

Abstract
Background
The coronavirus (CoV) spike (S) protein is critical for receptor binding, membrane fusion and internalization of the virus into the human cells. We have tried to search the epitopic component of the S-protein that might be served as crucial targets for the vaccine development and also tried to understand the molecular mechanism of epitopes and TLR4/MD-2 complex for adaptive immunity.
Material and methods
Here we identified the antigenicity and the epitopic divergence of S-protein via immunoinformatics approach. The study was performed to identify the epitopes, composition of amino acids and its distribution in epitopic regions, composition of amino acid between the identified epitopes, secondary structure architecture of epitopes, physicochemical and biochemical parameters and molecular interaction between the identified epitope and TLR4/MD-2 complex. The SARS-CoV-2 can be possibly recognised by TLR4 of host immune cells that are responsible for the adaptive immune response.
Results
We identified four SARS-CoV-2 S-protein 9mer antigenic epitopes and observed that they bind with the TLR4/MD-2 complex by varied stable molecular bonding interactions. Molecular interaction between these characterized epitopes with TLR4/MD-2 complex might be indicated the binding affinity and downstream signalling of adaptive immune response. Different physicochemical and biochemical parameters such as O-glycosylation and N-glycosylation, Hydrophobicity, GRAVY were identified within epitopic regions of S-protein. These parameters help to understand the protein-protein interaction between epitopes and TLR4/MD-2 complex. The study also revealed different epitopic binding pockets of TLR4/MD-2 complex.
Conclusions
The identified epitopes impart suitable prospects for the development of novel peptide-based epitopic vaccine for the control of COVID-19 infection.
Keywords: Spike protein, SARS-CoV-2, Antigenicity, Epitopes, TLR4/MD-2 complex
1. Introduction
Since the beginning of the 21st century three coronavirus (CoV) strains emerged as the causative agents of life threatening pneumonia-like symptoms in humans, namely Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) in the year 2002–03, Middle-East Respiratory Syndrome Coronavirus (MERS-CoV) in the year 2010–12 and SARS-CoV-2 in the year 2019–20 (Chakraborty et al., 2020a). Although among the family of coronaviruses SARS-CoV and MERS-CoV can cause severe pathological condition, only SARS-CoV-2 was declared pandemic by World Health Organization (WHO) (Chakraborty et al., 2020b). It also suggested that SARS-CoV-2 genome organization is closely related (79.7%) to SARS-CoV (Zhang et al., 2020; Zhou et al., 2020). COVID-19 has claimed more than 400,000 lives globally, till date. Thus, although the development of prophylactic vaccine and therapy against COVID-19 is of utmost importance, no vaccines or treatments for COVID-19 are available so far. However, the potential therapeutic candidates and vaccines are on the way to clinical trials (Amanat and Krammer, 2020), thus supportive therapies are the current choice for COVID-19 treatment (Le Thanh et al., 2020).
The SARS-CoV-2 proteome consist with numerous potent vaccine candidate proteins and drug targets. These proteins are significantly concerned within the host-virus interaction and pathogenesis. They comprise spike (S) glycoprotein, envelope (E), nucleocapsid (N) protein, membrane (M) protein and others associates open reading frames (ORFs) encoded part (Srivastava et al., 2019). S-protein of SARS-CoV-2 is a viral transmembrane structural glycoprotein, which has high binding affinity to ACE2 receptor of human host cells (Wan et al., 2020), and is also characterized by its capacity to activate immune system (Ou et al., 2020). The E-protein act a determining component in virus–host cell interaction and viral pathogenesis (Jimenez-Guardeno et al., 2014). While the M-protein part mediate budding and assemblage of viral particles that are critical for viral pathogenesis. The N-protein segments of viral genome is in the form of a helical ribonucleocapsid and is essentially involved into the viral self-assembly and pathogenesis (Schoeman and Fielding, 2019). All the mentioned proteins of SARS-CoV-2 are significantly implicated in viral infection, proliferation or host cell pathogenesis and consequently recognised as the potential targets for effective vaccine or drug development.
The Toll-like receptor (TLR) proteins identify pathogenic microbial elements and activate the pathway for functioning and regulation of adaptive immune response (Chakraborty et al., 2020c; Iwasaki and Medzhitov, 2004). Among TLRs family, TLR4 type receptor protein associates with the myeloid differentiation protein 2 (MD2) to form a complex that interacts with viral antigens (Choudhury and Mukherjee, 2020; Fransen et al., 2010). The TLR4 expressed CD4+ T-cell containing signalling pathway have facilitated the development of safe and effective clinical-grade vaccines in human (Duthie et al., 2011). Additionally, TLR4 served as the ligand-based adjuvants which are most innovative in the development of commercial vaccines and is important for the specific adaptive responses (Reed et al., 2016).
