70 | kjrEx21 (Punc-25::aos-1; Pmyo-2::NLS::gfp) |
| JRK71 | kjrEx11 (Psnb-1::ubc-9;Pmyo-2::NLS::gfp) |
| JRK72 | kjrEx22 (Punc-25::ubc-9;Pmyo-2::NLS::gfp) |
| JRK77 | kjrEx11; nuIs152 |
| JRK82 | kjrEx12; juIs1*2 |
| JRK85 | kjrEx21; juIs1*2 |
| JRK86 | kjrEx22; juIs1*2 |
| JRK89 | kjrEx22;nuIs152 |
| JRK102 | kjrEx29 (Punc-17::ubc-9;Pmyo-2::NLS::gfp)] |
| JRK110 | kjrEx31 (Punc-25::ubc-9(C93S); Pmyo-2::NLS::gfp)] |
| JRK134 | kjrEx34 (Punc-25::gfp::ubc-9;Pmyo-2::NLS::gfp) |
| JRK135 | kjrEx35 (Punc-25::gfp::smo-1(GG);Pmyo-2::NLS::gfp) line #1 |
| JRK136 | kjrEx36 (Punc-25::gfp::smo-1(GG);Pmyo-2::NLS::gfp) line #2 |
| JRK137 | kjrEx37 (Punc-25::ubc-9;Pttx-3::dsRed) |
| JRK138 | kjrEx35; kjrEx37 |
| JRK139 | kjrEx4; kjrEx34 |
| JRK141 | kjrEx4; kjrEx36 |
Plasmid and strain generation
The Psnb-1::ubc-9 (pJRK22) and Psnb-1::aos-1 (pJRK23) plasmids were made by inserting ubc-9 or aos-1 cDNA, respectively, into the PD49.26 plasmid56 containing the Psnb-1 promoter (pFJ19). To do this, the cDNAs were amplified from the Vidal Orfeome clones (F29B9.6 and C08B6.9) using forward and reverse primers engineered with NheI and KpnI restriction sites. The amplified PCR products and plasmid pFJ19 were digested with NheI and KpnI, then the ubc-9 or aos-1 cDNA was inserted into pFJ19 using T4 DNA ligase. To make Punc-25::aos-1 (pJRK39), pJRK23 was digested with SphI and BamHI to remove the Psnb-1 promoter. The Punc-25 promoter was isolated from the pJRK8 (Punc-25::yfp::emb-27) plasmid using the same enzymes, then ligated into pJRK23. The Punc-25::ubc-9 (pJRK43) and Punc-17::ubc-9 (pJRK44) plasmids were made by inserting the Punc-25 and Punc-17 promoters into the PD49.26 plasmid56 containing ubc-9 cDNA. To do this, plasmids pJRK39 and pJRK42 (Punc-17::aos-1) were digested with SphI and BamHI to remove the unc-25 and unc-17 promoters, and these were ligated between the SphI and BamHI sites in pJRK22 following removal of Psnb-1. To generate the Punc-25::ubc-9(C93S) plasmid (pJRK69), QuikChange site-directed mutagenesis (Agilent) was performed using primers designed to introduce a single basepair mutation into the ubc-9 sequence (forward primer: 5’-cccatcaggtaccgtgAgcttatctcttctgga-3’; reverse primer: 5’-tccagaagagataagcTcacggtacctgatggg-3) and performed according to the manufacturer’s instructions. The Punc-25::gfp::smo-1(GG) (pJRK81) plasmid was made by inserting gfp::smo-1(GG) amplified from pJRK72 [previously generated from plasmid #462]57into plasmid pJRK39 using gfp forward and smo-1 reverse primers containing NheI and KpnI restriction sites, respectively. The PCR products and pJRK39 were digested with NheI and KpnI. This removed aos-1 from pJRK39 and allowed for the subsequent insertion of gfp::smo-1 into pJRK39 using T4 DNA ligase. The final pJRK81 plasmid was confirmed by sequencing. The Punc-25::gfp::ubc-9 (pJRK85) plasmid was constructed in 2 steps. First, ubc-9 cDNA containing an internal NotI restriction site at the N-terminus was generated via PCR amplification of ubc-9 from pJRK43 using forward and reverse primers containing NheI and SacI restriction sites, respectively. Plasmid pJRK39 and the amplified [ubc-9(NotI)] products were then digested with NheI and SacI to remove the aos-1 cDNA from pJRK39 and to create restriction sites on the ubc-9(NotI) insert, and the two fragments were ligated to create Punc-25::ubc-9(NotI) (pJRK80). Second, pJRK80 and pJRK50 (Pfshr-1::gfp::fshr-1) were digested with NotI to remove gfp. Following heat inactivation, the digested pJRK80 was treated with calf intestinal alkaline phosphatase. pJRK85 was created by ligating gfp into the NotI site in pJRK80.
Transgenic strains with panneuronal, GABAergic, or cholinergic neuron-specific UBC-9, UBC-9(C93S), AOS-1, or SMO-1(GG) overexpression were isolated following standard microinjection of the plasmids into the gonads of gravid N2 adult worms as described previously and at concentrations that generated viable lines for each transgene.58 Psnb-1::ubc-9 (pJRK22) and Psnb-1::aos-1 (pJRK23) were each injected at a concentration of 1 ng/μL, along with 10 ng/μL of Pmyo-2::NLS::gfp (co-injection marker) to generate strains JRK71and JRK50, respectively. Punc-25::aos-1 (pJRK39) was injected at 25 ng/µL, along with 10 ng/µL of Pmyo-2::NLS::gfp to generate strain JRK70. Punc-25::ubc-9 (pJRK43) was injected at a concentration of 10 ng/μL, along with 10 ng/μL of Pmyo-2::NLS::gfp (co-injection marker) to generate strain JRK72 or with 50 ng/μL of Pttx3::dsRed to generate JRK137. Punc-17::ubc-9 (pJRK44) was injected at 20 ng/µL, along with 10 ng/µL of Pmyo-2::NLS::gfp to generate strain JRK102. The Punc-25::ubc-9(C93S) (pJRK69) plasmid, Punc-25::gfp::smo-1(GG) (pJRK81), and Punc-25::gfp::ubc-9 (pJRK85) were each injected at a concentration of 10 ng/μL, along with 10 ng/μL of Pmyo-2::NLS::gfp (co-injection marker) to generate strains JRK110, JRK136, and JRK134, respectively. The mock overexpression (OE) control strain (JRK64) was generated by injection of 10 ng/µL of Pmyo-2::NLS::gfp alone.
RNA interference
RNA interference (RNAi) was performed as follows using the feeding method in RNAi-enhanced strains. Panneuronal RNAi was performed using the uIs60;nre-1(hd20) lin-15b(hd126) strain, which expresses SID-1 RNA channels under a neuron-specific promoter.59,60 RNAi clones were obtained from a genome-wide library consisting of HT115(DE) E. coli carrying gene fragments for double-stranded RNA production in the T7-tailed L4440 vector.61 Bacteria were maintained in the presence of 50 μg/ml ampicillin and 15 μg/ml tetracycline for plasmid selection.62 RNAi feeding plates were prepared as follows. First, 3 ml overnight cultures of bacteria carrying an RNAi plasmid targeting either ubc-9, aos-1, or gfp (negative control) were grown in Luria broth (LB) containing 50 μg/ml ampicillin. Cultures were spotted (150 μl each) onto 35 mm NGM agar plates containing 50 μg/ml ampicillin and 5 mM IPTG. Plates were dried open for 2 hours, then closed and left overnight at room temperature. For panneuronal RNAi, a <1-generation protocol was used to avoid embryonic lethality caused by potential knockdown in non-neuronal tissues. Worms were synchronized by bleaching gravid adults to hatch and recover L1s, which were then plated onto RNAi-containing plates and grown at 20°C for 3 days. For all experiments, RNAi-treated worms were grown to the young adult stage for use in aldicarb assays.
Aldicarb assays
NGM agar plates (35 mm) containing 1mM of aldicarb (Sigma-Aldrich) were prepared, then spotted with 150 μL of OP50 E. coli. After 1 day, approximately 20 young adult worms were placed on each plate in 2-minute intervals, then measured for total paralysis every 25 minutes. Total paralysis was defined as no physical movement of the worms when prodded 3 times with a platinum wire on the nose.47 Three plates were tested for each strain of worms per experiment with the experimenter double-blinded to genotype. The average percentage of worms paralyzed for each strain at each time point ± s.e.m. was calculated using Microsoft Excel. Data from a total of 3 to 15 plates were pooled from experiments taken at the same time intervals over several days. Statistical analyses were performed using JMP 14 software to compare the average percentages of worms of each strain paralyzed at the timepoint with the largest differences for each experiment (75 or 100 minutes). All data were first confirmed to fall within a normal distribution using a Shapiro Wilk Goodness of Fit test, and equality of variances confirmed using the Analysis of Means (ANOM) of Variances test. One-way ANOVAs then were used to assess statistical significance of the differences in the means between groups (α < 0.05), followed by a Tukey’s post hoc test (α < 0.05 for all). Connecting letters reports, as well as p value results of ordered difference reports are provided.
