

Abstract
Botanical and other natural products (NPs) are often coconsumed with prescription medications, presenting a risk for cytochrome P450 (P450)-mediated NP-drug interactions. The NP goldenseal (Hydrastis canadensis) has exhibited antimicrobial activities in vitro attributed to isoquinoline alkaloids contained in the plant, primarily berberine, (−)-β-hydrastine, and to a lesser extent, hydrastinine. These alkaloids contain methylenedioxyphenyl rings, structural alerts with potential to inactivate P450s through formation of metabolic intermediate complexes. Time-dependent inhibition experiments were conducted to evaluate their ability to inhibit major P450 activities in human liver microsomes by using a cocktail of isozyme-specific substrate probes. Berberine inhibited CYP2D6 (dextromethorphan O-demethylation; KI = 2.7 μM, kinact = 0.065 minute−1) and CYP3A4/5 (midazolam 1′-hydroxylation; KI = 14.8 μM, kinact = 0.019 minute−1); (−)-β-hydrastine inhibited CYP2C9 (diclofenac 4′-hydroxylation; KI = 49 μM, kinact = 0.036 minute−1), CYP2D6 (KI > 250 μM, kinact > 0.06 minute−1), and CYP3A4/5 (KI = 28 μM, kinact = 0.056 minute−1); and hydrastinine inhibited CYP2D6 (KI = 37 μM, kinact = 0.049 minute−1) activity. Berberine additionally exhibited allosteric effects on midazolam hydroxylation, showing both positive and negative heterotropic cooperativity. Experiments with recombinant isozymes showed that berberine activated midazolam 1′-hydroxylation by CYP3A5, lowering Km(app), but showed mixed inhibition and negative cooperativity toward this reaction when catalyzed by CYP3A4. Berberine inactivated CYP3A4 at a much faster rate than CYP3A5 and was a noncompetitive inhibitor of midazolam 4-hydroxylation by CYP3A4 but a strong mixed inhibitor of the CYP3A5 catalyzed reaction. These complex kinetics should be considered when extrapolating the risk for NP-drug interactions involving goldenseal.
SIGNIFICANCE STATEMENT
Robust kinetic parameters were determined for the reversible and time-dependent inhibition of CYP2C9, CYP2D6, and CYP3A4/5 activities in human liver microsomes by major component isoquinoline alkaloids contained in the botanical natural product goldenseal. The alkaloid berberine also exhibited opposing, isozyme-specific allosteric effects on midazolam hydroxylation mediated by recombinant CYP3A4 (inhibition) and CYP3A5 (activation). These data will inform the development of a physiologically based pharmacokinetic model that can be used to predict potential clinically relevant goldenseal-drug interactions.
Introduction
Botanical natural products (NPs) include herbal and other dietary supplements (Paine and Roe, 2018). Many NPs occupy a growing niche in the contemporary health care market, presenting unique challenges in the context of concomitant drug therapy. Such challenges are partly because of a lack of product regulation and standardization as well as the fact that plant extracts often contain a complex mixture of constituents that can vary substantially in both content and concentration depending on the preparation (Paine et al., 2018; Kellogg et al., 2019). Although up to 25% of adults report taking a dietary supplement with a prescription medication (Asher et al., 2017), there is a paucity of literature data examining potential NP-drug interactions for even widely used NPs.
Goldenseal [Hydrastis canadensis L. (Ranunculaceae)], a plant indigenous to southeastern Canada and the northeastern United States, is used to self-treat a wide spectrum of maladies, including the common cold, upper respiratory tract infections, and gastrointestinal and menstrual disorders. Goldenseal produces a variety of isoquinoline alkaloids (Fig. 1A) that convey antimicrobial activity in vitro. The highest content alkaloids in goldenseal are berberine and (−)-β-hydrastine. The plant also contains a number of minor alkaloids, including hydrastinine, whose concentration in commercial extracts is highly dependent on the method of preparation (Weber et al., 2003; Ettefagh et al., 2011; Hermann and von Richter, 2012). Goldenseal appears unique among hydrastine-containing plants in that it produces only the (−)-enantiomer, whereas other plants produce primarily (+)-β-hydrastine (Gupta et al., 2015).
Fig. 1.

Structures of (A) major isoquinoline alkaloids isolated from goldenseal [Hydrastis canadensis L. (Ranunculaceae)] extract and (B) the steroid progesterone.
Several clinical studies have highlighted the potential for goldenseal extracts (GSEs) to precipitate NP-drug interactions by inhibiting cytochrome P450 (P450) enzymes. Pretreatment of human volunteers with a GSE (∼1 g by mouth 3× daily for 14 or 28 days) significantly inhibited both CYP2D6 and CYP3A4/5 activities (40%–50% decrease in metabolite/parent ratios) and had negligible effects on CYP2E1 and CYP1A2 activities (Gurley et al., 2005, 2008a,b). Berberine (200 mg by mouth 3× daily for 12 days) also inhibited CYP3A4/5-mediated cyclosporin A clearance (34.5% increase in area under the plasma concentration versus time curve) in renal transplant recipients (Wu et al., 2005). Finally, berberine pretreatment (300 mg by mouth 3× daily for 14 days) followed by administration of an oral cocktail of P450 substrate probes resulted in a 10-fold, 2-fold, and 1.6-fold increase in the parent/metabolite ratio for dextromethorphan (CYP2D6), losartan (CYP2C9), and midazolam (MDZ; CYP3A4/5), respectively (Guo et al., 2012).
In vitro experiments have evaluated the ability of GSE and/or one of the major alkaloids to reversibly inhibit human liver P450s. One GSE exhibited the lowest IC50 (0.03% of full extract) for inhibition of CYP3A4 activity among 21 herbal extracts tested (Budzinski et al., 2000), and other studies reported strong inhibition (>50%) by a GSE of CYP3A4 and CYP2D6 activities. There are conflicting reports as to whether goldenseal inhibits CYP1A2, 2C9, 2C19, or 2E1 activities in vitro. Berberine generally has been shown to be a weak to moderate inhibitor of the CYP1As as well as CYP2C8, 2D6, 2E1, and 3A4, and (−)-β-hydrastine has shown to be a weak to moderate inhibitor of 2C8, 2C9, 2D6, 2E1, and 3A4 (Chatterjee and Franklin, 2003; Foster et al., 2003; Etheridge et al., 2007; Raner et al., 2007; Lo et al., 2013).
Two studies have evaluated the potential for goldenseal alkaloids to inactivate P450s. Berberine exhibited time-dependent inhibition (TDI) of CYP2D6 and 3A4 (Zhao et al., 2015), whereas (−)-β-hydrastine showed TDI of CYP3A4 (Chatterjee and Franklin, 2003). Although both studies presented evidence for inactivation of specific P450s by berberine and/or (−)-β-hydrastine, the reported inactivation kinetic parameters were approximations.
The objective of the present study was to fully define the mode and kinetics of inhibition of the major liver P450s CYP2C9, 2D6, and 3A4/5 by the two major GSE component alkaloids, berberine and (−)-β-hydrastine, and the minor alkaloid, hydrastinine, which appears to be the third most abundant alkaloid present in some commercial extracts (Chatterjee and Franklin, 2003; Weber et al., 2003). Through comprehensive kinetic studies, robust parameters were recovered for both the reversible inhibition and TDI of CYP2C9, 2D6, and 3A4/5 by these three alkaloids. A comprehensive analysis of the unique kinetic allosteric interactions of berberine with CYP3A4 and CYP3A5 provided further insight into these complex NP-drug interactions. The kinetic parameters generated can be used to develop physiologically based pharmacokinetic (PBPK) models to predict the risk of goldenseal-drug interactions involving susceptible object drugs, aid in the design of clinical interaction studies, and help inform clinicians and consumers about managing these potentially adverse NP-drug interactions.