Current study aim to screen the potential multi-epitopes that are common for B-cells and T-cells from SARS-CoV-2 S-protein sequence. The epitopes are characterized as cytotoxic T lymphocyte (CTL) epitopes, and B-cell epitopes compare to human leukocyte antigen surface marker with potential to elicit cellular and humoral immune responses. Since the whole S-protein is not responsible to trigger B-cells and T-cells mediated immune response, here, we predicted B-cells and T-cells antigenic epitopes within the full length S-protein of SARS-CoV-2 that can bind to TLR4/MD-2 complex and might activate the adaptive immune response.
2. Materials and methods
2.1. Retrieval of proteomic data
The complete amino acid sequence (GenBank: QIH45053.1) of SARS-CoV-2 S-protein was retrieved in FASTA format from the National Centre for Biotechnology Information (NCBI) database (Coordinators, 2017). The obtained amino acid sequence was used for the prediction of antigenic epitopic that can be recognised by T-cells and B-cells.
2.2. Model protein structure generation of SARS-CoV-2 S-protein and its collection of PDB file
To identify the location of epitopes in 3D structure of S- protein we developed the 3D structure model of SARS-CoV-2 S-protein. We used I-TASSER server for this model development (Roy et al., 2010). Simultaneously we have extracted SARS-CoV-2 S-protein from PDB databank (6VYV) for further analysis (Berman et al., 2000).
2.3. Identification and selection of B-cell epitopes
B-cell epitopes are recognised by lymphocytes and activate B-lymphocytes to secrete antibodies. The Immune Epitope Database (IEDB) prediction server cooperated BepiPred Linear Epitope Prediction algorithm was used to predict the B-cells epitopes present on the SARS-CoV-2 S-protein using the retrieved S-protein amino acid sequence (Jespersen et al., 2017; Vita et al., 2015).
2.4. Identification of T-cell epitopes within selected B-cell epitopes and their antigenicity prediction
T-cell epitopes are known to trigger the cell-mediated immunity within the host cell. T-cell epitope prediction encompasses the identification of major histocompatibility complex (MHC)-I and MHC-II binding epitopes, consequently activate both the Cytotoxic T lymphocytes (CTL) and helper T-lymphocytes (HTL) mediated immune response. The MHC-I and type II epitopes were searched within B-cell epitopes to identify B-cell derived T-cell epitopes, using ProPred-I and Propred servers to predict the MHC-I and MHC-II binding epitopes, respectively (Singh and Raghava, 2003). These servers predict epitopes with high efficiency and might propose the interacting with allelic form probability with any of 47 MHC-I and 51 MHC-II alleles. The 9mer B-cell epitopes are further processed in Proped and Proped-I server to identify T cell as well as known common epitopes. Further, we predict the antigenic score to recognize the antigenicity of the common epitopes (both B-cells and T-cells) within the S-protein of SARS-CoV-2 using VaxiJen 2.0-Drug Design server (default threshold value of 0.45) (Bhattacharya et al., 2020; Doytchinova and Flower, 2007).
2.5. Composition of amino acids and its distribution of epitopic regions
To analyse the composition of amino acids distribution in four epitopic regions we have used ProtParam tool-ExPASy and plotted the amino acids distribution(Portal, 2011).
2.6. Secondary structure architecture of epitopes in S-protein of SARS-Cov-2
To analyse the secondary structure we retrieved PDBsum database and marked the particular epitopic region into the secondary structure of S-protein (Laskowski et al., 1997).
2.7. Glycosylation site prediction of S-protein
Glycosylation of protein is one type of post translational modification; and it is essential for functional characterization of proteins. Protein Glycosylation has been implicated in folding pattern, transport and interaction with other receptor protein. We have predicted the O-glycosylation and N-glycosylation sites of S-protein using the NetNGlyc 1.0 Server (Hamby and Hirst, 2008; Julenius et al., 2005).
2.8. Hydrophobicity prediction of S-protein
Hydrophobicity of protein plays a crucial role in biomolecular interactions (Xi et al., 2017). The hydrophobicity of S-protein was predicted based on Kyte-Doolittle algorithm by using ProtScale server (Kyte and Doolittle, 1982). This web server was also employed to create plots of percent accessible residues for each S-protein residues in linear weight variation model.