Pentylenetetrazole assays
NGM agar plates (35 mm) containing 10 mg/ml pentylenetetrazole (PTZ, Sigma-Aldrich) were prepared on the day of the experiment, allowed to dry open for 1 hour at room temperature, then spotted with 25 μL of OP50 E. coli. After 2 additional hours of drying, approximately 20 (and no less than 15) young adult worms were placed onto each plate in 4-minute intervals, then measured for anterior convulsions (“head bobs”) after 50 minutes.48,63 These anterior convulsions were accompanied by full body paralysis in some animals. Two plates were tested for each strain of worms per experiment with the experimenter double-blinded to genotype. One plate of worms of each genotype was also tested on plates containing 0 mg/ml PTZ to ensure the absence of convulsions. The average percentage of worms exhibiting anterior convulsions for each strain at each time point ± SD was calculated using Microsoft Excel. Data from a total of 8 to 10 plates were pooled from experiments taken at the same time intervals over several days. Statistical analyses were performed using JMP 14 software, with data tested for normality and equality of variances followed by a one-way ANOVA (α < 0.05) and Tukey’s post hoc test (α < 0.05 for all). Connecting letters reports and p value results of ordered difference reports are provided.
Fluorescence microscopy
For all imaging experiments, young adult worms were immobilized in 30 mg/ml 2, 3-butanedione monoxamine (Sigma-Aldrich) for 5 to 7 minutes, then mounted on 2% agarose pads. Their dorsal nerve cords halfway between the vulva and the tail were imaged with a Leica DMLB microscope (Leica Microsystems) with a 100x Plan Apochromat (1.4 NA) objective equipped with green and red fluorescence filters. Images were captured using an Exi Aqua cooled CCD camera (Qimaging) with Metamorph (v7.7) software (Molecular Devices) as described previously.48
Quantitative imaging of GFP::SNB-1 fluorescent puncta in the dorsal nerve cord of worms overexpressing UBC-9 in all neurons (kjrEx11) or only in GABA neurons (kjrEx22) was performed by generating maximum intensity projections of z-series stacks (0.2 μm steps, 1 μm total depth). Exposure settings, gain, and binning were set to fill a 12-bit dynamic range without saturation. These settings were identical for all images taken of a given fluorescent marker [i.e., GFP::SNB-1 in GABAergic (juIs1) or cholinergic (nuIs152) neurons] for each imaging set. Linescans of dorsal nerve cord puncta were generated using Metamorph (v7.1) software, and the linescan data were analyzed with Igor Pro (Wavemetrics) using custom written software as previously described.64 Mercury arc lamp output was normalized by measuring the intensities of 0.5 μm FluoSphere beads (Invitrogen Life Technologies) for each imaging day. Puncta intensities were calculated by dividing the average maximal peak intensity by the average bead intensity for the corresponding day. Interpunctal axonal intensities were similarly calculated using the average baseline fluorescence value within the dorsal nerve cord. Puncta widths were determined by measuring the width of each punctum at half the maximum peak fluorescence. Puncta densities were determined by quantifying the average number of puncta per 10 μm of the dorsal nerve cord. For all data, an average of the values for each worm in the data set ± s.e.m. is reported. Statistical significance of any differences between wild type and transgenic strain values was determined in Igor using a one-way ANOVA with Tukey’s post hoc test (P ⩽ .05). Graphs of puncta intensities, interpunctal axonal fluorescence, and width show data normalized to wild type values. Images were cropped and set to matching input levels using Adobe Photoshop.
For colocalization studies, z-stacks were obtained of worms co-expressing mCherry::SNB-1 and GFP::UBC-9 (kjrEx4;kjrEx34) or mCherry::SNB-1 and GFP::SMO-1 (kjrEx4;kjrEx36) over a 2 μm total depth (0.2 μm steps), and quantification of colocalization was performed using Metamorph 7.1. An overlaid z-stack image file was created by merging images taken in each of the red and green channels. Then, the individual plane in best focus from the merged stacks of both red and green channel images for each worm was selected for analysis. Regions containing in-focus puncta were selected from these single plane images, and the software’s linescan function was used to measure intensities across these portions of the image in both the red and green channels. Red peaks generated from the puncta were counted and recorded, and individual green peaks were counted and recorded separately. Then, the regions where red and green peaks overlapped, indicating colocalization of the two fluorophores, were counted. Only regions at least 5 a.u. greater than the background intensity value were considered as peaks, and red and green peaks that overlap at least 75% were counted for the colocalization analysis. The mean percentage of mCherry::SNB-1 peaks overlapping GFP::UBC-9 or GFP::SMO-1 and the percentage of GFP::UBC-9 or GFP::SMO-1 overlapping with mCherry::SNB-1 were determined from these counts. To control for potential bleed-through effects, worms overexpressing just mCherry::SNB-1 (kjrEx4), just GFP::UBC-9 (kjrEx34), or just GFP::SMO-1 (kjrEx36) were also imaged under both red and green filters using the same parameters as for the double-transgenic strains (see Supplemental Material). Images were cropped and set to matching input levels for each channel using Adobe Photoshop.
Results
We began by assessing whether SUMO pathway enzymes were required for neuromuscular signaling. To do this, we tested animals with reduced expression of the genes encoding individual SUMO-activating subunits (AOS-1 and UBA-2), the SUMO-conjugating enzyme (UBC-9), the E3 SUMO ligase GEI-17, or the SUMO proteases (ULP-1, -2, -3, -4 and -5) for paralysis responses induced by the acetylcholinesterase inhibitor, aldicarb. In these experiments reduction of gene expression was induced by either feeding RNAi (aos-1, uba-2, ubc-9, gei-17, ulp-2, and -4) or genetic mutation (ulp-1, -3, and -5). Individual inhibition of each of the 5 the proteases led to wild type aldicarb phenotypes. Wild type phenotypes were also observed for ulp-1;ulp-3 and ulp-1;ulp-5 genetic double mutant animals, potentially due to redundancy of protease function (data not shown). Whereas no consistent results were observed with knockdown of uba-2 and gei-17, modest aldicarb hypersensitivity was observed with knockdown of ubc-9 or aos-1 relative to control-treated worms (Figure 1), indicative of increased muscle contraction. We also observed weak but variable aldicarb hypersensitivity following cell type-specific knockdown65 of aos-1 or ubc-9 only in GABAergic but not in cholinergic neurons (data not shown), suggesting that ubc-9 activity may be required specifically in GABAergic neurons to prevent excess muscle excitation. Due to the modest effects and the variability inherent in doing cell type-specific RNAi, we centered our subsequent investigations on the possible effects of SUMO enzyme overexpression on neuromuscular function, with a particular focus on the SUMO-conjugating enzyme UBC-9.
Figure 1.

Knockdown of ubc-9 or aos-1 expression in neurons causes increased muscle excitation. Aldicarb assays were performed on animals neuronally sensitized to RNAi. Worms were fed RNAi-containing bacteria targeting gfp as a negative control, (A) ubc-9, or (B) aos-1 from the L1 stage. Approximately 20 young adult RNAi-treated worms per plate were exposed to 1 mM aldicarb and paralysis was measured in response to gentle prodding on the nose with a platinum wire. Representative assays testing 3 plates per strain are shown. Error bars denote s.e.m.
Since inhibition of SUMO enzyme expression led to increased muscle contraction, we expected overexpression would cause aldicarb resistance, consistent with decreased muscle contraction. However, overexpression of UBC-9 in all neurons or only in GABAergic neurons also led to increased muscle excitation, indicated by aldicarb hypersensitivity, relative to wild type worms or to negative control worms expressing green fluorescent protein (GFP) in pharyngeal cells as a co-injection marker (Figure 2a and b, Figure S1A). In contrast, a wild type aldicarb paralysis phenotype was observed for UBC-9 overexpression exclusively in cholinergic (ACh) neurons (Figure 2b, Figure S1B) Together, these data suggest that too much or too little UBC-9 expression in GABA neurons can shift the balance of neuromuscular signaling toward excitation. Similar results were observed for GABA neuron-specific overexpression of AOS-1, suggesting the effects may be due to global disruption or activation of the SUMOylation pathway at the NMJ (Figure 2c, Figure S1C).
Figure 2.

