

Abstract
On June 28, 2018, the Committee for Advanced Therapies and the Committee for Medicinal Products for Human Use adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Yescarta for the treatment of adult patients with relapsed or refractory diffuse large B‐cell lymphoma and primary mediastinal large B‐cell lymphoma, after two or more lines of systemic therapy. Yescarta, which was designated as an orphan medicinal product and included in the European Medicines Agency's Priority Medicines scheme, was granted an accelerated review timetable.
The active substance of Yescarta is axicabtagene ciloleucel, an engineered autologous T‐cell immunotherapy product whereby a patient's own T cells are harvested and genetically modified ex vivo by retroviral transduction using a retroviral vector to express a chimeric antigen receptor (CAR) comprising an anti‐CD19 single chain variable fragment linked to CD28 costimulatory domain and CD3‐zeta signaling domain. The transduced anti‐CD19 CAR T cells are expanded ex vivo and infused back into the patient, where they can recognize and eliminate CD19‐expressing cells.
The benefits of Yescarta as studied in ZUMA‐1 phase II (NCT02348216) were an overall response rate per central review of 66% (95% confidence interval, 56%–75%) at a median follow‐up of 15.1 months in the intention to treat population and a complete response rate of 47% with a significant duration. The most common adverse events were cytokine release syndrome, neurological adverse events, infections, pyrexia, diarrhea, nausea, hypotension, and fatigue.
Implications for Practice
Yescarta (axicabtagene ciloleucel) was the first chimeric antigen receptor T‐cell therapy to be submitted for evaluation to the European Medicines Agency and admitted into the “priority medicine” scheme; it was granted accelerated assessment on the basis of anticipated clinical benefit in relapsed/refractory diffuse large B‐cell lymphoma, a condition of unmet medical need. Indeed, Yescarta showed an overall response rate of 66% and a complete response rate of 47% with a significant duration and a manageable toxicity that compared very favorably with historical controls. Here the analysis of benefits and risks is presented, and specific challenges with this important novel product are highlighted, providing further insights and reflections for future medical research.
Keywords: Diffuse large B‐cell lymphoma, Primary mediastinal B‐cell lymphoma, Chimeric antigen receptor, Axicabtagene ciloleucel, Cytokine release syndrome, CAT, CHMP
Short abstract
This article summarizes the data leading to marketing authorization for the medicinal product axicabtagene ciloleucel for diffuse large B‐cell lymphoma.
Introduction
Diffuse large B‐cell lymphoma (DLBCL) is the most common type of non‐Hodgkin lymphoma (NHL), accounting for approximately 31% of all NHLs in Western countries and 37% of B‐cell malignancies worldwide. The median age at presentation is 70 years; however, it can occur at any age, with a slightly higher incidence in men; the incidence rate of DLBCL was 3.44 in 100,000 in Europe in 2014 1. Primary mediastinal B‐cell lymphoma (PMBCL) constitutes approximately 2% to 4% of all NHLs. This disease affects mainly young adults (median age of 35 years), predominantly women (female:male ratio, 1.7–2:1). There are also cases of PMBCL among children and adolescents.
The current standard of care for first‐line treatment for aggressive B‐cell NHL is a regimen of cyclophosphamide, doxorubicin, vincristine, and prednisolone (CHOP) in combination with an anti‐CD20 monoclonal antibody such as rituximab (R‐CHOP) 2. Although more effective than chemotherapy alone, first‐line R‐CHOP results in long‐term disease remission in 50%–70% of patients depending on disease stage and prognostic index 3, 4. Thus, patients with relapsed or refractory aggressive B‐cell DLBCL and PMBCL may comprise 50% or more of all patients with aggressive B‐cell NHL 4.
Patients with relapsed or refractory DLBCL and PMBCL typically are treated with a rituximab and platinum‐based chemotherapy regimen, followed by high‐dose chemotherapy (HD) and autologous stem cell transplant (ASCT) for those who are deemed eligible based on adequate performance status (defined by age and absence of major organ dysfunctions) 5. However, among patients suitable for HD‐ASCT, only about half will have a response to salvage therapy that is sufficient for them to proceed to HD‐ASCT 6. In addition, of those proceeding to HD‐ASCT, 60% of patients will relapse after transplant. Clinical studies, palliative chemotherapy, and in rare cases a second HD‐ASCT or allogeneic stem cell transplant are some of the options available for these patients 7.
Outcomes are particularly poor for patients who have primary refractory disease after first‐line therapies; furthermore, most of these patients are not eligible for transplant because of their chemotherapy‐resistant disease 8, 9, and primary refractory disease was found to be a significant risk factor for failing second‐line therapy. Outcomes are also poor for patients with aggressive B‐cell NHL that is refractory to second‐line therapy (overall response rate [ORR] of 18%).
Most patients with PMBCL will initially respond to therapy with a rapid decrease in the tumor mass, but rapid disease progression during treatment cycles can occur in 5%–10% of patients. Second‐line treatment strategies are like those used for DLBCL, attempting reinduction with non–cross‐resistant agents, followed by consolidation with HD‐ASCT in those with a chemosensitive disease. In general, the outcomes of these patients have been disappointing 10.
The human CD19 antigen is a 95‐kD transmembrane glycoprotein belonging to the immunoglobulin superfamily 11, 12. It is encoded by the cd19 gene located on the short arm of chromosome 16, 16p11.2 13. CD19 was first identified as the B4 antigen of human B lymphocytes through the use of anti‐B4 monoclonal antibody against CD19. It is specifically expressed in normal and neoplastic B cells, as well as follicular dendritic cells 14, 15, 16.
