

Abstract
Background
With the diffusion of SARS-CoV-2 around the world, human health is being threatened. As there is no effective vaccine yet, the development of the vaccine is urgently in progress.
Materials and methods
Immunoinformatics methods were applied to predict epitopes from the Spike protein through mining literature associated with B- and T-cell epitopes prediction published or preprinted since the outbreak of the virus till June 1, 2020. 3D structure of the Spike protein were obtained (PDB ID: 6VSB) for prediction of discontinuous B-cell epitopes and localization of epitopes in the hotspot regions.
Results
Methods provided by the Immune Epitope Database (IEDB) server were the most frequently used to predict epitopes. Sequence alignment of the epitopes extracted from literature with the Spike protein demonstrated that the epitopes in different studies converged to multiple short hotspot regions. There were three hotspot regions found in RBD of the Spike protein harboring B-cell linear epitopes (‘RQIAPGQTGKIADYNYKLPD’, ‘SYGFQPTNGVGYQ’ and ‘YAWNRKRISNCVA’) predicted to have high antigenicity score. Two T-cell epitopes (‘KPFERDISTEIYQ’ and ‘NYNYLYRLFR’) predicted to be highly antigenic in the original studies were discovered in the hotspot region. Toxicity and allergenicity analysis confirmed all the five epitopes are of non-toxin, and four of them are of non-allergen. The five epitopes identified in hotspot regions of RBD were found fully exposed based on the 3D structure of the Spike protein.
Conclusion
The five epitopes we discovered from literature mining may be potential candidates for diagnostics and vaccine development against SARS-CoV-2.
Keywords: SARS-CoV-2, Spike protein, T-cell epitope, B-cell epitope, Literature mining, Vaccine design
1. Introduction
SARS-CoV-2 has been spreading worldwide since December 2019. As of May 2020, the cumulative number of confirmed cases worldwide has exceeded 5 million, with more than 300,000 deaths. The number of confirmed cases in more than a dozen countries has exceeded 100,000. The World Health Organization has declared this outbreak as a public health emergency of international concern (PHEIC) since January 30, 2020 [1].
As a novel coronavirus, SARS-CoV-2 approximates to SARS-COV [2] and EBOV, which have caused large-scale infection and mortality. However, SARS-COV-2 is significantly more infectious than other coronaviruses [3], and it has been determined that this virus can be spread among humans through sneezing, coughing, and respiratory droplets [4]. The majority of infected people have no symptoms until symptom onset, and the incubation period can be up to 24 days. Therefore, many areas that seemingly to have contained the epidemic experienced a second outbreak. Under such circumstances, it is imperative to develop stable and effective vaccines.
Researchers all over the world conducted a series of immunoinformatics analyzes on the Spike protein of SARS-CoV-2 to search for B- and T-cell epitopes. The Immune Epitope Database (IEDB) [5] was widely utilized to forecast B- and T-cell epitopes. The ANNpred-based server ABCpred [6], and ProPred [7] were also optional methods in predicting B-cell linear epitopes. Disctope 2.0 [8] was usually used to predict discontinuous B-cell epitopes based on the 3D structure of the Spike protein. ProPred-I [9] and ProPred servers were able to identify B cell-derived T cell epitopes by searching for MHC I and II binding epitopes from predetermined linear B cell epitope regions. In addition, RECON [10] (real-time tumor antigen epitope calculation), CTL Pred [11], ProPred-I, ProPred, netMHCpan4 [12] and MARIA [13], netMHCpan and INEO-Pred [14] were also able to predict T-cell epitopes.
As the accuracy and specificity of the epitopes prediction methods were not well benchmarked yet, the methods were chosen in most studies purely based on the researchers’ experiences and preferences. A huge repertoire of epitopes were generated within a short period of time, but most in silico predicted epitopes were not validated by biological experiments, which made it difficult to select a few epitopes for diagnostics and vaccine development. The epitopes from regions predicted to harbor epitopes by various in-silico methods would be robust and more likely to induce immune response, as a result, they would be better vaccine candidates.