Materials and Methods
General Reagents
MDZ and 1′-OH-MDZ were obtained as 1 mg/ml and 100 μg/ml methanolic solutions, respectively, from Cerilliant (Round Rock, TX). Diclofenac sodium salt, berberine chloride, 1′-OH-MDZ-d4, dextrorphan tartrate, dextrorphan tartrate-d3, and 4′-hydroxydiclofenac-d4 were purchased from Toronto Research Chemicals, Inc. (North York, ON); dextromethorphan was procured from LKT Laboratories, Inc. (St. Paul, MN). Hydrastinine, (−)-β-hydrastine, 4-OH-MDZ, and all other chemicals were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO). All inhibitors and substrates tested in these studies were deemed to be ≥95% pure. Solvents were purchased from J.T. Baker, Inc. (Phillipsburg, NJ) or Fischer Scientific (Springfield, NJ). Recombinant CYP3A4 and CYP3A5 Supersomes, coexpressed in insect cells with cytochrome P450 oxidoreductase and cytochrome b5, were purchased from Corning Life Sciences, Inc. (Corning, NY). Pooled human liver microsomes (HLMs) of mixed sex were either prepared in-house from eight individual livers as reported previously (Sadeque et al., 1992; McDonald et al., 2015) or were purchased from Corning Life Sciences (Ultrapool HLM150) specifically for use in the berberine-MDZ allosterism experiments.
IC50 Shift Experiments for the Inhibition of P450 Activity by Berberine, (−)-β-Hydrastine, or Hydrastinine
Substrate Cocktail Assay.
IC50 shift experiments were carried out using a validated substrate cocktail method (McDonald et al., 2015). Wells 1–24 and 25–48 of a 96-well plate contained identical solutions of inhibitor (added from 12 different 200× concentrated stock solutions, yielding final incubation inhibitor concentrations ranging from 0 to 500 μM) and pooled HLMs (final concentration, 0.25 mg/ml microsomal protein) in 100 mM potassium phosphate (KPi buffer), pH 7.4. After a 3-minute equilibration at 37°C/50 rpm in a water bath, 2.5 μl NADPH stock (wells 1–24; final concentration, 1 mM) or buffer without NADPH (wells 25–48) were added; final incubation volumes were 250 μl. The plate was incubated at 37°C for 30 minutes and then 196 μl were removed from each well and were added to a second plate containing 2 μl of a 100× concentrated 3-substrate cocktail stock (prepared with 50% aqueous methanol; final concentrations: 4 μM diclofenac, 4 μM dextromethorphan, and 2 μM MDZ) per well, plus 2 μl of either buffer alone (wells 1–24) or buffer with NADPH (1 mM final concentration, wells 25–48). This plate was incubated for 5 minutes at 37°C prior to quenching with 20 μl ice-cold 15% aqueous ZnSO4. Ten picomoles each of 1′-OH-MDZ-d4 and dextrorphan-d3 and 20 pmol of 4′-hydroxydiclofenac-d4 were added as internal standards; then, precipitates were removed by centrifugation, and the supernatants were analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS) (described below).
MDZ Assay.
These experiments used the same methodology outlined above for the cocktail assay with the following exceptions: 1) either CYP3A4 or CYP3A5 Supersomes (3 pmol per incubation reaction) were substituted for HLMs as the enzyme source; 2) rather than using a substrate cocktail solution, a 100× concentrated stock solution containing MDZ (final substrate concentration of either 2 μM for the (−)-β-hydrastine experiments or 20 μM for the berberine experiments) was added to the second 96-well plate; and 3) 1′-OH-MDZ-d4 (10 pmol) was added to each reaction product as internal standard.
LC-MS/MS Protocol.
LC-MS/MS analysis was conducted with a Waters Xevo TQ-S Tandem Quadrupole Mass Spectrometer (Waters Co., Milford, MA) coupled to an ACQUITY Ultra Performance LC (UPLC) System with an integral autoinjector (Waters). The Xevo was operated in positive electrospray ionization mode at a source temperature of 150°C and a desolvation temperature of 350°C. The following mass transitions were monitored in separate ion channels for the substrate cocktail assay: m/z (mass to charge ratio) 258 → 157 (dextrorphan-d0), m/z 261 → 157 (dextrorphan-d3), m/z 312 → 230 (4′-hydroxydiclofenac-d0), m/z 316 → 234 (4′-hydroxydiclofenac-d4), m/z 342 → 324 (1′-OH-MDZ-d0), and m/z 346 → 328 (1′-OH-MDZ-d4) at cone voltages of 25, 23, and 30 V and collision energies of 35, 33, and 35 eV for dextrorphan, 4′-hydroxydiclofenac, and 1′-OH-MDZ, respectively.
Metabolites from the substrate cocktail incubations were separated on a Shimadzu Shim-Pack XR-ODS, 2.2 μ, 2.0 × 75 mm LC column (Shimadzu Scientific, Colombia, MD) using a binary solvent gradient (solvent A = 0.1% aqueous formic acid, and solvent B = methanol) with a constant flow rate of 0.35 ml/min and a column temperature of 50°C. From 0 to 1 minute, solvent was set at 20% B and increased linearly to 95% B from 1 to 5 minutes, which was maintained for 0.5 minutes and then re-equilibrated to 5% B over 0.2 minutes. Metabolites were quantified by comparison of their peak area ratios (relative to the 4′-hydroxydiclofenac-d4, 1′-OH-MDZ-d4, or dextrorphan-d3 peak areas) to calibration curves prepared with commercially obtained metabolite standards using linear regression analysis. Limits of quantitation for 4′-hydroxydiclofenac, 1′-OH-MDZ, and dextrorphan were, respectively, 3, 10, and 3 fmol of metabolite injected on the column.
Cytochrome P450 Kinetic Inactivation Experiments
Substrate Cocktail Assay.
Incubations were carried out in 1.2-ml sample tubes for each of six inhibitor concentrations [added from 200× concentrated methanolic stock solutions to yield final concentrations ranging from 0 to 50 μM for berberine or 0 to 500 μM for (−)-β-hydrastine and hydrastinine]. Each incubation contained 0.25 mg/ml pooled HLMs in 100 mM KPi buffer, pH 7.4. The tubes were equilibrated at 37°C/50 rpm for 3 minutes in a water bath prior to initiating the reactions with NADPH (1 mM in 1.1 ml final volume). At 0, 5, 10, 15, and 20 minutes, 198-μl aliquots were removed and added to 2 μl of a 100× concentrated substrate cocktail stock solution (in 50% aqueous methanol) to yield final concentrations of 40 μM diclofenac, 40 μM dextromethorphan, and 20 μM MDZ. The substrate reactions were incubated for 5 minutes at 37°C/50 rpm and then quenched with 20 μl 15% aqueous ZnSO4, after which 10 pmol each of 1′-OH-MDZ-d4 and dextrorphan-d3 and 20 pmol of 4′-hydroxydiclofenac-d4 were added as internal standards. Protein precipitate was removed by centrifugation, and metabolites within the supernatants were quantified by LC-MS/MS analysis. The LC-MS/MS protocol used for these experiments was identical to that used in the IC50 shift experiments (described above).
Results were plotted as percent enzyme activity remaining versus preincubation time, and slopes were determined for each inhibitor concentration by linear regression analysis. KI and kinact values were determined from plots of inverse slope (λ) versus inhibitor concentration.
Allosterism Experiments with Berberine and CYP3A4/5 Enzymes
The allosteric effect of berberine on MDZ metabolism (mediated by either HLMs or CYP3A4 or CYP3A5 Supersomes) was monitored at eight different substrate concentrations (0–250 μM MDZ) and five different inhibitor concentrations (0–100 μM berberine). Solutions of berberine and MDZ in 100 mM KPi buffer, pH 7.4, containing either 0.25 mg/ml HLMs or 2.5 pmol of CYP3A Supersomes, were pre-equilibrated in a 96-well plate at 37°C/50 rpm in a water bath for 3 minutes prior to initiating the reactions with the addition of a stock solution of NADPH (prewarmed to 37°C, 1 mM final concentration). After a 3-minute incubation, reactions were quenched with the addition of 20 μl 15% aqueous ZnSO4 solution, and 10 pmol of 1′-OH-MDZ-d4 were added. Protein precipitate was removed by centrifugation, and the supernatants were analyzed by LC-MS/MS.