Axicabtagene ciloleucel is an engineered autologous T‐cell immunotherapy product whereby a patient's own T cells are harvested and genetically modified ex vivo by retroviral transduction using an retroviral vector to express a chimeric antigen receptor (CAR) comprising an anti‐CD19 single chain variable fragment linked to CD28 and CD3‐zeta costimulatory domains. CD19 is expressed as a surface antigen in DLBCL and other aggressive B‐cell lymphomas. The transduced anti‐CD19 CAR T cells are expanded ex vivo and infused back into the patient, where they can recognize and eliminate CD19‐expressing target cells. Axicabtagene ciloleucel binds to CD19‐expressing cancer cells, normal B cells, and follicular dendritic cells. After anti‐CD19 CAR T‐cell engagement with CD19‐expressing target cells, the CD28 costimulatory domain and CD3‐zeta signaling domain activate downstream signaling cascades that lead to T‐cell activation, proliferation, acquisition of effector functions, and secretion of inflammatory cytokines and chemokines, eventually leading to elimination of CD19‐expressing target tumor cells (Fig. 1).
Figure 1.
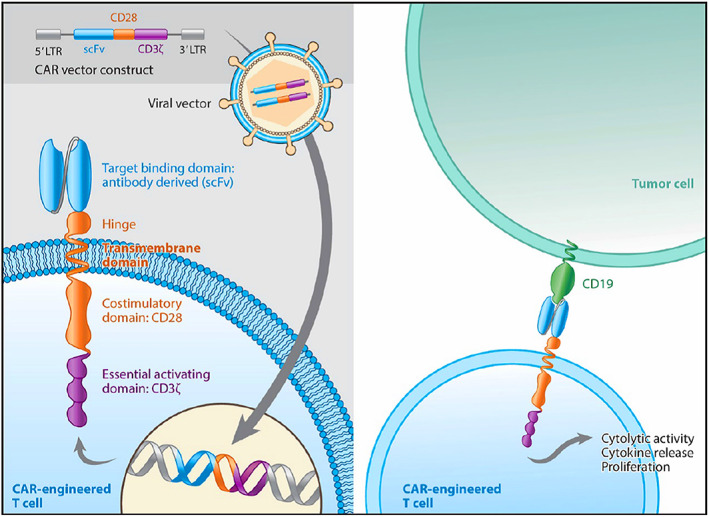
Axicabtagene ciloleucel CAR construct and mechanism of action. Source: Applicant's clinical overview.Abbreviations: CAR, chimeric antigen receptor; LTR, long terminal repeat; scFv, single chain variable fragment.
Table 1.
Summary of efficacy results for ZUMA‐1 phase II: 12‐month analysis, at the time of the authorization, updated with 24‐month analysis data provided postauthorization 27
| Category | All leukapheresed (ITT) Cohorts 1 and 2 (n = 111) | All treated (mITT) Cohorts 1 and 2 (n = 101) | ||
|---|---|---|---|---|
| 12‐month analysis | 24‐month analysis | 12‐month analysis | 24‐month analysis | |
| ORR, (95% CI), % | 66 (56–75) | 68 (58–76) | 72 (62–81) | 74 (65–82) |
| CR, % | 47 | 50 | 51 | 54 |
| Duration of response,a median (range), months | 14.0 (0.0–17.3) | NE (0.0–29.5) | 14.0 (0.0–17.3) | NE (0.0–29.5) |
| Duration of response,a CR, median (range), months | NE (0.4–17.3) | NE (0.4–29.5) | NE (0.4–17.3) | NE (0.4–29.5) |
| Overall survival, median (95% CI), months | 17.4 (11.6–NE) | 17.4 (11.6–NE) | NE (12.8–NE) | NE (12.8–NE) |
| 6‐month OS (95% CI), % | 81.1 (72.5–87.2) | 81.1 (72.5–87.2) | 79.2 (69.9–85.9) | 79.2 (69.9–85.9) |
| 9‐month OS (95% CI), % | 69.4 (59.9–77.0) | 69.4 (59.9–77.0) | 69.3 (59.3–77.3) | 69.3 (59.3–77.3) |
| 12‐month OS (95% CI), % | 59.3 (49.6–67.8) | 59.5 (49.7–67.9) | 60.4 (50.2–69.2) | 60.4 (50.2–69.2) |
| 24‐month OS (95% CI), % | Not applicable | 47.7 (38.2–56.7) | Not applicable | 50.5 (40.4–59.7) |
| Progression‐free survival, median (95% CI), months | 9.5 (6.1–12.9) | 9.5 (6.1–15.4) | 9.1 (5.8–12.5) | 9.1 (5.7–NE) |
The 12‐month analysis had a median follow‐up of 15.1 months. The 24‐month analysis (provided postauthorization) had a median follow‐up of 27.1 months. OS relates to the time from the leukapheresis date (ITT) or Yescarta infusion (mITT) to death from any cause.
Duration of response was censored at the time of stem cell transplant for patients who received stem cell transplant while in response.
Abbreviations: CI, confidence interval; CR, complete response; ITT, intention to treat; mITT; modified intention to treat; NE, not estimable (not reached); ORR, overall response rate; OS, overall survival.
Table 2.
