

Abstract
The lack of a vaccine or any effective treatment for the aggressive novel coronavirus disease (COVID-19) has created a sense of urgency for the discovery of effective drugs. Several repurposing pharmaceutical candidates have been reported or envisaged to inhibit the emerging infections of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), but their binding sites, binding affinities and inhibitory mechanisms are still unavailable. In this study, we use the ligand-protein docking program and molecular dynamic simulation to ab initio investigate the binding mechanism and inhibitory ability of seven clinically approved drugs (Chloroquine, Hydroxychloroquine, Remdesivir, Ritonavir, Beclabuvir, Indinavir and Favipiravir) and a recently designed α-ketoamide inhibitor (13b) at the molecular level. The results suggest that Chloroquine has the strongest binding affinity with 3CL hydrolase (Mpro) among clinically approved drugs, indicating its effective inhibitory ability for SARS-CoV-2. However, the newly designed inhibitor 13b shows potentially improved inhibition efficiency with larger binding energy compared with Chloroquine. We further calculate the important binding site residues at the active site and demonstrate that the MET 165 and HIE 163 contribute the most for 13b, while the MET 165 and GLN 189 for Chloroquine, based on residual energy decomposition analysis. The proposed work offers a higher research priority for 13b to treat the infection of SARS-CoV-2 and provides theoretical basis for further design of effective drug molecules with stronger inhibition.
Keywords: COVID-19, SARS-CoV-2, 3CL Mpro, α-ketoamide inhibitor, ligand-protein docking, binding free energy, drug design
Introduction
The recent outbreak of the novel coronavirus disease (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a serious global risk for health and economy. Unfortunately, no drug or vaccine has yet been approved to prevent the infection or to treat the patients, and more time is required to develop more specific strategy against this pathogen [1, 2]. Given the urgency of the situation, researchers have been trying a variety of clinically approved drugs in the hope that they hold promise to combat the COVID-19. Existing antivirals, including virus protease inhibitors (Indinavir, Ritonavir etc.) as well as some antiviral drugs that are still in the clinical trials (e.g. Remdesivir and Chloroquine) are being tested against SARS-CoV-2 [3–7]. Remdesivir is a nucleotide analog precursor that has been reported to be active against specific viruses such as SARS and Middle East respiratory syndrome (MERS) [8–11]. The efficacy of Remdesivir is suggested by a recent recovery of US patient infected with SARS-CoV-2 after intravenous treatment [12], while Chloroquine and Hydroxychloroquine are being reported to be potential inhibitors of SARS-CoV-2 in vitro [13, 14]. However, Remdesivir failed in a clinical trial against Ebola virus in 2019 [15], and there has been no clinical trial supporting its efficacy against SARS-CoV-2. Empirical trials often involve substantial trial-and-error costs, which require inevitable time consuming in drug development. To prevent the spread of COVID-19 timely, it is necessary to apply all approaches, including computational analysis, to prioritize the pharmaceutical candidates, so as to rapidly develop a strategy for COVID-19 treatment.
The SARS-CoV-2 genome is comprised of approximate 30 000 nucleotides, where two overlapping polyprotein genes (pp1a and pp1ab) encode replicases essential for viral replications and transcriptions. The 3C-like main protease (3CL Mpro) is a main protease of SARS-CoV-2, essential for the immune regulation and the releases of the polyproteins pp1a and pp1ab [16]. 3CL Mpro controls the functional polypeptides releases from the polyproteins by extensive proteolytic processing [17]. Its functional importance in the viral life cycle and the absence of closely related homologs in human make it an attractive target for anti-SARS-CoV-2 drugs. Therefore, the discovery or design of drug molecules that can bind to 3CL Mpro and inhibit its function is an effective way to combat the virus. In this study, we chose 3CL Mpro as the therapeutic target to ab initio investigate its inhibition mechanism and binding ability of these most promising drug molecules by ligand-protein docking program (Rosetta) and molecular dynamics (MD) simulations. The experiments and high-throughput screening researches have been going on around the clock in hope of finding a cure for the COVID-19. Based on pioneers’ studies, we selected seven most promising drugs (i.e. Chloroquine, Hydroxychloroquine, Remdesivir, Ritonavir, Beclabuvir, Indinavir and Favipiravir) and conducted high-precision quantum calculations, so as to find the most effective inhibitor. Recently, Zhang et al. [18] pursued a structure-based design of peptidomimetic α-ketoamides as inhibitors of 3CL Mpro, and demonstrated that the inhibitor 11r exhibited activity against coronavirus. Soon later, based on 11r, Zhang et al. [19] reported three structure modified inhibitors (i.e. 13a, 13b and 14b), and demonstrated that 13b is the most effective designed inhibitor, which showed good tropism to the lungs with a well-tolerated administration through inhalation in mice. Therefore, in addition to the seven clinically approved drug molecules, we also included 13b, as a candidate to investigate its binding ability with 3CL Mpro.
