

Abstract
Cannabis is used for both recreational and medicinal purposes. The most abundant constituents are the cannabinoids - cannabidiol (CBD, nonpsychoactive) and (−)-trans-Δ9-tetrahydrocannabinol (THC, psychoactive). Both have been reported to reversibly inhibit or inactivate cytochrome P450 (CYPs) enzymes. However, the low aqueous solubility, microsomal protein binding, and nonspecific binding to labware were not considered, potentially leading to an underestimation of CYPs inhibition potency. Therefore, the binding-corrected reversible (IC50,u) and irreversible (KI,u) inhibition potency of each cannabinoid toward major CYPs were determined. The fraction unbound of CBD and THC in the incubation mixture was 0.12 ± 0.04 and 0.05 ± 0.02, respectively. The IC50,u for CBD toward CYP1A2, 2C9, 2C19, 2D6, and 3A was 0.45 ± 0.17, 0.17 ± 0.03, 0.30 ± 0.06, 0.95 ± 0.50, and 0.38 ± 0.11 µM, respectively; the IC50,u for THC was 0.06 ± 0.02, 0.012 ± 0.001, 0.57 ± 0.22, 1.28 ± 0.25, and 1.30 ± 0.34 µM, respectively. Only CBD showed time-dependent inactivation (TDI) of CYP1A2, 2C19, and CYP3A, with inactivation efficiencies (kinact/KI,u) of 0.70 ± 0.34, 0.11 ± 0.06, and 0.14 ± 0.04 minutes−1 µM−1, respectively. A combined (reversible inhibition and TDI) mechanistic static model populated with these data predicted a moderate to strong pharmacokinetic interaction risk between orally administered CBD and drugs extensively metabolized by CYP1A2/2C9/2C19/2D6/3A and between orally administered THC and drugs extensively metabolized by CYP1A2/2C9/3A. These predictions will be extended to a dynamic model using physiologically based pharmacokinetic modeling and simulation and verified with a well-designed clinical cannabinoid-drug interaction study.
SIGNIFICANCE STATEMENT
This study is the first to consider the impact of limited aqueous solubility, nonspecific binding to labware, or extensive binding to incubation protein shown by cannabidiol (CBD) and delta-9-tetrahydrocannabinol (THC) on their true cytochrome P450 inhibitory potency. A combined mechanistic static model predicted a moderate to strong pharmacokinetic interaction risk between orally administered CBD and drugs extensively metabolized by CYP1A2, 2C9, 2C19, 2D6, or 3A and between orally administered THC and drugs extensively metabolized by CYP1A2, 2C9, or 3A.
Introduction
Cannabis (also known as marijuana) is the most commonly used recreational illicit substance in the United States, with an estimated 22.2 million users ((Bose et al., 2016). Cannabis and its main phytoconstituents, cannabinoids, are also used to treat a variety of ailments, including pain, nausea, loss of appetite, and childhood epilepsy (Grotenhermen, 2003; Cox et al., 2019). To date, cannabis has been legalized in 33 states, including the District of Columbia, for medicinal use and in 11 states for recreational use (https://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx). Among users in the United States, 53% consume cannabis for recreational purposes, 11% for medicinal purposes, and 36% for both recreational and medicinal purposes (Schauer et al., 2016). Because of the ever-increasing use of cannabis, especially in populations taking multiple medications, determining potential pharmacokinetic interactions between cannabinoids and coadministered medications is imperative.
The prevalent cannabinoids in cannabis products include the nonpsychoactive constituent, cannabidiol (CBD), and the psychoactive constituent, (−)-trans-Δ9-tetrahydrocannabinol (THC) (Fig. 1). CBD and THC have been shown to reversibly inhibit cytochrome P450 (CYP) 1A, 1B1, 2A6, 2B6, 2D6, 2J2, and 3A with varying degrees of potency; IC50 or Ki values ranged from 0.20 to 36 µM (Yamaori et al., 2010; Arnold et al., 2018; Cox et al., 2019). In addition, CBD showed time-dependent inactivation (TDI) of CYP1A1, 1A2, and 1B1, whereas THC showed TDI of CYP1A1 and 2A6 (Yamaori et al., 2010, 2011b). However, the inhibition potency of these cannabinoids was likely underestimated because their poor aqueous solubility and extensive binding to microsomal proteins and labware (Garrett and Hunt, 1974) were not considered.
Fig. 1.

Chemical structures of CBD, THC, 11-OH-THC, and COOH-THC.
THC is metabolized to an active metabolite, 11-hydroxy-THC (11-OH-THC), which is further metabolized to the inactive metabolite, 11-nor-9-carboxy-∆9-THC (COOH-THC) (Fig. 1). These metabolites have been detected in the systemic circulation at higher concentrations than THC after oral administration of THC (Frytak et al., 1984; Nadulski et al., 2005). Consequently, these metabolites should be considered when predicting the magnitude of a THC-mediated drug interaction as recommended by the US Food and Drug Administration (FDA) (https://www.fda.gov/media/134582/download). However, the potential for 11-OH-THC/COOH-THC to inhibit CYPs has not been previously determined.
Based on these knowledge gaps, the objective of this study was to predict the potential for CBD and THC to precipitate CYP-mediated pharmacokinetic drug interactions in vivo. The aims were to: 1) determine the average oral or inhalational doses of CBD and THC used for recreational or medicinal purposes, allowing estimation of the maximum plasma concentration of CBD and THC (and circulating THC metabolites) achieved at these doses; 2) determine the biorelevant gastrointestinal solubility of CBD and THC; and 3) determine the binding-corrected potency of CBD, THC, and circulating metabolites of THC to reversibly (IC50) and irreversibly (KI and kinact) inhibit CYP enzymes involved in the metabolism of a majority of drugs, specifically CYP1A2, 2C9, 2C19, 2D6, and 3A. A mechanistic static model was populated with the data to predict the ratio of the area under the plasma concentration-time curve (AUCR) of a CYP probe drug substrate in the presence to absence of cannabinoid. Both CBD and THC were predicted to precipitate pharmacokinetic interactions with several of the probe drugs after oral administration, warranting further investigation via physiologically based pharmacokinetic (PBPK) modeling and simulation and, potentially, clinical evaluation.
Materials and Methods
Biologic Materials, Chemicals, and Reagents
Pooled adult human liver microsomes (HLMs) (mixed gender; pool of 50 donors) were purchased from Corning Inc. (Corning, NY). Drug Enforcement Administration–exempt methanolic stock solutions of (-)-∆9-THC (1 mg/ml), (±) 11-OH-THC (0.1 mg/ml), and (±) (COOH-THC) (0.1 mg/ml) as well as tolbutamide were purchased from Cerilliant (Round Rock, TX). (-)-∆9-THC, CBD, omeprazole, testosterone, dextromethorphan, phenacetin, diclofenac, 5-hydroxy omeprazole, 6β-hydroxy testosterone, dextrorphan, acetaminophen, and 4-hydroxy diclofenac were purchased from Cayman Chemicals (Ann Arbor, MI). Micro ultracentrifuge polycarbonate tubes and Dulbecco’s phosphate-buffered saline were purchased from Thermo Scientific (Asheville, NC). Ultralow-binding microcentrifuge tubes, bovine serum albumin (BSA), acetonitrile, and formic acid were purchased from Fisher Scientific (Hampton, NH). β-nicotinamide adenine dinucleotide phosphate (NADP+), D-glucose 6-phosphate (G6P), and glucose-6-phosphate dehydrogenase were purchased from Sigma-Aldrich (St. Louis, MO). Milli-Q water was used for all preparations. All other chemicals and experimental reagents were obtained from reputable commercial sources.
Search Strategy to Determine Oral or Inhalational Doses of CBD and THC Used for Recreational and/or Medicinal Purposes
To predict the magnitude of pharmacokinetic CBD- or THC-drug interactions, the average and maximum oral and/or inhalational doses of each cannabinoid used for recreational and/or medicinal purposes is required. Epidiolex (CBD) and Marinol (THC) are FDA-approved drugs indicated for childhood epileptic seizures and chemotherapy-induced nausea, respectively. In addition, numerous clinical trials involving CBD and THC have been conducted to determine their efficacy for various medicinal purposes. FDA-recommended CBD and THC doses and their average doses used in clinical trials and case reports were collected and tabulated (Supplemental Tables 1 and 2). To estimate the average THC and CBD doses for recreational and/or medicinal use, a thorough search of the doses reported in social media, cannabinoid vendor websites, and newspapers was conducted. The doses were categorized as either low (CBD ≤ 200 mg or THC ≤ 50 mg) or high (CBD > 200 mg or THC > 50 mg) (Supplemental Tables 1 and 2). The cutoffs were based on natural clustering. Average low and high doses of CBD or THC were calculated. The maximum dose of CBD or THC for recreational and/or medicinal use was tabulated (Supplemental Tables 1 and 2).
Cannabinoid Biorelevant Solubility
Preparation of FaSSIF-v2 and FeSSIF-v2 Media.