GABAergic neuron-specific overexpression of UBC-9 or AOS-1 causes increased muscle excitation. Aldicarb assays were performed on wild type (WT) animals, animals overexpressing a control plasmid (Mock OE), and animals overexpressing (A) UBC-9 in all neurons (UBC-9 Neuron OE), (B) UBC-9 in GABAergic neurons only (UBC-9 GABA OE), UBC-9 in cholinergic neurons only (UBC-9 ACh OE), or (C) AOS-1 in GABAergic neurons only (AOS-1 GABA OE). (Upper panels) Representative timecourses showing the average percentage of young adult worms paralyzed per plate (15-20 worms per plate, n = 3 plates) on 1 mM aldicarb. (Lower panels) Bar graphs showing cumulative data for worms paralyzed at the 75 minute timepoint (indicated by * in upper panels). Error bars for all graphs denote s.e.m. of the corresponding dataset. n = total # plates per strain. All bars sharing common letters (a-b) were determined to be statistically similar using a one-way ANOVA, followed by a Tukey’s post hoc analysis. Neuronal UBC-9 overexpression F(2, 35) = 3.5487, P = .0395; Tukey’s post hoc WT versus Neuronal UBC-9 OE, P = .0362; Mock OE versus Neuronal UBC-9 OE, P = .1412; WT versus Mock OE, P = .834. GABA neuron-specific UBC-9 overexpression: F(2, 45) = 20.9361, P < .0001; Tukey’s post hoc WT versus UBC-9 GABA OE, P < .0001; Mock OE versus UBC-9 GABA OE, P < .0001; WT versus Mock OE, P = .6833. ACh neuron UBC-9 overexpression: F(2,24) = 11.7255, P = .0003; Tukey’s post hoc Mock OE versus UBC-9 ACh OE, P = .7794; Mock OE versus UBC-9 GABA OE, P = .0004; UBC-9 ACh OE versus UBC-9 GABA OE, P = .0023. GABA neuron-specific AOS-1 overexpression: F(2,24) = 10.093, P = .0007. Tukey’s post hoc WT versus AOS-1 GABA OE, P = .0004, Mock OE versus AOS-1 GABA OE, P = .0736; WT versus Mock OE, P = .0947.
Given the effects of neuronal UBC-9 expression on muscle function and the reported roles for SUMOylation on presynaptic proteins and synaptic vesicle release at other synapse types,10,27 we used quantitative fluorescence imaging of GFP-tagged SNB-1/synaptobrevin, a synaptic vesicle associated v-SNARE protein, to test whether neuronal UBC-9 overexpression alters synaptic vesicle localization in GABAergic or cholinergic motor neurons. Previous studies have shown that GFP::SNB-1 expressed under cell type-specific neuronal promoters marks presynaptic sites and correlates with the number of vesicles at synapses.66,67 For this reason, quantitative imaging of GFP::SNB-1 has been used extensively to measure synapse density, as well as serving as a readout of synaptic vesicle accumulation that typically correlates with changes in synaptic vesicle cycling.48–50,68 Worms with increased GFP::SNB-1 puncta intensity tend to have more vesicles clustered at synapses due to decreased release, whereas decreased GFP::SNB-1 puncta intensity (often coupled with increased interpunctal axonal fluorescence) correlates with increased vesicle exocytosis and more signaling.49,50,68,69 Compared to wild type worms, animals overexpressing UBC-9 in all neurons or only in GABA neurons had significantly brighter (~40-50%) GFP::SNB-1 synaptic puncta in GABAergic motor neurons, as well as increased (30-40%) interpunctal fluorescence, compared to wild type animals (Figure 3a). These effects were not seen for GFP::SNB-1 in cholinergic motor neurons in animals with panneuronal or GABAergic neuron-specific overexpression of UBC-9 (Figure 3b), confirming the cell autonomous effect of UBC-9 overexpression. Similar effects on GABAergic synaptic vesicles were seen with AOS-1 overexpression in GABA neurons (Figure 3c). Together, these results demonstrate overexpression of SUMO pathway enzymes causes synaptic vesicle accumulation in and between synapses in GABAergic neurons, potentially suggesting a decrease in GABA release that could explain the increased muscle contraction of the UBC-9 and AOS-1 overexpressing worms (Figure 2).
Figure 3.

Neuronal UBC-9 or AOS-1 overexpression causes synaptic vesicle accumulation at and between synaptic sites in GABAergic, but not cholinergic, motor neurons. Wild type (WT) worms or worms with neuronal UBC-9 or AOS-1 overexpression (Neuronal OE), GABAergic neuron-specific UBC-9 or AOS-1 overexpression (GABA Neuron OE) that also expressed GFP::SNB-1 in (A, C) GABAergic neurons (GABA) or in (B) cholinergic (ACh) neurons were imaged using a 100× objective. (Upper panels) Representative images of the dorsal nerve cords halfway between the vulva and the tail. (Lower panels) Quantification of puncta (synaptic) intensity, interpunctal (axonal) fluorescence, puncta width (µm), and puncta density (per 10 µm) ± s.e.m. Puncta intensity, interpunctal intensity, and puncta width are all shown normalized to wild type. For (A), n = 28 animals imaged for WT, n = 23 for Neuronal OE, n = 28 for GABA Neuron OE. For (B), n = 30 for WT, n = 25 for Neuronal OE, n = 28 for GABA Neuron OE. For (C), n = 37 for WT; n = 17 for Neuronal OE, n = 23 for GABA Neuron OE. Young adult worms were used for all images. *P < .05, **P ⩽ .01, ***P ⩽ .001 versus WT, one-way ANOVA with Tukey’s post hoc test. All other comparisons P > .05.
Since the aldicarb and imaging results were consistent with decreased GABA signaling, we tested for direct effects on GABA-dependent behaviors by assessing the UBC-9 and AOS-1 GABA neuron overexpressing worms for their sensitivity to the GABAA receptor antagonist, PTZ. PTZ is epileptogenic in mammals70,71 and induces seizures in C. elegans with reduced GABAergic transmission. Such seizures do not occur in wild type animals or in those with defects only in cholinergic signaling63,72 The combination of PTZ-induced seizures and aldicarb hypersensitivity is routinely used to identify mutants with GABAergic signaling defects.48,63,73 While we observed some background anterior “head bobbing” behavior in approximately 25% of PTZ-exposed wild type and Mock OE animals under our assay conditions, 95% of snb-1(md247) mutants, in which general synaptic transmission is reduced, showed convulsions on PTZ, as expected (Figure 4)63,73,74. Although not as robust as the response seen with snb-1 mutants, nearly 50% of animals with UBC-9 GABAergic neuron overexpression and 67% of worms with AOS-1 GABAergic neuron overexpression exhibited PTZ-induced convulsions (Figure 4)63. These effects are in line with the moderate hypersensitivity we observed with the UBC-9 and AOS-1 overexpressing animals in the aldicarb assay (Figure 2) and provide further support that animals with SUMO enzyme overexpression have reduced GABAergic signaling, likely due to decreased GABA release.73
Figure 4.
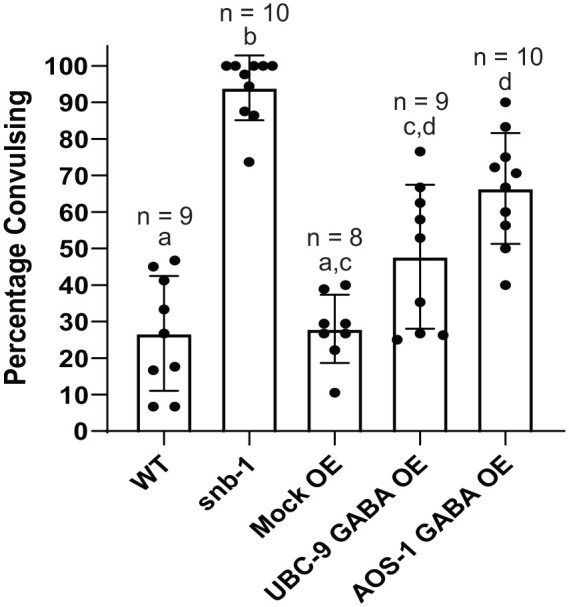
GABAergic neuron-specific overexpression of UBC-9 or AOS-1 causes PTZ-induced seizures, indicative of defects in GABAergic transmission. Wild type (WT) animals, snb-1(md427) mutants (defective in general synaptic transmission), animals overexpressing a control plasmid (Mock OE), animals overexpressing UBC-9 in GABAergic neurons only (UBC-9 GABA OE), or animals expressing AOS-1 in GABAergic neurons only (AOS-1 GABA OE) were exposed to plates containing 10 mg/ml PTZ for 50 minutes. Bar graphs show the mean percentage of young adult worms per plate exhibiting anterior convulsions (15-25 worms per plate). Each overlaid dot indicates the percentage of animals convulsing on one of the individual plates from which the mean was derived. n = # of plates per strain. Error bars denote ± SD. All bars sharing common letters (a-b) were determined to be statistically similar using a one-way ANOVA [F(4,41) = 36.2529, P < .0001] followed by a Tukey’s post hoc analysis: WT/Mock OE versus snb-1, P < .0001; WT versus UBC-9 GABA OE, P = .0271; Mock OE versus UBC-9 GABA OE, P = .0525; WT/Mock OE versus AOS-1 GABA OE P < .0001; UBC-9 GABA OE versus AOS-1 GABA OE, P = .0535.