The 3CL Mpro structure (PDB ID: 6 LU7) was obtained from the RCSB Protein Data Bank, which was recently released by Rao’s group [20, 21], while the 3D structures of the seven drug molecules were obtained from the PubChem database. Through molecular docking and kinetic analysis, we found that for repurposed drugs, the Chloroquine molecule has the strongest interaction with the 3CL Mpro, indicating that Chloroquine is the best potential inhibitor for SARS-CoV-2, followed by Hydroxychloroquine, Remdesivir, Ritonavir, Beclabuvir, Indinavir and Favipiravir. Although Chloroquine and Hydroxychloroquine share similar chemical structures and mechanisms of acting as a weak base and immunomodulator, we demonstrated that Chloroquine molecule has a stronger binding with 3CL Mpro than Hydroxychloroquine, based on the high-precision calculations of enthalpy and entropy. However, the newly designed inhibitor 13b represents slightly stronger binding affinity with 3CL Mpro compared with Chloroquine, suggesting that 13b may be more effective than the selected clinically approved drugs. We further analyzed the specific residues of 3CL Mpro that contribute significantly to the binding energy, and demonstrated that the residues MET165 and GLN189 in 3CL Mpro have the strongest affinities with Chloroquine molecule, with decomposition energies of −2.50 and −2.34 kcal/mol, respectively. For 13b, the residues MET165 and HIE163 in 3CL Mpro contribute most to the binding energy, with decomposition energies of −3.23 and −2.65 kcal/mol, respectively. The determination of dominant residues in 3CL Mpro provides a theoretical guidance for further drug molecule design with larger binding energies and stronger inhibitory abilities with target protein.
Results and Discussion
Since the complex structures between 3CL Mpro and inhibitors have not been identified, we used the ligand-protein docking program (Rosetta3) to determinate their 3D binding conformation. Based on the total energy of complex and interaction energy between ligand and protein calculated by Rosetta, we generated 10 000 conformations for each inhibitor and selected the most likely combination based on Rosetta score function. The total Rosetta score, the binding energy between protein and ligand and whether the ligand is touching the protein are considered for the conformational selection. Table S8 in Supplementary Information shows the total Rosetta energy score and important energy terms of the selected conformations for the seven clinically approved drugs from docking studies. The results show that the selected conformation of Hydroxychloroquine has the lowest Rosetta score, while Indinavir has the lowest binding energy. The docking binding energies of all seven clinically approved drugs with 3CL Mpro are lower than −10 kcal/mol, with the total energy score lower than −642 kcal/mol. The virtual docking results show that all seven inhibitors have high binding affinities to 3CL Mpro.
To investigate the binding ability and the interactions between inhibitors and 3CL Mpro, the most favorable complex conformations were used for molecular dynamic simulations. After the equilibrium run of 1.2 ns, the ligand-protein complex system became stable with the convergence of temperature, density and total energy. Then a 40 ns production run was performed while keeping the system stable. The root-mean-square deviation (RMSD) was analyzed to detect the stability of the ligand-protein system during simulation. Figure 1a shows the RMSD of all non-hydrogen atoms for the complex of 3CL Mpro and 13b over the entire production simulation. The result indicates that the ligand-protein complex mostly remains stable from 15 to 40 ns at 2.5–3 Å. Besides, the root means square fluctuation (RMSF) was analyzed to detect the fluctuation and flexibility of protein residues during the MD simulation. Figure 1b shows the RMSFs of all non-hydrogen atoms of 3CL Mpro over the entire simulation, where the LEU50, PRO52, ASN51, ASN53, MET49, GLN189, ARG222, ASP48, SER46, ASN277, GLU55 and TYR154 are the most flexible residues in the protein middle region while 13b binds to 3CL Mpro. The RMSDs and RMSFs of seven clinically approved drug molecules are plotted in Figures S3, S6, S9, S12, S15, S18 and S21 of supporting information, respectively. From these figures, the RMSFs of the 3CL Mpro are similar when the eight molecules (Chloroquine, Hydroxychloroquine, Remdesivir, Ritonavir, Beclabuvir, Indinavir and Favipiravir and 13b) bind to the protein pocket.