LC-MS/MS Protocol.
LC-MS/MS analyses were conducted on the same Xevo TQ-S Mass Spectrometer (Waters) and ACQUITY UPLC system (Waters) described above for the substrate cocktail assay. The Xevo instrument was operated in positive electrospray ionization mode at the same source and desolvation temperatures, and the following mass transitions were monitored in separate ion channels: m/z 342 → 324 (1′-OH-MDZ-d0), m/z 346 → 328 (1′-OH-MDZ-d4), and m/z 342 → 234 (4-OH-MDZ). The cone voltage was set to 30 V for all analytes, with collision energies of 35 eV for 1′-OH-MDZ (labeled and unlabeled) and 20 eV for 4-OH-MDZ.
MDZ hydroxylation products from berberine coincubations were separated on an ACQUITY BEH Phenyl, 1.7 μ, 2.1 × 150 mm UPLC column (Waters) using a binary solvent gradient (solvent A = 0.1% aqueous formic acid, and solvent B = acetonitrile) at a constant flow rate of 0.35 ml/min and a column temperature of 50°C. An isocratic gradient of 25% B was maintained for 5.5 minutes and then increased linearly to 95% B over 0.5 minutes, at which time the gradient was maintained for 1 minute prior to column re-equilibration. Under these conditions, 4-OH-MDZ, 1′-OH-MDZ-d4, 1′-OH-MDZ-d0, MDZ, and berberine had retention times of approximately 3.7, 4.6, 4.7, 5.4, and 6.3 minutes, respectively. Metabolites were quantified through comparison of their peak area ratios (relative to the 1′-OH-MDZ-d4 peak area) to calibration curves, prepared from synthetic metabolite standards, using linear regression analysis. Limits of quantitation for both metabolites were 20 fmol injected on the column.
Data Analysis
Mass spectral data analyses were conducted using Windows-based Micromass MassLynxNT software, version 4.1. GraphPad Prism v7 (GraphPad Software, San Diego, CA) was used to estimate all kinetic parameters using reaction velocities calculated as the mean of duplicate technical replicates. Unless otherwise stated, at least three biologic replicates were conducted for all experiments. Goodness-of-fit of the various metabolic/inhibition equations (Michealis-Menten, IC50 determination, reversible inhibition, TDI, and substrate inhibition) were evaluated based on visual inspection, the extra sum-of-squares F test, Akaike information criterion, and randomness of the residuals. Tabulated data are presented as means ± S.D.
Preliminary In Vitro–In Vivo Prediction of Goldenseal-Drug Interactions
A preliminary risk assessment of potential clinical goldenseal-drug interactions due to inhibition of hepatic CYP2C9, CYP2D6, or CYP3A4 activity was made using basic static models (eqs. 1 and 2) (U.S. Food and Drug Administration, 2020). Specifically, the ratios of intrinsic clearance values (R) for a probe drug in the absence to presence of an interacting reversible or time-dependent inhibitor were estimated. Equation 1 predicts the change in clearance because of reversible inhibition:
| (1) |
where Imax.u (maximum unbound plasma concentration × fraction unbound in plasma) is the maximum unbound plasma concentration of the inhibitor (goldenseal alkaloid), and Ki,u [Ki × fraction unbound in human liver microsomes (fu,HLM)] is the experimentally determined reversible inhibition constant (conservatively estimated as one-half of the IC50 value determined in the absence of NADPH in the preincubation step, Table 1) corrected for nonspecific binding to HLMs (fu,HLM). The fraction unbound in plasma and fu,HLM (at 0.25 mg/ml microsomal protein) were predicted using GastroPlus (v9.7; Simulations Plus, Lancaster, CA); values for berberine were 0.75 and 1.0, respectively, and those for hydrastine were 0.10 and 0.79, respectively. Imax values were obtained from the literature (1.75 and 470 nM for berberine and hydrastine, respectively; Gupta et al., 2009). Equation 2 predicts the change in probe drug clearance because of TDI:
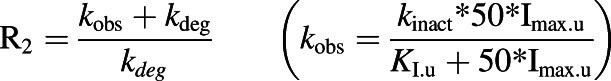 |
(2) |
where KI,u (KI × fu,HLM) and kinact are the TDI-binding (corrected for nonspecific binding to HLMs) and inactivation rate constants (Table 2), and kdeg is the degradation rate constant for a given P450. For hepatic CYP2C9, CYP2D6, and CYP3A4, kdeg has been estimated as 0.00026, 0.000226, and 0.000321 minute−1, respectively (Obach et al., 2007).
TABLE 1.
Results of IC50 shift experiments showing TDI of CYP2C9, CYP2D6, and CYP3A4/5 activities in HLMs by goldenseal component alkaloids
Values denote means ± S.D. of three separate experiments, with the exception of the tienilic acid, paroxetine, and troleandomycin inhibitors, which were each run as a singlet. These three compounds were chosen as positive controls for the IC50 shift experiments because they are known to exhibit strong isozyme-specific TDI of CYP2C9, CYP2D6, and CYP3A4/5 activities, respectively.
| Inhibitor | Probe Substrate | Inhibited Enzyme | IC50 (μM)a | Shift Ratiob | |
|---|---|---|---|---|---|
| (−) NADPH | (+) NADPH | ||||
| Berberine | Dextromethorphan | CYP2D6 | 9.9 ± 1.3 | 1.6 ± 0.3 | 6.0 |
| Berberine | MDZ | CYP3A4/5 | Activation | 180 ± 25 | — |
| (−)-β-Hydrastine | Diclofenac | CYP2C9 | 120 ± 30 | 31 ± 3.2 | 3.9 |
| (−)-β-Hydrastine | Dextromethorphan | CYP2D6 | 270 ± 21 | 80 ± 18 | 3.4 |
| (−)-β-Hydrastine | MDZ | CYP3A4/5 | 58 ± 7.6 | 9.9 ± 1.5 | 5.9 |
| Hydrastinine | Dextromethorphan | CYP2D6 | 65 ± 21 | 8.4 ± 1.1 | 7.7 |
| Tienilic Acid | Diclofenac | CYP2C9 | 2.8 | 0.2 | 13 |
| Paroxetine | Dextromethorphan | CYP2D6 | 0.9 | 0.1 | 9 |
| Troleandomycin | MDZ | CYP3A4/5 | 61 | 1.5 | 40 |
Prior to the addition of substrate, HLMs were preincubated with inhibitor for 30 minutes in the presence (+) or absence (−) of NADPH cofactor.
Shift ratio = IC50 (−) NADPH/IC50 (+) NADPH.
TABLE 2.
Kinetic parameters for the inactivation of CYP2C9, CYP2D6, and CYP3A4/5 in HLMs by goldenseal component alkaloids
Values denote means ± S.D. of three separate experiments.
| Inhibitor | Inhibited Enzyme | KI (μM) | kinact (min−1) | kinact/KIa (mM−1 min−1) |
|---|---|---|---|---|
| Berberine | CYP2D6 | 2.68 ± 0.26 | 0.065 ± 0.006 | 24.3 |
| Berberine | CYP3A4/5 | 14.8 ± 2.6 | 0.019 ± 0.005 | 1.3 |
| (−)-β-Hydrastine | CYP2C9 | 49 ± 16 | 0.036 ± 0.007 | 0.7 |
| (−)-β-Hydrastine | CYP2D6 | >250 | >0.06 | <0.2 |
| (−)-β-Hydrastine | CYP3A4/5 | 28 ± 12 | 0.056 ± 0.005 | 2.0 |
| Hydrastinine | CYP2D6 | 37 ± 13 | 0.049 ± 0.009 | 3.8 |
Reported kinact/KI values (mM−1 min−1) for the TDI of specific P450 activities, in either HLMs (paroxetine, troleandomycin) or human hepatocytes (tienilic acid), for clinically relevant time-dependent inhibitors: tienilic acid (CYP2C9) = 25; paroxetine (CYP2D6) = 35; and troleandomycin (CYP3A4/5) = 13.3 (Bertelsen et al., 2003; Zhao et al., 2005; McGinnity et al., 2006).