Serious adverse events in more than one patient, ZUMA‐1 safety analysis set, 12‐month analysis
| MedDRA preferred term | Phase I and II combined (n = 108), n (%) |
|---|---|
| Patients with and serious treatment‐emergent adverse event | 59 (55) |
| Encephalopathy | 19 (18) |
| Lung infection | 8 (7) |
| Pyrexia | 8 (7) |
| Pneumonia | 6 (6) |
| Confusional state | 5 (5) |
| Febrile neutropenia | 5 (5) |
| Aphasia | 4 (4) |
| Atrial fibrillation | 4 (4) |
| B‐cell lymphoma | 4 (4) |
| Cardiac arrest | 4 (4) |
| Urinary tract infection | 4 (4) |
| Acute kidney injury | 3 (3) |
| Agitation | 3 (3) |
| Ejection fraction decreased | 3 (3) |
| Hypotension | 3 (3) |
| Hypoxia | 3 (3) |
| Neutropenia | 3 (3) |
| Somnolence | 3 (3) |
| Atrial flutter | 2 (2) |
| Delirium | 2 (2) |
Preferred terms are sorted in descending order of total frequency count. Adverse events are coded using MedDRA version 19.0 and graded per Common Terminology Criteria for Adverse Events version 4.03. Percentages are calculated using n in each column as the denominator.
Abbreviation: MedDRA, Medical Dictionary for Regulatory Activities.
Nonclinical Studies
In vitro data sufficiently demonstrated specific activity of axicabtagene ciloleucel (company code, KTE‐C19) against its target antigen CD19. The specificity and potency of the anti‐CD19 CAR T cells were evaluated by measuring their ability to produce cytokines in response to co‐culture with either CD19+ or CD19− target cells. Results showed that production of cytokines (e.g., IFN‐γ) by anti‐CD19 CAR T cells was dependent on both the presence of the anti‐CD19 CAR T cells and the co‐culture with CD19+ target cells. For control cultures containing either transduced or nontransduced T cells co‐cultured with CD19− target cells or no target cells, only minimal cytokine production was observed. Specificity, potency, and polyfunctionality of the anti‐CD19 CAR T cells derived from 15 patients with advanced NHL were evaluated for cytokine production during co‐culture with CD19+ or negative control target cell lines. The analysis demonstrated that the CD19 CAR T cells released all 17 tested cytokines, chemokines, and effector molecules when specifically co‐cultured with CD19+ target cells. Because the 17 analytes comprised markers for immune cell homeostasis, proliferation, proinflammatory activity, immune‐modulating activity, chemokines, and effector molecules, CD19 CAR T cells seem to be polyfunctional at the end of the manufacturing process. The phenotypic evaluation demonstrated that CD4+ and CD8+ T cells were transduced at a similar ratio. Manufactured axicabtagene ciloleucel is a diverse product that is clinically effective across a broad range of CD4‐to‐CD8 ratios and comprises phenotypically naive, central memory, effector, and effector memory T cells 17.
Importantly, the in vivo antilymphoma activity of murine T cells transduced with the vector encoding the antimurine CD19 CAR was confirmed in a syngeneic mouse lymphoma model and led to prolonged survival. However, total body irradiation conditioning (causing lymphodepletion) prior to CAR T‐cell therapy was required for achieving tumor clearance in this immunocompetent mouse model. Lymphoma and normal B‐cell depletion was CD19‐ and CAR‐specific 18. Elimination of normal B cells was persistent; B‐cell aplasia is an expected on‐target/off‐tumor effect (see the Clinical Pharmacology section).
The combination of both the use of a γ‐retroviral vector with full‐length viral long terminal repeats and the high proliferative potential of the transduced T cells provides a certain risk of insertional oncogenesis that has previously been addressed through a Committee for Medicinal Products for Human Use (CHMP) Scientific Advice procedure. Literature data reported an exceptionally high resistance of mature mouse T cells against transformation induced by genomic integration of γ‐retroviral vectors. Moreover, there were no reported cases of insertional oncogenesis (see the Clinical Safety section) of either axicabtagene ciloleucel itself or T cells that were transduced with γ‐retroviral vectors encoding other transgenes. The experience so far with mouse and human T cells suggests that T‐cell transformation caused by genomic integration of γ‐retroviral vectors is, if it happens at all, a very rare event.
Clinical Pharmacology
Results from the National Cancer Institute (NCI) study 09‐C‐0082 (NCT00924326), a phase I open‐label study of the safety and feasibility of anti‐CD19 CAR T cells in patients with advanced B‐cell malignancies 19, and ZUMA‐1 20, showed that peak levels of anti‐CD19 CAR T cells occurred within the first 7–14 days after axicabtagene ciloleucel infusion. In ZUMA‐1 the median peak level of anti‐CD19 CAR T cells in the blood (Cmax) was 41.9 cells per microliter (range, 0.8–1513.7 cells/μL), which decreased to a median of 2.1 cells per microliter by 1 month (range, 0–167.4 cells/μL) and to a median of 0.4 cells per microliter by 3 months (range, 0–15.8 cells/μL) after the infusion.
The extent of T‐cell expansion did not appear to be related to the total dose of CAR T cells with respect to few patients within NCI‐09‐C‐0082 and ZUMA‐1 who received a higher dose (up to 6 × 106 anti‐CD19 CAR T cells/kg instead of the target dose of 2 × 106 anti‐CD19 CAR T cells/kg). Overall, there was no overt relationship between the dose of anti‐CD19 CAR T cells and their expansion and persistence in the peripheral blood. Likewise, to date, there is no apparent relationship between the axicabtagene ciloleucel dose in large B‐cell lymphoma, the anti‐CD19 CAR T‐cell persistence in the blood, and the clinical response or the toxicities related to this therapy, respectively.
The number of anti‐CD19 CAR T cells in blood was positively associated with objective response (complete response [CR] or partial response [PR]). The median anti‐CD19 CAR T‐cell Cmax levels in responders (n = 73) were 205% higher compared with the corresponding level in nonresponders (n = 23; 43.6 cells/μL vs. 21.2 cells/μL). Median area under the curve (AUC)Day 0–28 in responding patients (n = 73) was 251% of the corresponding level in nonresponders (n = 23; 557.1 days × cells/μL vs. 222.0 days × cells/μL).