Figure 1.
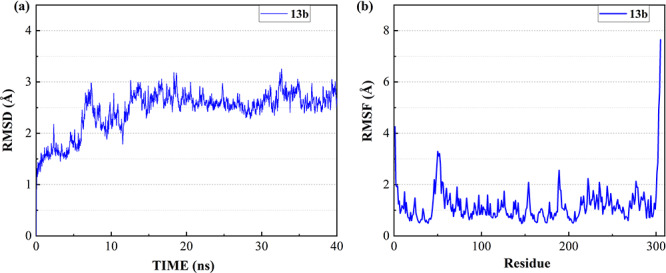
The RMSD and RMSF of the complex of 3CL Mpro and 13b over 40 ns MD simulation. (a) RMSD of all the non-hydrogen atoms of the complex. (b) RMSF of all non-hydrogen atoms of 3CL Mpro over the entire simulation.
Table 1 shows the binding free energies of eight potential inhibitors to 3CL Mpro, where the total enthalpies (ΔETOT) were calculated by MD simulation and the total binding free energy (ΔGbind) were obtained by the Molecular Mechanics-Generalized Born Surface Area (MM/GBSA)-binding free energy. The total enthalpies include the components of gas-phase and solvation energies but without the entropy, which sometimes plays an important role for ligand-protein interaction. So far, many effective methods have been applied to calculate the binding free energy of proteins and ligands. MM/GBSA is a time-saving and highly accurate method, which is often used to compute the absolute binding free energy and shows great success in elucidating the protein–protein and protein-inhibitor interactions [22–25]. Based on the MM-GBSA method, we calculated the binding energy 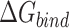 between inhibitors and 3CL Mpro, consisting of van der Waals energy, electrostatic energy, polar solvation energy nonpolar solvation energy and entropic contribution.
between inhibitors and 3CL Mpro, consisting of van der Waals energy, electrostatic energy, polar solvation energy nonpolar solvation energy and entropic contribution.
Table 1.
The enthalpies and binding free energies of eight ligands to 3CL Mpro, where ΔETOT-gas, ΔETOT-sol, ΔETOT, TΔSnmode and ΔGbind represent the gas phase enthalpy, solvation enthalpy, total enthalpy, entropy contribution and total binding free energy, respectively. All values of energy are shown in kcal/mol
| Conformations |
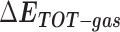
|
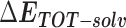
|
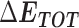
|
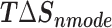
|
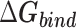
|
|---|---|---|---|---|---|
| 13b | -100.2815 | 53.0246 | -47.2569 | -29.5070 | -17.7499 |
| Chloroquine | -49.1969 | 14.0119 | -35.1849 | -19.0655 | -16.1194 |
| Hydroxychloroquine | -52.3765 | 17.7180 | -34.6585 | -20.9370 | -13.7215 |
| Remdesivir | -88.4507 | 49.1173 | -39.3334 | -28.4566 | -10.8768 |
| Ritonavir | 16.8060 | -47.8443 | -31.0383 | -26.2617 | -4.7766 |
| Beclabuvir | -49.6568 | 18.8371 | -30.8197 | -23.0698 | -7.7499 |
| Indinavir | -60.1888 | 28.1835 | -32.0052 | -25.4817 | -6.5235 |
| Favipiravir | -20.7864 | 12.9465 | -7.8399 | -12.0494 | 4.2095 |
From Table 1, the calculated total enthalpy 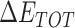 by MD simulation (the green column) shows that for clinically approved drugs, Remdesivir has the strongest binding energy as −39.33 kcal/mol. However, the total enthalpy does not consider the contribution of entropy, which depends on the alternation of motional freedom induced by inhibitor binding and contributes partly to the total binding energy [26]. After taking into account of entropy contribution, Table 1 shows that Chloroquine exhibits the lowest binding free energy (−16.119 kcal/mol) among the seven clinically approved drugs (orange column), followed by Hydroxychloroquine (−13.7215 kcal/mol), Remdesivir (−10.876 kcal/mol), Ritonavir (−4.78 kcal/mol), Beclabuvir (−7.75 kcal/mol), Indinanir (−6.52 kcal/mol) and Favipiravir (4.21 kcal/mol). This finding is consistent with the recent in vitro study [13, 14] showing that Chloroquine, Hydroxychloroquine and Remdesivir are the most effective in controlling SARS-CoV-2 infection among a number of clinically approved drugs. For patients with acute porphyria or porphyria cutanea tarda, Chloroquine has some side effects of causing fever, serum aminotransferase elevations and even jaundice. Hydroxychloroquine, a less toxic metabolite of Chloroquine, does not cause these adverse side effects and was found to have partial beneficial effects in porphyria [27]. However, we demonstrated that Chloroquine has a stronger affinity and inhibitory ability to 3CL Mpro than Hydroxychloroquine. In addition, from Table 1, the enthalpy and binding free energy of the newly proposed inhibitor 13b are higher than those of all selected clinically approved drug molecules, indicating the strongest binding affinity of 13b with 3CL Mpro.