To provide B- and T-cell epitope candidates with full potentials for vaccine development, we performed literature mining by collecting published and pre-printed literature on the SARS-COV-2 vaccine development since the outbreak of the epidemic till June 1, 2020. In terms of methodology, we discovered that the Immune Epitope Database (IEDB) server were the most frequently used to predict epitopes. After comprehensively analyzing those epitopes extracted from literature, we found that many epitopes predicted in different studies came from five short hotspot regions of the Spike protein. We investigated the antigenicity and locations of the epitopes in the five short hotspot regions by integrating the amino acid sequences of the five epitopes and 3D structure of the Spike protein, demonstrating that the five epitopes with potentially high antigenicity located in the fully exposed Receptor Binding Domain (RBD) region of the Spike protein. In short, the epitopes identified here in the hotspot regions may serve as potential candidates for diagnostics and vaccine development to confront SARS-CoV-2.
2. Materials and methods
2.1. Literature collection and mining
The Spike protein of SARS-COV-2 is regarded as the target due to its formation of characteristic crown of the virus and protruding from the viral envelope [15]. In order to determine the distribution of epitopes that may be used for vaccine construction, we integrated 24 published articles or preprints [14,[16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38]] about epitope predictions from January 29, 2020 to June 1, 2020. Based on the results of these researches, we collected all the results about Spike protein for statistical analysis. 214 B-cell epitopes and 150 T-cell epitopes were obtained. Experimentally known 3D structure of the SARS-CoV-2 Spike protein was obtained from Protein Data Bank (PDB ID: 6VSB) [39].
2.2. The analysis of B- and T-cell epitopes derived from curated literature
We use Blastp to get the location of each epitope with the parameters num_threads-8, -p-value 100. The positions were used to evaluate the distribution of the epitopes on Spike protein. The RBD region and Spike protein receptor-binding motif binding to human ACE2 are drawn to observe the distribution of epitopes on Spike protein, respectively.
2.3. Locations of B-cell epitopes on Spike protein
All the epitopes were evaluated according to the frequency of occurrence in different literature and the distribution of epitopes on Spike protein. We defined the regions with epitopes reported in multiple studies as hotspot regions. The epitopes in the hotspot regions locating in the RBD region were considered as the potential epitopes. Pymol 2.3.4 was used to plot the 3D structure of the potential epitopes [40].
2.4. Antigenicity prediction for B-cell linear epitopes
We submitted B-cell linear epitopes in the hotspot regions to the VaxiJen v.2.0 server [41], with the virus as the target field, to analyze the antigenicity of epitopes derived from the coronavirus Spike protein.
2.5. Allergenicity and toxicity of selected B-cell and T-cell epitopes
The B-cell and MHC class I/II binding T cell epitopes were evaluated for their allergenicity and toxicity. Allergenicity of B-cell and T-cell epitopes were predicted by Allergen FP 1.0 [42]. Toxicity, hydrophobicity, hydropathicity, hydrophilicity and charge of B-cell and T-cell epitopes were assessed by ToxinPred [43].
3. Results
3.1. The usage landscape of in silico B-cell epitope prediction methods
To determine epitope candidates in the design of the COVID-19 vaccine, researchers all over the world conducted a series of immunoinformatics analysis to predict B- and MHC class I/II T-cell epitopes from the Spike protein, M protein, E protein and N protein of the SARS-CoV-2 virus. We collected 24 published and pre-printed literature [14,[16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38]] about the SARS-COV-2 epitopes from the outbreak of the epidemic to June 1, 2020 in total (Supplementary Table 1). We discovered that the most frequently used B-cell linear epitope prediction methods were those provided on the Immune Epitope Database (IEDB) server including Bepipred [44], Kolaskar and Tongaonkar’s antigenic prediction [45], Emini’s surface accessibility prediction [46] (Fig. 1 A). Particularly, the Bepipred/Bepipred2.0 was the top frequently applied method appearing in 13 literature (Fig. 1A). Besides, several other inmunoinformatics tools were used to predict potential epitopes. The 3D structure of the Spike protein and the DiscoTope server [47] were applied to predict discontinuous B-cell epitopes. The most popular method for predicting B-cell linear and discontinuous antibody epitopes on the basis of the 3D structure of protein antigens was Ellipro server [48], with seven users. Four publications used ABCpred [6], a server that predicts linear B cell epitope regions in an antigen sequence by an artificial neural network, to locate epitope regions. RECON [10], RapidPeptidesGenerator [49], OptMAVEn [50], Bcpred [51] and Bcepred [52] were chosen by a few researchers.