Several cytokines were observed to increase after infusion of axicabtagene ciloleucel, peaking within 14 days of infusion and generally decreasing toward the baseline levels within 1 month. Notably, IL‐15 was induced after conditioning chemotherapy, whereas all other cytokines were induced after the cell infusion. IFN‐γ was not induced after conditioning chemotherapy but showed a meaningful increase after the cell infusion.
Analyses performed to identify associations between cytokine levels and incidence of cytokine release syndrome (CRS) or neurologic events showed that higher levels (peak and AUC at 1 month) of IL‐15, as well as IL‐6, were associated with grade 3 or higher neurologic events and grade 3 or higher CRS. The AUCs of IL‐15 and IL‐6 were measured throughout the first 4 weeks after infusion of axicabtagene ciloleucel in patients who developed grade 3 or higher neurologic events. The measurements were compared with levels in patients who had grade 2 or lower events. Bonferroni stepdown corrected p values, using a prespecified group of serum analytes, for IL‐15 and IL‐6 with neurologic events, were .0003 and <.0001, respectively. Similarly, the AUC of IL‐2Rα (p = .0829) and IL‐10 (p = .0123) were likewise associated with grade 3 or higher neurologic events as compared with patients with grade 2 or lower events. The association between peak levels of IL‐15, IL‐6, IL‐2Rα, and IL‐10 and grade 3 or higher neurologic events was also significant.
Peak and cumulative levels of IL‐2 and ferritin were associated with grade 3 or higher neurologic events (p < .05 after multiplicity adjustment) and were not associated with CRS.
The following serum markers were associated with both grade 3 or higher neurologic events and with grade 3 or higher CRS: the cytokines TNF‐α and IFN‐γ, the chemokines IP‐10 and IL‐8, the proinflammatory marker IL‐1ra, the immune effector molecule granzyme B, and the angiogenic factor VCAM‐1 (all p < .05 by Bonferroni stepdown).
Because of the on‐target/off‐tumor effect of Yescarta, a period of B‐cell aplasia is expected after treatment (see also the Clinical Safety section). Among 73 patients with evaluable samples at baseline, 40% had detectable B cells; the B‐cell aplasia observed in the majority of patients at baseline was attributed to prior therapies. After Yescarta treatment, the proportion of patients with detectable B cells decreased: 20% had detectable B cells at month 3, and 22% had detectable B cells at month 6. The initiation of B‐cell recovery was first noted at month 9, when 56% of patients had detectable B cells. This trend of B‐cell recovery continued over time, as 64% of patients had detectable B cells at month 18, and 77% of patients had detectable B cells at month 24 (24‐month updates were provided postauthorization).
Efficacy Data
The efficacy results (see Table 1) were primarily coming from the single‐arm phase II part of the ZUMA‐1 trial, which enrolled 111 adult patients with refractory or relapsed DLBCL, including patients with DLBCL arising from follicular lymphoma and PMBCL, into two cohorts. Cohort 1 was to be analyzed independently, whereas cohort 2 was only to be analyzed in combination with cohort 1.
In ZUMA‐1 phase II, among 111 leukapheresed patients, 110 lots of axicabtagene ciloleucel were successfully manufactured, and 101 patients (91%) received axicabtagene ciloleucel. The median time from leukapheresis to delivery of Yescarta to the treatment facility was 17 days (range, 14–51 days), and the median time from leukapheresis to infusion was 24 days (range, 16–73 days). Bridging chemotherapy between leukapheresis and lymphodepleting chemotherapy was not permitted. The median dose was 2.0 × 106 anti‐CD19 CAR T cells per kilogram. All patients were hospitalized for observation and management of adverse reactions for a minimum of 7 days after axicabtagene ciloleucel infusion.
The ORR based on the intention to treat (ITT) population 21 and on the central review assessment was 66% (95% confidence interval [CI], 56–75) with a median follow‐up of 15.1 months and a complete response rate of 47%. In the modified ITT (mITT) set, which included only patients who actually received axicabtagene ciloleucel, ORR for cohorts 1 and 2 combined was 72% (95% CI, 62–81) with a complete response rate of 51%. The estimated median duration of response was 14.0 months (95% CI, 8.3–not estimable) and was not yet reached in patients who achieved CR with a median follow‐up of 11.3 months. ORRs for cohorts 1 and 2 combined were further analyzed based on investigators’ assessment in the modified ITT set, by baseline demographic and disease characteristics, product characteristics, and use of tocilizumab and systemic steroids. Mean ORRs for each subset were comparable to the overall ORR (83%; 95% CI, 74%–90%; mITT, investigators’ assessment). No significant impact of subsets based on age, sex, disease type (DLBCL or PMBCL) and refractory subgroups, primary refractory status, refractory status to two or more consecutive lines of therapy, disease stage, International Prognostic Index (IPI) risk score, tumor burden, CD4:CD8 ratio (>1 or ≤ 1), and the use of tocilizumab or steroids (yes or no) became apparent. Responses also were consistent in 82 evaluable patients whose tumors were retrospectively assessed as CD19+ (92% of patients; ORR, 85%) or CD19− (ORR, 75%). However, the small sample size limits the interpretability of this subgroup ORR analysis.
A retrospective, patient‐level, pooled analysis of outcomes in refractory aggressive NHL (n = 636) was conducted (SCHOLAR‐1 22). Of 636 SCHOLAR‐1 evaluable patients, 389 patients came from two randomized phase III clinical studies (170 patients in CORAL, 219 patients in LY12), whereas 247 patients (University of Iowa Mayo Clinic, n = 82; MD Anderson Cancer Center, n = 165) came from retrospective databases. SCHOLAR‐1 was developed as a companion study to ZUMA‐1 to provide context for interpreting the ZUMA‐1 results. The analysis included patients who had not responded (stable disease or progressive disease) to their last line of therapy or had relapsed within 12 months after ASCT.