Results
IC50 Shift Experiments: Inhibition of CYP2C9, CYP2D6, and CYP3A4/5 Activities in HLMs by Goldenseal Isoquinoline Alkaloids
Substrate Cocktail Assay.
Berberine, (−)-β-hydrastine, and hydrastinine were tested by IC50 shift assay as time-dependent inhibitors of CYP2C9, CYP2D6, and CYP3A4/5 activities in HLMs using a cocktail of specific substrate probes. Additionally, tienilic acid (CYP2C9), paroxetine (CYP2D6), and troleandomycin (CYP3A4) were tested as positive controls for clinically relevant isozyme-specific TDI (Bertelsen et al., 2003; Zhao et al., 2005; McGinnity et al., 2006).
Each isoquinoline alkaloid exhibited an IC50 shift ratio >1.5 (the predefined cutoff for TDI) for at least one of the P450 activities tested. Berberine had no effect on CYP2C9-mediated diclofenac-4′-hydroxylation (data not shown) but was a relatively strong time-dependent inhibitor of CYP2D6-mediated dextromethorphan O-demethylation (Fig. 2A) and showed both reversible activation and TDI of CYP3A4/5-mediated MDZ-1′-hydroxylation (Fig. 2B). (−)-β-Hydrastine inhibited CYP2C9 (Fig. 2C), CYP2D6 (Fig. 2D), and CYP3A4/5 (Fig. 2E) activities in a time-dependent manner. Hydrastinine had no effect on CYP2C9 or CYP3A4/5 activities (data not shown) but was a moderate time-dependent inhibitor of CYP2D6 (Fig. 2F; Table 1).
Fig. 2.

Results from IC50 shift experiments showing TDI of (A) CYP2D6-mediated dextromethorphan O-demethylation by berberine, (B) CYP3A4/5-mediated MDZ 1′-hydroxylation by berberine, (C) CYP2C9-mediated diclofenac 4′-hydroxylation by (−)-β-hydrastine, (D) CYP2D6-mediated dextromethorphan O-demethylation by (−)-β-hydrastine, (E) CYP3A4/5-mediated MDZ 1′-hydroxylation by (−)-β-hydrastine, and (F) CYP2D6-mediated dextromethorphan O-demethylation by hydrastinine in HLMs. Mean IC50 values for each set of experiments (repeated in triplicate) are shown in Table 1.
Cytochrome P450 Kinetic Inactivation by Goldenseal Isoquinoline Alkaloids in HLMs (Determination of KI and kinact)
TDI experiments were conducted to determine the kinetics of inactivation of CYP2C9, CYP2D6, and CYP3A4/5 activities in HLMs by each alkaloid. The same isozyme-specific substrate probes used in the IC50 shift substrate cocktail experiments were used at 10× their reported Km values (McDonald et al., 2015). (−)-β-Hydrastine was a weak to moderate inactivator of the three enzyme activities (Fig. 3; Table 2). Berberine had no effect on CYP2C9 activity (data not shown), was a relatively strong inactivator of CYP2D6, and demonstrated both weak reversible activation and modest TDI of CYP3A4/5 activity (Table 2). Hydrastinine showed moderate TDI of CYP2D6 (Table 2) and had no effect on either CYP2C9 or CYP3A4/5 activity (data not shown).
Fig. 3.

P450 inactivation kinetics showing inhibition of (A) CYP2C9-mediated diclofenac 4′-hydroxylation by (−)-β-hydrastine, (B) CYP2D6-mediated dextromethorphan O-demethylation by (−)-β-hydrastine, (C) CYP3A4/5-mediated MDZ 1′-hydroxylation by (−)-β-hydrastine, (D) CYP2D6-mediated dextromethorphan O-demethylation by berberine, (E) CYP3A4/5-mediated MDZ 1′-hydroxylation by berberine, and (F) CYP2D6-mediated dextromethorphan O-demethylation by hydrastinine in HLMs as a function of enzyme/NADPH preincubation time. KI and kinact values (see Table 2) were determined from plots [shown as insets in graphs (A–F)] of inverse slope (λ), for the individual lines in each graph, vs. inhibitor concentration. Berb, berberine.
Preliminary Hepatic P450-Mediated Goldenseal-Drug Interaction Predictions
The risk of each alkaloid to precipitate clinical pharmacokinetic interactions with drugs metabolized by hepatic CYP2C9, CYP2D6, and CYP3A4/5 was predicted using basic static equations. As reversible inhibitors, the R1 value for all alkaloids involving all P450 substrates was <1.02, suggesting low interaction risk. U.S. Food and Drug Administration (FDA) guidance recommends further investigation, either through a clinical study or using mechanistic models for time-dependent inhibitors with R2 ≥1.25 (U.S. Food and Drug Administration, 2020). The R2 values for berberine involving CYP2D6 substrates and for (−)-β-hydrastine involving all P450 substrates exceeded this cutoff; the R2 for berberine involving TDI of CYP3A4/5 substrates was near the minimum FDA cutoff value (Table 6).
TABLE 6.
Preliminary hepatic P450-mediated goldenseal-drug interaction predictions
R2, ratio of intrinsic clearance values for a probe drug in the absence to presence of an interacting time-dependent P450 inhibitor (predicted using a basic static model; U.S. Food and Drug Administration, 2020).
| Inhibitor | R2 Value | ||
|---|---|---|---|
| CYP2C9 | CYP2D6 | CYP3A4 | |
| Berberine | 1.00 | 7.90 | 1.26 |
| (−)-β-Hydrastine | 8.94 | ∼4 | 17.8 |
Allosteric Effects of Berberine on CYP3A4/5-Mediated MDZ Metabolism
Rates of MDZ hydroxylation at both the 1′ and 4 positions mediated by pooled HLMs (Corning Life Sciences) or recombinant CYP3A4 or CYP3A5 Supersomes were monitored across a range of substrate and effector concentrations. A short (3-minute) incubation time was used to minimize CYP3A4/5 enzyme inactivation by berberine. The commercial Ultrapool 150 HLM preparation contained 57 pmol CYP3A4/mg microsomal protein and 25 pmol/mg CYP3A5 according to Western Blot analysis (Corning).
In the absence of effector, MDZ 1′-hydroxylation showed substrate inhibition kinetics with both HLMs and CYP3A4 Supersomes. The addition of berberine lowered Ki(app) by ∼50% with both enzyme sources (i.e., from 900 to 420 μM with recombinant CYP3A4 and from 315 to 130 μM with HLMs), at which point this binding effect appeared to reach saturation. The addition of high concentrations of berberine (≥50 μM) appeared to inhibit MDZ 1′-hydroxylation mediated by recombinant CYP3A4 but not HLMs, causing both an increase in Km(app) and decrease in Vmax (Fig. 4; Tables 3 and 4). Instead, berberine activated 1′-OH-MDZ formation in HLMs by lowering the Km(app) for MDZ without obvious effect on Vmax. In the absence of effector, the kinetics with CYP3A5 Supersomes shifted from substrate inhibition (observed with recombinant CYP3A4) to a simple unienzyme Michaelis-Menten profile. When berberine was added to the CYP3A5 Supersome incubations, an activation effect was observed because of a lowering of Km apparent for MDZ (Table 5).
Fig. 4.