by MD simulation (the green column) shows that for clinically approved drugs, Remdesivir has the strongest binding energy as −39.33 kcal/mol. However, the total enthalpy does not consider the contribution of entropy, which depends on the alternation of motional freedom induced by inhibitor binding and contributes partly to the total binding energy [26]. After taking into account of entropy contribution, Table 1 shows that Chloroquine exhibits the lowest binding free energy (−16.119 kcal/mol) among the seven clinically approved drugs (orange column), followed by Hydroxychloroquine (−13.7215 kcal/mol), Remdesivir (−10.876 kcal/mol), Ritonavir (−4.78 kcal/mol), Beclabuvir (−7.75 kcal/mol), Indinanir (−6.52 kcal/mol) and Favipiravir (4.21 kcal/mol). This finding is consistent with the recent in vitro study [13, 14] showing that Chloroquine, Hydroxychloroquine and Remdesivir are the most effective in controlling SARS-CoV-2 infection among a number of clinically approved drugs. For patients with acute porphyria or porphyria cutanea tarda, Chloroquine has some side effects of causing fever, serum aminotransferase elevations and even jaundice. Hydroxychloroquine, a less toxic metabolite of Chloroquine, does not cause these adverse side effects and was found to have partial beneficial effects in porphyria [27]. However, we demonstrated that Chloroquine has a stronger affinity and inhibitory ability to 3CL Mpro than Hydroxychloroquine. In addition, from Table 1, the enthalpy and binding free energy of the newly proposed inhibitor 13b are higher than those of all selected clinically approved drug molecules, indicating the strongest binding affinity of 13b with 3CL Mpro.
Figure 2a shows the complex structure of the binding between 3CL Mpro and 13b, with similar binding position to that of the analyzed clinically approved drugs. The complex structure of 3CL Mpro bounded by clinically approved drugs with the highest Rosetta score are shown in the Supplementary Information. According to the calculated results, 13b molecule as well as the clinically approved drugs binds tightly to 3CL Mpro, with the binding position close to that of N3 inhibitor proposed by Rao group [28]. Such binding pocket is also similar to the previous report for SARS 3CL Mpro with inhibitors [29]. Figure 2b shows the ball-and-stick model for the binding pocket of five residues with dominant binding contributions of 3CL Mpro with 13b, where the blue, red, golden, yellow and orange molecules represent the residues MET 165, HIE 163, HIE 164, CYS 145 and ASN 142, respectively. Table 2 lists out the top five residues with dominant binding contributions of 3CL Mpro with the eight selected potential inhibitors. Figures S16, S17, S19 and S20 show the docking conformation and ball-and-stick model of 3CL Mpro with Chloroquine and Hydroxychloroquine. Although Chloroquine and Hydroxychloroquine share similar chemical structures, the small difference makes the top five residues with dominant binding contributions of Hydroxychloroquine (CYS 145, MET 165, ASN 142, GLY 143 and SER 144 as shown in Figure S17 and Table 2) very different from those of Chloroquine (MET 165, GLN 189, MET 49, ASP 187 and ARG 188 as shown in Figure S20 and Table 2).
Figure 2.
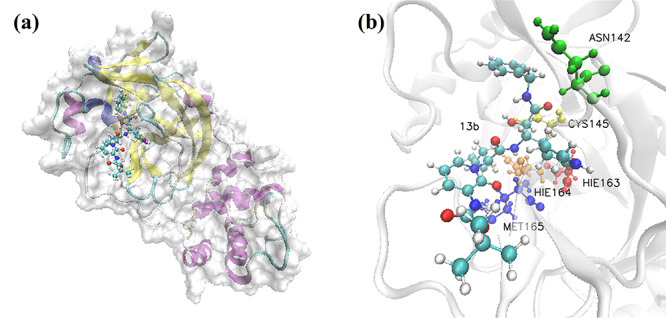
Visualization of the docking conformation of 3CL Mpro and 13b. (a) The solid surface model of binding pocket between 13b molecule and 3CL Mpro. (b) The ball-and-stick model, which shows the binding pocket of five residues with dominant binding contributions of 3CL Mpro with 13b.