The ORR was 26% (95% CI, 21–31), and the CR rate was 7% (95% CI, 3–15), with a median overall survival (OS) of 6.3 months. A reanalysis as “worst case” scenario was performed by excluding patients with Eastern Cooperative Oncology Group (ECOG) performance status 2–4, patients with unknown ECOG performance status, and patients whose baseline assessment was more than 3 months before relapsed/refractory disease was diagnosed. With these patients, a head to head comparison of response (CR + PR), CR, and OS comparing SCHOLAR‐1 and ZUMA‐1 was conducted by the applicant. In this comparison, the difference in response rates between SCHOLAR‐1 (“worst case” subset) and ZUMA‐1 (mITT set) was 53.1% (95% CI, 43.6%–62.5%), the difference in CR rates was 46.9% (95% CI, 36.4%–57.4%), and the hazard ratio for the reduction in the risk of death for patients in the ZUMA‐1 study was 0.4 (95% CI, 0.29–0.56).
Using the ITT set and central review, the difference for ORR between SCHOLAR‐1 (ORR; 30.1%) and ZUMA‐1 (ORR; 66%) was 35.9%; the difference for CR between SCHOLAR‐1 (CR; 11.5%) and ZUMA‐1 (CR; 47%) was 35.5%. As expected, these numbers were substantially lower than the comparison provided by the applicant based on the mITT set and local investigators’ assessments. Yet a difference of more than 35% in both ORR and CR makes chance findings or pure bias sufficiently unlikely.
Clinical Safety
Data from 108 patients of the ZUMA‐1 trial (7 patients in phase I and 101 patients in cohorts 1 and 2 of phase II) were considered as principal source of safety information (see Table 2). The expansion cohort 3 of ZUMA‐1 introduced a revised CRS and neurotoxicity management algorithm, including prophylactic use of tocilizumab and levetiracetam as well as the reactive use of corticosteroids, and was originally not intended to support the primary analysis of ZUMA‐1.
The most serious and frequently occurring adverse reactions were CRS (93%), encephalopathy (58%), and infections (38%). Four patients died from an adverse event, two of which were considered related to Yescarta.
Cytokine Release Syndrome
CRS occurred predominantly in the first 2 weeks after infusion and eventually subsided. The median time to onset was 2 days (range, 1–12), and the median duration was 7 days (range, 2–29). Grade 3 or higher CRS was experienced by 12% of patients.
Most of the reported acute adverse events, such as pyrexia, chills, tachycardia, serum electrolyte changes, headache, and myalgia, as well as consequences to the function of important organs, including hypoxia, hypotension, cardiac rhythm disturbances, and acute kidney injury, are likely to be associated with CRS. All the events associated with the cytokine release syndrome resolved except for one event of grade 5 hemophagocytic lymphohistiocytosis. Another event of grade 5 cardiac arrest occurred in a patient with cytokine release syndrome.
Empirical treatment recommendations 23 taking the severity grades of CRS into account have been provided. Tocilizumab is an anti–IL‐6 monoclonal antibody that can result in rapid resolution of CRS toxicities without loss of CAR T‐cell expansion or efficacy. Tocilizumab doses of 4–8 mg/kg (maximum dose 800 mg) can be repeated as needed for patients with persistent signs and symptoms of CRS 24. Forty‐three percent of patients received tocilizumab, and 27% received glucocorticoids for the management of CRS, neurologic events, or both.
Neurological Adverse Reactions
Neurological adverse reactions, occurring in 65% of patients appeared to be mostly nonlocalizing, that is, encephalopathy with the symptoms and signs such as changes in consciousness levels, disturbance of attention, somnolence, agitation, confusion, and attention disturbance. Some potentially localizing symptoms and signs, including ataxia, dyskinesia, speech disorders, and aphasia, did, however, also occur. Ninety‐eight percent (98%) of all patients recovered from neurologic adverse reactions. Grade 3 or higher neurological adverse reactions occurred in 31% of patients. The median time to onset was 5 days (range, 1–17 days). The median duration was 13 days, with a range of 1 to 191 days, but it should be noted that the 191 days reflects a single patient with grade 1 memory impairment. In an expansion cohort 3 of the ongoing ZUMA‐1 trial, one case of cerebral edema with a fatal outcome 9 days after Yescarta infusion was reported in a patient with high levels of proinflammatory, marked cell adhesion or vascular damage, and chemokines in serum prior to chemotherapy conditioning and Yescarta infusion.
The relationship of CRS and the neurological adverse reaction still requires further clarification. A pathophysiological explanation of the observed encephalopathy seems rather obscure at present. Similar neurotoxicity has been observed with other forms of CD19 directed immunotherapies such as blinatumomab, so there is a reason to believe that this is caused either by the intended pharmacological effect, that is, an off‐target immune bystander effect; cytotoxicity and cytokine release by CAR T cells to their natural CD19 positive target (B cells); CD19 expression on other cells of the central nervous system; or cross‐reactivity to a yet unknown target. However, it may also be caused by systemic changes in cytokine levels and inflammatory activity in a population that is prone to this neurological adverse event.
Hypogammaglobinemia
B‐cell aplasia leading to hypogammaglobulinemia can occur in patients receiving treatment with Yescarta. Hypogammaglobulinemia has been very commonly observed patients treated with Yescarta. In ZUMA‐1, hypogammaglobulinemia occurred in 17% of patients. However, because 49 of 81 patients (60%) with data that could be evaluated had no detectable B cells at study entry, the estimates for the incidence of B‐cell aplasia and hypogammaglobulinemia were confounded. Immunoglobulin levels should be monitored after treatment with Yescarta and managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement.