Plots showing allosteric effects of berberine on the kinetics of MDZ 1′-hydroxylation mediated by HLMs or CYP3A4 or CYP3A5 Supersomes. With both HLMs and CYP3A4 Supersomes, the kinetics were consistent with a substrate inhibition model, with an inhibitory allosteric effect exhibited with the addition of berberine (i.e., decreased Ki). There also appears to be some mixed inhibition of CYP3A4 at high berberine concentrations. In contrast, the kinetics of MDZ 1′-hydroxylation by CYP3A5 were described best by a simple, unienzyme Michaelis-Menten model. Berberine activated MDZ 1′-hydroxylation in both HLMs and CYP3A5 Supersomes by lowering Km(app) (Tables 2–4).
TABLE 3.
Allosteric effects of berberine on MDZ hydroxylation in HLMs
Values denote means ± S.D. of four separate experiments. Km and Ki are given in micromolar; Vmax is in picomoles per minute per milligram microsomal protein; Vmax/Km is in microliters per minute per milligram protein.
| Berberine (μM) | 1′-Hydroxy MDZ | 4-Hydroxy MDZ | |||||
|---|---|---|---|---|---|---|---|
| Km | Vmax | Vmax/Km | Ki | Km | Vmax | Vmax/Km | |
| 0 | 2.29 ± 0.29 | 2110 ± 380 | 930 ± 220 | 315 ± 92 | 21.1 ± 4.4 | 1260 ± 220 | 60.8 ± 9.4 |
| 5 | 1.63 ± 0.41 | 2080 ± 380 | 1330 ± 370 | 301 ± 86 | 25.3 ± 6.2 | 1150 ± 230 | 48.3 ± 16.5 |
| 25 | 1.58 ± 0.12 | 2280 ± 330 | 1450 ± 210 | 202 ± 28 | 26.6 ± 4.5 | 960 ± 180 | 37.1 ± 10.1 |
| 50 | 1.22 ± 0.18 | 2010 ± 220 | 1680 ± 300 | 156 ± 17 | 28.8 ± 6.5 | 680 ± 130 | 26.6 ± 7.9 |
| 100 | 1.06 ± 0.15 | 1880 ± 120 | 1810 ± 310 | 130 ± 17 | 23.2 ± 4.3 | 450 ± 60 | 20.0 ± 3.6 |
TABLE 4.
Allosteric effects of berberine on MDZ hydroxylation by recombinant CYP3A4
Values denote means ± S.D. of four separate experiments. Km and Ki are given in micromolar; Vmax is in picomoles per minute per picomole CYP3A4; Vmax/Km is in microliters per minute per picomole CYP3A4.
| Berberine (μM) | 1′-Hydroxy MDZ | 4-Hydroxy MDZ | |||||
|---|---|---|---|---|---|---|---|
| Km | Vmax | Vmax/Km | Ki | Km | Vmax | Vmax/Km | |
| 0 | 1.38 ± 0.22 | 28.0 ± 4.6 | 20.7 ± 4.1 | 900 ± 250 | 25.0 ± 6.9 | 26.4 ± 7.5 | 1.13 ± 0.40 |
| 5 | 1.28 ± 0.25 | 31.3 ± 3.7 | 24.7 ± 2.6 | 610 ± 190 | 27.4 ± 8.9 | 27.1 ± 5.5 | 1.05 ± 0.29 |
| 25 | 1.34 ± 0.20 | 24.7 ± 4.0 | 18.6 ± 2.9 | 560 ± 120 | 40.1 ± 17 | 19.5 ± 6.3 | 0.56 ± 0.25 |
| 50 | 1.63 ± 0.39 | 23.4 ± 2.6 | 15.2 ± 4.9 | 420 ± 160 | 30.0 ± 3.4 | 14.6 ± 5.4 | 0.50 ± 0.20 |
| 100 | 2.04 ± 0.55 | 21.4 ± 2.4 | 11.0 ± 2.7 | 470 ± 70 | 32.0 ± 6.2 | 11.5 ± 4.2 | 0.37 ± 0.13 |
TABLE 5.
Allosteric effects of berberine on MDZ hydroxylation by recombinant CYP3A5
Values denote means ± S.D. of three separate experiments. Km and Ki are given in micromolar; Vmax is in picomoles per minute per picomole CYP3A5; Vmax/Km is in microliters per minute per picomole CYP3A5.
| Berberine | 1′-Hydroxy MDZ | 4-Hydroxy MDZ | ||||
|---|---|---|---|---|---|---|
| (μM) | Km | Vmax | Vmax/Km | Km | Vmax | Vmax/Km |
| 0 | 1.55 ± 0.18 | 59.3 ± 12.5 | 38.5 ± 12.9 | 12.6 ± 2.7 | 14.9 ± 2.6 | 1.20 ± 0.09 |
| 5 | 1.55 ± 0.50 | 58.7 ± 8.9 | 41.6 ± 19.3 | 20.5 ± 4.1 | 14.1 ± 2.3 | 0.69 ± 0.07 |
| 25 | 1.11 ± 0.10 | 58.2 ± 14.3 | 51.5 ± 9.7 | 39.4 ± 4.9 | 12.4 ± 2.1 | 0.32 ± 0.09 |
| 50 | 1.01 ± 0.15 | 56.3 ± 12.1 | 54.7 ± 10.8 | 60.5 ± 5.8 | 11.4 ± 2.8 | 0.19 ± 0.05 |
| 100 | 0.91 ± 0.11 | 53.9 ± 11.5 | 58.9 ± 15.4 | 100 ± 28 | 10.0 ± 3.3 | 0.10 ± 0.02 |
In the absence of effector, the kinetics of 4-OH-MDZ formation in both HLMs and CYP3A4 Supersomes were described best by a simple unienzyme Michaelis-Menten model, showing a similar Km for MDZ (20–25 μM); upon addition of berberine, noncompetitive inhibition of MDZ 4-hydroxylation was observed. MDZ 4-hydroxylation by CYP3A5 Supersomes also were described best by a simple unienzyme Michaelis-Menten kinetic model, exhibiting a Km and Vmax that were both approximately 50% of those for recombinant CYP3A4 (Tables 2–4). When berberine was added to incubations containing CYP3A5 Supersomes, strong mixed inhibition of MDZ 4-hydroxylation was observed (Fig. 5).
Fig. 5.

The kinetics of MDZ 4-hydroxylation by HLMs or by recombinant CYP3A4 or CYP3A5 Supersomes were described best by a simple unienzyme Michaelis-Menten model. Berberine acted as a noncompetitive inhibitor of CYP3A4 and HLM-mediated MDZ 4-hydroxylation as well as a mixed inhibitor of this reaction when catalyzed by CYP3A5 (Tables 2–4).
In the absence of effector, CYP3A4 Supersomes produced a 1′-OH/4-OH-MDZ ratio of 7–9 at the lowest MDZ concentration tested, CYP3A5 Supersomes produced a ratio of 35–40, and HLMs produced a ratio of 10–12. The addition of berberine (up to 100 μM) resulted in a modest increase (1.35–1.6-fold) in the maximum metabolite ratio observed for CYP3A4-mediated MDZ hydroxylation while causing an increase of up to 30-fold in the CYP3A5-mediated metabolite ratio and an increase of 3- to 4-fold in the HLMs-mediated metabolite ratio (Fig. 6).
Fig. 6.

Effect of berberine on the metabolite ratio for MDZ hydroxylation (i.e., 1′-OH/4-OH-MDZ) produced by HLMs or CYP3A4 or CYP3A5 Supersomes. Metabolite ratios were highest at the lowest concentrations of substrate tested and decreased markedly at concentrations exceeding the Km for MDZ 4-hydroxylation (∼12–25 μM). When berberine was added as effector, an increase in the maximum metabolite ratio was observed with all three enzyme sources; however, this increase was modest for CYP3A4 (1.35–1.6-fold at 100 μM berberine) compared with CYP3A5 (∼30-fold), whereas the 3.5-fold increase in the maximum metabolite ratio produced by berberine in HLMs lay in between these values.