2.9. Grand average of hydropathicity (GRAVY) analysis of S-protein
For GRAVY analysis we selected each epitopic region of S-protein and as well as the full S-protein (Kyte and Doolittle, 1982). To analyse the protein structure hydrophobicity value is important. GRAVY is calculating using hydrophobicity.
2.10. Molecular docking of SARS-CoV-2 S-protein epitopes and human TLR4/MD-2 complex
Molecular docking between the identified common B-cell and T-cell epitopes of SARS-CoV-2 S-protein and human TLR4/MD-2 complex (RCSB PDB: 4G8A) was performed using PatchDock molecular docking server (Schneidman-Duhovny et al., 2005). This server customs a geometric complimentary based algorithm to perform the molecular docking. The PatchDock server practices consecutive steps as docking of rigid-body, lowest energy structure clustering, and refinement of predicted structure by energy minimization. For the analysis PDB file of common B-cell and T-cell epitopes were generated through Distill 2.0, a suite of web server (Baú et al., 2006; Yang et al., 2011). The top docked complex solutions (epitopes and TLR4/MD-2) were achieved based on the lowest weighted score of energy and the docking efficiency. Further, visualization and interaction analysis of the protein docked complex was performed using the Chimera v1.14 and LigPlot+146, correspondingly (Laskowski and Swindells, 2011; Pettersen et al., 2004).
Finally to understand the total materials and methods we have provided a flow diagram in Fig. 1 as a bird's eye view.
Fig. 1.

Flow diagram of our complete methodology to understand the predicted molecular interaction between multi-epitopic regions of SARS-CoV-2 S-protein with TLR4/MD-2 complex.
3. Results
3.1. Identification and selection of B-cell and T-cell epitopes, prediction of common epitopes and their antigenicity
The sequential linear B-cell epitopes of varying lengths were identified from the IEDB server within SARS-CoV-2 strain. Specific B-cell epitopes were selected based on their positional value, sequence, and length. The epitopic calculation by IEDB applied BepiPred prediction method to support the B-cell epitopic and non-epitopic part with targeted protein chain.
T-cell mediated immune response and its cascade impart to interact with peptide-MHC complexes, that is a pivotal factor for specific cellular immunogenicity. As these epitopes are selected from the B-cell epitopic region of S-protein, these can serve as both B-cell and T-cell epitopes. Here, the SARS-CoV-2 S-protein is characterized accordingly of their antigenicity and their binding affinity towards MHC-I and MHC-II alleles. We observed that the S-protein of SARS-CoV-2 consists of our 9mer multi-epitopic regions (VRQIAPGQT: 0.8675, YQAGSTPCN: 0.4992, FQPTNGVGF: 0.5711, ILPDPSKPS: 1.2217) that have high antigenicity score (>threshold score 0.45) and are considered as the probable potent antigen for both B and T cell (Fig. 2A–D, Table S1).
Fig. 2.


Different diagrams and model of S-protein and its identified epitopes (A) Schematic diagram of identified epitopes location in S-protein such as VRQIAPGQT (407-415aa), YQAGSTPCN (473-481aa), FQPTNGVGF (497-505aa), ILPDPSKPS (805–813) (B) 3D of S-protein (ribbon diagram), (C) 3D model illustrated different epitopic regions in S-protein, (D) 3D model of different epitopes such as VRQIAPGQT, YQAGSTPCN, FQPTNGVGF, ILPDPSKPS.
3.2. Composition of amino acids and its distribution of epitopic regions
Compositions of amino acids play an important role in protein-protein interaction (Kringelum et al., 2013). We have analysed the composition of amino acid distribution of four epitopic regions (VRQIAPGQT, YQAGSTPCN, FQPTNGVGF, ILPDPSKPS) and compare the amino acid distribution of four regions (Fig. 3A–D).
Fig. 3.

Amino acids composition and its distribution of different epitopic regions.
(A) Epitope VRQIAPGQT, (B) epitope YQAGSTPCN, (C) epitope FQPTNGVGF, (D) epitope ILPDPSKPS.
3.3. Secondary structure architecture of epitopes in S-protein of SARS-Cov-2
Secondary structure architecture plays an important role in protein-protein interaction and surface accessibility (Kringelum et al., 2013). Therefore, secondary structure has a significant role in interaction between epitopic regions of S-protein and TLR4/MD-2 complex. Component of secondary structure architecture and motif structure such as α helix, β hairpin, β turn as well as γ turn were recorded in all four epitopic region (Fig. 4A–D).