Synaptic proteins, as well as many transcription factors and other nuclear proteins, are known to be SUMOylated.8,16,17 To test whether UBC-9 might be able to act directly at synapses, we imaged animals expressing either GFP::UBC-9 or the processed form of C. elegans SUMO GFP::SMO-1(GG)57 along with our synaptic vesicle marker mCherry::SNB-148 and performed colocalization analyses. We first observed that both GFP::UBC-9 and GFP::SMO-1 localized in punctate distributions within the nerve cord, similar to that seen for mCherry::SNB-1 (Figure 5). To determine whether these GFP::UBC-9 and GFP::SMO-1 puncta colocalize with mCherry::SNB-1-labeled synapses, we isolated a single focal plane from z-stacks taken of the dorsal nerve cords of worms co-expressing mCherry::SNB-1 and either GFP::UBC-9 or GFP::SMO-1 and performed quantitative linescan analyses to determine the percentage of GFP::UBC-9 or GFP::SMO-1 puncta that showed peak overlap with mCherry::SNB-1 puncta. We found that 85.3 % of green GFP::UBC-9 and 87.9 % of green GFP::SMO-1 peaks colocalized with red mCherry::SNB-1 peaks (Figure 5). Similarly, 84% and 83.4% of red mCherry::SNB-1 peaks colocalized with green GFP::UBC-9 and green GFP::SMO-1 peaks, respectively. This strong colocalization was not due to bleed-through of fluorophores between channels (Figure S2). Thus, while there was not perfect colocalization, well over three quarters of UBC-9 and SMO-1 puncta in the dorsal nerve cord were found with SNB-1-labeled synapses; likewise, the vast majority of SNB-1 sites contained UBC-9 and SMO-1.
Figure 5.

UBC-9 and SMO-1 localize to synaptic sites in GABAergic motor neurons. Young adult worms expressing both mCherry::SNB-1 and either (A) GFP::UBC-9 or (B) GFP::SMO-1 in GABAergic motor neurons were immobilized and imaged on a Leica DMLB fluorescence microscope at 100X magnification. (Upper panels) Representative single plane images from 2 µm total depth z-stacks taken of the dorsal nerve cords where neuromuscular synapses are found. White arrowheads mark sites of colocalization; yellow arrows denote sites with only one marker localized. (Lower panels) Quantification of the mean percentage of colocalization of peaks of mCherry::SNB-1 colocalization with GFP::UBC-9 and GFP::SMO-1 ± SD generated from linescans of the dorsal nerve cords. Box and whisker plots show the minima and maxima (whiskers), as well as the first and third quartiles of data (boxes), divided by the median line. The “X” denotes the mean value of the data set, and circles show individual data points. (n = 27 UBC-9/SNB-1 and n = 26 SMO-1/SNB-1 animals imaged).
Since UBC-9 and SMO-1 both localized to synapses where they could interact to regulate synaptic protein SUMOylation, we next asked whether the catalytic activity of UBC-9 was necessary for the NMJ effects observed in worms overexpressing UBC-9. To do this, we mutated the active site of UBC-9 at cysteine 93, which forms a thioester bond with the activated SUMO75, to serine (C93S) and compared the aldicarb sensitivity of worms overexpressing wild type UBC-9 and UBC-9(C93S) in GABAergic neurons to each other and to that of wild type animals. The C93S mutation has been shown to render UBC-9 catalytically inactive by preventing thioester bond formation with activated SUMO.76,77 Worms overexpressing UBC-9(C93S) in GABA neurons showed hypersensitivity to aldicarb-induced paralysis compared to mock overexpressing worms and wild type animals; this hypersensitive phenotype was similar to that seen with wild type UBC-9 overexpression (Figure 6a; Figure S3A).
Figure 6.

UBC-9 overexpression effects on NMJ signaling are independent of UBC-9 catalytic activity and are non-additive with co-expression of SMO-1. Aldicarb assays were performed on wild type (WT) animals, on animals overexpressing a control plasmid (Mock OE), and animals overexpressing (A) UBC-9 or UBC-9(C93S) or (B) UBC-9, SMO-1(GG), or UBC-9+SMO-1(GG) in GABA neurons. (Upper panels) Representative timecourses showing the average percentage of young adult worms paralyzed per plate (15-20 worms per plate, n = 3 plates) on 1 mM aldicarb. (Lower panels) Bar graphs quantifying cumulative data for worms paralyzed at a single timepoint (indicated by * in upper panels). Error bars for all graphs denote s.e.m. of the corresponding dataset. n = total # plates per strain. All bars sharing common letters (a-b) were determined to be statistically similar using a one-way ANOVA, followed by a Tukey’s post hoc analysis. (A) ANOVA: F(3,32) = 17.2912, P < .0001; Tukey’s post hoc WT versus UBC-9(C93S), P < .0001; Mock OE versus UBC-9(C93S), P < .0001; WT versus UBC-9, P = .0002; Mock OE versus UBC-9, P = .0002; UBC-9 versus UBC-9(C93S), P = .9184; WT versus Mock OE, P = .4934). (B) ANOVA: F(3,40) = 8.4909, P = .0002; Tukey’s post hoc WT versus UBC-9+SMO-1, P < .0001; UBC-9 versus UBC-9+SMO-1, P = .0589; SMO-1 versus UBC-9+SMO-1, P = .2976.
Because overexpression of UBC-9 lacking its SUMO-conjugating ability resulted in the same neuromuscular phenotype as did overexpression of wild type UBC-9, the active site of UBC-9 may not be necessary for the increased muscle contraction observed in UBC-9 overexpressing worms. On the other hand, because SUMO levels are limiting in cells.78 overexpressing UBC-9 alone may lead to SUMO-limiting effects where proteins are bound to excess UBC-9 in the presence of too little free SUMO, causing insufficient SUMOylation and a UBC-9 dominant negative phenotype. To mitigate any SUMO-limiting effects, we co-overexpressed wild type UBC-9 with SMO-1(GG) and again performed aldicarb assays. Co-overexpression of wild type UBC-9 with SMO-1 in GABA neurons yielded a hypersensitive phenotype relative to wild type worms (Figure 6b, Figure S3B). The phenotype is slightly, but not significantly, higher than the levels observed in worms overexpressing only wild type UBC-9 or only SMO-1. The more modest effects observed with UBC-9 overexpression in this experiment compared to previous experiments may be due to small differences in expression levels, as different lines of worms had to be used to accommodate the co-expression. Overall, however, this non-additive phenotype suggests UBC-9 and SMO-1 may be acting in the same pathway in GABAergic neurons to impact muscle contraction and that ultimately either too much or too little SUMO enzyme activity may cause E:I imbalance leading to excess muscle excitation.
Discussion
SUMOylation is an essential part of synaptic transmission for all animals, as it controls the activity of many proteins necessary for synapse formation and for pre- and post-synaptic function8,17. Most studies to date, however, have focused on the role of the SUMO pathway at excitatory glutamatergic synapses. As a result, potential functions for SUMO enzymes in controlling signaling at different synapse types and the effects of SUMOylation on E:I balance remain largely unexplored, despite links between several known SUMOylated neuronal proteins and diseases involving E:I imbalances. Here, we show the SUMO-conjugating enzyme UBC-9 can act in inhibitory motor neurons, where it localizes with SUMO to presynaptic sites and controls synaptic vesicle localization, to modulate E:I balance and muscle excitation at the C. elegans NMJ.
Tight regulation of UBC-9 activity is required in GABAergic neurons to maintain E:I balance
Overall, our data show that misregulation of UBC-9 and SUMO expression in GABAergic neurons disrupts neuromuscular activity. Based on our results, we favor a model in which UBC-9 acts, likely via its SUMO-conjugation activity, to regulate GABAergic vesicle release and prevent excess muscle excitation. Several lines of evidence support our proposed model. First, our over- and under-expression experiments suggest tight control of neuronal UBC-9 expression is critical to maintain proper muscle excitation. In our initial screen, neuronal knockdown of the SUMO enzyme genes ubc-9 or aos-1 led to increased muscle contraction compared to wild type worms (Figure 1), indicating ubc-9 and aos-1 are necessary to inhibit muscle excitation. Similar increases in muscle contraction were seen in animals with panneuronal or GABAergic neuron-specific overexpression of UBC-9, but not with UBC-9 overexpression in cholinergic neurons (Figure 2), supporting a specific effect of these enzymes in GABAergic neurons. Second, overexpression of UBC-9 in all neurons or just in GABAergic neurons caused synaptic vesicle accumulation at GABAergic motor neuron presynapses, as well as sensitivity to the GABAA receptor antagonist, PTZ (Figures 3 and 4). These results are consistent with decreased GABA release and with the increased muscle contraction observed in these animals.48-50 Third, both UBC-9 and SMO-1 co-localized with synaptic vesicle markers in GABAergic neurons (Figure 5), implying UBC-9 may act locally at presynaptic sites where it could SUMOylate known or novel synaptic targets to control synaptic vesicle release. Fourth, the increased muscle contraction seen with overexpression of wild type UBC-9 or the UBC-9(C93S) mutant indicates the active site of UBC-9 is not necessary for the NMJ effects of UBC-9 overexpression (Figure 6a). Given these are overexpression experiments and that levels of free SUMO in cells are reportedly low,78 both UBC-9 variants may cause loss of function phenotypes due to binding and sequestration of UBC-9 targets unable to be SUMOylated because of catalytic inactivity [e.g., UBC-9(C93S)] or insufficient free SUMO (e.g. wild type UBC-9).78,79 The fact that overexpression of AOS-1 in GABAergic neurons also led to increased muscle contraction, synaptic vesicle accumulation and PTZ-induced convulsions (Figures 2–4) further supports this model in which SUMO:enzyme stoichiometry is disrupted in the overexpression context rather than a specific SUMO-independent effect of UBC-9. Fifth and finally, the non-additive increases in muscle excitation caused by UBC-9 and SMO-1 co-overexpression suggest these proteins act in the same pathway to impact neuromuscular function and that too much, as well as too little, SUMOylation can increase signaling for muscle contraction (Figure 6b). Together, these findings support a role for UBC-9 in GABAergic neurons to control GABA release, prevent excess muscle contraction, and maintain E:I balance. While this is the first study implicating UBC-9 and the SUMO system at inhibitory presynapses, recent work showed GABAergic neuron-specific effects of several ubiquitin ligase enzymes in neuromuscular regulation,48,80,81 highlighting the importance of related protein modification systems in inhibitory neuronal signaling.