FaSSIF-v2 (fasted state simulated intestinal fluid v2) and FeSSIF-v2 (fed state simulated intestinal fluid v2) media were prepared fresh on the day of an experiment according to the manufacturer’s protocol (https://biorelevant.com/fassif-fessif-fassgf/buy/). A blank buffer (50 ml) for FaSSIF-v2 composed of sodium hydroxide (0.07 g), maleic acid (0.11 g), and sodium chloride (0.20 g) was prepared, and the pH was adjusted to 6.5 with sodium hydroxide (1 N). FaSSIF powder (0.09 g) was added to 25 ml of blank buffer. A similar procedure was used for FeSSIF-v2, of which a blank buffer composed of sodium hydroxide (0.16 g), maleic acid (0.32 g), and sodium chloride (0.37 g) was adjusted to pH 5.8 with sodium hydroxide (1 N). FeSSIF v2 powder (0.49 g) was added to 25 ml of blank buffer. Both FaSSIF and FeSSIF solutions were stirred to dissolve the powder. The volume of each solution was increased to 50 ml using respective blank buffer. The solutions were allowed to stand at room temperature for 1 hour prior to experimentation.
Determination of Cannabinoid Solubility.
The maximum gastrointestinal solubility of CBD and THC was determined using FaSSIF-v2 or FeSSIF-v2 medium (prepared as described above). Aliquots from CBD or THC stock solutions in methanol (1 mg/ml) were added to FaSSIF-v2 or FeSSIF-v2 medium in low-binding microcentrifuge tubes to achieve a maximum concentration of 100 µM. The tubes were incubated in an air incubator maintained at 37°C with continuous shaking and protected from light. The incubation was carried out for 24 hours to achieve a presumptive thermodynamic equilibrium. At the end of the incubation, the tubes were centrifuged at 15,000g for 5 minutes at 4°C. The supernatant was subjected to liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis (described below).
Reversible CYP Inhibition by Cannabinoids
Because THC exhibits low aqueous solubility (2.8 µg/ml) and extensive binding (70%–97%) to protein and labware (Garrett and Hunt, 1974), microsomal incubation conditions were optimized to prevent underestimation of inhibitory potency (IC50 or KI). To reduce nonspecific binding and adsorption to labware, low-binding microcentrifuge tubes were used, and BSA (0.2%) was included in the incubation mixtures. The latter also served to increase cannabinoid solubility.
A previously validated CYP cocktail assay was modified to simultaneously evaluate inhibition of CYP1A2, 2C9, 2C19, 2D6, and 3A by each cannabinoid (Dixit et al., 2007; Dinger et al., 2014; Chen et al., 2016). The cocktail consisted of the probe substrates phenacetin (CYP1A2; 50 μM), diclofenac (CYP2C9; 5 μM), omeprazole (CYP2C19; 10 μM), dextromethorphan (CYP2D6; 5 μM), and testosterone (CYP3A; 10 μM) at concentrations less than reported Km values (Spaggiari et al., 2014; Dahlinger et al., 2016). The cocktail was optimized for linearity of metabolite formation with respect to time and microsomal protein concentration. The probe substrates showed minimal interaction with each other (data not shown).
To determine the inhibitory effects of each cannabinoid on CYP activity, reaction mixtures (200 μl) were prepared in low-binding Eppendorf tubes that consisted of HLMs (0.1 mg/ml), CYP cocktail, and THC (0.003–100 μM), CBD (0.003–100 μM), 11-OH-THC (0.1–50 μM), or COOH-THC (0.1–50 μM) in 0.1 M potassium phosphate buffer (pH 7.4) containing 0.2% BSA. Stock solutions of probe substrates were prepared in DMSO. The final concentration of DMSO was 0.4% in each incubation mixture. Mixtures were equilibrated for 10 minutes at 37°C in a heating block with constant stirring (300 rpm). After 10 minutes, the NADPH regenerating system (1.3 mM NADP+, 3.3 mM G6P, 3.3 mM MgCl2, and 0.4 U/ml G6P dehydrogenase) was added to initiate the reaction. After an additional 10 minutes (with THC) or 15 minutes (with CBD, 11-OH-THC, or COOH-THC), reactions were quenched with 200 µl ice-cold acetonitrile containing internal standard (125 nM tolbutamide) and centrifuged at 18,000g for 10 minutes to precipitate microsomal proteins. Supernatants were analyzed using LC-MS/MS (described below). Four independent experiments were conducted, each in duplicate, with THC or CBD; three independent experiments were conducted, each in duplicate, with 11-OH-THC or COOH-THC. IC50 was determined by nonlinear regression analysis (GraphPad Prism 6.01; Graphpad Software Inc., San Diego, CA) using the following equation:
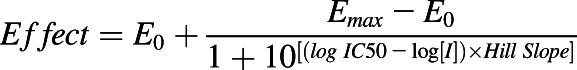 |
where I represents inhibitor concentration, and E0 and Emax represent minimum and maximum effect, respectively.
TDI of CYP Activity by Cannabinoids
Each primary incubation mixture (200 μl) consisted of potassium phosphate buffer (100 mM; pH 7.4), HLMs (0.5 mg/ml protein), and THC, 11-OH THC, COOH-THC (10 µM), CBD (0.5, 1, 2.5, 5, 10, 20, 40, or 60 μM), or DMSO (0.2% v/v; vehicle) as specified in each figure legend. The mixture was equilibrated for 5 minutes at 37°C in a shaking heat block. Reactions were initiated by adding an NADPH regenerating system described earlier and incubating at 37°C for 0, 4, 8, 10, 12, 16, 20, or 30 minutes as specified in each figure legend. An aliquot (10 µl) of the primary incubation mixture was transferred to a prewarmed secondary incubation mixture (190 µl) containing phosphate buffer (pH 7.4), CYP cocktail (phenacetin; 50 μM, diclofenac; 5 μM, omeprazole; 20 μM, dextromethorphan; 5 μM, and testosterone; 20 μM), and NADPH regenerating system. The secondary incubation mixture was incubated at 37°C for 10 or 15 minutes as specified in each figure legend. Reactions were quenched with 200 µl ice-cold acetonitrile containing internal standard (125 nM tolbutamide). Reaction mixtures were processed and analyzed as described for reversible inhibition experiments. Three or four independent experiments were conducted, each in duplicate.
The observed first-order rate constants for inactivation (kobs) were calculated as described previously (Cheong et al., 2017). The maximal inactivation rate constant (kinact) and half-maximal inactivation concentration (KI) were estimated by nonlinear least-squares regression analysis (GraphPad Prism 6.01; Graphpad Software Inc., San Diego, CA) of the kobs versus inactivator concentration ([I]) data using the following equation:
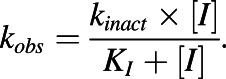 |
Cannabinoid Protein Binding in Incubation Mixture
Cannabinoid binding to proteins in the incubation mixture (fu,inc) was determined using the tube adsorption method (Patilea-Vrana and Unadkat, 2019). In brief, HLMs (0.1 mg/ml) were incubated with BSA (0.2%) and CBD (0.1 and 5 μM) for 10 minutes or THC (0.5 and 5 μM) or 11-OH-THC (0.1 μM) for 15 minutes. Three independent experiments were conducted, each in quadruplicate. Microsomal protein concentrations in the incubation mixtures for inactivation experiments (0.5 mg/ml) were five-times greater than that used for reversible inhibition experiments (0.1 mg/ml); BSA concentration (0.2%) was same in both experiments. As we previously reported, binding to incubation proteins is predominate to BSA, not to microsomal protein (Patilea-Vrana and Unadkat, 2019). Therefore, the fu,inc of CBD or THC, assumed to be independent of the HLMs, was used to compute the binding-corrected IC50 and KI values.
LC-MS/MS Analysis
Acetaminophen, 4-hydroxy diclofenac, 5-hydroxy omeprazole, dextrorphan, 6β-hydroxy testosterone, and tolbutamide were quantified using an ACQUITY ultra-high-performance LC (UPLC) system (Waters, Milford, MA) coupled to an SCIEX 6500 QTRAP mass spectrometer (SCIEX, Framingham, MA). Chromatographic separation was achieved on an ACQUITY UPLC BEH C18 column (2.1 × 50 mm, 1.7 µm) with an ACQUITY UPLC BEH C18 VanGuard precolumn (2.1 × 5 mm, 1.7 µm). The column and the autosampler compartment were maintained at 45 and 4°C, respectively. The flow rate was 0.5 ml/min, and the sample injection volume was 10 µl. The mobile phases were water containing 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B). The gradient conditions were optimized as follows: 5% B at 0.0 to 1.0 minute, linear increase from 5% to 95% B at 1.0 to 2.0 minutes, 95% B at 2.0–2.5 minutes, linear decrease from 95% to 5% B at 2.5–2.6 minutes, and 5% B at 2.6–3.2 minutes. The total run time was 3.2 minutes.
The mass spectrometer was operated in the positive electrospray ionization mode. Compound-dependent mass spectrometric parameters were optimized to achieve maximal ion intensities in the multiple reaction monitoring mode. Acetaminophen, 4-hydroxy diclofenac, 5-hydroxy omeprazole, dextrorphan, 6β -hydroxy testosterone, and tolbutamide were quantified in the multiple reaction monitoring mode using the mass transition of m/z 152.0 → 110.0, 312.0 → 231.0, 362.0 → 214.0, 258.1 → 157.0, 305.3 → 269.1, and 271.3 → 155.0, respectively. The ion source parameters were as follows: spray voltage, 5500 V; ion source temperature, 600°C; curtain gas, 30 psi; ion source gas 1, 50 psi; and ion source gas 2, 50 psi.
CBD, THC, and THC metabolites were quantified using LC-MS/MS method as reported previously (Patilea-Vrana and Unadkat, 2019).