Fig. 4.

Secondary structure architecture of different epitopes regions.
(A) VRQIAPGQT. (B) YQAGSTPCN, (C) FQPTNGVGF, (D) ILPDPSKPS.
3.4. Glycosylation site of S-protein
Our result shown three O-glycosylation and 17 N-glycosylation sites are available within the complete amino acids chain of S-protein. While the epitopic (ILPDPSKPS) residues 805–813 sites having single N-glycosylation site. Hence the N-glycosylation residues containing epitopic region influence different parameters of substrate protein folding, which may plays a crucial role in interactions with receptor molecules of TLr4/MD-2 complex (Fig. S2). Total number of O-glycosylation and N-glycosylation sites has been noted in Fig. 5A.
Fig. 5.

Physicochemical parameters and biochemical parameters of S-protein and its epitopic regions. (A) Number of O-Glycosylation site N-Glycosylation site in S-protein. (B) Hydrophobicity in the residues of S-protein. (C) GRAVY of S-protein and different epitopes.
3.5. Hydrophobicity of S-protein
Hydrophobicity is considered as a vital property of protein and we have presented the pattern of hydrophobicity of S-protein residues. The pattern of S-protein hydrophobicity was also found to confirmed higher peak in epitope ILPDPSKPS (0.5–1.0) above the “0.0” midline as compared with the hydrophobicity score of others epitopes. Even though, epitope VRQIAPGQT and FQPTNGVGF shown positive hydrophobicity score (Fig. 5B).
3.6. GRAVY of S-protein and selected epitopic region
GRAVY is linked with protein solubility. It also observed that positive GRAVY values are associated with hydrophobicity and negative GRAVY values are associated with hydrophilicity. These characters are linked with protein-protein interaction. We have noted GRAVY value of S-protein, VRQIAPGQT, YQAGSTPCN, FQPTNGVGY and ILPDPSKPS are −0.079, −0.411, −0.833, −0.488, 0.611 respectively (Fig. 5C).
3.7. Molecular interaction and binding affinity evaluation of TLR4/MD-2 complex with multi-epitopes (VRQIAPGQT, YQAGSTPCN, FQPTNGVGF, ILPDPSKPS)
Considering the binding affinity of viral glycoprotein with human TLR4/MD-2 complex (Olejnik et al., 2018), we explored the molecular interaction of the four identified antigenic epitopes SARS-CoV-2 S-protein with TLR4/MD-2 complex of human. Molecular docking has shown the binding of SARS-CoV-2 S-protein epitopic regions to TLR4/MD-2 complex. The 9mer epitope VRQIAPGQT binds with TLR4/MD-2 complex, might signified through their own atomic contact energy (ACE) value. The ACE value of epitope VRQIAPGQT-TLR4/MD-2 complex was −283.96Kcal/mol, top 20 docked complexes considered for their high ranking as well as their high negative ACE value. The epitope: VRQIAPGQT and TLR4/MD-2 complex formed maximum (five) number hydrogen bond (average bond length > 2 Å). Subsequently, four amino acid residues of TLR4 and one from MD-2 involved to the epitope VRQIAPGQT (Fig. 6A, Table 1 ).
Fig. 6.

Detained molecular binding modes structure (rotate view) of SARS-CoV-2 S-protein multi-epitope (A) VRQIAPGQT (cyan) and TLR4/MD-2 complex, (B) ILPDPSKPS (pink) and TLR4/MD-2 complex, (C) FQPTNGVGF (yellow) and TLR4/MD-2 complex. (D) YQAGSTPCN (pink) and TLR4/MD-2 complex. The intramolecular H-bonds are depicted and for clarity the non-polar bonds are omitted. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Amino acids residues comprise in hydrogen bond formation, bond length and ACE value between the docking complex of SARS-CoV-2 S-protein epitopes and human TLR4/MD-2 complex protein.