Fig. 1.
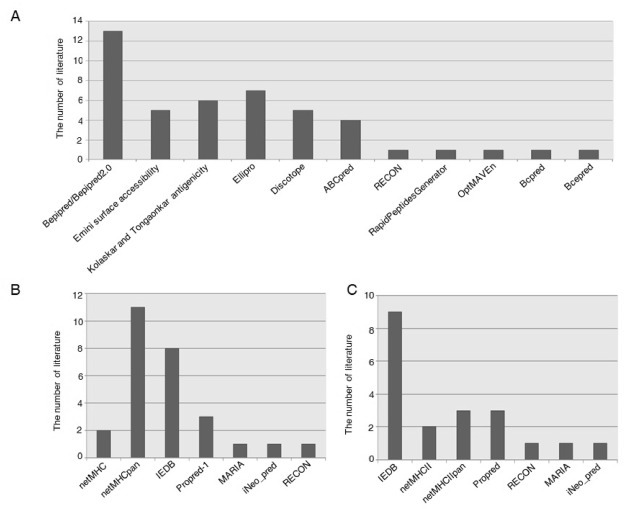
Usage frequency of different prediction methods among the literature. A: B-cell epitope prediction. B: MHC-I epitope prediction. C: MHC-II epitope prediction.
3.2. The usage landscape of in silico T-cell epitope prediction methods
MHC class I/II molecules play a decisive role in the processing of intracellular and extracellular antigens and presentation of antigens to CD8+ and CD4+ T cells to stimulate specific immune responses. The tools on IEDB server that predict IC50 values for peptides binding to specific MHC molecules were most frequently used by the researchers for MHC-I and MHC-II epitopes prediction (Fig. 1B and C) (Supplementary Table 1). The netMHCpan [12] with Eluted Ligand Prediction option is the default MHC- I prediction method selection of IEDB. NetMHCpan can make prediction to any custom MHC class I molecule and peptides of any length, while netMHC [53] predicts peptide-MHC class I binding for HLA-A0101, HLA-A0201, HLA-A0301, HLA-A2402, HLA-A2601, HLA-B0702, HLA-B0801, HLA-B2705, HLA-B3901, HLA-B4001, HLA-B5801 and HLA-B1501. Moreover, ProPred-I [9], a graphical network tool to predict MHC class I binding regions in antigenic protein sequences, had the highest usage rate among the other prediction methods, such as RECON, iNeo_pred and MARIA [13]. IEDB default algorithm to predict MHC-II epitopes uses the consensus approach, combining NN-align, SMM-align, CombLib and Sturniolo. Alternatively, NetMHCIIpan is also provided by IEDB. NetMHCII 2.3 server predicts binding of peptides to HLAs. Similarly, Propred [7] was more frequently selected than RECON, iNeo_pred and MARIA.
3.3. Identification of B-cell linear epitope candidates from the Spike protein
The anti-infective function of B cells is started by epitope recognition of viral antigen proteins. A total of 302 potential B-cell linear epitopes were obtained from the literature we collected, and were then aligned against the genome of the Spike protein (Supplementary Table 2). We discovered that B-cell linear epitopes predicted in multiple studies converged to three hotspot regions in the RBD of the Spike protein with the highest peak consisting of B-cell linear epitopes from a single study (Fig. 2 ). The three hotspot regions harbored three B-cell linear epitopes including ‘RQIAPGQTGKIADYNYKLPD’, ‘SYGFQPTNGVGYQ’ and ‘YAWNRKRISNCVA’. We estimated the antigenicity score of the three B-cell epitopes with VaxiJen v.2.0, and found that ‘RQIAPGQTGKIADYNYKLPD’ had the highest antigenicity of 1.41, followed by ‘SYGFQPTNGVGYQ’ of 0.76 and ‘YAWNRKRISNCVA’ of 0.39. The three B-cell epitopes were then illustrated on the 3D structure of the Spike protein, showing the most exposed region of the Spike protein (Fig. 3 A and B).