How likely is it that GABAergic neurons are the primary site of UBC-9 action at the NMJ? Our data suggest UBC-9 is necessary and sufficient in all neurons, and specifically in GABAergic neurons, to control muscle excitation, where it is also sufficient to cause PTZ-induced seizures and to alter synaptic vesicle localization in a manner consistent with decreased GABA release (Figures 1, 2, 3, 4, 6). While is it possible the knockdown phenotype we observed is due to inhibition of ubc-9 or aos-1 in non-neuronal cells, the strain used for the RNAi experiments expresses high levels of SID-1 dsRNA channels in neurons, which biases RNAi effects toward these cells.59 When examining specific neuronal subtypes relevant for NMJ control, we found that overexpression of UBC-9 in GABAergic neurons increased muscle excitation, whereas there was no effect of UBC-9 overexpression in cholinergic neurons. The reason for this GABAergic neuron expression effect remains unknown; however, our data do not rule out a role in excitatory cells. One possible explanation for the cell type-specific effects is that differing levels of UBC-9 expression are achieved with each of the cell type-specific promoters used for the overexpression studies. Similar variability in expression also could explain the lack of ubc-9 knockdown effects in animals engineered to allow RNAi only in cholinergic versus only in GABAergic neurons in our preliminary studies (data not shown). Alternatively, it is possible there is more compensation for effects on neuromuscular transmission in the cholinergic neurons. Unlike GABAergic neurons, which only synapse with postsynaptic muscle, cholinergic motor neurons directly signal to both GABAergic motor neurons and to muscle cells to maintain E:I balance.82 Similar GABAergic neuron-specific effects to those we observed were seen with inhibition of the anaphase promoting complex and EEL-1 ubiquitin ligases, the F-box protein MEC-15 FBXW9, as well as the cell adhesion molecule CASY-1, in prior studies at this synapse.48,80,81,83 These collective results demonstrate GABAergic neuron-specific regulation is possible, and even common, and that the aldicarb paralysis assay may be particularly sensitive to defects in inhibitory neurons.50
We found that GABAergic neuron-specific overexpression not only of wild type UBC-9 but also the C93S mutant increased muscle excitation (Figure 6a), suggesting UBC-9’s effects may be independent of its catalytic activity; however, because of the overexpression context, we believe this does not necessarily reflect a SUMOylation-independent role for endogenous UBC-9 at this synapse. Rather, the increased muscle contraction seen with overexpression of either wild type UBC-9 or UBC-9(C93S) may actually be due to a loss of function of the enzyme in both cases due to sequestration of UBC-9 targets unable to be SUMOylated. In support of this idea, a yeast two-hybrid assay performed by Eloranta and Hurst (2002) showed the human Ubc9(C93S) mutant is severely compromised in its interaction with SUMO-1 but can still interact with its SUMOylation target, the AP-2 transcription factor, as efficiently as wild type UBC9, leading the authors to suggest a dominant negative mechanism.79 Similarly, because free SUMO levels are reported to be limiting in cells,78 it possible that, when overexpressed alone, wild type UBC-9 binds to a greater number of its protein targets but lacks sufficient free SUMO to SUMOylate those proteins efficiently, resulting in a similar dominant negative effect. Further supporting this possibility, we also found that UBC-9 and SMO-1 co-expression in GABAergic neurons caused non-additive increases in muscle contraction (Figure 6b), indicating these proteins likely exert their effects on NMJ function through a common pathway, which would be expected to involve SUMOylation. Our additional observation that neuronal knockdown or GABAergic neuron overexpression of the E1 SUMO-activating enzyme subunit AOS-1 also resulted in increased muscle contraction, synaptic vesicle accumulation, and PTZ sensitivity (Figures 1–4) further indicates that the expression and activity of multiple SUMO enzymes must be precisely regulated for proper NMJ function. Because the Aos1-Uba2 heterodimeric E1 enzyme must bind Ubc9 to enforce SUMO transfer to the E2,32 the UBC-9 and AOS-1 overexpression phenotypes in our system also could be due to an imbalance in UBC-9:AOS-1-UBA-2 stoichiometry again leading to dominant negative-type substrate sequestration effects. Together, while SUMOylation-independent effects of UBC-9 have been previously demonstrated84–87 and cannot fully be ruled out, these data suggest the phenotypes seen with wild type UBC-9 overexpression in our study are most likely due to SUMO-limiting effects and/or incorrect stoichiometry of SUMO pathway enzymes rather than indicating an endogenous non-catalytic UBC-9 function at this synapse.
When considering our data in light of a model involving UBC-9’s SUMOylation activity, it appears that either too much or too little SUMOylation can lead to increased muscle excitation. Too little SUMOylation could be occurring in the case of ubc-9 knockdown and in the case of UBC-9 or UBC-9(C93S) expression alone through the dominant negative and substrate sequestration mechanisms discussed above. Conversely, if UBC-9 acts in a common pathway with SMO-1 when the 2 are overexpressed together, it is likely due to excess SUMOylation, since conjugation-independent effects of SMO-1 and UBC-9 would not necessarily be expected to depend on one another. Previous work in C. elegans showed that although muscle-specific overexpression of the non-conjugatable SMO-1(GA) or human SUMO-2(GA) caused some defects in C. elegans body morphology, these were less severe than the defects seen with wild type SMO-1 or SUMO-1 overexpression.45 This suggests the majority of effects of SMO-1 overexpression result from increased target SUMOylation, which has been detected with SUMO overexpression in other systems,78 rather than non-specific or indirect effects. Other studies described additional cases in which either the excess or absence of synaptic protein SUMOylation causes similar defects in synapse formation or function. For example, SUMOylation of the active zone protein, Rim1α, is required for action potential-evoked synaptic vesicle exocytosis and calcium entry, whereas non-SUMOylated Rim1α aids in synaptic vesicle docking and priming. Thus, either too much or too little Rim1α SUMOylation can decrease synaptic vesicle release.10 Likewise, SUMOylated syntaxin1A slows endocytosis, generally under intense stimulation, while non-SUMOylated syntaxin1A is a critical part of the SNARE complex required for membrane fusion during exocytosis.9 As our experiments focused on steady state neuromuscular activity in adult organisms rather than muscle excitation achieved under specific stimulation conditions, it is possible subtle differences in the nature of the defects caused by excess or reduced SUMOylation, if both occurred in our system, all manifested as increased muscle excitation. Future studies examining the SUMOylation levels of specific proteins in GABAergic neurons under knockdown and overexpression conditions and using electrophysiology or calcium imaging to more directly measure synaptic transmission will be needed to fully delineate the mechanisms by which UBC-1 and SMO-1 affect muscle excitation.