| Epitope | Residues of epitope involved in H-bond | Residues of TLR4 involved in H-bond | Residues of MD-2 involved H-bond | Bond length (Å) | ACE value (Kcal/mol) |
|---|---|---|---|---|---|
| VRQIAPGQT | THR9 | LYS130 | 2.473 | −283.96 | |
| THR9 | LYS130 | – | 2.801 | ||
| ARG2 | LEU180 | – | 2.712 | ||
| ARG2 | SER207 | – | 1.918 | ||
| GLN8 | – | ARG106 | 3.114 | ||
| YQAGSTPCN | PRO7 | LYS230 | 3.557 | −184.83 | |
| FQPTNGVGF | GLY8 | – | CYS133 | 3.272 | −432.73 |
| ILPDPSKPS | ASP4 | CYS133 | 2.691 | −291.68 |
The molecular interaction between the epitope YQAGSTPCN and TLR4/MD-2 complex was established based on the minimal ACE value of protein-protein docked composite structure. The ACE value of epitope YQAGSTPCN-TLR4/MD-2 complex was −184.83Kcal/mol. The epitope: YQAGSTPCN and TLR4/MD-2 complex formed only one hydrogen bond (bond length 3.557 Å). Within this molecular interaction the single amino acid residue of TLR4 involved into the H-bond of epitope VRQIAPGQT (Fig. 6B, Table 1).
The epitope FQPTNGVGF and TLR4/MD-2 complex was complementarily interacting to require the minimal ACE value. The ACE value of epitope FQPTNGVGF and TLR4/MD-2 complex was −432.73Kcal/mol. The epitope: FQPTNGVGF and TLR4/MD-2 complex shaped only one hydrogen bond (bond length 3.272 Å). Inside this molecular interaction the single amino acid residue of MD-2 was involved to the H-bond with FQPTNGVGF epitope (Fig. 6C, Table 1).
The epitope ILPDPSKPS and TLR4/MD-2 complex was bind with lowest ACE value −291.68Kcal/mol. The docked complex also formed one hydrogen bond (bond length 2.691 Å), and within molecular interaction the single amino acid residue of MD-2 was involved to the H-bond of ILPDPSKPS epitope (Fig. 6D, Table 1).
The molecular interface area was 941.10Å2 of epitope VRQIAPGQT and TLR4/MD-2 complex binding pocket, and transformation score of rotational angles of ligand was 1.11, −0.75, 2.16. Conversely, the molecular interface area score of epitope FQPTNGVGF was 783.10Å2 and MD-2 receptor of TLR4/MD-2 complex binding pocket, and transformation score of rotational angles of ligand was 1.55, 0.57, 0.95. In case of epitope ILPDPSKPS and MD-2 receptor of TLR4/MD-2 complex binding pocket the molecular interface area was 912.90Å2, and transformation score of rotational angles of ligand was 1.79, 0.49, 2.45.
The four multi-epitopes of S-protein and TLR4/MD-2 complex intermolecular interactions including hydrophobic and hydrogen bonding interactions on the most representative structure obtained from cluster analysis was performed by Ligplot (Fig. S1, A–D).
4. Discussion
Here, we identified multi-epitopes of SARS-CoV-2 S-protein that are common of both B-cells and T cells. Our observation indicated that the multi-epitopes derived from SARS-CoV-2 S-protein bind to human TLR4/MD2 complex. It is possible that the interaction of SARS-CoV-2 S-protein with TLR4/MD2 complex might induce TLR4-mediated signalling cascade that is responsible for adaptive immune response in host body after SARS-CoV-2 infection. Therefore, we studied the interaction of identified multi-epitopic region of S-protein with human TLR4/MD2 complex, which might be responsible for adaptive immune response associated with SARS-CoV-2 infection.
It is known that TLR4 can recognize the viral proteins (Lester and Li, 2014). The interaction of TLR4 viral epitopes has been associated with the viral infection and immune responses (Olejnik et al., 2018). Georgel et al. (2007) demonstrated that vesicular stomatitis virus glycoprotein induces CD14/TLR4 dependent signalling pathway (Georgel et al., 2007). It has been shown that the envelop glycoprotein of mouse mammary tumor virus (MMTV) binds to and activate TLR4 (Burzyn et al., 2004). In addition, it has been demonstrated that TLR4 can also recognize the respiratory syncytial viral protein (Kurt-Jones et al., 2000). Modhiran et al. (2015) describe that TLR4 sense the NS1 antigen of dengue virus (Modhiran et al., 2015).
The S-proteins epitopic regions are subjected to the antigenicity of the viral pathogen (Berry et al., 2010). Zhang et al. (2020) identified the 11 epitopes, among which one was in conserved region, from SARS-CoV-2 whole genome. He translated S-protein reference sequence based on the surface accessibility using bioinformatics tools (Zheng and Song, 2020). Our computational analysis predicted four multi-epitopic regions that are common for both B-cells and T-cells, having binding affinity of MHC (class I, II) allele or human leukocyte antigen (HLA-DRB1) gene within S-protein sequence of SARS-CoV-2. These epitopic regions have high antigenicity score, HLA-promiscuous regions and therefore might be able to induce adaptive immune responses (Singh and Raghava, 2001).