Fig. 2.
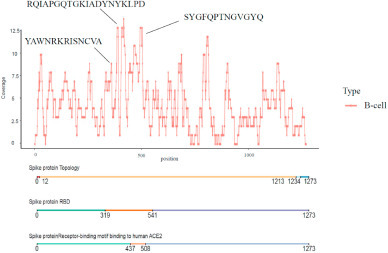
The coverage of potential B-cell linear epitopes on the Spike protein. ‘RQIAPGQTGKIADYNYKLPD’, ‘SYGFQPTNGVGYQ’ and ‘YAWNRKRISNCVA’ are the B-cell linear epitopes in hotspot regions in the RBD of the Spike protein.
Fig. 3.
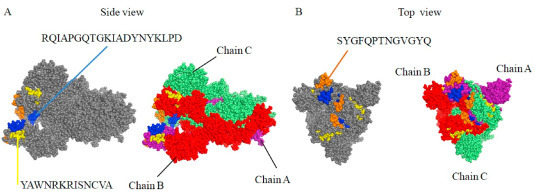
Location of three predicted B-cell epitopes on the 3D structure of the SARS-CoV-2 S protein (PDB ID 6VSB). A. Location of ‘RQIAPGQTGKIADYNYKLPD’ and ‘YAWNRKRISNCVA’. B. Location of ‘SYGFQPTNGVGYQ’. Chain A, B, and C are shown in purple, red and lime color, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.4. Identification of T-cell epitope candidates from the Spike protein
A total of 189 T-cell epitope candidates were curated from all literature we collected (Supplementary Table 3). Two T-cell epitopes (‘KPFERDISTEIYQ’ and ‘NYNYLYRLFR’) predicted to have high antigenicity in most studies were discovered to locate in the RBD region of the Spike protein (Fig. 4 ). ‘KPFERDISTEIYQ’ was able to bind with MHC-I alleles containing HLA-A:0206,2402, 2403,2403, 3207, 6601,6802, 6823, HLA-B:0802, 1402, 1502,1503, 2720, 3503,4002, 4013, 4201,4506, 4801, 8301, HLA-C:0401, 0401, 0702,1203, 1402, 1402, and MHC-II alleles including DRB1: 0401, 0701, 0801, 1101, 1602, DPA10-DPB10:201-501. ‘NYNYLYRLFR’ were able to bind with MHC-I allele HLA∗33:01. After mapping the two T-cell epitopes to the 3D structure of SARS-CoV-2 Spike protein, we discovered that they located in the fully exposed region of RBD, though T-cell epitopes can be from any region of the Spike protein (Fig. 5 A and B).
Fig. 4.
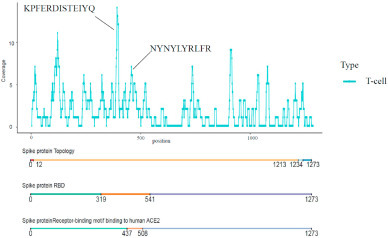
The coverage of potential T-cell epitopes on the Spike protein. ‘KPFERDISTEIYQ’ and ‘NYNYLYRLFR’ are the T-cell epitopes in hotspot regions in the RBD of the Spike protein.
Fig. 5.
