

Abstract
Cytokine storm can result from cancer immunotherapy or certain infections, including COVID-19. Though short-term immune-related adverse events are routinely described, longer-term immune consequences and sequential immune monitoring are not as well defined. In 2006, six healthy volunteers received TGN1412, a CD28 superagonist antibody, in a first-in-man clinical trial and suffered from cytokine storm. After the initial cytokine release, antibody effect-specific immune monitoring started on Day + 10 and consisted mainly of evaluation of dendritic cell and T-cell subsets and 15 serum cytokines at 21 time-points over 2 years. All patients developed problems with concentration and memory; three patients were diagnosed with mild-to-moderate depression. Mild neutropenia and autoantibody production was observed intermittently. One patient suffered from peripheral dry gangrene, required amputations, and had persistent Raynaud’s phenomenon. Gastrointestinal irritability was noted in three patients and coincided with elevated γδT-cells. One had pruritus associated with elevated IgE levels, also found in three other asymptomatic patients. Dendritic cells, initially undetectable, rose to normal within a month. Naïve CD8+ T-cells were maintained at high levels, whereas naïve CD4+ and memory CD4+ and CD8+ T-cells started high but declined over 2 years. T-regulatory cells cycled circannually and were normal in number. Cytokine dysregulation was especially noted in one patient with systemic symptoms. Over a 2-year follow-up, cognitive deficits were observed in all patients following TGN1412 infusion. Some also had signs or symptoms of psychological, mucosal or immune dysregulation. These observations may discern immunopathology, treatment targets, and long-term monitoring strategies for other patients undergoing immunotherapy or with cytokine storm.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02725-2) contains supplementary material, which is available to authorized users.
Keywords: Cytokine storm, Cytokine release syndrome, TGN1412, Immunotherapy, Immune monitoring, Immune-related adverse events (irAEs)
Introduction
Antibody or cell-based immunotherapeutics for cancer carry inherent risks of bystander immune activation, in addition to the effects of treating the intended tumor target [1–5]. The most severe of these immune-related adverse events (irAEs) are cytokine release syndrome (CRS) and neurotoxicity, most prominently described in patients treated with CAR-T-cells [6–11]. Treatment for CRS and associated neurotoxicity primarily targets IL-6, likely arising from monocytes or activated endothelium [1, 6, 11–17]. The site of cell activation is unclear, and may depend on disease burden, treatment target or how CRS is induced; antibody and cell therapies target tumor sites, whereas infections, such as SARS-CoV-2 causing COVID-19, target points of pathogen entry, e.g. mucosal sites of the nasopharynx, lung and gastrointestinal tract [1, 11–19]. Immune checkpoint blockade induces irAEs during short-term follow-up of some patients with cancer, though robust immune biomarkers correlating with adverse effects of therapy are still being investigated [1, 4, 7, 20–25]. For long-term survivors who received immunotherapy or following CRS, immune monitoring is not yet standard of care, despite evidence that such patients may continue to experience secondary effects of treatment and irAEs [1, 3].
In 2006, a superagonist anti-CD28 humanized monoclonal antibody (TGN1412) was infused into six healthy young male volunteers in a first-in-man clinical trial [26]. The antibody had specificity for the C’’D loop of the CD28 glycoprotein and had the unique capacity to activate T-cells solely through CD28 (signal 2) without the usual ligation of the T-cell receptor (signal 1) [27]. All six volunteers suffered from CRS, first noted clinically within 90 min of infusion. Although the expected effect from pre-clinical studies was for selective T-regulatory cell (Treg) expansion, without cytokine release, the first effects observed in humans were those of cytokine storm. Patients displayed early high TNF-α release within an hour of infusion. This was associated with fever, delirium, nausea, vomiting and diarrhea, multi-organ failure (starting with the lung and rapidly progressive hypoxemia), disseminated intravascular coagulation, and a discrete absence of T-cells and monocytes from the peripheral blood [26]. The acute effects of the TGN1412-induced cytokine storm and the first 30 days of follow-up have already been reported [26]. Herein, we present the clinical and linked immunological data from 10 days sequentially to 2 years following TGN1412 infusion. All patients survived; dysfunction in cognitive, psychological, gastrointestinal, integumentary, and immune regulatory systems persisted for years following the event. We believe that the features described herein may help to identify the immunopathology of similar illnesses, such as irAEs and CRS due to other immunotherapies (e.g. checkpoint inhibitors, CAR-T-cells) or COVID-19 [4, 18, 19], and to inform discussions on the type and length of clinical and immune monitoring warranted in such patients.