IC50 Shift Experiments with CYP3A4/5 Supersomes: Inhibition of MDZ 1′-Hydroxylation by Berberine and (−)-β-Hydrastine
Additional studies were conducted to determine the relative ability of berberine and (−)-β-hydrastine to inactivate recombinant CYP3A4 and CYP3A5 separately by using MDZ as a probe substrate. The final MDZ concentration in the incubation mixtures was either 2 μM (the reported Km for CYP3A4) for experiments involving (−)-β-hydrastine or 20 μM when berberine was used as the inhibitor. In the latter case, the higher concentration of substrate used was an attempt to minimize the observed activation effect of berberine on CYP3A5-mediated MDZ 1′-hydroxylation.
There was essentially no reversible inhibition of CYP3A5-mediated MDZ 1′-hydroxylation by berberine (up to 500 μM) and only weak to moderate TDI (shifted IC50 = 83 μM; shift ratio >6). Berberine was a weak reversible inhibitor of CYP3A4 activity (IC50 = ∼500 μM) but a stronger time-dependent inhibitor of this enzyme (shifted IC50 = 9.9 μM; shift ratio = ∼50) (Supplemental Fig. 1). By contrast, there was no observable difference for the TDI of CYP3A4 versus CYP3A5 by (−)-β-hydrastine (reversible IC50 = ∼30 μM; shifted IC50 = 2.5 μM; shift ratio = 12 for both isozymes) (Supplemental Fig. 2).
Discussion
Botanical NP usage continues to increase worldwide as a means to self-treat various illnesses and/or supplement prescribed pharmacotherapeutic regimens (Paine and Roe, 2018; Smith et al., 2019). Coconsuming NPs with prescribed medications can increase the risk of adverse NP-drug interactions, which can lead to suboptimal therapeutic outcomes. Mechanisms of pharmacokinetic NP-drug interactions include inhibition of the P450s, which are responsible for the metabolic elimination of numerous clinically used drugs, including those with a narrow therapeutic window (Guengerich, 2008; Zanger and Schwab, 2013). Goldenseal is a widely used NP that contains several isoquinoline alkaloids, the most prominent of which are berberine and (−)-β-hydrastine (Weber et al., 2003). Despite clinical studies demonstrating goldenseal or berberine to precipitate pharmacokinetic interactions with the probe substrates MDZ (CYP3A4/5), debrisoquine (CYP2D6), dextromethorphan (CYP2D6), and losartan (CYP2C9) (Gurley et al., 2008a,b; Guo et al., 2012), a comprehensive characterization of the inhibition kinetics of major isoquinoline alkaloids has not been reported. Robust kinetic parameters are needed to develop PBPK models, which can be used to predict the magnitude and time course of potential clinical interactions with critical object drugs (i.e., drugs with narrow therapeutic windows) as well as the risk of these interactions in vulnerable populations (e.g., elderly, hepatically impaired, renally impaired, pregnant women). Based on these knowledge gaps, the reversible inhibition and TDI kinetics of berberine, (−)-β-hydrastine, and hydrastinine against CYP2C9, CYP2D6, and CYP3A4/5 activities were determined using HLMs.
These isoquinoline alkaloids were predominantly weak reversible inhibitors of the P450 activities tested (at probe substrate concentrations equal to the respective Km values), generating IC50 values in the absence of an NADPH preincubation step of >50 μM (a lone exception was berberine, which was a moderate reversible inhibitor of CYP2D6 activity in HLMs, IC50 = 9.9 μM) (Table 1). Gupta et al. (2009) reported maximum human plasma concentrations of 1.75 and 470 nM, respectively, for berberine and hydrastine after administration of a single 2.14 g oral dose of a GSE containing 132 mg berberine (6.2% of extract) and 77 mg hydrastine (3.6% of extract). Although no standard dosing protocol exists for GSEs, recommended doses generally range from 250 to 1000 mg 3× daily. The alkaloid percentages contained in the extract used in the Gupta study were within the expected ranges determined for a selection of commercial GSEs (Brown and Roman, 2008). Using these reported Imax values for berberine and hydrastine, we conclude that reversible inhibition is an unlikely mechanism of the reported clinical P450-mediated goldenseal-drug interactions at the level of the liver. However, reversible inhibition of CYP3A4/5 by either alkaloid at the level of the intestine cannot be ruled out based on estimated luminal concentrations ranging from 0.8 to 1.6 mM (dose/0.25 l per FDA guidance) (U.S. Food and Drug Administration, 2020).
Based on these observations, any clinical NP-drug interaction caused by GSE via inhibition of hepatic CYP2C9, CYP2D6, or CYP3A4/5 metabolism is likely due to TDI of enzyme activity by one or more of the component GSE alkaloids. All of these alkaloids contain a methylenedioxyphenyl ring, a known structural alert for the inactivation of P450 enzymes primarily via formation of metabolic intermediate (MI) complexes (Taxak et al., 2013). MI complexes typically exhibit a distinct absorption maximum at ∼455 nm, and evidence for MI complex formation between (−)-β-hydrastine and CYP2C9, 2D6, and 3A4 has been described (Chatterjee and Franklin, 2003). It is, therefore, not surprising that we observed TDI of the tested P450s in HLMs by (−)-β-hydrastine or berberine (Fig. 3; Table 2).
Using a basic static model, we predicted clinical goldenseal-drug interactions may result from inactivation of hepatic CYP2C9, CYP2D6, and CYP3A4 by (−)-β-hydrastine and from the inactivation of hepatic CYP2D6 and CYP3A4 by berberine (Table 6). The inactivation parameters used to predict these drug interactions were estimated using a standard Michaelis-Menten model. Recent work by Barnaba et al. suggested that the TDI kinetics of MI complex formation reflects a more complicated “quasi-irreversible intermediate” model, wherein an initial intermediate MI complex is reversibly formed and subsequently progresses to an irreversible MI complex (Barnaba et al., 2016). If the rate at which the second irreversible complex forms is significantly slower than the formation rate of the initial complex, estimating TDI kinetics using a Michaelis-Menten model can cause an overprediction of the kinact value, thus resulting in a corresponding overprediction of in vivo drug interaction risk.
Berberine not only showed TDI but also reversibly activated CYP3A4/5-mediated MDZ 1′-hydroxylation in HLMs. We further evaluated this allosteric relationship by monitoring the effects of berberine on the regiochemistry and kinetics of MDZ hydroxylation by HLMs and recombinant CYP3A4 and CYP3A5 Supersomes, using methodology similar to that employed in the study of fluconazole’s allosteric effects on MDZ hydroxylation (Yang et al., 2012). Berberine showed weak mixed inhibition along with negative heterotropic cooperativity [lowering Ki(app) for substrate inhibition] for MDZ 1′-hydroxylation by CYP3A4 but activated this reaction [lowering Km(app)] when catalyzed by CYP3A5. Berberine’s allosteric effects on MDZ 1′-hydroxylation in HLMs were an amalgamation of the individual effects observed for recombinant CYP3A4 and CYP3A5, exhibiting positive and negative heterotropic cooperativity via lowering of both Km(app) and Ki(app). Berberine acted as a noncompetitive inhibitor of MDZ 4-hydroxylation by both HLMs and recombinant CYP3A4 but was a mixed inhibitor of this reaction when catalyzed by CYP3A5. The berberine effect on CYP3A4 kinetics for this reaction was dominant in HLMs because, based on relative intrinsic clearance rates (Tables 4 and 5), the alkaloid was a stronger inhibitor of MDZ 4-hydroxylation by CYP3A5.