Hematological Reactions
Overall, most patients had any‐grade cytopenias according to baseline laboratory values: 93% had anemia, 34% had thrombocytopenia, and 15% had neutropenia. Grade 3 or higher neutropenia, anemia, and thrombocytopenia occurred in 93%, 63%, and 56% of patients, respectively. Prolonged neutropenia (still present at day 30 or beyond) occurred in 31%, 27%, and 17% of patients, respectively. Febrile neutropenia was observed in 35% of patients.
Infections
Infections were overall also very common. Infections occurred in 38% of patients in ZUMA‐1. Grade 3 or higher (severe, life threatening, or fatal) occurred in 25% of patients. Grade 3 or higher unspecified pathogen, bacterial, and viral infections occurred in 19%, 8%, and 6% of patients, respectively. The most common site of infection was in the respiratory tract. Two deaths caused by infection were reported in the supportive data set; one was associated with a chemotherapy‐related thrombocytopenia.
Discussion and Benefit‐Risk Balance
Within ZUMA‐1 phase II, 101 of 111 patients who underwent leukapheresis received axicabtagene ciloleucel; 10 patients were not treated. Nine patients did not receive Yescarta primarily because of progressive disease or serious adverse events after enrollment and prior to cell delivery, and one patient did not receive Yescarta because of manufacturing failure. The ITT set was defined as all patients who underwent leukapheresis, and the mITT set was defined as all patients who received Yescarta.
By the data cutoff date of August 11, 2017, the ORR based on the ITT population and central review was 66% (95% CI, 56%–75%) with a CR rate of 47%. Baseline patients and disease characteristics appear very consistent between the ITT and mITT populations (Panel 1).
Panel 1: ITT Versus mITT Population.
Although the application mainly focused on the 101 treated patients (mITT population), the Committee for Advanced Therapies (CAT) and CHMP laid a special focus on the ITT population. The ITT population is based on all enrolled patients and takes events that happen between enrollment and product application into account; analyses based on the ITT set include leukapheresis, which is considered to be part of the treatment algorithm, and take the time and success of manufacturing into account. Hence, only the ITT population allows a valid comparison with historic controls, the current standard of care, and other novel products.
Treatment with axicabtagene ciloleucel at target doses of 2 × 106 anti‐CD19 CAR T cells per kilogram of body weight shows high rates of durable responses also when viewed in the context of the results of the retrospective global patient‐level pooled study SCHOLAR‐1. The ORR of ZUMA‐1 based on the ITT population and central review was about 66%, whereas the ORR in SCHOLAR‐1 was about 26% (Panel 2).
Panel 2: Assessing Single‐Arm Trials with Historic Controls (ZUMA‐1 Versus SCHOLAR‐1).
SCHOLAR‐1 was developed as a companion study to ZUMA‐1 to provide context for interpreting the ZUMA‐1 results. The analysis included patients who had not responded (stable disease or progressive disease) to their last line of therapy or had relapsed within 12 months after ASCT.
Comparisons of single‐arm trials to historic controls are particularly problematic for a variety of reasons: (a) Patient populations might differ in known or unknown prognostic factors. (b) The choice of historic control data (studies, registers, etc.) might not be complete, and important studies might be missing for various reasons (such as unavailability of data). (c) The standard of care treatment obtained in the historic control might have improved over time. (d) The follow‐up routine (number and timing of visits, extent and standardization of patient observation at visits, gathered information at baseline and follow‐up visits) might substantially differ between a clinical trial and routine care.
Statistical methods such as weighting or matching of patients based on baseline characteristics exist to potentially reduce the bias. Nevertheless, especially differences in the follow‐up routine (d) and unknown or unmeasured confounders (a) can never be appropriately addressed. Hence, it is crucial to thoroughly assess these points and to obtain an idea of the variability of differences between the active arm and historic controls. In the current situation, CAT and CHMP focused on the data of the two randomized phase III clinical studies to overcome issue (d) discussed above. Further standardization was attempted by focusing on response rates per central review. Additionally, worst case analyses were requested to reduce the impact of baseline differences in prognostic factors and to avoid overly optimistic findings. Finally, patient‐level data were requested from the applicant to allow the assessors to conduct further analyses, for example, to assess the sensitivity of the results. In this regard, overall and subgroup specific Kaplan‐Meier estimates were derived from the patient‐level data, and the concordance of local and central investigators was recomputed. This helped in the assessment to gain a deeper understanding of possible differences between the three presented subtypes of NHL (DLBCL, PMBCL, and transformed follicular lymphoma [TFL]) and to judge the differences between the tumor assessments.
Although most of the patients in ZUMA‐1 had DLBCL (76%), 16% had TFL, and 8% had PMBCL. All patients had a baseline ECOG score of 0 or 1, the median number of prior therapies was three (range, 1–10), two patients received study treatment for primary refractory disease, 26% had a history of primary refractory disease, 77% were refractory to second or later line of therapy, and 21% had a relapse within 12 months after ASCT.
The initial indication proposed by the applicant was as follows: “YESCARTA is indicated for the treatment of adult patients with relapsed/refractory diffuse large B‐cell lymphoma (DLBCL), primary mediastinal B‐cell lymphoma (PMBCL), and transformed follicular lymphoma (TFL) who are ineligible for autologous stem cell transplant (ASCT).” The indication agreed upon is for treatment of adult patients with relapsed or refractory diffuse large B‐cell lymphoma (DLBCL) and primary mediastinal large B‐cell lymphoma, after two or more lines of systemic therapy.