UBC-9 can control GABAergic vesicle localization potentially through local presynaptic effects
Although we did not explore specific SUMOylation targets in this study, our imaging data provide some clues as to the potential mechanism by which UBC-9 misexpression impacts GABAergic signaling to control E:I balance. Using quantitative imaging of GFP::SNB-1, we found worms with panneuronal or GABAergic neuron-specific UBC-9 overexpression accumulated synaptic vesicles relative to wild type worms in GABAergic but not in cholinergic motor neurons (Figure 3a). Increased GFP::SNB-1 puncta intensity typically reflects decreased exocytosis of vesicles into the synaptic cleft and correlates with decreased signaling.48–50 Conversely, increased interpunctal fluorescence often coincides with endocytic defects, as this fluorescence is indicative of GFP::SNB-1-containing vesicles that have fused with the membrane, allowing the GFP::SNB-1 to diffuse away from the synapse.49 The fact that we observed increases in both punctal and interpunctal GFP::SNB-1 fluorescence could indicate that the UBC-9 overexpressing animals actually have increased release and/or increased levels of GABA-containing vesicles. Alternatively, it is possible UBC-9 overexpression affects aspects of both synaptic vesicle exo- and endocytosis. While we cannot exclude either of these possibilities, or more complicated multicellular effects on both cholinergic and GABAergic signaling, a net decrease in GABA release is most consistent with the increased muscle contraction and the GABA-dependent seizure behaviors we observed in the aldicarb and PTZ assays, respectively, following UBC-9 overexpression (Figures 2 and 4). Accordingly, as loss of function of either the calcium channel subunit UNC-2 or the anaphase-promoting complex ubiquitin ligase, both of which are required for synaptic vesicle release from motor neurons, exhibit similar increases in both GFP::SNB-1 punctal and interpunctal fluorescence,48,88 it is possible the increases we observed in UBC-9 overexpressing animals resulted from synaptic vesicle buildup that led to spillover beyond synaptic sites. Such spillover may or may not be due to overall increases in the total number of vesicles and/or to defects in vesicle trafficking or maintenance at synapses.48
Importantly, these presynaptic effects may be accompanied by postsynaptic changes in GABA receptor levels or sensitivity, as mutants lacking UNC-49 GABAA receptors are both aldicarb hypersensitive and exhibit PTZ-induced convulsions.50,72 Increases in GABAA receptors have been observed in response to decreased GABA release in C. elegans lacking the function of the anaphase promoting complex, and the opposing homeostatic responses to increases in GABA release have been reported in other systems.48,89 While it is possible increased GABA release could be leading to decreased GABA receptor expression or sensitivity (or vice versa, if there are cell non-autonomous effects of our neuronally expressed SUMO enzymes) to cause the behavioral responses we observe, such changes would be expected to maintain relatively wild type levels of GABAergic signaling. Thus, while further exploration of the homeostatic effects of neuronal UBC-9 overexpression on muscle excitation, as well as additional roles for SUMO enzymes within the muscle cells themselves, will be of interest to explore, our current data most directly support a presynaptic role for the overexpression effects.
Not only do our data suggest UBC-9 expression levels critically regulate synaptic vesicle localization and potentially release, but by imaging worms co-expressing GFP::UBC-9 or GFP::SMO-1 with mCherry::SNB-1 in GABAergic neurons, we observed strong localization of UBC-9 and SMO-1 at presynaptic sites (Figure 4). Given that we also observe GFP::UBC-9 and GFP::SMO-1 in the cell bodies (data not shown), as well as at synapses and more diffusely along the dorsal and ventral nerve cords, our results do not preclude the function of overexpressed UBC-9 in the nucleus or elsewhere within the motor neurons in this context. However, the fact that UBC-9 and SMO-1 can localize to synapses is consistent with a model in which UBC-9 and SMO-1 regulate overall neuromuscular activity and synaptic vesicles in GABAergic neurons by acting directly at presynaptic sites through effects on synaptic proteins. This model is supported by prior studies demonstrating stimulation-dependent localization of mammalian Aos1, Ubc9, as well as other SUMOylation and deSUMOylation machinery, to hippocampal presynaptic sites, where increased SUMOylation of synaptic proteins and effects on synaptic vesicle release are also observed.14,15,25 In hippocampal spines, activity-dependent Ubc9 localization occurs in response to an increase in its Protein Kinase C (PKC)-phosphorylated substrates and is dependent upon its catalytic cysteine.13 It will be of interest to determine whether activity-dependent localization of C. elegans SUMOylation machinery to GABAergic presynapses also occurs and what domains are required for this localization. Mammalian Ubc9 is also known to be regulated by acetylation and SUMOylation, modifications that can alter its substrate specificity and thus potentially its localization.30,90 Whether C. elegans UBC-9 is similarly regulated remains to be explored.
As noted above, work done at glutamatergic synapses in other systems uncovered several SUMOylated presynaptic proteins, including Rim1α, synaptotagmin-1, synapsin1A, and syntaxin1A, which could mediate the effects of UBC-9 at GABAergic presynapses to regulate synaptic vesicle release, supporting the relevance of UBC-9 activity to synaptic plasticity9,10,27,28 and to learning and memory.37 Defects in synaptic protein SUMOylation are also linked to neurological and neurodegenerative diseases in which E:I imbalances occur.16,91 For example, the A548T mutation in synapsin1A, which is strongly associated with autism and epilepsy,91 causes reduced synapsin1A SUMOylation leading to a reduction in the releasable synaptic vesicle pool following neuronal stimulation in mouse hippocampal neuron cultures. It will be of interest to determine whether the defects in vesicle exocytosis due to loss of synapsin1A SUMOylation contribute to the disease phenotype in patients with the A548T mutation and whether SUMOylation of synapsin1A or other synaptic vesicle-associated proteins is critical for transmission at GABAergic synapses.
Conclusions and Future Directions
Overall, our data are consistent with a role for the SUMO pathway, and specifically, the SUMO-conjugating enzyme UBC-9, in promoting E:I balance at the C. elegans NMJ by acting locally at presynaptic sites in GABA neurons to control GABAergic signaling to prevent excess muscle excitation. Although additional studies are required to determine the precise mechanism underlying UBC-9 and other SUMO enzyme effects at this synapse, to our knowledge, this work is the first to demonstrate presynaptic function and localization of both UBC-9 and SMO-1 at inhibitory GABAergic synapses in any organism. It also further supports the increasingly prevalent notion that tight regulation of the SUMO enzyme system is critical for E:I balance within the nervous system. Given that AOS-1 overexpression causes similar effects to that seen with UBC-9, additional investigation into the effects of other SUMOylating and de-SUMOylating enzymes will be of interest to ascertain the full extent of SUMO pathway influence at the NMJ. Future studies identifying targets of SUMO-dependent and/or independent functions of UBC-9 at this synapse, associated E3 SUMO ligases and/or deubiquitinating enzymes, and potential regulation of UBC-9 and other SUMO pathway components by activity, oxidative stress, and hormone signaling, all of which can impact neuronal SUMOylation,16,53,92 will be critical for understanding how UBC-9 regulates presynaptic function in GABAergic neurons. Such studies will inform further investigation into the roles Ubc9 and the SUMO system play in mammalian GABAergic neurons and, in so doing, will contribute to our understanding of E:I balance misregulation in neurological and neurodegenerative diseases in which the SUMO pathway is disrupted.
Supplemental Material
Supplemental material, SupplementalFigure1_NeuronalOE_AChGABAOE_AllData for UBC-9 Acts in GABA Neurons to Control Neuromuscular Signaling in C. elegans by Victoria A Kreyden, Elly B Mawi, Kristen M Rush and Jennifer R Kowalski in Neuroscience Insights
Supplemental material, SupplementalFigure2_BleedthroughColoc for UBC-9 Acts in GABA Neurons to Control Neuromuscular Signaling in C. elegans by Victoria A Kreyden, Elly B Mawi, Kristen M Rush and Jennifer R Kowalski in Neuroscience Insights
Supplemental material, SupplementalFigure3_C93S_SMO1c for UBC-9 Acts in GABA Neurons to Control Neuromuscular Signaling in C. elegans by Victoria A Kreyden, Elly B Mawi, Kristen M Rush and Jennifer R Kowalski in Neuroscience Insights
Acknowledgments
We would like to thank Peter Juo (Tufts University School of Medicine), Richard Nass (Indiana University School of Medicine), Federico Pelisch and Ronald Hay (University of Dundee) for strains and reagents, Morgan Harrison for assistance with subcloning, Torey Kazeck for help with SMO-1 and UBC-9 experiments, and Lindsay Lewellyn (Butler University) for critical reading of this manuscript.
Footnotes
Declaration of Conflicting Interests:The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). Funding sources for this project include Holcomb Awards Committee Butler Summer Institute Awards to V.A.K., K.M.R., and E.B.M. and Butler Holcomb Awards Committee Faculty Research Awards (2011-2012, 2012-2013, 2013-2014, 2014-2015, 2015-2016, 2016-2017) and Indiana Academy of Sciences Senior Research Grants (2011-2012, 2014-2015, 2017-2018) to J.R.K.
ORCID iD: Jennifer R Kowalski  https://orcid.org/0000-0003-1807-6831
https://orcid.org/0000-0003-1807-6831
Supplemental material: Supplemental material for this article is available online.