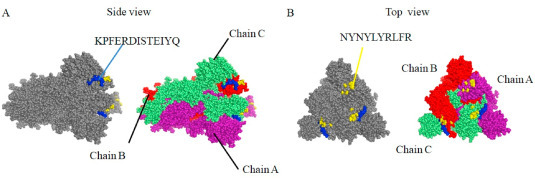
Location of three predicted B-cell linear epitopes on the 3D structure of the SARS-CoV-2 S protein (PDB ID 6VSB). A. Location of ‘KPFERDISTEIYQ’. B. Location of ‘NYNYLYRLFR’. Chain A, B, and C are shown in purple, red and lime color, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.5. Allergenicity and toxicity of B- and T-cell epitopes
Allergen FP 1.0 was used to access the allergenicity of all five epitopes. Four B-cell epitope was predicted to be non-allergenic with only one allergenic (Table 1 ). Toxicity, hydrophobicity, hydropathicity, hydrophilicity and charge of B-cell epitopes were examined through ToxinPred, which based on machine learning technique and quantitative matrix. The result demonstrates that all the epitopes were predicted to be non-toxin (Table 1).
Table 1.
Allergenicity, toxicity, hydro and physiochemical properties of all epitopes.
| Cell Type | Peptide | Toxicity | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Mol wt | Allergenicity |
|---|---|---|---|---|---|---|---|---|
| B-cell | RQIAPGQTGKIADYNYKLPD | Non-Toxin | −0.24 | −1.03 | 0.21 | 1 | 2248.82 | NA |
| B-cell | SYGFQPTNGVGYQ | Non-Toxin | −0.07 | −0.8 | −0.62 | 0 | 1417.69 | NA |
| B-cell | YAWNRKRISNCVA | Non-Toxin | −0.3 | −0.62 | −0.1 | 3 | 1581 | A |
| T-cell | KPFERDISTEIYQ | Non-Toxin | −0.3 | −1.15 | 0.52 | −1 | 1625.98 | NA |
| T-cell | NYNYLYRLFR | Non-Toxin | −0.31 | −0.95 | −0.66 | 2 | 1421.76 | NA |
3.6. Identification of discontinuous B-cell epitopes using the 3D structure of the Spike protein
Discotope 2.0 was the most popular tool to predict discontinuous B-cell epitopes in the majority of studies. We also predicted discontinuous B-cell epitopes using A, B, and C chain of the 3D structure of the Spike protein, respectively. The locations of discontinuous epitopes were illustrated on the surface of the Spike protein 3D structure. We then extracted the surface glycoprotein amino acid positions that were included in the three B-cell linear epitopes (‘RQIAPGQTGKIADYNYKLPD’, ‘SYGFQPTNGVGYQ’ and ‘YAWNRKRISNCVA’) in the hotspot region. The relevant amino acid positions were mapped onto the 3D structure of the Spike protein to identify the predicted epitope residue/regions (415, 420, 494,496, 498, 500, 503, 505) in the surface glycoprotein (Fig. 6 A and B).
Fig. 6.
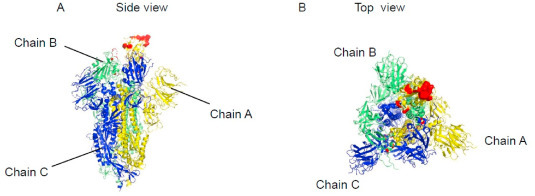
Location of discontinuous B-cell epitopes included in the three B-cell linear epitopes on the 3D structure of the SARS-CoV-2 S protein (PDB ID 6VSB). A. Side-view. B. Top-view.Chain A, B, and C are shown in yellow, lime and yellow color, respectively. Color should be used for Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6 in print. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
Since the end of 2019, SARS-CoV-2 began to spread around the world, causing huge panic. Until now, there are already an enormous number of people infected with SARS-CoV-2 with a continuous increase and intimidating death rate. In order to overcome this common human problem, researchers worldwide are making tremendous efforts to develop the SARS-CoV-2 vaccine or antidote. In previous work, various screening and verification methods were applied to obtain epitopes in each work, however, those predicted epitopes were not complete consistent and lack of evidences from in-vitro and in-vivo experiments. Therefore, it is necessary and useful to identify the most robust regions predicted to harbor epitopes through literature mining previous work. The epitopes from those robust regions would be more likely to be vaccine candidates.