The differences in MDZ metabolic kinetics between CYP3A4 and CYP3A5 resulted in large variability in the maximum MDZ 1′-OH/4-OH ratios produced by the two isozymes. In the absence of effector, CYP3A4 produced a maximum observed metabolite ratio of ∼8, whereas that produced by CYP3A5 was ∼5-fold higher (the ratio decreased substantively as MDZ concentrations approached and surpassed the Km for 4-hydroxylation). Adding berberine increased the ratio produced by all three enzyme sources, albeit to different degrees. Because berberine inhibited CYP3A4-mediated MDZ hydroxylation at both the 1′ and 4 positions, a relatively modest increase (∼1.4-fold) was observed in the maximum metabolite ratio. In contrast, berberine not only activated CYP3A5-mediated MDZ 1′-hydroxylation but also more strongly inhibited MDZ 4-hydroxylation. This synergistic effect led to more than a 30-fold increase in the observed ratio produced by CYP3A5. As expected, the 3- to 4-fold increase in MDZ metabolite ratio effected by berberine in HLMs (which contain both CYP3A4 and CYP3A5) lay between the values observed for the individual isozymes. In a similar experiment, fluconazole decreased the maximum MDZ metabolite ratio produced by recombinant CYP3A4 by >50%, as it more strongly inhibited hydroxylation at the 1′ position of MDZ compared with the 4 position (Yang et al., 2012).
These allosteric data highlight a potential issue with using MDZ 1′-hydroxylation as a probe to monitor the effect of berberine or GSE on CYP3A4/5 activity in vivo. Because berberine is a stronger time-dependent inhibitor of CYP3A4 (IC50 shift ratio ∼50) compared with CYP3A5 (shift ratio ∼6), the possibility exists, especially after prolonged treatment with the alkaloid, that activation of CYP3A5-mediated MDZ 1′-hydroxylation could mask some degree of CYP3A4 inactivation. Therefore, ifMDZ is used as an in vivo CYP3A4/5 probe in clinical studies involving GSE or berberine, monitoring MDZ 4-hydroxylation should be considered, as an increase over control in the MDZ 1′-OH/4-OH metabolite ratio would suggest an increased involvement of CYP3A5 in MDZ metabolism.
Small structural differences between CYP3A4 and CYP3A5 cause a marked alteration in how each isozyme binds to berberine, which, in turn, leads to the observed opposing allosteric interactions on MDZ hydroxylation effected by the alkaloid. We propose that berberine may activate MDZ 1′-hydroxylation by CYP3A5 through occupation of the peripheral allosteric site most often associated with progesterone binding (Williams et al., 2004). Berberine is similar in size and overall structure to progesterone (Fig. 1), a steroid known to promote metabolism of several different CYP3A4/5 probe substrates (Niwa et al., 2003; Polic and Auclair, 2017). In a similar fashion to the berberine activation reported here, progesterone increased CYP3A4/5-mediated nifedipine oxidation in HLMs by lowering Km(app) without effect on Vmax (Niwa et al., 2003). We postulate that berberine binds more strongly to the progesterone site of CYP3A5 but preferentially binds within the active site of CYP3A4, leading to negative heterotropic cooperativity with MDZ along with increased mechanism-based inactivation of that enzyme. Given the report of a (ritonavir bound) CYP3A5 X-ray crystal structure (Hsu et al., 2018), testing this theory through a docking site comparison of berberine within the CYP3A4 and CYP3A5 active and peripheral binding sites is warranted.
TDI of metabolic drug elimination has clinical implications for the management of drug therapies. Both the onset and offset of the inhibitory effect must be fully characterized to anticipate when and by how much an object drug dose should be adjusted. This scenario is most relevant for narrow therapeutic window drugs such as the calcineurin inhibitors, which are metabolized by CYP3A4/5 (Hesselink et al., 2005), and several antidepressants that are metabolized by CYP2D6 (Kwadijk-de Gijsel et al., 2009). Although inactivation of P450 enzymes might occur rapidly under optimal conditions, a return to the original uninhibited state will take days because of its dependence on the half-life of the P450 enzyme (∼24–48 hours). The KI and kinact values generated in this study can be used in conjunction with expected concentration-time profiles of inhibitors at the site(s) of metabolism to simulate the time course of enzyme inactivation and changes in substrate exposure in blood by using PBPK modeling, and they can inform the clinical dosing decision process.
In summary, we report robust kinetic parameters for the reversible inhibition and TDI of CYP2C9, CYP2D6, and CYP3A4/5 activities in HLMs by the goldenseal component isoquinoline alkaloids berberine, (−)-β-hydrastine, and hydrastinine. These data are a necessary precursor to the development of a PBPK model that can be used to predict potential adverse clinical NP-drug interactions resulting from inhibition of P450 metabolism by goldenseal. We also investigated the allosteric interactions observed with berberine on CYP3A4/5 activity, with the alkaloid exhibiting isozyme-specific positive (CYP3A5) and negative (CYP3A4) heterotropic cooperativity with respect to MDZ 1′-hydroxylation. These complex interactions should be considered in the design and analysis of future clinical studies that monitor the effect of berberine or goldenseal on CYP3A4/5-mediated drug metabolism.
Acknowledgments
The authors would like to thank James Nguyen, with the Department of Pharmaceutical Sciences at Washington State University, for his valuable contributions in predicting the fraction unbound values for the goldenseal alkaloids.
Abbreviations
- FDA
U.S. Food and Drug Administration
- fu,HLM
fraction unbound in human liver microsomes
- GSE
goldenseal extract
- HLM
human liver microsome
- KPi
potassium phosphate
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- MDZ
midazolam
- MI
metabolic intermediate m/z mass to charge ratio
- NmP
natural product
- P450
cytochrome P450
- PBPK
physiologically based pharmacokinetic
- TDI
time-dependent inhibition
- UPLC
ultraperformance liquid chromatography
Authorship Contributions
Participated in research design: McDonald, Tian, Thummel, Paine, Rettie.
Conducted experiments: McDonald.
Performed data analysis: McDonald.
Wrote or contributed to the writing of the manuscript: McDonald, Tian, Thummel, Paine, Rettie.