It is acknowledged that DLBCL, PMBLC, and TFL all are classified as large‐cell lymphoma because of their similarities with respect to pathogenesis, treatment, and outcome. PMBCL and TFL are typically treated along a DLBCL treatment paradigm. In accordance with the updated World Health Organization (WHO) classification (revised 4th edition 25), TFL is not recognized as an entity but as DLBCL, which is nevertheless a heterogenous group; therefore, it was considered redundant to specifically mention TFL in the indication. The indication has further been revised reflecting this revision of the WHO classification of B‐cell lymphoma subtypes.
CRS and cytopenias are transient adverse drug reactions (ADRs) that appeared to be amenable to treatment. Two of the reported deaths were likely a consequence of CRS, which occurs predominantly in the first 2 weeks after infusion and eventually subsides. Empirical treatment recommendations, taking the severity grades of CRS into account, have been developed. The availability of tocilizumab at all hospitals and associated centers must be ensured to mitigate the CRS‐related safety risks. Axicabtagene ciloleucel will, furthermore, only be supplied to hospitals and associated centers that are qualified and where the health care professionals involved in the treatment of a patient have completed the educational program.
A regulatory hurdle in recommending tocilizumab as part of the management of CRS in the product information for Yescarta was that at the time there was no formal approval of tocilizumab in this indication in the European Union. In order to ensure availability of tocilizumab and therefore safe use of CAR T‐cell therapies and to avoid off‐label use, the CHMP coordinated efforts so that an application was filed by the Marketing Authorization Holder for RoActemra (tocilizumab) and reviewed in an expedited way for a final outcome to be in place by the time Yescarta received a positive opinion by the CHMP.
Neurological adverse reactions also appear to be transient, with only one case judged as not resolved (mild memory impairment) at the cutoff point, but resolved later.
Furthermore, despite targeting of CD19 and the expected induction of B‐cell aplasia, the frequency of late‐onset grade 3 or worse serious infections was low, and 77% of assessable patients with ongoing responses showed evidence of B‐cell recovery by 24 months; initiation of B‐cell recovery was noted in some patients at 9 months. Additional exploration of the importance of tumor‐reactive CAR T‐cell persistence and its association with B‐cell recovery is warranted.
Conclusion
Based on the review of data on quality, safety, and efficacy, it was considered by consensus that the benefit‐risk balance of axicabtagene ciloleucel for the treatment of adult patients with relapsed or refractory DLBCL and PMBCL, after two or more lines of systemic therapy, is favorable.
The safety database is considered rather limited in terms of size and duration. The follow‐up of patients was short, and the median follow‐up time in lymphoma data set was 5.9 months, maximum 17.9 months. Longer‐term data (median follow‐up of 27.1 months) have been submitted as a part of a variation procedure. Safety data from cohort 3—still recruiting at the time of the assessment—were agreed with the Marketing Authorization Holder to be forthcoming (postauthorization).
A postauthorization safety study using a registry is planned with the purpose of additional characterization of identified risks, further evaluation of potential risks, and missing information with special focus on long‐term safety to assess whether administration is associated with subsequent neoplasm.
A number of opportunities and challenges arise from using existing registries to support CAR T‐cell therapy benefit‐risk evaluations and postauthorization follow‐up, especially given the requirement for long‐term follow‐up. In order to explore these possibilities, the European Medicines Agency hosted a stakeholder workshop in February 2018 26. Discussions focused around registry governance, patient consent, data sharing, data quality, registry interoperability, and core common data elements needed by stakeholders; as an outcome, agreement was reached on implementable recommendations to advance CAR T‐cell therapy evaluation and monitoring.
Additional risk minimization measures were agreed upon by CAT and CHMP, including the availability of tocilizumab and site qualification to minimize the risks of CRS associated with the treatment. Hospitals and their associated centers that dispense axicabtagene ciloleucel should be specially qualified in accordance with the agreed control distribution program and ensure on‐site, immediate access to four doses of tocilizumab for each patient as CRS management medication prior to treating patients.
Author Contributions
Conception/design: Irene Papadouli, Frank Petavy, Kyriaki Tzogani, Francesco Pignatti
Provision of study material or patients: Jan Mueller‐Berghaus, Claire Beuneu, Benjamin Hofner, Anne Miermont, Koenraad Norga, Olga Kholmanskikh, Tim Leest
Collection and/or assembly of data: Irene Papadouli, Jan Mueller‐Berghaus, Claire Beuneu, Benjamin Hofner, Frank Petavy, Anne Miermont, Koenraad Norga, Olga Kholmanskikh, Tim Leest, Francesco Pignatti
Data analysis and interpretation: Irene Papadouli, Jan Mueller‐Berghaus, Claire Beuneu, Sahra Ali, Benjamin Hofner, Frank Petavy, Kyriaki Tzogani, Anne Miermont, Koenraad Norga, Olga Kholmanskikh, Tim Leest, Martina Schuessler‐Lenz, Tomas Salmonson, Christian Gisselbrecht, Jordi Llinares Garcia, Francesco Pignatti
Manuscript writing: Irene Papadouli
Final approval of manuscript: Martina Schuessler‐Lenz, Tomas Salmonson, Francesco Pignatti
Disclosures
The authors indicated no financial relationships.