We observed that the identified multi-epitopic regions of SARS-CoV-2 S-protein can bind with the TLR4/MD-2 complex. The binding interaction between epitope: VRQIAPGQT and TLR4/MD-2 complex formed five H-bonds and had higher molecular interface area compared to other recognised three epitopic regions. The highest value of molecular interface area between VRQIAPGQT and TLR4/MD-2 complex binding pocket, and maximum transformation score of ligand rotational angles implies that the strength of VRQIAPGQT and TLR4/MD-2 complex molecular docked interface is the most stabilised structure compared to other identified epitopic regions. On the contrary the epitopes: FQPTNGVGF and ILPDPSKPS were not able to form any molecular interaction with TLR4, however these epitopes bind with the MD-2 region of TLR4/MD-2 complex. Both the epitope shown lower value of molecular interface area with the MD-2 receptor of TLR4/MD-2 complex.
The O-glycosylation and N-glycosylation sites prediction of S-protein showed no such significant consequences of studied multi-epitopic regions, except ILPDPSKPS epitope, that may play a critical role in folding of S-protein and molecular interactions with receptor molecules of TLR4/MD-2 complex. Moreover, the residues of S-protein epitope ILPDPSKPS showed strong hydrophobic interactions with the residues of TLR4/MD-2 complex compared to the epitopes VRQIAPGQT and FQPTNGVGF.
In current study, we intended to identify novel multi-epitopic regions of S-protein (SARS-CoV-2) containing more effective antigenic epitopes-rich domains. The molecular binding activity of these short protein chains were assessed using bioinformatics tools to identify the stable molecular interaction between the epitopes candidate and the immunoreceptors (TLR4) that might elicits adaptive immune response. Finally we have also developed different model about the molecular interaction of different epitopic regions and TLR4/MD-2 complex (Fig. 7A–D). We also projected a mechanism for the probable activation of adaptive immune response through the epitopic interaction with TLR4/MD-2 complex (Fig. 8 ). Therefore, our analysis support that the recognised four multi-epitope from the SARS-CoV-2 S-protein might serve as potent peptide vaccine candidate against COVID-19, by using the techniques of reverse vaccinology in platforms of immunoinformatics.
Fig. 7.

Proposed model for molecular interaction of different epitopic region and TLR4/MD-2 complex.
A model for interaction (A) VRQIAPGQT and TLR4/MD-2 complex.
(B) ILPDPSKPS and TLR4/MD-2 complex.
(C) FQPTNGVGF and TLR4/MD-2 complex.
(D) YQAGSTPCN and TLR4/MD-2 complex.
Fig. 8.
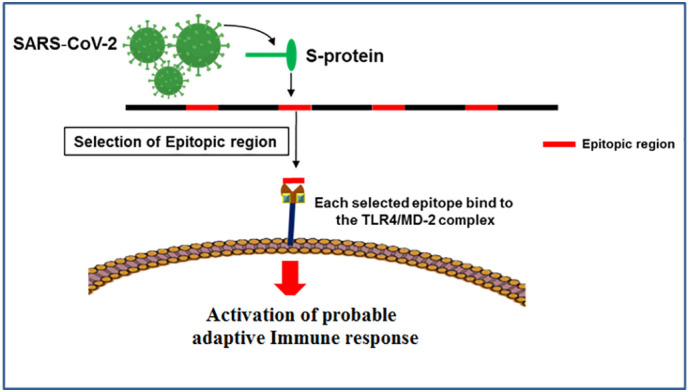
Proposed mechanism for probable activation of adaptive immune response through the interaction of epitopes and TLR4/MD-2 complex.
5. Conclusion
The 2019-nCoV is a novel pathogenic virus that becomes a severe public health emergency of global concern. Our computational analysis of S-protein sequence of SARS-CoV-2 predicted the level of antigenicity and numbers of epitopes for B and T cells. The four multi-epitopic part of SARS-CoV-2 S-protein has been found to bind with TLR4/MD-2 complex. Hence, the stable molecular interaction of S-protein 9mer multi-epitopic parts and TLR4/MD-2 possibly develop adaptive immune response. Understanding the interaction of TLR4/MD-2 complex with the screened epitopes present within S-protein of SARS-CoV2 as immunogenic target might be beneficial in the development of multi-epitopic based peptide vaccine by inducing the cell-mediated immune response. Thus, we suggest that these recognised multi-epitopes might be exploited as peptide based vaccine against SARS-CoV-2 infection. However, the proposed B and T cell epitopes-based peptide vaccine quickly needs to validate clinically that certify its safety and immunogenic profile to support on stopping this epidemic before it leads to devastating global outbreaks.