Prediction of Cannabinoids to Precipitate Pharmacokinetic Drug Interactions
Reversible inhibition (IC50,u) and inactivation parameters (KI,u and kinact) of CBD, THC, 11-OH-THC, or COOH-THC were incorporated into a previously developed mechanistic static model (Fahmi et al., 2008; Cheong et al., 2017) to predict the net effect of reversible inhibition and inactivation of CYPs in both the liver and intestine (eq. 1). As stated earlier, CYP probe substrate concentrations used to determine IC50 values were < Km; IC50,u was assumed to be equivalent to Ki,u (Cheng and Prusoff, 1973). AUCR represents the ratio of area under the plasma concentration-time curve of the object (probe) drug in the presence (AUC’PO) to absence (AUCPO) of the cannabinoid CYP inactivator/inhibitor.
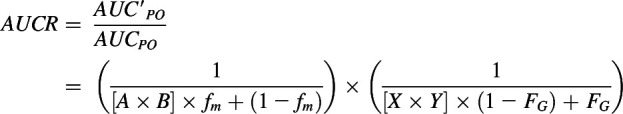 |
(1) |
where A is the term that describes TDI of a CYP enzyme observed in the liver:
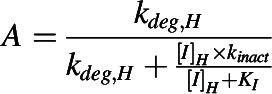 |
(2) |
B is the term that describes reversible inhibition of a CYP enzyme observed in the liver:
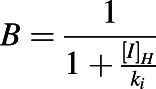 |
(3) |
X is the term that describes TDI of a CYP enzyme observed in the intestine:
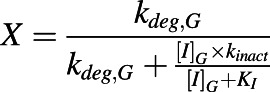 |
(4) |
Y is the term that describes reversible inhibition of a CYP enzyme observed in the intestine:
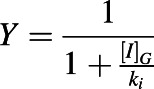 |
(5) |
and [I]H is the unbound in vivo maximum plasma concentration of an inhibitor/inactivator in the liver. [I]G is the in vivo concentration of an inactivator/inhibitor available to the enzyme in the intestine. kdeg,H and kdeg,G are the degradation rate constants of the CYP in the liver and intestine, respectively. fm is the fraction of the object drug metabolized by a given CYP, and FG is the fraction of the object drug escaping intestinal metabolism.
Each CYP probe drug except phenacetin and testosterone was used to predict the magnitude of the various potential CYP-mediated drug interactions in the liver or intestine precipitated by orally administered or inhaled CBD or THC. Because phenacetin is not used clinically and testosterone is an endogenous steroid hormone, theophylline and midazolam, respectively, were used as the object drugs to predict the magnitude of CYP1A2- and 3A4-mediated drug interactions. The fm and FG values of object drugs and kdeg value of CYP enzymes are given in Supplemental Table 1.
The maximum magnitude of a cannabinoid-drug interaction in the liver after oral cannabinoid administration was predicted using the following two different approaches: 1) unbound in vivo maximum plasma concentration of inhibitor/inactivator in hepatic portal vein ([I]inlet,max,u) was set equal to [I]H in eqs. 2 and 3 to predict interactions precipitated by inactivator/inhibitor, and 2) unbound in vivo maximum plasma concentration of inhibitor/inactivator ([I]max,u) was set equal to [I]H in eq. 2 and [I]inlet,max,u as [I]H in eq. 3 to predict interactions precipitated by inactivator (term A) and reversible inhibitor (term B), respectively. To predict the potential of orally administered CBD or THC to precipitate CYP2C9- and 3A-mediated interactions in the intestine, the calculated maximum intestinal fluid solubility of CBD or THC was set equal to [I]G in eqs. 4 and 5. To predict drug interactions precipitated by 11-OH-THC and COOH-THC, the [I]max,u of each metabolite was set equal to [I]H. To predict drug interactions precipitated by THC after inhalation, [I]max,u was set equal to [I]H in eqs. 2 and 3.
The orally administered CBD doses used to predict in vivo hepatic and intestinal CYP-mediated drug interactions were 70 mg (average low dose), 700 mg (average high dose), and 2000 mg (maximum dose used in the clinical studies) (Supplemental Table 1). For THC, oral doses of 20 mg (average low dose), 130 mg (average high dose), and 160 mg (maximum dose used in a clinical study) were used (Supplemental Table 2). The inhaled doses of THC were 25 mg (average low dose), 70 mg (average high dose), and 100 mg (maximum dose consumed for recreational use).
Results
Average and Maximum Doses and Plasma Concentrations of CBD and THC after Oral or Inhalational Administration when Used for Recreational or Medicinal Purposes.
The recommended dose of Epidiolex (CBD) is 2.5–10 mg/kg twice daily (https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210365lbl.pdf). However, in clinical trials, CBD has been tested at 0.5–25 mg/kg twice daily, with an average oral dose of 7.5 mg/kg twice daily, the equivalent of 600 mg twice daily for an average adult weighing 80 kg (Millar et al., 2019). In contrast, over-the-counter CBD products are used at much lower oral doses depending on the medical condition, including sleep disorders (20–80 mg twice daily), chronic pain (1.25–10 mg twice daily), movement problems (350 mg twice daily), schizophrenia (20–640 mg twice daily), and glaucoma (20–40 mg twice daily) (https://cbdoilreview.org/cbd-cannabidiol/cbd-dosage). These products are commonly available in 1000–10,000 mg packs of 30–300 mg CBD per dose (https://www.amazon.com/Pack-10000mg-Hemp-Relief-Stress/dp/B07VL3B6MG). Despite that CBD has a long terminal half-life (56–61 hours), minimal drug accumulation is reported following multiple doses (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210365Orig1s000ClinPharmR.pdf). Therefore, single doses of CBD were used to predict the magnitude of oral CBD-drug interactions instead of multiple daily doses. Low and high single oral doses of CBD averaged 70 and 700 mg, respectively (Supplemental Table 2). A maximum CBD dose of 2000 mg used in previous clinical studies was also tested (Devinsky et al., 2016; Hess et al., 2016; Rosenberg et al., 2017; Warren et al., 2017).
THC is consumed orally at doses of 1–2.5 mg (by inexperienced users) and 50–100 mg (by experienced users) for both recreational and medicinal purposes (https://www.leafly.com/news/cannabis-101/cannabis-edibles-dosage-guide-chart). In clinical studies, THC has been administered orally at doses ranging from 8 to 156 mg. The recommended dose of prescription THC (Marinol) is 10 mg twice daily. Based on these data, low and high single oral THC doses averaged 20 and 130 mg, respectively (Supplemental Table 3). The maximum oral dose of THC consumed for recreational or medicinal use was approximately 160 mg (Ware et al., 2015) and was used to predict the magnitude of oral THC-drug interactions.
THC is used recreationally at approximately 30–100 mg per joint, bong, or vaporizer (https://www.latimes.com/projects/la-me-weed-101-thc-calculator/), whereas the inhaled dose for medicinal use is lower at approximately 6–80 mg (Kahan et al., 2014; Ware et al., 2015). Based on these data, low and high single inhaled doses of THC averaged 25 and 70 mg, respectively (Supplemental Table 3). The maximum inhaled dose of THC was 100 mg (https://www.leafly.com/news/cannabis-101/cannabis-edibles-dosage-guide-chart) and was used to predict the magnitude of THC-drug interactions.
Based on the dose-normalized Cmax of CBD, THC, 11-OH-THC, and COOH-THC for various routes of administration of CBD and THC and their fraction unbound in plasma (fu,p) (Cox et al., 2019), [I]max,u of CBD, THC, and the THC metabolites were determined after oral or inhalational administration of CBD and THC (Table 1). The [I]inlet,max,u of CBD and THC was estimated as reported previously (Ito et al., 1998) (Table 1). These concentrations were incorporated in the mechanistic static model to predict the magnitude of cannabinoid-drug interactions.
TABLE 1.
Oral and inhalational doses and estimated maximum plasma concentrations of CBD, THC, and THC metabolites used for predicting the magnitude of pharmacokinetic cannabinoid-drug interactions
| Cannabinoid | CBD or THC Dose (mg) | Route of Administration | [I]max,u (nM)a | [I]inlet,max,u (µM)b |
|---|---|---|---|---|
| CBD | 70 | Oral | 2.94 | 0.12 |
| 700 | Oral | 29.4 | 1.24 | |
| 2000 | Oral | 84.0 | 3.55 | |
| THC | 20 | Oral | 0.33 | 0.01 |
| 130 | Oral | 2.15 | 0.03 | |
| 160 | Oral | 2.64 | 0.04 | |
| 25 | Inhalation | 2.72 | NA | |
| 70 | Inhalation | 7.62 | NA | |
| 100 | Inhalation | 10.89 | NA | |
| 11-OH-THC | 20 | Oral | 0.26 | – |
| 130 | Oral | 1.72 | – | |
| 160 | Oral | 2.11 | – | |
| 25 | Inhalation | 0.20 | NA | |
| 70 | Inhalation | 0.55 | NA | |
| 100 | Inhalation | 0.78 | NA | |
| COOH-THC | 20 | Oral | 3.19 | – |
| 130 | Oral | 20.75 | – | |
| 160 | Oral | 25.54 | – | |
| 25 | Inhalation | 1.12 | NA | |
| 70 | Inhalation | 3.13 | NA | |
| 100 | Inhalation | 4.48 | NA |
NA, not applicable; –, not estimated as data on fraction of dose metabolized to these metabolites by the intestine vs. liver are not available.
fu,p (CBD) = 0.07 (Taylor et al., 2019), fu,p (THC) = 0.011 (Patilea-Vrana and Unadkat, 2019), fu,p (11-OH-THC) = 0.012 (Patilea-Vrana and Unadkat, 2019); fu,p (COOH-THC) was not available and assumed to be the same as of 11-OH-THC, and Cmax/dose of CBD and THC after oral administration or inhalation were taken from Cox et al. (2019).