Table 2.
Top five residues with dominant binding contributions of 3CL Mpro with different potential inhibitors
| Conformations | Important residues | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| 13b | MET 165 | HIE 163 | HIE 164 | CYS 145 | ASN 142 |
| Chloroquine | MET 165 | GLN 189 | MET 49 | ASP 187 | ARG 188 |
| Hydroxychloroquine | CYS 145 | MET 165 | ASN 142 | GLY 143 | SER 144 |
| Remdesivir | ASN 142 | MET 165 | GLU 166 | GLN 189 | HIE 41 |
| Ritonavir | GLN 189 | MET 49 | HIE 41 | MET 165 | CYS 44 |
| Beclabuvir | GLN 189 | MET 49 | MET 165 | HIE 41 | LEU 50 |
| Indinavir | GLN 189 | MET 49 | MET 165 | THR 24 | ARG 188 |
| Favipiravir | TYR 126 | SER 139 | GLY 138 | PHE 140 | ALA 116 |
To reasonably design drugs and select appropriate ligands, we further analyzed the contribution of each residue of 3CL Mpro when binding with ligands. Figure 3a shows the top 15 residues with dominant binding attractions for 13b, while Figure 3b shows the contributions of the binding free energy components for the top five residues, which are decomposed into van der Waals energy (vdW), electrostatic energy (Ele), polar solvation energy (polar) and nonpolar solvation energy (nonpolar). In Figure 3a, the major binding attractions of 3CL Mpro with 13b molecule come from the residues MET 165, HIE 163, HIE 164, CYS 145 and ASN 142, which contribute more than −1.5 kcal/mol to the binding energy. The decomposed energy distribution in Figure 3b shows that the dominant favorable interactions of the above-mentioned residues come from the electrostatic energy (the purple bars). For Chloroquine (Figure S20), the top five residues (MET 165, GLN 189, MET 49, ASP 187 and ARG 188) also contribute more than −1.5 kcal/mol and the dominant favorable interactions of these residues are van der Waals interactions.
Figure 3.

Decomposing the binding free energy of key residues. (a) The top 15 residues with dominant binding contributions of 3CL Mpro to 13b. (b) Decomposing the energy of top five residues and Chloroquine pairs into four energy terms, which are van der Waals interaction (vdW), electrostatic interaction (ele), polar solvation energy (polar) and nonpolar energy (nonpolar).
Furthermore, MET 165 also forms strong hydrophobic interaction with 13b and Chloroquine molecules, but not with Hydroxychloroquine (Figure S17), which makes the former two bind stronger to Mpro than the latter. When 13b binding with 3CL Mpro, the electrostatic interaction contributes the most for residues HIE 163 and HIE 164, while the van der Waals interaction is beneficial to residues MET 165, CYS 145 and ASN 142, as shown in Figure 3b. In summary, the configuration and environment of ligands impact significantly on the interaction of ligand and residue pairs at the binding site, which leads to the binding free energy difference. In this work, residues MET 165 and HIE 163 in 3CL Mpro make significant contributions when binding with α-ketoamide inhibitor 13b, in which the van der Waals interaction and electrostatic interaction play the most important roles. The discussions of binding free energy decomposing of key residues for Chloroquine, Hydroxychloroquine, Remdesivir, Ritonavir, Beclabuvir, Indinavir and Favipiravir are shown in Supplementary Information. Such results can be used for inhibitor molecule design with stronger binding affinity with 3CL Mpro of SARS-CoV-2.
Conclusions
In conclusion, based on the molecular dynamic simulation, we ab initio determine that for clinically approved drugs, Chloroquine, a widely used antimalarial and autoimmune disease drug possesses the strongest binding affinity with 3CL Mpro. However, the newly designed inhibitor 13b exhibits stronger binding ability with 3CL Mpro, thus it is more likely to become a pharmaceutical inhibitor against SARS-CoV-2. The proposed work suggests that the α-ketoamide inhibitor 13b is expected to be a promising candidate to treat the infection of SARS-CoV-2 and provides theoretical guidance for further design of drug molecular structure with stronger inhibitory effect with target protein.