The following are the supplementary data related to this article.
Common B-cell and T-cell epitopes, position of epitopic regions and antigenicity score of SARS-CoV-2 S-protein.
Supplementary Figure S1 and S2
Declaration of competing interest
All of the authors declare that there is no competing interest in this work.
Acknowledgments
Acknowledgments
This research was supported by Hallym University Research Fund and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2017R1A2B4012944 & NRF-2020R1C1C1008694).
Author contributions
All authors conceived of the research, designed the study, interpreted data and wrote, edited the final manuscript.
References
- Amanat F., Krammer F. SARS-CoV-2 vaccines: status report. Immunity. 2020;52:583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baú D., Martin A.J., Mooney C., Vullo A., Walsh I., Pollastri G. Distill: a suite of web servers for the prediction of one-, two-and three-dimensional structural features of proteins. BMC Bioinformatics. 2006;7:402. doi: 10.1186/1471-2105-7-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry J.D., Hay K., Rini J.M., Yu M., Wang L., Plummer F.A., Corbett C.R., Andonov A. Taylor & Francis; 2010. Neutralizing Epitopes of the SARS-CoV S-Protein Cluster Independent of Repertoire, Antigen Structure or mAb Technology, MAbs; pp. 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M., Sharma A.R., Patra P., Ghosh P., Sharma G., Patra B.C., Lee S.S., Chakraborty C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): immunoinformatics approach. J. Med. Virol. 2020;92:618–631. doi: 10.1002/jmv.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D., Rassa J.C., Kim D., Nepomnaschy I., Ross S.R., Piazzon I. Toll-like receptor 4-dependent activation of dendritic cells by a retrovirus. J. Virol. 2004;78:576–584. doi: 10.1128/JVI.78.2.576-584.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty C., Sharma A., Sharma G., Bhattacharya M., Lee S. SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): diagnostic and proposed therapeutic options. Eur. Rev. Med. Pharmacol. 2020;24:4016–4026. doi: 10.26355/eurrev_202004_20871. [DOI] [PubMed] [Google Scholar]
- Chakraborty C., Sharma A.R., Bhattacharya M., Sharma G., Lee S.-S. The 2019 novel coronavirus disease (COVID-19) pandemic: a zoonotic prospective. Asian Pac. J. Trop. Med. 2020;13(6):242–246. [Google Scholar]
- Chakraborty C., Sharma A.R., Bhattacharya M., Sharma G., Lee S.S., Agoramoorthy G. Consider TLR5 for new therapeutic development against COVID-19. J. Med. Virol. 2020;92(11):2314–2315. doi: 10.1002/jmv.25997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury A., Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020;92(10):2105–2113. doi: 10.1002/jmv.25987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coordinators N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2017;45:D12. doi: 10.1093/nar/gkw1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova I.A., Flower D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthie M.S., Windish H.P., Fox C.B., Reed S.G. Use of defined TLR ligands as adjuvants within human vaccines. Immunol. Rev. 2011;239:178–196. doi: 10.1111/j.1600-065X.2010.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen F., Stenger R.M., Poelen M.C., Van Dijken H.H., Kuipers B., Boog C.J., Van Putten J.P., van Els C.A., van der Ley P. Differential effect of TLR2 and TLR4 on the immune response after immunization with a vaccine against Neisseria meningitidis or Bordetella pertussis. PLoS One. 2010;5 doi: 10.1371/journal.pone.0015692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgel P., Jiang Z., Kunz S., Janssen E., Mols J., Hoebe K., Bahram S., Oldstone M.B., Beutler B. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology. 2007;362:304–313. doi: 10.1016/j.virol.2006.12.032. [DOI] [PubMed] [Google Scholar]
- Hamby S.E., Hirst J.D. Prediction of glycosylation sites using random forests. BMC Bioinformatics. 2008;9:500. doi: 10.1186/1471-2105-9-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A., Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Jespersen M.C., Peters B., Nielsen M., Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45:W24–W29. doi: 10.1093/nar/gkx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Guardeno J.M., Nieto-Torres J.L., DeDiego M.L., Regla-Nava J.A., Fernandez-Delgado R., Castaño-Rodriguez C., Enjuanes L. The PDZ-binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julenius K., Molgaard A., Gupta R., Brunak S. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology. 2005;15:153–164. doi: 10.1093/glycob/cwh151. [DOI] [PubMed] [Google Scholar]