Fa (CBD and THC) = 1 (estimated based on FDA guidance), ka (CBD) = 0.0048 min−1 (estimated from Epidiolex (CBD) pharmacokinetic data using Phoenix WinNonlin (Phoenix WinNonlin 8.1; Certara USA, Princeton, NJ), ka (THC) = 0.0045 min−1 (Wolowich et al., 2019), QH (hepatic blood flow) = 1500 ml/min, and RB (THC) (blood to plasma ratio) = 0.4 (Schwilke et al., 2009); RB (CBD) value is not available and was assumed to be same as that for THC.
Biorelevant Gastrointestinal Solubility of CBD and THC.
The solubility of CBD and THC in FaSSIF buffer was 34 ± 7.5 and 28 ± 5.9 μM, respectively, and was similar to that in FeSSIF buffer (40 ± 2.5 and 36 ± 3.6 μM, respectively). Therefore, the latter was used for all oral cannabinoid-drug interaction predictions.
Nonspecific and HLM Incubation Binding of CBD, THC, and 11-OH-THC.
Nonspecific binding of CBD (0.1 and 5 µM), THC (0.5 and 5 µM), and 11-OH-THC (0.1 µM) to low-binding tubes was 69% ± 3%, 80% ± 9%, and 66% ± 5%, respectively. fu,inc of CBD (0.1 and 5 µM), THC (0.5 and 5 µM), and 11-OH-THC (0.1 µM) was 0.12 ± 04, 0.05 ± 02, and 0.16 ± 05, respectively. These results indicated that the nonspecific and microsomal protein binding of cannabinoids was concentration-independent.
Reversible Inhibition of CYPs by CBD, THC, and THC Metabolites.
CBD and THC inhibited CYP activity in a concentration-dependent manner (Figs. 2 and 3). Binding-corrected IC50 values (IC50,u) were determined using fu,inc. Compared with CBD, THC was an approximately 7.5- and 14-times more potent inhibitor of CYP1A2 and 2C9, respectively (Table 2), but was an approximately 3.5-times less potent inhibitor of CYP3A activity (Table 2). CBD and THC showed comparable inhibitory potency toward CYP2C19 and 2D6. 11-OH THC was a strong inhibitor of CYP2C9 and a relatively weak inhibitor of CYP2C19, 2D6, and 3A (Table 2). Compared with THC, 11-OH-THC was a weak inhibitor of CYP2C9, 2C19, 2D6, and 3A (Table 2). COOH-THC was a weak inhibitor of all CYPs tested (Table 2).
Fig. 2.

Concentration-dependent reversible inhibition of CYP activity in HLMs by CBD. The order of CBD inhibition potency was CYP2C9 > 2C19 ≈ 3A ≈ 1A2 > 2D6 (Table 2). Pooled HLMs (0.1 mg/ml protein) were incubated (37°C, 15 minutes) with a CYP cocktail consisting of phenacetin (CYP1A2; 50 μM), diclofenac (CYP2C9; 5 μM), omeprazole (CYP2C19; 10 μM), dextromethorphan (CYP2D6; 5 μM), and testosterone (CYP3A; 10 μM) and varying concentrations of CBD (0.003–100 µM) or vehicle (0.2% v/v DMSO). Data represent mean ± S.D. of three independent experiments, each conducted in duplicate. Solid lines represent model fit to the data.
Fig. 3.

Concentration-dependent reversible inhibition of CYP activity in HLMs by THC. The order of THC inhibition potency was CYP2C9 > 1A2 > 2C19 > 2D6 ≈ 3A (Table 2). THC was a more potent inhibitor of CYP2C9 and 1A2 than CBD, but it was a less potent inhibitor of CYP3A (Table 2). Pooled HLMs (0.1 mg/ml protein) were incubated (37°C, 10 minutes) with a CYP cocktail consisting of phenacetin (CYP1A2; 50 μM), diclofenac (CYP2C9; 5 μM), omeprazole (CYP2C19; 10 μM), dextromethorphan (CYP2D6; 5 μM), and testosterone (CYP3A; 10 μM) and varying concentrations of THC (0.003–100 µM) or vehicle (0.2% v/v DMSO). Data represent mean ± S.D. of three independent experiments, each conducted in duplicate. Solid lines represent model fit to the data.
TABLE 2.
IC50 and IC50,u values for CBD, THC, 11-OH-THC, and COOH-THC against select CYP activities in HLMs
Data represent means ± S.D. of three independent experiments, each conducted in duplicate.
| Enzyme | CBD | THC | 11-OH-THC | COOH-THCa | |||
|---|---|---|---|---|---|---|---|
| IC50 (μM) | IC50,u (μM) | IC50 (μM) | IC50,u (μM) | IC50 (μM) | IC50,u (μM) | IC50 (μM) | |
| CYP1A2 | 3.76 ± 1.44 | 0.45 ± 0.17 | 1.26 ± 0.31 | 0.06 ± 0.02 | 13.49 ± 1.19 | 2.16 ± 0.19 | >50 |
| CYP2C9 | 1.43 ± 0.28 | 0.17 ± 0.03 | 0.23 ± 0.03 | 0.012 ± 0.001 | 2.68 ± 0.92 | 0.43 ± 0.15 | >50 |
| CYP2C19 | 2.58 ± 0.46 | 0.30 ± 0.06 | 11.44 ± 4.44 | 0.57 ± 0.22 | 13.1 ± 1.91 | 2.1 ± 0.31 | >50 |
| CYP2D6 | 7.88 ± 4.14 | 0.95 ± 0.50 | 25.5 ± 4.9 | 1.28 ± 0.25 | 39.2 ± 7.72 | 6.27 ± 1.24 | >50 |
| CYP3A | 3.16 ± 0.96 | 0.38 ± 0.11 | 26.09 ± 6.78 | 1.30 ± 0.34 | >50 | >8 | >50 |
Unable to determine because of inability to inhibit by >50% at concentration range tested.
TDI of CYPs by CBD, THC, and THC Metabolites.
CBD (10 µM), after preincubation for 30 minutes, showed TDI of CYP1A2, 2C19, and 3A as evidenced by a decrease in activity by 83%, 75%, and 85%, respectively, as compared with the vehicle-treated control group (0 minutes) (Fig. 4). THC and its metabolites did not show TDI of any of the CYPs tested (Fig. 4). Based on these data, TDI parameters for CBD toward CYP1A2, 2C19, and 3A were determined using a shorter time period to minimize CBD depletion. These parameters (Kinact, KI,u, and kinact/KI,u) indicated that CBD was a time-dependent inhibitor of CYP1A2, 2C19, and 3A (Table 3). The efficiency (kinact/KI,u) of CBD to inactivate CYP1A2 was 5- to 6-fold greater than that for CYP2C19 or CYP3A (Fig. 5).
Fig. 4.