Methods
Target identification
In this study, 3CL Mpro was selected as the target of docking research and binding analysis, due to its functional importance in the life cycle of SARS-CoV-2. The 3D crystal structure of 3CL Mpro (PDB ID: 6 LU7) was taken from RCSB Protein Data Bank, which was resolved by Rao’s group [20] with a resolution of 2.16 Å.
Compounds selection
For docking and binding analysis, seven clinically approved inhibitors (Chloroquine, Hydroxychloroquine, Remdesivir, Ritonavir, Beclabuvir, Indinavir and Favipiravir) were selected from previous reported virtual screening works or were found to be highly effective in the control of SARS-CoV-2 infection in vitro [6, 12]. And a newly designed α-ketoamide inhibitor (13b) was selected for comparative analysis, which was a co-crystallized ligand of the protein-ligand complex (PDB ID: 6Y2F) [19].
Ligand-protein docking
In this study, we applied ligand-protein docking by Rosetta program [30–33], which attempted to find the conformation and relative orientation of each ligand to minimize the Rosetta score function. To explore the binding pocket, the initial position of ligand was set near the protein target. After Monte Carlo minimization, we obtained 1000 conformations for 3CL Mpro and ligand and selected the 5% candidates with the lowest energies according to their energy scores from the ligand-protein contact structures. The Rosetta total energy score is the weighted sum of energy terms, including physical forces (e.g. electrostatics), and statistical terms (e.g. possibility of finding torsion angles in the Ramachandran space). Then, according to the sorting of binding energy of ligand and protein, the structure with the lowest binding energy was selected for MD simulation. The presented binding energy calculation included van der Waals energy, electrostatic energy, polar solvation energy nonpolar solvation energy and entropic contribution; therefore, it considered the core differences between the ligand and protein target and eliminated some irrelevant noise in the protein variation.
Molecular dynamic simulation
Amber16 software was used to perform MD simulation and binding energy calculation between drug molecules and 3CL Mpro [34]. The AM1-BCC charges of drug molecules were computed by sqm program in amber16. The ff14SB force field [35] and General Amber force field (GAFF) [36] were used for parameters’ generation of proteins and drug molecules, respectively. The ligand-protein complexes were immersed in a rectangular TIP3P water box (12A), with counterions (Na+ and Cl−) to neutralize the systems. The energy minimization and MD simulation were performed with sander program in amber16, during which a cutoff for nonbonded interactions of 8 Å was used and the time step was set to 2 fs. After 30 000 steps of energy minimization, the solvated ligand-protein complex was gradually heated from 0 to 300 K in the NVT ensemble using Langevin thermostat during 100 ps, followed by 100 ps of density equilibration with weak restraints on the complex (10 kcal mol−1 Å−2). Then 1 ns equilibration run was performed in NPT ensemble with temperature of 300 K and pressure pf 1 atm. At last, we performed a 40 ns production run in the NPT ensemble for each solvated complex system with a collection interval of 100 ps, and 300 frames were collected for further energy calculation. All hydrogen atoms were constrained with the SHAKE algorithm.
Binding free energy calculation
The binding free energy for small drug molecules and protein was calculated by the Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) [22–25] method using MMPBSA.py script based on the MD trajectory. The MM-GBSA free energy 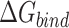 was calculated by the following equation:
was calculated by the following equation:
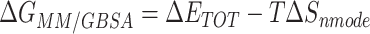 |
(1) |
where 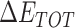 is the total enthalpy from the generalized Born model, including internal and solvation energies, and
is the total enthalpy from the generalized Born model, including internal and solvation energies, and 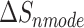 is the entropy calculated by normal mode [37, 38]. Here, 150 frames were used for
is the entropy calculated by normal mode [37, 38]. Here, 150 frames were used for 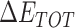 calculation while 50 frames were used for
calculation while 50 frames were used for 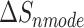 calculation. The enthalpy
calculation. The enthalpy 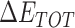 can be calculated by:
can be calculated by:
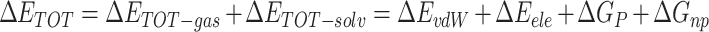 |
(2) |
where 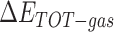 is the gas phase free energy,
is the gas phase free energy, 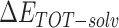 is the solvation free energy,
is the solvation free energy, 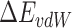 is the van der Waals energy,
is the van der Waals energy, 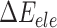 is the electrostatic energy,
is the electrostatic energy, 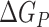 and
and 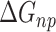 are the polar and nonpolar solvation free energies, respectively. The binding free energy
are the polar and nonpolar solvation free energies, respectively. The binding free energy 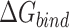 was determined by:
was determined by:
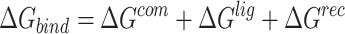 |
(3) |
where 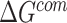 ,
, 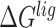 and
and 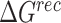 were MM/GBSA free energies, corresponding to complex, ligand and protein target, respectively.
were MM/GBSA free energies, corresponding to complex, ligand and protein target, respectively.