We collected published or preprinted articles on possible epitope predictions of SARS-CoV-2 since the outbreak of the virus till June 1 2020, and made a comprehensive summary of their methods and results. In these studies, researchers applied immunoinformatics-based methods on SARS-CoV-2 viral genomic data with different strict standards to identify B-cell and T-cell epitopes that mainly target the Spike protein. The multiple physical and chemical properties of the predicted epitope, for instance, antigenicity, allergenicity and toxicity, were evaluated to determine the feasibility of constructing a vaccine. In B-cell epitope prediction, Bepipred and Ellipro supplied by the IEDB server were the most used, while netMHC and netMHC II are most used in T-cell epitope prediction. The obtained possible epitopes need to be verified on other aspects such as antigenicity verification. Vaxijen 2.0 was mostly used for antigenicity verification and can be used to filter out epitopes with poor antigenicity. The remaining epitope may be used for vaccine construction.
Through mining the literature, we identified the hotspot regions in the Spike protein from which epitopes were predicted in multiple studies, suggesting the importance of these regions. These hotspot regions located in the full exposed RBD of the Spike protein, further demonstrating the high potential of possessing antigenicity. Epitopes from the hotspot regions are potential stable candidates for vaccine development in the near future.
5. Conclusion
We identified hotspot regions in the Spike protein harboring epitopes reported in multiple studies through mining literature on the SARS-CoV-2 epitopes published and pre-printed since the outbreak of the epidemic till June 1, 2020. Five epitopes in these hotspot regions were discovered locating in the RBD region of the Spike protein. The five epitopes including three B-cell epitopes (‘RQIAPGQTGKIADYNYKLPD’, ‘SYGFQPTNGVGYQ’ and ‘YAWNRKRISNCVA’), and two T-cell epitopes (‘KPFERDISTEIYQ’ and ‘NYNYLYRLFR’), were predicted as non-toxic epitope candidates with high potential for diagnostics and vaccine development.
Author contributions
Jing Zhang: Conceptualization, Supervision, Writing-Original draft preparation, Reviewing and Editing. Yubo Fan: Conceptualization, Writing-Reviewing and Editing. Wendong Li: Immune-informatics analysis, Visualization, Writing-: Original draft preparation. Lin Li: Immune-informatics analysis, Visualization, Writing-Reviewing. Ting Sun: Data curation, Writing-Reviewing and Editing. Yufei He: Data curation, Writing-Reviewing and Editing. Guang Liu: Data curation, Writing-Reviewing and Editing. Zixuan Xiao: Data curation, Writing-Reviewing and Editing.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (NSFC No. 11421202, and 11827803 to YBF), the Youth Thousand Scholar Program of China (J.Z.) and Beijing Advanced Innovation Center for Biomedical Engineering, BUAA (J.Z.)
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.medntd.2020.100048.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Phelan Al K.R., Gostin L.O. The novel coronavirus originating in Wuhan, China: challenges for global health governance. Jama. 2020 doi: 10.1001/jama.2020.1097. [DOI] [PubMed] [Google Scholar]
- 2.Zhou P., Y X L., Wang X.-G. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020:1–4. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Response., E.W.G.f.N.E., The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China. . Chin J Epidemiol., 2020. 41(2): p. 145-151.