Footnotes
This work was supported by National Institutes of Health National Center for Complementary and Integrative Health [Grant U54 AT008909]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Asher GN, Corbett AH, Hawke RL. (2017) Common herbal dietary supplement-drug interactions. Am Fam Physician 96:101–107. [PubMed] [Google Scholar]
- Barnaba C, Yadav J, Nagar S, Korzekwa K, Jones JP. (2016) Mechanism-Based Inhibition of CYP3A4 by Podophyllotoxin: Aging of an Intermediate is Important for in Vitro/in Vivo Correlations. Molecular Pharmaceutics 13:2833–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen KM, Venkatakrishnan K, Von Moltke LL, Obach RS, Greenblatt DJ. (2003) Apparent mechanism-based inhibition of human CYP2D6 in vitro by paroxetine: comparison with fluoxetine and quinidine. Drug Metab Dispos 31:289–293. [DOI] [PubMed] [Google Scholar]
- Brown PN, Roman MC. (2008) Determination of hydrastine and berberine in goldenseal raw materials, extracts, and dietary supplements by high-performance liquid chromatography with UV: collaborative study. J AOAC Int 91:694–701. [PMC free article] [PubMed] [Google Scholar]
- Budzinski JW, Foster BC, Vandenhoek S, Arnason JT. (2000) An in vitro evaluation of human cytochrome P450 3A4 inhibition by selected commercial herbal extracts and tinctures. Phytomedicine 7:273–282. [DOI] [PubMed] [Google Scholar]
- Chatterjee P, Franklin MR. (2003) Human cytochrome p450 inhibition and metabolic-intermediate complex formation by goldenseal extract and its methylenedioxyphenyl components. Drug Metab Dispos 31:1391–1397. [DOI] [PubMed] [Google Scholar]
- Etheridge AS, Black SR, Patel PR, So J, Mathews JM. (2007) An in vitro evaluation of cytochrome P450 inhibition and P-glycoprotein interaction with goldenseal, Ginkgo biloba, grape seed, milk thistle, and ginseng extracts and their constituents. Planta Med 73:731–741. [DOI] [PubMed] [Google Scholar]
- Ettefagh KA, Burns JT, Junio HA, Kaatz GW, Cech NB. (2011) Goldenseal (Hydrastis canadensis L.) extracts synergistically enhance the antibacterial activity of berberine via efflux pump inhibition. Planta Med 77:835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BC, Vandenhoek S, Hana J, Krantis A, Akhtar MH, Bryan M, Budzinski JW, Ramputh A, Arnason JT. (2003) In vitro inhibition of human cytochrome P450-mediated metabolism of marker substrates by natural products. Phytomedicine 10:334–342. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. (2008) Cytochrome p450 and chemical toxicology. Chem Res Toxicol 21:70–83. [DOI] [PubMed] [Google Scholar]
- Guo Y, Chen Y, Tan ZR, Klaassen CD, Zhou HH. (2012) Repeated administration of berberine inhibits cytochromes P450 in humans. Eur J Clin Pharmacol 68:213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PK, Barone G, Gurley BJ, Fifer EK, Hendrickson HP. (2015) Hydrastine pharmacokinetics and metabolism after a single oral dose of goldenseal (Hydrastis canadensis) to humans. Drug Metab Dispos 43:534–552. [DOI] [PubMed] [Google Scholar]
- Gupta PK, Hubbard M, Gurley B, Hendrickson HP. (2009) Validation of a liquid chromatography-tandem mass spectrometric assay for the quantitative determination of hydrastine and berberine in human serum. J Pharm Biomed Anal 49:1021–1026. [DOI] [PubMed] [Google Scholar]
- Gurley BJ, Gardner SF, Hubbard MA, Williams DK, Gentry WB, Khan IA, Shah A. (2005) Clin Pharmacol Ther 77 (5):415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley BJ, Swain A, Hubbard MA, Hartsfield F, Thaden J, Williams DK, Gentry WB, Tong Y. (2008a) Supplementation with goldenseal (Hydrastis canadensis), but not kava kava (Piper methysticum), inhibits human CYP3A activity in vivo. Clin Pharmacol Ther 83:61–69. [DOI] [PubMed] [Google Scholar]
- Gurley BJ, Swain A, Hubbard MA, Williams DK, Barone G, Hartsfield F, Tong Y, Carrier DJ, Cheboyina S, Battu SK. (2008b) Clinical assessment of CYP2D6-mediated herb-drug interactions in humans: effects of milk thistle, black cohosh, goldenseal, kava kava, St. John’s wort, and Echinacea. Mol Nutr Food Res 52:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann R, von Richter O. (2012) Clinical evidence of herbal drugs as perpetrators of pharmacokinetic drug interactions. Planta Med 78:1458–1477. [DOI] [PubMed] [Google Scholar]
- Hesselink DA, van Gelder T, van Schaik RH. (2005) The pharmacogenetics of calcineurin inhibitors: one step closer toward individualized immunosuppression? Pharmacogenomics 6:323–337. [DOI] [PubMed] [Google Scholar]
- Hsu MH, Savas U, Johnson EF. (2018) The x-ray crystal structure of the human mono-oxygenase cytochrome P450 3A5-ritonavir complex reveals active site differences between P450s 3A4 and 3A5. Mol Pharmacol 93:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg JJ, Paine MF, McCune JS, Oberlies NH, Cech NB. (2019) Selection and characterization of botanical natural products for research studies: a NaPDI center recommended approach. Nat Prod Rep 36:1196–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwadijk-de Gijsel S, Bijl MJ, Visser LE, van Schaik RHN, Hofman A, Vulto AG, van Gelder T, Ch Stricker BH. (2009) Variation in the CYP2D6 gene is associated with a lower serum sodium concentration in patients on antidepressants. Br J Clin Pharmacol 68:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SN, Chang YP, Tsai KC, Chang CY, Wu TS, Ueng YF. (2013) Inhibition of CYP1 by berberine, palmatine, and jatrorrhizine: selectivity, kinetic characterization, and molecular modeling. Toxicol Appl Pharmacol 272:671–680. [DOI] [PubMed] [Google Scholar]
- McDonald MG, Au NT, Rettie AE. (2015) P450-based drug-drug interactions of amiodarone and its metabolites: diversity of inhibitory mechanisms. Drug Metab Dispos 43:1661–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnity DF, Berry AJ, Kenny JR, Grime K, Riley RJ. (2006) Evaluation of time-dependent cytochrome P450 inhibition using cultured human hepatocytes. Drug Metab Dispos 34:1291–1300. [DOI] [PubMed] [Google Scholar]
- Niwa T, Shiraga T, Yamasaki S, Ishibashi K, Ohno Y, Kagayama A. (2003) In vitro activation of 7-benzyloxyresorufin O-debenzylation and nifedipine oxidation in human liver microsomes. Xenobiotica 33:717–729. [DOI] [PubMed] [Google Scholar]
- Obach RS, Walsky RL, Venkatakrishnan K. (2007) Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug-drug interactions. Drug Metab Dispos 35:246–255. [DOI] [PubMed] [Google Scholar]
- Paine MF, Shen DD, McCune JS. (2018) Recommended approaches for pharmacokinetic natural product-drug interaction research: a NaPDI Center commentary. Drug Metab Dispos 46:1041–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine MF, Roe AL. (2018) “Green medicine”: the past, present, and future of botanicals. Clin Pharmacol Ther 104:410–415. [DOI] [PubMed] [Google Scholar]
- Polic V, Auclair K. (2017) Allosteric activation of cytochrome P450 3A4 via progesterone bioconjugation. Bioconjug Chem 28:885–889. [DOI] [PubMed] [Google Scholar]
- Raner GM, Cornelious S, Moulick K, Wang Y, Mortenson A, Cech NB. (2007) Effects of herbal products and their constituents on human cytochrome P450(2E1) activity. Food Chem Toxicol 45:2359–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeque AJ, Eddy AC, Meier GP, Rettie AE. (1992) Stereoselective sulfoxidation by human flavin-containing monooxygenase. Evidence for catalytic diversity between hepatic, renal, and fetal forms. Drug Metab Dispos 20:832–839. [PubMed] [Google Scholar]
- Smith T, Gillespie M, Eckl V, Knepper J, Morton Reynolds C. (2019) Market report: herbal supplement sales increase by 9.4% in 2018. HerbalGram 62–73. [Google Scholar]
- Taxak N, Patel B, Bharatam PV. (2013) Carbene generation by cytochromes and electronic structure of heme-iron-porphyrin-carbene complex: a quantum chemical study. Inorg Chem 52:5097–5109. [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration (2020) In vitro drug interaction studies - cytochrome P450 enzyme- and transporter-mediated drug interactions: guidance for industry.
- Weber HA, Zart MK, Hodges AE, Molloy HM, O’Brien BM, Moody LA, Clark AP, Harris RK, Overstreet JD, Smith CS. (2003) Chemical comparison of goldenseal (Hydrastis canadensis L.) root powder from three commercial suppliers. J Agric Food Chem 51:7352–7358. [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305:683–686. [DOI] [PubMed] [Google Scholar]
- Wu X, Li Q, Xin H, Yu A, Zhong M. (2005) Effects of berberine on the blood concentration of cyclosporin A in renal transplanted recipients: clinical and pharmacokinetic study. Eur J Clin Pharmacol 61:567–572. [DOI] [PubMed] [Google Scholar]
- Yang J, Atkins WM, Isoherranen N, Paine MF, Thummel KE. (2012) Evidence of CYP3A allosterism in vivo: analysis of interaction between fluconazole and midazolam. Clin Pharmacol Ther 91:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanger UM, Schwab M. (2013) Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 138:103–141. [DOI] [PubMed] [Google Scholar]
- Zhao P, Kunze KL, Lee CA. (2005) Evaluation of time-dependent inactivation of CYP3A in cryopreserved human hepatocytes. Drug Metab Dispos 33:853–861. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Hellum BH, Liang A, Nilsen OG. (2015) Inhibitory mechanisms of human P450s by three alkaloids isolated from traditional Chinese herbs. Phytother Res 29:825–834. [DOI] [PubMed] [Google Scholar]