Key Points
Ab initio simulation is used to investigate the inhibitory ability between eight potential clinical pharmaceutical molecules and 3CL hydrolase in SARS-CoV-2.
Theoretical calculation demonstrates that the α-ketoamide 13b is the most promising inhibitor for SARS-CoV-2, while Chloroquine is the best among the clinically approved drugs.
The MET 165 and HIE 163 contribute the most based on residual energy decomposition analysis.
Supplementary Material
Y. Han and Z. Wang are currently PhD candidates in Shanghai Jiao Tong University and supervised by Professor Jinjin Li.
J. Ren is currently a master student in Shanghai Jiao Tong University and supervised by Professor Jinjin Li.
Z. Wei is an associate professor in Clinical and Translational Research Center of Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine.
J. Li is a professor in National Key Laboratory of Science and Technology on Micro/Nano Fabrication; Key Laboratory for Thin Film and Microfabrication of Ministry of Education, Department of Micro/Nano Electronics, Shanghai Jiao Tong University.
Contributor Information
Yanqiang Han, Shanghai Jiao Tong University.
Zhilong Wang, Shanghai Jiao Tong University.
Jiahao Ren, Clinical and Translational Research Center of Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine.
Zhiyun Wei, National Key Laboratory of Science and Technology on Micro/Nano Fabrication; Key Laboratory for Thin Film and Microfabrication of Ministry of Education, Department of Micro/Nano Electronics, Shanghai Jiao Tong University.
Jinjin Li, National Key Laboratory of Science and Technology on Micro/Nano Fabrication, Shanghai Jiao Tong University, Shanghai, 200240, China.
Data Availability
The data that support the findings of this study are available from the corresponding author, Professor Jinjin Li (email: lijinjin@sjtu.edu.cn) upon reasonable request.
Funding
Research reported in this publication was partially supported by the National Natural Science Foundation of China (Nos 51672176 and 21901157), and the SJTU Global Strategic Partnership Fund (2020 SJTU-HUJI).
Competing Interest
The authors declare no competing interests.
References
- 1. Song Z, Xu Y, Bao L, et al. . From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses 2019;11:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dong N, Yang X, Ye L, et al. . Genomic and protein structure modelling analysis depicts the origin and infectivity of 2019-nCoV, a new coronavirus which caused a pneumonia outbreak in Wuhan, China. bioRxiv 2020. doi: 10.1101/2020.01.20.913368. [DOI] [Google Scholar]
- 3. Vanhaelen Q, Mamoshina P, Aliper AM, et al. . Design of efficient computational workflows for in silico drug repurposing. Drug Discov Today 2017;22:210–22. [DOI] [PubMed] [Google Scholar]
- 4. Karaman B, Sippl W. Computational drug repurposing: current trends. Curr Med Chem 2019;26:5389–409. [DOI] [PubMed] [Google Scholar]
- 5. Zhavoronkov A, Aladinskiy V, Zhebrak A, et al. . Potential 2019-nCoV 3C-like protease inhibitors designed using generative deep learning approaches. 2020. doi: 10.26434/chemrxiv.11829102.v1. [DOI]
- 6. Chang Y-C, Tung Y-A, Lee K-H, et al. . Potential therapeutic agents for COVID-19 based on the analysis of protease and RNA polymerase docking. 2020. doi: 10.20944/preprints202002.0242.v2. [DOI]
- 7. Elmezayen AD, Al-Obaidi A, Şahin AT, et al. . Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J Biomol Struct Dyn 2020. doi: 10.1080/07391102.2020.1758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sheahan TP, Sims AC, Leist SR, et al. . Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat Commun 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sheahan TP, Sims AC, Graham RL, et al. . Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med 2017;9:eaal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Agostini ML, Andres EL, Sims AC, et al. . Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio 2018;9:e00221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wit E, Feldmann F, Cronin J, et al. . Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc Natl Acad Sci 2020. doi: 10.1073/pnas.1922083117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holshue ML, DeBolt C, Lindquist S, et al. . First case of 2019 novel coronavirus in the United States. N Engl J Med 2020;382:929–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu J, Cao R, Xu M, et al. . Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov 2020;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang M, Cao R, Zhang L, et al. . Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 2020;30:269–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lucey DR. New treatments for Ebola virus disease. BMJ 2019;366:I5371. [DOI] [PubMed] [Google Scholar]