- 4.Qun Li M.M., Xuhua Guan PhD., Peng Wu PhD., Xiaoye Wang M.P.H., Lei Zhou M.Med, Yeqing Tong PhD., Ruiqi Ren M.Med, Kathy S.M., Leung PhD., Eric H.Y., Lau PhD., Jessica Y., Wong PhD., Xuesen Xing PhD., Nijuan Xiang M.Med. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med. 2020;382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.R Vita J.O., Greenbaum J.A., Ponomarenko J., Clark J.D., Cantrell J.R., Wheeler D.K., Gabbard J.L., Hix D., Sette A., Peters B. Nucleic Acids Res.; 2014. The immune epitope database (iedb) 3.0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saha S., Raghava G.P. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins. 2006;65(1):40–48. doi: 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- 7.Raghava, H.S.a.G.P.S., ProPred: prediction ofHLA-DR binding sites. Bioinformatics, 2001. [DOI] [PubMed]
- 8.Kringelum J.V. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol. 2012;8(12) doi: 10.1371/journal.pcbi.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh H., Raghava G.P. ProPred1: prediction of promiscuous MHC Class-I binding sites. Bioinformatics. 2003;19(8):1009–1014. doi: 10.1093/bioinformatics/btg108. [DOI] [PubMed] [Google Scholar]
- 10.Abelin J.G. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46(2):315–326. doi: 10.1016/j.immuni.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhasin M., R G. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccines (Basel) 2004;22(23–24):3195–3204. doi: 10.1016/j.vaccine.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Vanessa Jurtz S.P., Massimo Andreatta, Paolo Marcatili, Bjoern Peters, Morten Nielsen. Netmhcpan-4.0: improved peptide–mhc class i interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. 2017;199(9):3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen B. Predicting HLA class II antigen presentation through integrated deep learning. Nat Biotechnol. 2019;37(11):1332–1343. doi: 10.1038/s41587-019-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng Y. Multi-epitope vaccine design using an immunoinformatics approach for 2019 novel coronavirus in China (SARS-CoV-2) bioRxiv. 2020 [Google Scholar]
- 15.Chung M., B A., Mei X. CT imaging features of 2019 novel coronavirus. Radiology(2019-nCoV) 2020 doi: 10.1148/radiol.2020200230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yazdani Z. Design an efficient multi-epitope peptide vaccine candidate against SARS-CoV-2: an in silico analysis. bioRxiv. 2020 doi: 10.2147/IDR.S264573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yarmarkovich M. A SARS-CoV-2 vaccination strategy focused on population-scale immunity. Cell Rep Med. 2020 doi: 10.1016/j.xcrm.2020.100036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vashi Y., Jagrit V., Kumar S. Understanding the B and T cells epitopes of spike protein of severe respiratory syndrome coronavirus-2: a computational way to predict the immunogens. bioRxiv. 2020 doi: 10.1016/j.meegid.2020.104382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srivastava S. Structural basis to design multi-epitope vaccines against Novel Coronavirus 19 (COVID19) infection, the ongoing pandemic emergency: an in silico approach. bioRxiv. 2020 [Google Scholar]
- 20.Singh A. Designing a multi-epitope peptide-based vaccine against SARS-CoV-2. bioRxiv. 2020 doi: 10.1038/s41598-020-73371-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sardar R. Comparative analyses of SAR-CoV2 genomes from different geographical locations and other coronavirus family genomes reveals unique features potentially consequential to host-virus interaction and pathogenesis. bioRxiv. 2020 doi: 10.1016/j.heliyon.2020.e04658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saha R., Prasad B.V.L.S. In silico approach for designing of a multi-epitope based vaccine against novel Coronavirus(SARS-COV-2) bioRxiv. 2020 [Google Scholar]
- 23.Rehman H.M. Preprints; 2020. A putative prophylactic solution for COVID-19: development of novel multiepitope vaccine candidate against SARS-COV-2 by comprehensive immunoinformatic and molecular modelling approach. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramaiah A., Arumugaswami V. Insights into cross-species evolution of novel human coronavirus 2019-nCoV and defining immune determinants for vaccine development. bioRxiv. 2020 [Google Scholar]
- 25.Rahman M.S. Epitope-based chimeric peptide vaccine design against S, M and E proteins of SARS-CoV-2 etiologic agent of global pandemic COVID-19: an in silico approach. biorxiv. 2020 doi: 10.7717/peerj.9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poran A. Sequence-based prediction of vaccine targets for inducing T cell responses to SARS-CoV-2 utilizing the bioinformatics predictor RECON. bioRxiv. 2020 [Google Scholar]