Materials and methods
Clinical trial
TGN1412 was produced by TeGenero AG (Wϋrzberg, Germany), manufactured by Boehringer Ingelheim (Germany), and the clinical trial was conducted by the contract research organization, PAREXEL International (Waltham, MA, USA) in their leased UK clinical trial site at Northwick Park Hospital, London, UK. Details of the clinical trial and the first 30 days of clinical follow-up have been reported [26]. The patients presented herein as A, B, C, D, E and F correlate with those previously identified as patients 2, 1, 5, 6, 4 and 3, respectively [26]. None of the authors of this work were involved with the conduct of the clinical trial or any of the pre-clinical testing of TGN1412.
Patients and sources of data
All six patients were followed clinically (off-trial) and assessed as a cohort following the serious adverse event (SAE). Overall care and immune monitoring was coordinated and interpreted by the lead clinician (NP) who made appropriate referrals to subspecialists (CJM, MH, NA) or requests for specific immune monitoring tests as determined by clinical need. From 6 months post-event, all patients were assessed in a specialist cognitive disorders clinic by an expert neurologist (CJM) and psychiatrist (MH). All patient blood samples were anonymized and the scientists performing immunologic tests were not aware of clinical symptoms, signs, or clinical laboratory data (NEM, AJS, HOA-H, CLP, MK, AM and SCK). Blood was procured for immune, hematological and biochemical monitoring at each assessment by the lead clinician beginning 10 days after TGN1412 infusion and ending 2 years post-drug administration. In parallel, blood samples were obtained from healthy, male volunteers (n = 24; after written informed consent) as comparative controls. All six TGN1412 trial participants were male, with median age of 29.5 years (range 19–34). The healthy control volunteers were male, with a median age of 30 years (range 19–42). None of the patients had a notable medical history and all were well during the 2 weeks preceding the clinical trial. Patients B and C were lost to immunological follow-up after 15 and 22 months, respectively. Patients provided written informed consent to data publication.
Immune monitoring
Immune monitoring started on Day + 10 and was continued every 3–4 days for 2 weeks, then weekly for 4 weeks, then every 4 weeks for 3 months, and every 6 weeks to month 8 (time-points 1–17). In the second year, patients were evaluated every 3 months (time-points 18–21). For the first 6 months, whole blood was assessed for T-cell and dendritic cell (DC) subset numbers, phenotype and function by measuring intracellular and serum cytokines (Supplemental Fig. 1 and 2). After 6 months, tests were rationalized to those that were most informative. Correlates of immune function or potential were explored, including T-cell receptor Vβ repertoire, antigen recall assay with purified protein derivative (PPD), and T-cell homing for gut and skin based on expression of β7 integrin and cutaneous leukocyte antigen (CLA), respectively. These studies were conducted in a laboratory operating under GLP principles, undertaking exploratory research and using established protocols that were MIATA compliant (Supplementary MIATA information). The assays and reagents employed were previously validated and tested for performance during the course of standard general investigative research.
Flow cytometry
Whole blood was obtained by venipuncture into sodium-heparin Vacutainer™ tubes (Becton–Dickinson) and labeled with fluorochrome-conjugated monoclonal antibodies (mAb; Supplementary Table 1) for surface and intracellular cytokine detection, as previously described [28], and acquired on a FACSCalibur flow cytometer using CellQuest software (Becton–Dickinson). The Vβ repertoire kit was kindly donated by Beckman Coulter. The human FoxP3 staining kit (including anti-FoxP3 mAb; PCH101-APC) was from eBioscience (cat no. 77-5776-40). Analyses were performed using WinList software (Verity Software House, Maine, USA) using off-line compensation to ensure objective analysis of data. Absolute cell numbers were determined by reference to a known quantity of Flow-Count™ Fluorospheres (Beckman Coulter, cat no. 7547053) added to each aliquot of cells immediately prior to acquisition.