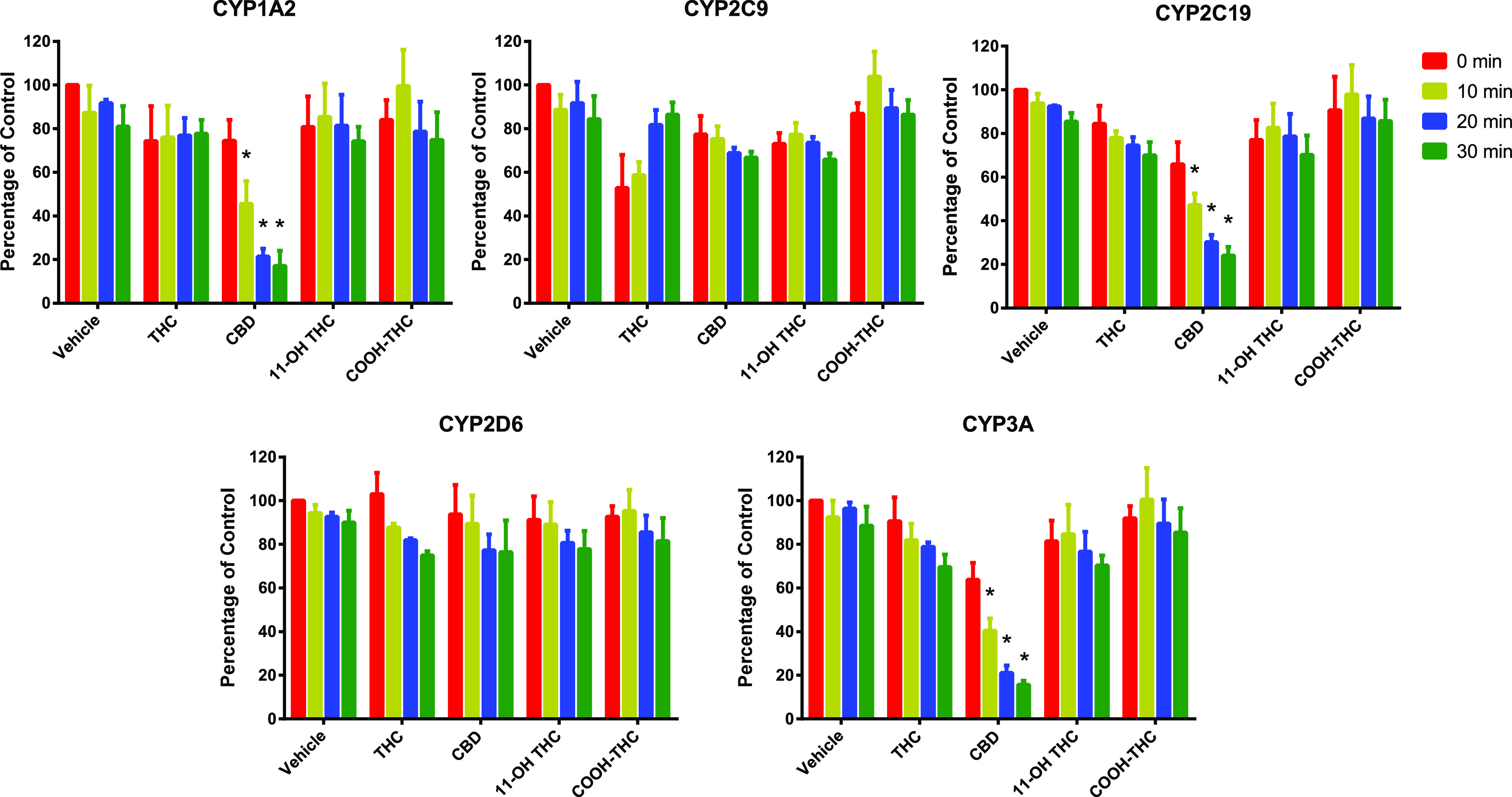
TDI of CYPs by CBD, THC, 11-OH-THC, and COOH-THC. When the cannabinoids were preincubated with HLMs only, CBD showed TDI of CYP1A2, 2C19, and 3A. Pooled HLMs (0.5 mg/ml protein) were preincubated with NADPH regenerating system, cannabinoid (THC, CBD, 11-OH-THC or COOH-THC; 10 μM), or vehicle (0.2% v/v DMSO) at 37°C for 0, 10, 20, or 30 minutes. Then, an aliquot (10 μl) of this mixture was incubated with NADPH regenerating system and a CYP substrate cocktail consisting of phenacetin (CYP1A2; 50 μM), diclofenac (CYP2C9; 5 μM), omeprazole (CYP2C19; 20 μM), dextromethorphan (CYP2D6; 5 μM), and testosterone (CYP3A; 20 μM) for 15 minutes. Data represent percent of activity in the vehicle-treated control group (0 minutes) that was not subjected to preincubation and are shown as means ± S.D. for three independent experiments. *P < 0.05, significantly different from the vehicle-treated control group (two-way analysis of variance test).
TABLE 3.
CBD inactivation kinetics of select CYPs in HLMs
Data represent means ± S.D. of four independent experiments.
| Enzyme | KI (μM) | KI,u (μM) | kinact (min−1) | kinact/KI,u (min−1 μM−1) |
|---|---|---|---|---|
| CYP1A2 | 0.95 ± 0.42 | 0.11 ± 0.05 | 0.07 ± 0.01 | 0.70 ± 0.34 |
| CYP2C19 | 3.33 ± 2.01 | 0.40 ± 0.24 | 0.04 ± 0.01 | 0.11 ± 0.06 |
| CYP3A | 4.83 ± 2.10 | 0.58 ± 0.25 | 0.08 ± 0.02 | 0.14 ± 0.04 |
Fig. 5.
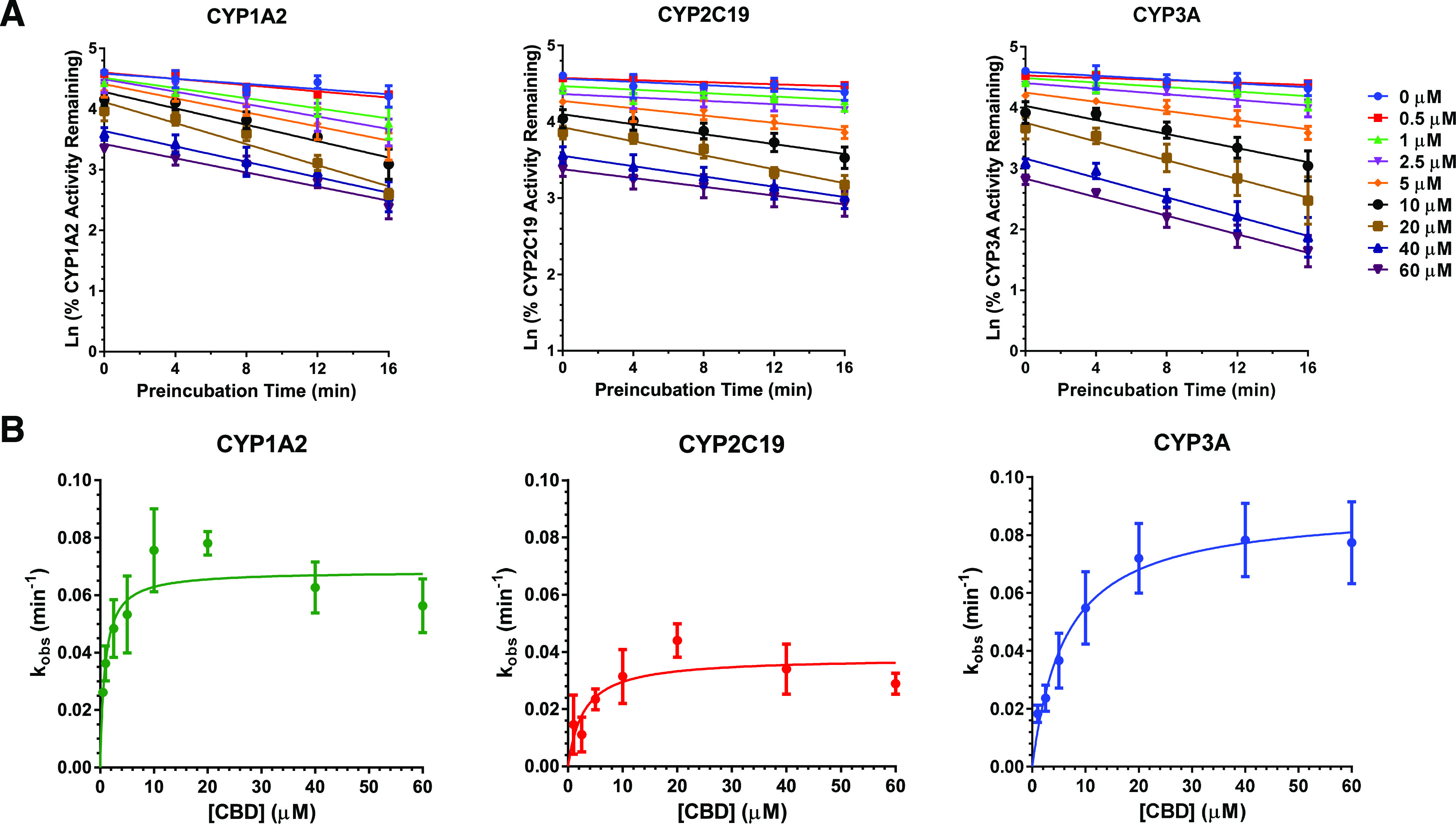
TDI of CYP1A2, 2C19, and 3A by various concentrations of CBD. The order of CBD inactivation potency was CYP1A2 > 2C19 ≈ CYP3A (Table 3). (A) Pooled HLMs (0.5 mg/ml protein) were preincubated with NADPH regenerating system, CBD (0.5, 1, 2.5, 5, 10, 20, 40, or 60 μM), or vehicle (0.2% v/v DMSO) at 37°C for 0, 4, 8, 12, or 16 minutes. Then, an aliquot (10 μl) of this mixture was incubated with NADPH regenerating system and a CYP substrate cocktail consisting of phenacetin (CYP1A2; 50 μM), diclofenac (CYP2C9; 5 μM), omeprazole (CYP2C19; 20 μM), dextromethorphan (CYP2D6; 5 μM), and testosterone (CYP3A; 20 μM) for 15 minutes. Data represent percent of activity in the vehicle-treated control group (0 minutes) that was not subjected to preincubation and are shown as means ± S.D. for four independent experiments. (B) Nonlinear regression model fits of the kobs data to estimate kinact and KI.
Prediction of In Vivo CYP-Mediated Cannabinoid-Drug Interactions.
Based on the combined hepatic and gut AUCR values (≥1.2) using [I]inlet,max,u for [I]H in eqs. 2 and 3 (approach 1), all tested CBD oral doses (70, 700, and 2000 mg) were predicted to precipitate pharmacokinetic interactions with theophylline, diclofenac, omeprazole, and midazolam, which are predominately metabolized by CYP1A2, 2C9, 2C19, and 3A4, respectively; the two higher doses were predicted to precipitate interactions with dextromethorphan, which is predominately metabolized by CYP2D6 (Table 4). CBD was predicted to precipitate strong interactions (AUCR > 5) with CYP2C19 and 3A substrates, even at the lowest average dose (Table 4). THC was predicted to precipitate interactions with CYP1A2, 2C9, and 3A4 substrates (AUCR > 1.2) only at high oral doses (130 and 160 mg). THC at the high average and maximum oral dose was predicted to precipitate strong interactions (AUCR > 5) with CYP2C9 substrates.