- 27.Poh C.M. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat Commun. 2020;11(1) doi: 10.1038/s41467-020-16638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nazneen Akhand M.R. Genome based evolutionary study of SARS-CoV-2 towards the prediction of epitope based chimeric vaccine. bioRxiv. 2020 [Google Scholar]
- 29.Li L. Epitope-based peptide vaccine design and target site characterization against novel coronavirus disease caused by SARS-CoV-2. bioRxiv. 2020 doi: 10.1016/j.virusres.2020.198082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ismail S., Ahmad S., Azam S.S. Immuno-informatics characterization SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. bioRxiv. 2020 doi: 10.1016/j.molliq.2020.113612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herst C.V. An effective CTL peptide vaccine for ebola zaire based on survivors’ CD8+ targeting of a particular nucleocapsid protein epitope with potential implications for COVID-19 vaccine design. bioRxiv. 2020 doi: 10.1016/j.vaccine.2020.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta E., Mishra R.K., Niraj R.R.K. Identification of potential vaccine candidates against SARS-CoV-2, A step forward to fight novel coronavirus 2019-nCoV: a Reverse Vaccinology Approach. bioRxiv. 2020 [Google Scholar]
- 33.Grifoni A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe. 2020;27(4):671–680 e2. doi: 10.1016/j.chom.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fast E., Altman R.B., Chen B. Potential T-cell and B-cell epitopes of 2019-nCoV. bioRxiv. 2020 [Google Scholar]
- 35.Blanco-Míguez, A., Release of potential pro-inflammatory peptides from SARS-CoV-2 spike glycoproteins in neutrophil-extracellular traps. bioRxiv.
- 36.Bhattacharya M. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): immunoinformatics approach. J Med Virol. 2020 doi: 10.1002/jmv.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Behbahani M. In silico Design of novel Multi-epitope recombinant Vaccine based on Coronavirus surface glycoprotein. bioRxiv. 2020 [Google Scholar]
- 38.Ahmed S.F., Quadeer A.A., McKay M.R. Viruses; 2020. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARSCoV immunological studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wrapp D. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020 doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.L W., Pymol D. An open-source molecular graphics tool. CCP4 Newslett Protein Crystallogr. 2002;40(1):82–92. [Google Scholar]
- 41.Doytchinova I.A., Flower D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ivan Dimitrov L.N. Irini doytchinova and ivan bangov, AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30(6):846–851. doi: 10.1093/bioinformatics/btt619. [DOI] [PubMed] [Google Scholar]
- 43.Gupta S K.P., Chaudhary K. In silico approach for predicting toxicity of peptides and proteins. PloS One. 2013;8(9) doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jespersen M.C. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45(W1):W24–W29. doi: 10.1093/nar/gkx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolaskar A.S., C T P. 1990. A semi-empirical method for prediction of antigenic dete~inants on protein antigens. [DOI] [PubMed] [Google Scholar]
- 46.Emilio A. Emini, J.V.H., DEBRA S PERLOW, AND Joshua BOGER, Induction of hepatitis A virus-neutralizing antibody by a VirusSpecific synthetic peptide. J Virol, 1985. [DOI] [PMC free article] [PubMed]
- 47.Andersen P.H., M.N.a.O.L. Prediction of residues in discontinuous B cell epitopes using protein 3D structures. Protein Sci. 2006;15:2558–2567. doi: 10.1110/ps.062405906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ponomarenko J. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinf. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maillet N. Rapid Peptides Generator: fast and efficient in silico protein digestion. NAR Genom Bioinf. 2020;2(1) doi: 10.1093/nargab/lqz004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ratul Chowdhury M.F.A., Maranas Costas D. OptMAVEn-2.0: de novo design of variable antibody regions against targeted antigen epitopes. Antibodies. 2018;7:23. doi: 10.3390/antib7030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.El-Manzalawy Y., Dobbs D., Honavar V. Predicting linear B-cell epitopes using string kernels. J Mol Recogn. 2008;21(4):243–255. doi: 10.1002/jmr.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saha, S.a.G.P.S.R., BcePred: prediction of continuous B-cell epitopes in antigenic sequences using physico-chemical properties. Berlin, Heidelberg: Springer Berlin Heidelberg. Artificial Immune Systems, 2004.: p. 197-204.
- 53.Andreatta M., Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.