- Kringelum J.V., Nielsen M., Padkjær S.B., Lund O. Structural analysis of B-cell epitopes in antibody: protein complexes. Mol. Immunol. 2013;53:24–34. doi: 10.1016/j.molimm.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt-Jones E.A., Popova L., Kwinn L., Haynes L.M., Jones L.P., Tripp R.A., Walsh E.E., Freeman M.W., Golenbock D.T., Anderson L.J., Finberg R.W. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Swindells M.B. ACS Publications; 2011. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Hutchinson E.G., Michie A.D., Wallace A.C., Jones M.L., Thornton J.M. PDBsum: a Web-based database of summaries and analyses of all PDB structures. Trends Biochem. Sci. 1997;22:488–490. doi: 10.1016/s0968-0004(97)01140-7. [DOI] [PubMed] [Google Scholar]
- Le Thanh T., Andreadakis Z., Kumar A., Gomez Roman R., Tollefsen S., Saville M., Mayhew S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020;19:305–306. doi: 10.1038/d41573-020-00073-5. [DOI] [PubMed] [Google Scholar]
- Lester S.N., Li K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014;426:1246–1264. doi: 10.1016/j.jmb.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modhiran N., Watterson D., Muller D.A., Panetta A.K., Sester D.P., Liu L., Hume D.A., Stacey K.J., Young P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015;7 doi: 10.1126/scitranslmed.aaa3863. 304ra142. [DOI] [PubMed] [Google Scholar]
- Olejnik J., Hume A.J., Mühlberger E. Toll-like receptor 4 in acute viral infection: too much of a good thing. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1007390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;11:1–12. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Portal E.B.R. 2011. ProtParam Tool. [Google Scholar]
- Reed S.G., Hsu F.-C., Carter D., Orr M.T. The science of vaccine adjuvants: advances in TLR4 ligand adjuvants. Curr. Opin. Immunol. 2016;41:85–90. doi: 10.1016/j.coi.2016.06.007. [DOI] [PubMed] [Google Scholar]
- Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeman D., Fielding B.C. Coronavirus envelope protein: current knowledge. Virol. J. 2019;16:69. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H., Raghava G.P. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236–1237. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- Singh H., Raghava G. ProPred1: prediction of promiscuous MHC Class-I binding sites. Bioinformatics. 2003;19:1009–1014. doi: 10.1093/bioinformatics/btg108. [DOI] [PubMed] [Google Scholar]
- Srivastava S., Kamthania M., Kumar Pandey R., Kumar Saxena A., Saxena V., Kumar Singh S., Kumar Sharma R., Sharma N. Design of novel multi-epitope vaccines against severe acute respiratory syndrome validated through multistage molecular interaction and dynamics. J. Biomol. Struct. Dyn. 2019;37:4345–4360. doi: 10.1080/07391102.2018.1548977. [DOI] [PubMed] [Google Scholar]
- Vita R., Overton J.A., Greenbaum J.A., Ponomarenko J., Clark J.D., Cantrell J.R., Wheeler D.K., Gabbard J.L., Hix D., Sette A. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43:D405–D412. doi: 10.1093/nar/gku938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020:94. doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi E., Venkateshwaran V., Li L., Rego N., Patel A.J., Garde S. Hydrophobicity of proteins and nanostructured solutes is governed by topographical and chemical context. Proc. Natl. Acad. Sci. 2017;114:13345–13350. doi: 10.1073/pnas.1700092114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Faraggi E., Zhao H., Zhou Y. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics. 2011;27:2076–2082. doi: 10.1093/bioinformatics/btr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Wu Q., Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020;30 doi: 10.1016/j.cub.2020.03.063. 1346-1351.e1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M., Song L. Novel antibody epitopes dominate the antigenicity of spike glycoprotein in SARS-CoV-2 compared to SARS-CoV. Cell. Mol. Immunol. 2020;17:536–538. doi: 10.1038/s41423-020-0385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Common B-cell and T-cell epitopes, position of epitopic regions and antigenicity score of SARS-CoV-2 S-protein.
Supplementary Figure S1 and S2