TABLE 4.
Prediction of the maximum magnitude of a drug interaction when CBD or THC is administered orally or by inhalation (THC only) with the indicated object drug based on the ability of the cannabinoids to inhibit CYPs in a time-dependent and/or reversible manner
| Precipitant | CYP Enzyme | Object Drug | Predicted AUCR after Oral Administration (Inhibition of Hepatic and Gut Metabolism) | Predicted AUCR after Inhalation (Inhibition of Hepatic Metabolism) | |
|---|---|---|---|---|---|
| Iinlet,max,ua | Imax,u and Iinlet,max,ub | Imax,uc | |||
| CBD (70 mg, oral) | 1A2 | Theophylline | 3.9 | 3.0 | – |
| 2C9 | Diclofenac | 2.6 | 2.6 | – | |
| 2C19 | Omeprazole | 6.2 | 1.9 | – | |
| 2D6 | Dextromethorphan | 1.1 | 1.1 | – | |
| 3A | Midazolam | 13.5 | 4.4 | – | |
| CBD (700 mg, oral) | 1A2 | Theophylline | 4.0 | 3.9 | – |
| 2C9 | Diclofenac | 11.1 | 11.1 | – | |
| 2C19 | Omeprazole | 7.5 | 6.4 | – | |
| 2D6 | Dextromethorphan | 2.3 | 2.3 | – | |
| 3A | Midazolam | 15.0 | 13.4 | – | |
| CBD (2000 mg, oral) | 1A2 | Theophylline | 4.0 | 4.0 | – |
| 2C9 | Diclofenac | 23.6 | 23.6 | – | |
| 2C19 | Omeprazole | 7.6 | 7.4 | – | |
| 2D6 | Dextromethorphan | 4.8 | 4.8 | – | |
| 3A | Midazolam | 15.1 | 14.8 | – | |
| THC (20 mg, oral) | 1A2 | Theophylline | 1.1 | ─ | – |
| 2C9 | Diclofenac | 2.2 | ─ | – | |
| 2C19 | Omeprazole | 1.0 | ─ | – | |
| 2D6 | Dextromethorphan | 1.0 | ─ | – | |
| 3A | Midazolam | 1.8 | ─ | – | |
| THC (130 mg, oral) | 1A2 | Theophylline | 1.4 | ─ | – |
| 2C9 | Diclofenac | 5.6 | ─ | – | |
| 2C19 | Omeprazole | 1.1 | ─ | – | |
| 2D6 | Dextromethorphan | 1.0 | ─ | – | |
| 3A | Midazolam | 1.8 | ─ | – | |
| THC (160 mg, oral) | 1A2 | Theophylline | 1.4 | ─ | – |
| 2C9 | Diclofenac | 6.5 | ─ | – | |
| 2C19 | Omeprazole | 1.1 | ─ | – | |
| 2D6 | Dextromethorphan | 1.0 | ─ | – | |
| 3A | Midazolam | 1.8 | ─ | – | |
| THC (25 mg, inhaled) | 1A2 | Theophylline | – | ─ | 1.0 |
| 2C9 | Diclofenac | – | ─ | 1.9 | |
| 2C19 | Omeprazole | – | ─ | 1.0 | |
| 2D6 | Dextromethorphan | – | ─ | 1.0 | |
| 3A | Midazolam | – | ─ | 1.8 | |
| THC (70 mg, inhaled) | 1A2 | Theophylline | – | ─ | 1.1 |
| 2C9 | Diclofenac | – | ─ | 2.5 | |
| 2C19 | Omeprazole | – | ─ | 1.0 | |
| 2D6 | Dextromethorphan | – | ─ | 1.0 | |
| 3A | Midazolam | – | ─ | 1.8 | |
| THC (100 mg, inhaled) | 1A2 | Theophylline | – | ─ | 1.1 |
| 2C9 | Diclofenac | – | ─ | 2.9 | |
| 2C19 | Omeprazole | – | ─ | 1.0 | |
| 2D6 | Dextromethorphan | – | ─ | 1.0 | |
| 3A | Midazolam | – | ─ | 1.8 | |
─, time-dependent inhibition was not evident; –, not predicted.
Estimated unbound plasma concentration of inhibitor/inactivator in portal vein.
Imax,u for inactivation and Iinlet,max,u for reversible inhibition.
Unbound systemic plasma concentration of inhibitor/inactivator.
The predictions of orally administered CBD-drug interaction using [I]max,u for [I]H in inactivation term A in eq. 2 and [I]inlet,max,u for [I]H in reversible inhibition term B in eq. 3 (approach 2) yielded, as expected, lower AUCR values, but the differences were modest (except interactions between omeprazole and midazolam at 70 mg), as compared with the values predicted using approach 1 (Table 4). Inhalational THC was predicted to precipitate systemic interactions only with CYP2C9 substrates (AUCR > 1.9) (Table 4). THC metabolites were predicted to have no interactions with any of the probe substrates (data not shown).
Discussion
Previous CYP inhibition studies involving CBD and THC as precipitants (Yamaori et al., 2010, 2011b; Arnold et al., 2018) did not consider the limited aqueous solubility, nonspecific binding to labware, or extensive binding to incubation protein for each cannabinoid. Therefore, the reported CYP inhibition potencies (IC50 or KI) of these cannabinoids are likely underestimated. The current work is the first to consider these properties to estimate true CYP inhibition potencies. BSA (0.2%) was added to the incubation mixtures to increase CBD or THC solubility and reduce nonspecific binding to plastic tubes and tips (Patilea-Vrana and Unadkat, 2019). Low-binding Eppendorf tubes were used to further reduce nonspecific binding. Under these optimal experimental conditions, the extent of nonspecific binding of CBD and THC in the incubation mixture (fu,inc) was used to determine binding-corrected inhibition potency (IC50,u or KI,u).
CBD or THC demonstrated reversible inhibition of CYP1A2, 2C9, 2C19, 2D6, and 3A activities in HLMs (Figs. 2 and 3; Table 2), consistent with previous reports (Yamaori et al., 2011a,b, 2012; Jiang et al., 2013). In the present study, the rank order of reversible inhibition (based on IC50,u values) of the tested CYPs by CBD and THC was CYP2C9 > 2C19 ≈ 3A ≈ 1A2 > 2D6 and CYP2C9 > 1A2 > 2C19 > 2D6 ≈ 3A, respectively (Table 2). The previously reported IC50 or Ki values (Yamaori et al., 2011a, 2012) are approximately 2- to 6-fold higher than those determined in the current study (Table 2), supporting low aqueous solubility and nonspecific binding of CBD and THC (Garrett and Hunt, 1974) as plausible explanations for the higher values. Another plausible explanation could reflect CYP3A5 genotype of a given lot of HLMs, as studies using recombinant enzymes suggest that CBD is a more potent inhibitor of CYP3A5 than CYP3A4 (Yamaori et al., 2011a).
The FDA recommends assessing the inhibitory effects of metabolites on CYP enzymes if the metabolite AUC exceeds parent AUC by ≥25% (https://www.fda.gov/media/134582/download). Despite the fact that the AUC of 11-OH-THC (930 µg/minute/l) and COOH-THC (14,600 µg/minute/l) is ∼2.5- and ∼40-fold higher, respectively, than that of THC (360 µg/minute/l) after oral administration of 10 mg THC (Nadulski et al., 2005), CYP inhibition potency of these metabolites has not been determined. The present study is the first to determine the IC50,u of 11-OH THC and COOH-THC against CYP enzymes. 11-OH THC was a reversible inhibitor of CYP2C9, 2C19, 2D6, and CYP3A, but the IC50,u values were much greater than those for THC, whereas COOH-THC did not inhibit any of the CYPs by an appreciable extent.
The efficiency of CBD to inactivate multiple CYPs in HLMs was determined for the first time. CBD was a time-dependent inhibitor of three CYPs, showing the strongest inhibition against CYP1A2, followed by 2C19 and 3A (Fig. 5). Previously, only the efficiency of TDI (kinact/KI) of CYP1A2 by CBD had been reported (0.19 minutes−1/μM−1) (Yamaori et al., 2010), which was ∼3.5-times lower than that determined in the present study (0.70 minutes−1/μM−1; Table 3), a difference likely attributed to CBD nonspecific binding and poor aqueous solubility. Comparatively, kinact/KI for CBD (0.074 minutes−1/μM−1) and kinact/KI,u for CBD (0.11 minutes−1/μM−1) in the present study were comparable to that of furafylline (kinact/KI = 0.12 minutes−1/μM−1) (Obach et al., 2007) and ticlopidine (kinact/KI,u = 0.17 minutes−1/μM−1) (Nishiya et al., 2009), the prototypic time-dependent inhibitors of CYP1A2 and 2C19, respectively. In contrast, CBD (kinact/KI = 0.017 minutes−1/μM−1) was approximately 90-times less efficient than ritonavir (kinact/KI = 1.55 minutes−1/μM−1) as a time-dependent inhibitor of CYP3A (Obach et al., 2007). These comparisons should ideally be made using the unbound KI. However, we were unable to find these values for furafylline and ritonavir in the literature.