References
- 1. Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry. 2019;24:1248-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vico Varela E, Etter G, Williams S. Excitatory-inhibitory imbalance in Alzheimer’s disease and therapeutic significance. Neurobiol Dis. 2019;127:605-615. [DOI] [PubMed] [Google Scholar]
- 3. Gatto CL, Broadie K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front Synaptic Neurosci. 2010;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nelson PG, Jia M, Li MX. Protein kinases and Hebbian function. Neuroscientist. 2003;9:110-116. [DOI] [PubMed] [Google Scholar]
- 5. Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9:826-838. [DOI] [PubMed] [Google Scholar]
- 6. Kowalski JR, Juo P. The role of deubiquitinating enzymes in synaptic function and nervous system diseases. Neural Plast. 2012;2012:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zareba-Koziol M, Figiel I, Bartkowiak-Kaczmarek A, Wlodarczyk J. Insights into protein S-Palmitoylation in synaptic plasticity and neurological disorders: potential and limitations of methods for detection and analysis. Front Mol Neurosci. 2018;11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Henley JM, Carmichael RE, Wilkinson KA. Extranuclear SUMOylation in neurons. Trends Neurosci. 2018;41:198-210. [DOI] [PubMed] [Google Scholar]
- 9. Craig TJ, Anderson D, Evans AJ, Girach F, Henley JM. SUMOylation of syntaxin1A regulates presynaptic endocytosis. Sci Rep. 2015;5:17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Girach F, Craig TJ, Rocca DL, Henley JM. RIM1alpha SUMOylation is required for fast synaptic vesicle exocytosis. Cell Rep. 2013;5:1294-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kantamneni S, Wilkinson KA, Jaafari N, et al. Activity-dependent SUMOylation of the brain-specific scaffolding protein GISP. Biochem Biophys Res Commun. 2011;409:657-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaur I, Yarov-Yarovoy V, Kirk LM, et al. Activity-Dependent palmitoylation controls SynDIG1 stability, localization, and function. J Neurosci. 2016;36:7562-7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Loriol C, Casse F, Khayachi A, et al. mGlu5 receptors regulate synaptic sumoylation via a transient PKC-dependent diffusional trapping of Ubc9 into spines. Nat Commun. 2014;5:5113. [DOI] [PubMed] [Google Scholar]
- 14. Loriol C, Khayachi A, Poupon G, Gwizdek C, Martin S. Activity-dependent regulation of the sumoylation machinery in rat hippocampal neurons. Biol Cell. 2013;105:30-45. [DOI] [PubMed] [Google Scholar]
- 15. Schorova L, Pronot M, Poupon G, et al. The synaptic balance between sumoylation and desumoylation is maintained by the activation of metabotropic mGlu5 receptors. Cell Mol Life Sci. 2019; 76:3019-3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Henley JM, Craig TJ, Wilkinson KA. Neuronal SUMOylation: mechanisms, physiology, and roles in neuronal dysfunction. Physiol Rev. 2014;94:1249-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schorova L, Martin S. Sumoylation in synaptic function and dysfunction. Front Synaptic Neurosci. 2016;8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilkinson KA, Nakamura Y, Henley JM. Targets and consequences of protein SUMOylation in neurons. Brain Res Rev. 2010;64:195-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355-382. [DOI] [PubMed] [Google Scholar]
- 20. Nayak A, Viale-Bouroncle S, Morsczeck C, Muller S. The SUMO-specific isopeptidase SENP3 regulates MLL1/MLL2 methyltransferase complexes and controls osteogenic differentiation. Mol Cell. 2014;55:47-58. [DOI] [PubMed] [Google Scholar]
- 21. Bao J, Qin M, Mahaman YAR, et al. BACE1 SUMOylation increases its stability and escalates the protease activity in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2018;115:3954-3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dorval V, Fraser PE. SUMO on the road to neurodegeneration. Biochim Biophys Acta. 2007;1773:694-706. [DOI] [PubMed] [Google Scholar]
- 23. Marcelli S, Ficulle E, Iannuzzi F, Kovari E, Nistico R, Feligioni M. Targeting SUMO-1ylation contrasts synaptic dysfunction in a mouse model of Alzheimer’s Disease. Mol Neurobiol. 2017;54:6609-6623. [DOI] [PubMed] [Google Scholar]
- 24. Feligioni M, Nistico R. SUMO: a (oxidative) stressed protein. Neuromolecular Med. 2013;15:707-719. [DOI] [PubMed] [Google Scholar]
- 25. Feligioni M, Nishimune A, Henley JM. Protein SUMOylation modulates calcium influx and glutamate release from presynaptic terminals. Eur J Neurosci. 2009;29:1348-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chao HW, Hong CJ, Huang TN, Lin YL, Hsueh YP. SUMOylation of the MAGUK protein CASK regulates dendritic spinogenesis. J Cell Biol. 2008;182:141-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang LT, Craig TJ, Henley JM. SUMOylation of synapsin Ia maintains synaptic vesicle availability and is reduced in an autism mutation. Nat Commun. 2015;6:7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsuzaki S, Lee L, Knock E, et al. SUMO1 Affects synaptic function, spine density and memory. Sci Rep. 2015;5:10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dustrude ET, Perez-Miller S, Francois-Moutal L, Moutal A, Khanna M, Khanna R. A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels (Austin). 2017;11:316-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hsieh YL, Kuo HY, Chang CC, et al. Ubc9 acetylation modulates distinct SUMO target modification and hypoxia response. EMBO J. 2013;32:791-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klug H, Xaver M, Chaugule VK, et al. Ubc9 sumoylation controls SUMO chain formation and meiotic synapsis in saccharomyces cerevisiae. Mol Cell. 2013;50:625-636. [DOI] [PubMed] [Google Scholar]
- 32. Pichler A, Fatouros C, Lee H, Eisenhardt N. SUMO conjugation - a mechanistic view. Biomol Concepts. 2017;8:13-36. [DOI] [PubMed] [Google Scholar]
- 33. Tomasi ML, Ramani K, Ryoo M. Ubiquitin-Conjugating enzyme 9 phosphorylation as a novel mechanism for potentiation of the inflammatory response. Am J Pathol. 2016;186:2326-2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones D, Crowe E, Stevens TA, Candido EP. Functional and phylogenetic analysis of the ubiquitylation system in Caenorhabditis elegans: ubiquitin-conjugating enzymes, ubiquitin-activating enzymes, and ubiquitin-like proteins. Genome Biol. 2002;3:RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nacerddine K, Lehembre F, Bhaumik M, et al. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev Cell. 2005; 9:769-779. [DOI] [PubMed] [Google Scholar]
- 36. Yasugi T, Howley PM. Identification of the structural and functional human homolog of the yeast ubiquitin conjugating enzyme UBC9. Nucleic Acids Res. 1996;24:2005-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwartz S, Truglio M, Scott MJ, Fitzsimons HL. Long-Term memory in drosophila is influenced by histone deacetylase HDAC4 interacting with SUMO-Conjugating enzyme Ubc9. Genetics. 2016;203:1249-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martin S, Nishimune A, Mellor JR, Henley JM. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447:321-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghosh H, Auguadri L, Battaglia S, et al. Several posttranslational modifications act in concert to regulate gephyrin scaffolding and GABAergic transmission. Nat Commun. 2016;7:13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu J, Tan P, Li H, et al. Direct SUMOylation of M1 muscarinic acetylcholine receptor increases its ligand-binding affinity and signal transduction. FASEB J. 2019;33:3237-3251. [DOI] [PubMed] [Google Scholar]
- 41. Li Y, Zhang Q, Wei Q, Zhang Y, Ling K, Hu J. SUMOylation of the small GTPase ARL-13 promotes ciliary targeting of sensory receptors. J Cell Biol. 2012;199:589-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Long X, Griffith LC. Identification and characterization of a SUMO-1 conjugation system that modifies neuronal calcium/calmodulin-dependent protein kinase II in Drosophila melanogaster. J Biol Chem. 2000;275:40765-40776. [DOI] [PubMed] [Google Scholar]
- 43. Berdnik D, Favaloro V, Luo L. The SUMO protease Verloren regulates dendrite and axon targeting in olfactory projection neurons. J Neurosci. 2012;32:8331-8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steffan JS, Agrawal N, Pallos J, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100-104. [DOI] [PubMed] [Google Scholar]
- 45. Rytinki MM, Lakso M, Pehkonen P, et al. Overexpression of SUMO perturbs the growth and development of Caenorhabditis elegans. Cell Mol Life Sci. 2011;68:3219-3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Richmond JE, Jorgensen EM. One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci. 1999;2:791-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahoney TR, Luo S, Nonet ML. Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat Protoc. 2006;1:1772-1777. [DOI] [PubMed] [Google Scholar]