Acknowledgments
The scientific assessment summarized in this report is based on important contributions from the rapporteur and co‐rapporteur assessment teams, Committee for Medicinal Products for Human Use (CHMP), Committee for Advanced Therapies (CAT) members, and additional experts after the application for a marketing authorization from the company. This publication is a summary of the European Public Assessment Report (EPAR; EMA/481168/2018; https://www.ema.europa.eu/en/medicines/human/EPAR/yescarta) and CHMP and CAT assessment. The EPAR is published on the European Medicines Agency (EMA) Web site (www.ema.europa.eu). For the most current information on this marketing authorization, please refer to the EMA Web site. The authors of this article remain solely responsible for the opinions expressed in this publication.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1.Gatta G, Capocaccia R, Botta L, Mallone S, De Angelis R, Ardanaz E. Burden and centralised treatment in Europe of rare tumours: Results of RARECAREnet–a population‐based study. Lancet Oncol 2017;18:1022–1039. [DOI] [PubMed] [Google Scholar]
- 2. Flowers CR, Sinha R, Vose JM. Improving outcomes for patients with diffuse large B‐cell lymphoma. CA Cancer J Clin 2010;60:393–408. [DOI] [PubMed] [Google Scholar]
- 3. Pfreundschuh M, Kuhnt E, Trümper L et al. CHOP‐like chemotherapy with or without rituximab in young patients with good‐prognosis diffuse large‐B‐cell lymphoma: 6‐year results of an open‐label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–1022. [DOI] [PubMed] [Google Scholar]
- 4. Coiffier B, Sarkozy C. Diffuse large B‐cell lymphoma: R‐CHOP failure—what to do? Hematology Am Soc Hematol Educ Program 2016;2016:366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.NCCN Guidelines Insights: B‐Cell Lymphomas, Version 3.2019. J Natl Compr Canc Netw. 2019;17(6):650–661. 10.6004/jnccn.2019.0029 [DOI] [PubMed] [Google Scholar]
- 6. Gisselbrecht C, Glass B, Mounier N et al. Salvage regimens with autologous transplantation for relapsed large B‐cell lymphoma in the rituximab era. J Clin Oncol 2010;28:4184–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tilly H, Gomes da Silva M, Vitolo U et al. Diffuse large B‐cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2015;26(suppl 5):116–125. [DOI] [PubMed] [Google Scholar]
- 8. Crump M, Kuruvilla J, Couban, S et al. Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem‐cell transplantation for relapsed and refractory agressive lymphomas: NCIC‐CTG LY.12. J Clin Oncol 2014;32:3490–3496. [DOI] [PubMed] [Google Scholar]
- 9. Hitz F, Connors JM, Gascoyne RD et al. Outcome of patients with primary refractory diffuse large B cell lymphoma after R‐CHOP treatment. Ann Hematol 2015;94:1839–1843. [DOI] [PubMed] [Google Scholar]
- 10. Kuruvilla J, Pintilie M, Tsang R et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B‐cell lymphoma compared with diffuse large B‐cell lymphoma. Leuk Lymphoma 2008;49:1329–1336. [DOI] [PubMed] [Google Scholar]
- 11. Carter RH, Barrington RA. Signaling by the CD19/CD21 complex on B cells. Curr Dir Autoimmun 2004;7:4–32. [DOI] [PubMed] [Google Scholar]
- 12. Thierry‐Mieg D, Thierry‐Mieg J. AceView: A comprehensive cDNA‐supported gene and transcripts annotation. Genome Biol 2006;7(suppl 1):11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou LJ, Ord DC, Omori SA et al. Structure of the genes encoding the CD19 antigen of human and mouse B lymphocytes. Immunogenetics 1992;35:102–111. [DOI] [PubMed] [Google Scholar]
- 14. Tedder TF. CD19: A promising B cell target for rheumatoid arthritis. Nat Rev Rheumatol 2009;5:572–577. [DOI] [PubMed] [Google Scholar]
- 15. Haas KM, Tedder TF. Role of the CD19 and CD21/35 receptor complex in innate immunity, host defense and autoimmunity. Adv Exp Med Biol 2005;560:125–139. [DOI] [PubMed] [Google Scholar]
- 16. Bradbury LE, Kansas GS, Levy S et al. The CD19/CD21 signal transducing complex of human B lymphocytes includes the target of antiproliferative antibody‐1 and Leu‐13 molecules. J Immunol 1992;149:2841–2850. [PubMed] [Google Scholar]
- 17. Neelapu SS, Locke FL, Bartlett NL et al. Axicabtagene ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J Med 2017;377:2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kochenderfer JN, Yu Z, Frasheri D et al. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 2010;116:3875–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kochenderfer JN, Dudley ME, Kassim SH et al. Chemotherapy‐refractory diffuse large B‐cell lymphoma and indolent B‐cell malignancies can be effectively treated with autologous T cells expressing an anti‐CD19 chimeric antigen receptor J Clin Oncol 2015;33:540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Locke FL, Neelapu SS, Bartlett NL et al. Phase 1 results of ZUMA‐1: A multicenter study of KTE‐C19 Anti‐CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther 2017;25:285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neelapu SS, Locke FL, Bartlett NL et al. Axicabtagene ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J Med 2017;377:2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crump M, Sattva S, Neelapu U et al. Outcomes in refractory diffuse large B‐cell lymphoma: Results from the international SCHOLAR‐1 study. Blood 2017;130:1800–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Le RQ, Li L, Yuan W et al. FDA approval summary: Tocilizumab for treatment of chimeric antigen receptor T cell‐induced severe or life‐threatening cytokine release syndrome. The Oncologist 2018;23:943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee DW, Gardner R, Porter DL et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014;124:188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Swerdlow SH, Campo E, Pileri SA et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127:2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Report on CAR T‐Cell Therapy Registries Workshop 9 February 2018 . London, UK: European Medicines Agency, May 15, 2018. Available at https://www.ema.europa.eu/en/documents/report/report‐car‐t‐cell‐therapy‐registries‐workshop_en.pdf. Accessed August 10, 2019.
- 27. Locke FL, Ghobadi A, Jacobson CA et al. Long‐term safety and activity of axicabtagene ciloleucel in refractory large B‐cell lymphoma (ZUMA‐1): A single‐arm, multicentre, phase 1‐2 trial. Lancet Oncol 2019;20:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]