Unlike CBD, THC, 11-OH-THC, and COOH-THC showed no inactivation of any of the CYPs tested, suggesting a role for the hydroxy group of the resorcinol moiety in CBD [absent in THC, 11-OH-THC, or COOH-THC (Fig. 1)] in the TDI of CYP1A2, 2C19, and 3A. The mechanism(s) of CYP TDI by CBD is not known but could involve the formation of CBD-hydroxyquinone as reported for TDI of murine Cyp3a11 (Bornheim and Grillo, 1998).
Knowledge of the unbound concentration of an inhibitor/inactivator [I] at the target enzyme is needed to predict a drug interaction accurately. Because this metric cannot be measured directly, [I]max,u or [I]inlet,max,u was used as a surrogate of [I] in the liver. The drug interaction potential of each cannabinoid after oral administration was predicted using [I]inlet,max,u (for both reversible and time-dependent inhibition) and using both [I]inlet,max,u and [I]max,u (for both reversible as well as time-dependent inhibition, respectively). The latter approach has been shown to better predict the magnitude of drug interactions for inhibitors that are reversible and time-dependent inhibitors of CYPs (Ito et al., 2004; Obach et al., 2006, 2007). After THC inhalation, AUCR predictions were made using [I]max,u (Table 4). To predict the magnitude of cannabinoid-drug interactions in the intestine, the maximum intestinal fluid solubility ([I]G), determined using FeSSIF buffer, was used as a surrogate for the concentration available in the intestine to inhibit CYP2C9 and 3A.
CBD was predicted to precipitate strong drug interactions (AUCR ≥ 5) mediated by CYP2C9, 2C19, and 3A and moderate drug interactions (1.2 ≤ AUCR < 5) mediated by CYP1A2 and 2D6 based on the AUCR cutoffs recommended by the FDA (https://www.fda.gov/media/134582/download). These predictions are largely consistent with clinical CBD- or THC-drug interactions reported in the literature. For example, oral administration of CBD (5–25 mg/kg per day) with the anticonvulsant clobazam led to a marked increase (about fivefold) in plasma concentrations of the metabolite N-desmethylclobazam, which is metabolized predominantly by CYP2C19 (Geffrey et al., 2015; Gaston and Szaflarski, 2018). Oral administration of CBD (750 mg twice daily) with clobazam and stiripentol (a CYP2C19 substrate) led to a 3.4- and 1.6-fold increase in N-desmethylclobazam and stiripentol AUC, respectively (Morrison et al., 2019). Likewise, oral CBD (600 mg/day for 5–12 days) increased the AUC (by 51%) of oral hexobarbital, which is partially cleared by CYP2C19 (Benowitz et al., 1980). In addition, one case report described an increased international normalized ratio when CBD was coadministered with warfarin (Grayson et al., 2017), which is cleared largely by CYP2C9. In contrast, although a strong interaction between CBD and oral midazolam was predicted, there was minimal change in the AUC of midazolam after chronic administration of CBD (250 mg/day on days 1–11, 750 mg twice/day on days 12–25) (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210365Orig1s000ClinPharmR.pdf). Possible induction of CYP3A4 (mRNA) by CBD in human hepatocytes (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210365Orig1s000ClinPharmR.pdf) may explain this discrepancy.
After oral administration, THC was predicted to produce strong CYP2C9-mediated (AUCR ≥ 5) but weak CYP1A2- and 3A-mediated (AUCR < 2) drug interactions. After inhalation, THC was predicted to produce drug interactions only with drugs extensively metabolized by CYP2C9. A case report involving THC and warfarin supports this prediction (Yamreudeewong et al., 2009), in which the patient’s international normalized ratio increased to 11.55 because of frequent cannabis smoking. In contrast, regardless of the inhaled THC dose, neither 11-OH-THC nor COOH-THC was predicted to precipitate interactions with CYP2C9, 2C19, 2D6, or 3A substrates (Table 4).
There are several limitations to the current work. First, the depletion of CBD or THC during the incubations was not considered when estimating IC50 or KI values. Thus, these observed values may be higher than the true IC50 or KI values. Second, when determining the IC50 for CBD, the possibility of simultaneous TDI of CYP1A2, 2C19, or 3A during the incubation period cannot be discounted. This scenario could result in estimation of lower IC50 values than the true values. Third, our drug interaction predictions are based on maximum cannabinoid plasma concentrations, which remain static. In humans, plasma concentrations of CBD and THC decrease rapidly after inhalation, whereas after oral administration, the decrease is more gradual (Ohlsson et al., 1986; Huestis, 2007). As such, PBPK modeling and simulation of cannabinoid-drug interactions is underway to capture these dynamic changes. These PBPK models should lead to improved predictions of interactions mediated by the formation of 11-OH-THC and COOH-THC during first pass. Although the majority of marijuana products on the market are either CBD rich or THC rich, there are some products that contain both CBD and THC. Based on the data presented here, drug interaction when both are simultaneously present can be predicted using PBPK modeling and simulation. Previously, CBD was reported to inhibit CYP2B6, 2C8 (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210365Orig1s000ClinPharmR.pdf; Yamaori et al., 2011a), and non-CYP enzymes such as UGT1A9 and 2B7 (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210365Orig1s000ClinPharmR.pdf). Potential interactions between CBD and drugs metabolized by these enzymes as well as drug transporters such as P-glycoprotein and breast cancer–resistance protein should also be evaluated (Alsherbiny and Li, 2018).
In conclusion, a combined mechanistic static model predicted a moderate to strong pharmacokinetic interaction risk between orally administered CBD and drugs extensively metabolized by CYP1A2, 2C9, 2C19, 2D6, or 3A and between orally administered THC and drugs extensively metabolized by CYP1A2, 2C9, or 3A. With respect to inhalational administration, THC was predicted to produce interactions only with drugs extensively metabolized by CYP2C9. These predictions need to be verified by a well designed clinical drug interaction study using prototypic CYP substrates.
Abbreviations
- AUC
area under the plasma concentration versus time curve
- AUCR
ratio of AUC of object drug in the presence to absence of inhibitor
- BSA
bovine serum albumin
- CBD
cannabidiol
- FaSSIF
fasted state simulated intestinal fluid
- FeSSIF
fed state simulated intestinal fluid
- G6P
D-glucose 6- phosphate
- FDA
US Food and Drug Administration
- HLM
human liver microsome
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- CYP
cytochrome P450
- PBPK
physiologically based pharmacokinetic
- TDI
time-dependent inhibition
- THC
(−)-trans-Δ9-tetrahydrocannabinol
- UPLC
ultra-high-performance liquid chromatography
- 11-OH-THC
11-hydroxy-THC
- COOH-THC
11-nor-9-carboxy-∆9-THC
Authorship Contributions
Participated in research design: Bansal, Maharao, Unadkat.
Conducted experiments: Bansal, Maharao.
Performed data analysis: Bansal, Maharao.
Wrote or contributed to the writing of the manuscript: Bansal, Maharao, Paine, Unadkat.
Footnotes
This work was supported by National Institutes of Health National Center for Complementary and Integrative Health [Grant U54 AT008909] (to M.F.P.) and in part by a grant from National Institute for Drug Abuse [Grant P01 DA032507] (to J.D.U.).