- 16. Zhou P, Yang X-L, Wang X-G, et al. . A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hegyi A, Ziebuhr J. Conservation of substrate specificities among coronavirus main proteases. J Gen Virol 2002;83:595–9. [DOI] [PubMed] [Google Scholar]
- 18. Zhang L, Lin D, Kusov Y, et al. . α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem 2020;63:4562–78. [DOI] [PubMed] [Google Scholar]
- 19. Zhang L, Lin D, Sun X, et al. . Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020;368:409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jin Z, Du X, Xu Y, et al. . Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020;289–93. [DOI] [PubMed] [Google Scholar]
- 21. Berman HM, Battistuz T, Bhat TN, et al. . The protein data bank. Acta Crystallogr D Biol Crystallogr 2002;58:899–907. [DOI] [PubMed] [Google Scholar]
- 22. Still WC, Tempczyk A, Hawley RC, et al. . Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc 1990;112:6127–9. [Google Scholar]
- 23. Miller BR, McGee TD, Swails JM, et al. . MMPBSA.Py: an efficient program for end-state free energy calculations. J Chem Theory Comput 2012;8:3314–21. [DOI] [PubMed] [Google Scholar]
- 24. Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct Funct Bioinforma 2004;55:383–94. [DOI] [PubMed] [Google Scholar]
- 25. Feig M, Onufriev A, Lee MS, et al. . Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem 2004;25:265–84. [DOI] [PubMed] [Google Scholar]
- 26. Chen J, Wang X, Zhu T, et al. . A comparative insight into amprenavir resistance of mutations V32I, G48V, I50V, I54V, and I84V in HIV-1 protease based on thermodynamic integration and MM-PBSA methods. J Chem Inf Model 2015;55:1903–13. [DOI] [PubMed] [Google Scholar]
- 27. Chloroquine LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases, 2012. [PubMed] [Google Scholar]
- 28. Liu X, Zhang B, Jin Z, et al. . The crystal structure of 2019-NCoV main protease in complex with an inhibitor N3. RCSB Protein Data Bank 2020.
- 29. Nukoolkarn V, Lee VS, Malaisree M, et al. . Molecular dynamic simulations analysis of ritronavir and lopinavir as SARS-CoV 3CLpro inhibitors. J Theor Biol 2008;254:861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davis IW, Raha K, Head MS, et al. . Blind docking of pharmaceutically relevant compounds using RosettaLigand. Protein Sci 2009;18:1998–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davis IW, Baker D. RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol 2009;385:381–92. [DOI] [PubMed] [Google Scholar]
- 32. Kufareva I, Rueda M, Katritch V, et al. . Status of GPCR modeling and docking as reflected by community-wide GPCR dock 2010 assessment. Structure 2011;19:1108–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guseynov A-AD, Pisarev SA, Shulga DA, et al. . Computational characterization of the glutamate receptor antagonist perampanel and its close analogs: density functional exploration of conformational space and molecular docking study. J Mol Model 2019;25:312. [DOI] [PubMed] [Google Scholar]
- 34. Case DA, Betz RM, Cerutti DS, et al. . AMBER 2016. San Francisco: University of California, 2016, 810. [Google Scholar]
- 35. Maier JA, Martinez C, Kasavajhala K, et al. . ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 2015;11:3696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang J, Wolf RM, Caldwell JW, et al. . Development and testing of a general amber force field. J Comput Chem 2004;25:1157–74. [DOI] [PubMed] [Google Scholar]
- 37. Xu B, Shen H, Zhu X, et al. . Fast and accurate computation schemes for evaluating vibrational entropy of proteins. J Comput Chem 2011;32:3188–93. [DOI] [PubMed] [Google Scholar]
- 38. Genheden S, Kuhn O, Mikulskis P, et al. . The normal-mode entropy in the MM/GBSA method: effect of system truncation, buffer region, and dielectric constant. J Chem Inf Model 2012;52:2079–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, Professor Jinjin Li (email: lijinjin@sjtu.edu.cn) upon reasonable request.