- 48. Kowalski JR, Dube H, Touroutine D, et al. The Anaphase-Promoting Complex (APC) ubiquitin ligase regulates GABA transmission at the C. elegans neuromuscular junction. Mol Cell Neurosci. 2014;58:62-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sieburth D, Ch’ng Q, Dybbs M, et al. Systematic analysis of genes required for synapse structure and function. Nature. 2005;436:510-517. [DOI] [PubMed] [Google Scholar]
- 50. Vashlishan AB, Madison JM, Dybbs M, et al. An RNAi screen identifies genes that regulate GABA synapses. Neuron. 2008;58:346-361. [DOI] [PubMed] [Google Scholar]
- 51. Broday L, Kolotuev I, Didier C, et al. The small ubiquitin-like modifier (SUMO) is required for gonadal and uterine-vulval morphogenesis in Caenorhabditis elegans. Genes Dev. 2004;18:2380-2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kaminsky R, Denison C, Bening-Abu-Shach U, Chisholm AD, Gygi SP, Broday L. SUMO regulates the assembly and function of a cytoplasmic intermediate filament protein in C. elegans. Dev Cell. 2009;17:724-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Drabikowski K, Ferralli J, Kistowski M, Oledzki J, Dadlez M, Chiquet-Ehrismann R. Comprehensive list of SUMO targets in Caenorhabditis elegans and its implication for evolutionary conservation of SUMO signaling. Sci Rep. 2018;8:1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yao X, Maity S, Gandhi S, Imielenski M, Vogel C. iSUMO - integrative prediction of functionally relevant SUMOylation events. bioRxiv. 2017:056564. [Google Scholar]
- 55. Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fire A, Harrison SW, Dixon D. A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene. 1990;93:189-198. [DOI] [PubMed] [Google Scholar]
- 57. Pelisch F, Sonneville R, Pourkarimi E, et al. Dynamic SUMO modification regulates mitotic chromosome assembly and cell cycle progression in Caenorhabditis elegans. Nat Commun. 2014;5:5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Calixto A, Chelur D, Topalidou I, Chen X, Chalfie M. Enhanced neuronal RNAi in C. elegans using SID-1. Nat Methods. 2010;7:554-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schmitz C, Kinge P, Hutter H. Axon guidance genes identified in a large-scale RNAi screen using the RNAi-hypersensitive Caenorhabditis elegans strain nre-1(hd20) lin-15b(hd126). Proc Natl Acad Sci U S A. 2007; 104(3): 834-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313-321. [DOI] [PubMed] [Google Scholar]
- 62. Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001;2:RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Locke C, Berry K, Kautu B, Lee K, Caldwell K, Caldwell G. Paradigms for pharmacological characterization of C. elegans synaptic transmission mutants. J Vis Exp. 2008;18:e837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron. 2002;35:107-120. [DOI] [PubMed] [Google Scholar]
- 65. Firnhaber C, Hammarlund M. Neuron-specific feeding RNAi in C. elegans and its use in a screen for essential genes required for GABA neuron function. PLoS Genet. 2013;9:e1003921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jorgensen EM, Hartwieg E, Schuske K, Nonet ML, Jin Y, Horvitz HR. Defective recycling of synaptic vesicles in synaptotagmin mutants of Caenorhabditis elegans. Nature. 1995;378:196-199. [DOI] [PubMed] [Google Scholar]
- 67. Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dittman JS, Kaplan JM. Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc Natl Acad Sci U S A. 2006;103:11399-11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fernandez-Alfonso T, Ryan TA. The kinetics of synaptic vesicle pool depletion at CNS synaptic terminals. Neuron. 2004;41:943-953. [DOI] [PubMed] [Google Scholar]
- 70. Huang RQ, Bell-Horner CL, Dibas MI, Covey DF, Drewe JA, Dillon GH. Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: mechanism and site of action. J Pharmacol Exp Ther. 2001;298:986-995. [PubMed] [Google Scholar]
- 71. Rocha L, Briones M, Ackermann RF, et al. Pentylenetetrazol-induced kindling: early involvement of excitatory and inhibitory systems. Epilepsy Res. 1996;26:105-113. [DOI] [PubMed] [Google Scholar]
- 72. Williams SN, Locke CJ, Braden AL, Caldwell KA, Caldwell GA. Epileptic-like convulsions associated with LIS-1 in the cytoskeletal control of neurotransmitter signaling in Caenorhabditis elegans. Hum Mol Genet. 2004;13:2043-2059. [DOI] [PubMed] [Google Scholar]
- 73. Thapliyal S, Babu K. Pentylenetetrazole (PTZ)-induced convulsion assay to determine GABAergic defects in Caenorhabditis elegans. Bio Protoc. 2018;8:e2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Locke CJ, Williams SN, Schwarz EM, Caldwell GA, Caldwell KA. Genetic interactions among cortical malformation genes that influence susceptibility to convulsions in C. elegans. Brain Res 2006;1120:23-34. [DOI] [PubMed] [Google Scholar]
- 75. Desterro JM, Thomson J, Hay RT. Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 1997;417:297-300. [DOI] [PubMed] [Google Scholar]
- 76. Gong L, Kamitani T, Fujise K, Caskey LS, Yeh ET. Preferential interaction of sentrin with a ubiquitin-conjugating enzyme, Ubc9. J Biol Chem. 1997;272:28198-28201. [DOI] [PubMed] [Google Scholar]
- 77. Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108:345-356. [DOI] [PubMed] [Google Scholar]
- 78. Ungureanu D, Vanhatupa S, Kotaja N, et al. PIAS proteins promote SUMO-1 conjugation to STAT1. Blood. 2003;102:3311-3313. [DOI] [PubMed] [Google Scholar]
- 79. Eloranta JJ, Hurst HC. Transcription factor AP-2 interacts with the SUMO-conjugating enzyme UBC9 and is sumolated in vivo. J Biol Chem. 2002;277:30798-30804. [DOI] [PubMed] [Google Scholar]
- 80. Opperman KJ, Mulcahy B, Giles AC, et al. The HECT family ubiquitin ligase EEL-1 regulates neuronal function and development. Cell Rep.2017;19:822-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sun Y, Hu Z, Goeb Y, Dreier L. The F-Box Protein MEC-15 (FBXW9) promotes synaptic transmission in GABAergic motor neurons in C. elegans. PLoS One. 2013;8:e59132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jorgensen EM, Nonet ML. Neuromuscular junctions in the nematode C. elegans. Semin Dev Biol. 1995;6:207-220. [Google Scholar]
- 83. Thapliyal S, Vasudevan A, Dong Y, Bai J, Koushika SP, Babu K. The C-terminal of CASY-1/Calsyntenin regulates GABAergic synaptic transmission at the Caenorhabditis elegans neuromuscular junction. PLoS Genet. 2018;14:e1007263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Guo Y, Yang MC, Weissler JC, Yang YS. Modulation of PLAGL2 transactivation activity by Ubc9 co-activation not SUMOylation. Biochem Biophys Res Commun. 2008;374:570-575. [DOI] [PubMed] [Google Scholar]
- 85. Kurtzman AL, Schechter N. Ubc9 interacts with a nuclear localization signal and mediates nuclear localization of the paired-like homeobox protein Vsx-1 independent of SUMO-1 modification. Proc Natl Acad Sci U S A. 2001;98:5602-5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu LB, Omata W, Kojima I, Shibata H. The SUMO conjugating enzyme Ubc9 is a regulator of GLUT4 turnover and targeting to the insulin-responsive storage compartment in 3T3-L1 adipocytes. Diabetes. 2007;56:1977-1985. [DOI] [PubMed] [Google Scholar]
- 87. Zhu S, Sachdeva M, Wu F, Lu Z, Mo YY. Ubc9 promotes breast cell invasion and metastasis in a sumoylation-independent manner. Oncogene. 2010;29:1763-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ch’ng Q, Sieburth D, Kaplan JM. Profiling synaptic proteins identifies regulators of insulin secretion and lifespan. PLoS Genet. 2008;4:e1000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wenner P. Mechanisms of GABAergic homeostatic plasticity. Neural Plast. 2011;2011:489470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Knipscheer P, Flotho A, Klug H, et al. Ubc9 sumoylation regulates SUMO target discrimination. Mol Cell. 2008;31:371-382. [DOI] [PubMed] [Google Scholar]
- 91. Fassio A, Patry L, Congia S, et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet. 2011;20:2297-2307. [DOI] [PubMed] [Google Scholar]
- 92. Lai YJ, Liu L, Hu XT, He L, Chen GJ. Estrogen modulates ubc9 expression and synaptic redistribution in the brain of APP/PS1 mice and cortical neurons. J Mol Neurosci. 2017;61:436-448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, SupplementalFigure1_NeuronalOE_AChGABAOE_AllData for UBC-9 Acts in GABA Neurons to Control Neuromuscular Signaling in C. elegans by Victoria A Kreyden, Elly B Mawi, Kristen M Rush and Jennifer R Kowalski in Neuroscience Insights
Supplemental material, SupplementalFigure2_BleedthroughColoc for UBC-9 Acts in GABA Neurons to Control Neuromuscular Signaling in C. elegans by Victoria A Kreyden, Elly B Mawi, Kristen M Rush and Jennifer R Kowalski in Neuroscience Insights
Supplemental material, SupplementalFigure3_C93S_SMO1c for UBC-9 Acts in GABA Neurons to Control Neuromuscular Signaling in C. elegans by Victoria A Kreyden, Elly B Mawi, Kristen M Rush and Jennifer R Kowalski in Neuroscience Insights