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Alsherbiny MA, Li CG. (2018) Medicinal cannabis-potential drug interactions. Medicines (Basel) 6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold WR, Weigle AT, Das A. (2018) Cross-talk of cannabinoid and endocannabinoid metabolism is mediated via human cardiac CYP2J2. J Inorg Biochem 184:88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL, Nguyen TL, Jones RT, Herning RI, Bachman J. (1980) Metabolic and psychophysiologic studies of cannabidiol-hexobarbital interaction. Clin Pharmacol Ther 28:115–120. [DOI] [PubMed] [Google Scholar]
- Bornheim LM, Grillo MP. (1998) Characterization of cytochrome P450 3A inactivation by cannabidiol: possible involvement of cannabidiol-hydroxyquinone as a P450 inactivator. Chem Res Toxicol 11:1209–1216. [DOI] [PubMed] [Google Scholar]
- Bose J, Hedden SL, Lipari RN, Park-LeeKey E. (2016) Substance Use and Mental Health Indicators in the United States: Results from the 2015 National Survey on Drug Use and Health. https://www.samhsa.gov/data/sites/default/files/NSDUH-FFR1-2015/NSDUH-FFR1-2015/NSDUH-FFR1-2015.pdf
- Chen Z-H, Zhang S-X, Long N, Lin L-S, Chen T, Zhang F-P, Lv X-Q, Ye P-Z, Li N, Zhang K-Z. (2016) An improved substrate cocktail for assessing direct inhibition and time-dependent inhibition of multiple cytochrome P450s. Acta Pharmacol Sin 37:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108. [DOI] [PubMed] [Google Scholar]
- Cheong EJY, Goh JJN, Hong Y, Venkatesan G, Liu Y, Chiu GNC, Kojodjojo P, Chan ECY. (2017) Application of static modeling --in the prediction of in vivo drug-drug interactions between rivaroxaban and antiarrhythmic agents based on in vitro inhibition studies. Drug Metab Dispos 45:260–268. [DOI] [PubMed] [Google Scholar]
- Cox EJ, Maharao N, Patilea-Vrana G, Unadkat JD, Rettie AE, McCune JS, Paine MF. (2019) A marijuana-drug interaction primer: precipitants, pharmacology, and pharmacokinetics. Pharmacol Ther 201:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlinger D, Duechting S, Nuecken D, Sydow K, Fuhr U, Frechen S. (2016) Development and validation of an in vitro, seven-in-one human cytochrome P450 assay for evaluation of both direct and time-dependent inhibition. J Pharmacol Toxicol Methods 77:66–75. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Marsh E, Friedman D, Thiele E, Laux L, Sullivan J, Miller I, Flamini R, Wilfong A, Filloux F, et al. (2016) Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol 15:270–278. [DOI] [PubMed] [Google Scholar]
- Dinger J, Meyer MR, Maurer HH. (2014) Development and validation of a liquid-chromatography high-resolution tandem mass spectrometry approach for quantification of nine cytochrome P450 (CYP) model substrate metabolites in an in vitro CYP inhibition cocktail. Anal Bioanal Chem 406:4453–4464. [DOI] [PubMed] [Google Scholar]
- Dixit V, Hariparsad N, Desai P, Unadkat JD. (2007) In vitro LC-MS cocktail assays to simultaneously determine human cytochrome P450 activities. Biopharm Drug Dispos 28:257–262. [DOI] [PubMed] [Google Scholar]
- Fahmi OA, Maurer TS, Kish M, Cardenas E, Boldt S, Nettleton D. (2008) A combined model for predicting CYP3A4 clinical net drug-drug interaction based on CYP3A4 inhibition, inactivation, and induction determined in vitro. Drug Metab Dispos 36:1698–1708. [DOI] [PubMed] [Google Scholar]
- Frytak S, Moertel CG, Rubin J. (1984) Metabolic studies of delta-9-tetrahydrocannabinol in cancer patients. Cancer Treat Rep 68:1427–1431. [PubMed] [Google Scholar]
- Garrett ER, Hunt CA. (1974) Physiochemical properties, solubility, and protein binding of delta9-tetrahydrocannabinol. J Pharm Sci 63:1056–1064. [DOI] [PubMed] [Google Scholar]
- Gaston TE, Szaflarski JP. (2018) Cannabis for the treatment of epilepsy: an update. Curr Neurol Neurosci Rep 18:73. [DOI] [PubMed] [Google Scholar]
- Geffrey AL, Pollack SF, Bruno PL, Thiele EA. (2015) Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 56:1246–1251. [DOI] [PubMed] [Google Scholar]
- Grayson L, Vines B, Nichol K, Szaflarski JP, UAB CBD Program (2017) An interaction between warfarin and cannabidiol, a case report. Epilepsy Behav Case Rep 9:10–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotenhermen F. (2003) Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet 42:327–360. [DOI] [PubMed] [Google Scholar]
- Hess EJ, Moody KA, Geffrey AL, Pollack SF, Skirvin LA, Bruno PL, Paolini JL, Thiele EA. (2016) Cannabidiol as a new treatment for drug-resistant epilepsy in tuberous sclerosis complex. Epilepsia 57:1617–1624. [DOI] [PubMed] [Google Scholar]
- Huestis MA. (2007) Human cannabinoid pharmacokinetics. Chem Biodivers 4:1770–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Brown HS, Houston JB. (2004) Database analyses for the prediction of in vivo drug-drug interactions from in vitro data. Br J Clin Pharmacol 57:473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. (1998) Prediction of pharmacokinetic alterations caused by drug-drug interactions: metabolic interaction in the liver. Pharmacol Rev 50:387–412. [PubMed] [Google Scholar]
- Jiang R, Yamaori S, Okamoto Y, Yamamoto I, Watanabe K. (2013) Cannabidiol is a potent inhibitor of the catalytic activity of cytochrome P450 2C19. Drug Metab Pharmacokinet 28:332–338. [DOI] [PubMed] [Google Scholar]
- Kahan M, Srivastava A, Spithoff S, Bromley L. (2014) Prescribing smoked cannabis for chronic noncancer pain: preliminary recommendations. Can Fam Physician 60:1083–1090. [PMC free article] [PubMed] [Google Scholar]
- Millar S.A., Stone N.L., Bellman Z.D., Yates A.S., England T.J., O’Sullivan S.E. (2019) A systematic review of cannabidiol dosing in clinical populations. Br J Clin Pharmacol 85 (9):1888–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison G, Crockett J, Blakey G, Sommerville K. (2019) A phase 1, open-label, pharmacokinetic trial to investigate possible drug-drug interactions between clobazam, stiripentol, or valproate and cannabidiol in healthy subjects. Clin Pharmacol Drug Dev 8:1009–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadulski T, Pragst F, Weinberg G, Roser P, Schnelle M, Fronk E-M, Stadelmann AM. (2005) Randomized, double-blind, placebo-controlled study about the effects of cannabidiol (CBD) on the pharmacokinetics of Delta9-tetrahydrocannabinol (THC) after oral application of THC verses standardized cannabis extract. Ther Drug Monit 27:799–810. [DOI] [PubMed] [Google Scholar]
- Nishiya Y, Hagihara K, Kurihara A, Okudaira N, Farid NA, Okazaki O, Ikeda T. (2009) Comparison of mechanism-based inhibition of human cytochrome P450 2C19 by ticlopidine, clopidogrel, and prasugrel. Xenobiotica 39:836–843. [DOI] [PubMed] [Google Scholar]
- Obach RS, Walsky RL, Venkatakrishnan K. (2007) Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug-drug interactions. Drug Metab Dispos 35:246–255. [DOI] [PubMed] [Google Scholar]
- Obach RS, Walsky RL, Venkatakrishnan K, Gaman EA, Houston JB, Tremaine LM. (2006) The utility of in vitro cytochrome P450 inhibition data in the prediction of drug-drug interactions. J Pharmacol Exp Ther 316:336–348. [DOI] [PubMed] [Google Scholar]
- Ohlsson A, Lindgren JE, Andersson S, Agurell S, Gillespie H, Hollister LE. (1986) Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed Environ Mass Spectrom 13:77–83. [DOI] [PubMed] [Google Scholar]
- Patilea-Vrana GI, Unadkat JD. (2019) Quantifying hepatic enzyme kinetics of (-)-∆9-Tetrahydrocannabinol (THC) and its psychoactive metabolite, 11-OH-THC, through in vitro modeling. Drug Metab Dispos 47:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg EC, Louik J, Conway E, Devinsky O, Friedman D. (2017) Quality of Life in Childhood Epilepsy in pediatric patients enrolled in a prospective, open-label clinical study with cannabidiol. Epilepsia 58:e96–e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer Gillian L., King Brian A., Bunnell Rebecca E., Promoff Gabbi, McAfee Timothy A. (2016) Toking, Vaping, and Eating for Health or Fun. American Journal of Preventive Medicine 50 (1):1–8. [DOI] [PubMed] [Google Scholar]
- Schwilke EW, Karschner EL, Lowe RH, Gordon AM, Cadet JL, Herning RI, Huestis MA. (2009) Intra- and intersubject whole blood/plasma cannabinoid ratios determined by 2-dimensional, electron impact GC-MS with cryofocusing. Clin Chem 55:1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaggiari D, Geiser L, Daali Y, Rudaz S. (2014) Phenotyping of CYP450 in human liver microsomes using the cocktail approach. Anal Bioanal Chem 406:4875–4887. [DOI] [PubMed] [Google Scholar]
- Taylor L, Crockett J, Tayo B, Morrison G. (2019) A phase 1, open-label, parallel-group, single-dose trial of the pharmacokinetics and safety of cannabidiol (CBD) in subjects with mild to severe hepatic impairment. J Clin Pharmacol 59:1110–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware MA, Wang T, Shapiro S, Collet J-P, COMPASS Study Team (2015) Cannabis for the management of pain: assessment of safety study (COMPASS). J Pain 16:1233–1242. [DOI] [PubMed] [Google Scholar]
- Warren PP, Bebin EM, Nabors LB, Szaflarski JP. (2017) The use of cannabidiol for seizure management in patients with brain tumor-related epilepsy. Neurocase 23:287–291. [DOI] [PubMed] [Google Scholar]
- Wolowich WR, Greif R, Kleine-Brueggeney M, Bernhard W, Theiler L. (2019) Minimal physiologically based pharmacokinetic model of intravenously and orally administered delta-9-tetrahydrocannabinol in healthy volunteers. Eur J Drug Metab Pharmacokinet 44:691–711. [DOI] [PubMed] [Google Scholar]
- Yamaori S, Ebisawa J, Okushima Y, Yamamoto I, Watanabe K. (2011a) Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci 88:730–736. [DOI] [PubMed] [Google Scholar]
- Yamaori S, Koeda K, Kushihara M, Hada Y, Yamamoto I, Watanabe K. (2012) Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metab Pharmacokinet 27:294–300. [DOI] [PubMed] [Google Scholar]
- Yamaori S, Kushihara M, Yamamoto I, Watanabe K. (2010) Characterization of major phytocannabinoids, cannabidiol and cannabinol, as isoform-selective and potent inhibitors of human CYP1 enzymes. Biochem Pharmacol 79:1691–1698. [DOI] [PubMed] [Google Scholar]
- Yamaori S, Okamoto Y, Yamamoto I, Watanabe K. (2011b) Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab Dispos 39:2049–2056. [DOI] [PubMed] [Google Scholar]
- Yamreudeewong W, Wong HK, Brausch LM, Pulley KR. (2009) Probable interaction between warfarin and marijuana smoking. Ann Pharmacother 43:1347–1353. [DOI] [PubMed] [Google Scholar]