Two DC subsets (Supplementary Fig. 1) and major CD3+ T-cell subsets were identified (Supplementary Fig. 2). The CD3+/CD8− (hereafter CD3+/CD4+) and CD3+/CD8+ T-cells were assessed for CD28+/CD25+ subsets, enriched for Tregs. Later analysis employed a mAb against FoxP3 (in lieu of CD28 mAb) to identify CD3+/CD4+ Tregs within the CD25+ population. This analysis verified that Tregs identified by CD28+/CD25+ contained the FoxP3+ subset, despite the absence of high CD25 expression (Supplementary Fig. 2). DC maturation markers (CD80/CD86/CD40) were not informative and were removed from the protocol after 4 months (Day + 133). At this time, patients were evaluated on 2 separate days, rather than all six on 1 day, allowing for faster laboratory processing of samples (with less cell death), but resulting in significant disparity in total cell numbers in certain T-cell subsets versus analyses prior to Day + 133 for both the patient and healthy-control samples.
Cytokine determinations
All sera (patient and healthy control) were stored at − 80 °C and thawed for re-aliquoting once prior to assessment. To ensure consistency, cytokines were assessed on a single run by one operator with a single multi-channel pipette (freshly calibrated) after a single freeze–thaw cycle for time points 1–19, and then a second run for the last two time points. Enzyme-linked immunosorbent assay (ELISA) with an ELx808TM absorbance microplate reader (BioTek Instruments Inc, USA) or a cytokine bead array was used for quantitative determination of cytokines as per manufacturer’s instructions (Supplementary Table 2). ELISA determinations were done in duplicate and on different plates to account for plate-to-plate variation. Method controls and normal control samples were included on each test plate in addition to standard controls for calibration. Normal controls and method controls were also included in cytokine bead array with samples tested once.
Statistical analysis
The statistical analysis was performed by scientists (MK, AM) not involved in acquisition of data, except for serum cytokine determinations by ELISA, and without knowledge of clinical outcomes. SigmaStat (Systat Software UK Ltd, London, UK, version 3.5) was used for three-way analysis of variance (ANOVA) to elucidate the effects of treatment (exposure to TGN1412) on cellular parameters over time within the patient group and compared with controls. Time-points were only included when data from all six patients and six controls were available. Two different hypotheses were tested: 1. whether exposure to TGN1412 affected cytokine levels over time, i.e. patients vs controls and, 2. whether the numbers of each cell subtype, intracellular cytokine-expressing cells, and level of serum cytokines differed amongst the six patients over time. The criterion for the implementation of the ANOVA tests was the normality assumption [29]. Due to the small sample size (no replicates), the homogeneity of variance, expectedly, was not satisfied only for the second hypothesis. The level of significance was accepted at p < 0.05.
Results
The clinical characteristics and recovery of blood counts in the first 30 days following TGN1412-infusion have been described [26]. Briefly, this 30-day period was broken down into 4 phases. Phase 1 was the “cytokine storm” starting within an hour of infusion with rapid induction of type 1 and 2 cytokines, associated with high fever, severe headache, delirium, nausea, vomiting, diarrhea, diffuse erythema, hypotension, tachycardia severe lymphopenia and monocytopenia and lasted for 2–3 days. Phase 2 was the “reactive phase”, overlapping with Phase 1 from Day + 1 from infusion through Day + 3, and consisted of end-organ damage with renal failure, pulmonary infiltrates, respiratory failure and disseminated intravascular coagulation. Phase 3 was the “recovery phase”—renal and pulmonary function normalized, with an accompanying reactive thrombocytosis, increase in alanine aminotransferase levels, and recovery of monocyte and lymphocyte counts. This phase started on Day + 3 and lasted to Day + 15 (or Day + 20 in the two sickest patients, C and D, requiring prolonged intensive care). Phase 4 was the “plateau” or “steady-state” beginning Day + 15–20 after TGN1412-infusion and consisted of normalization of blood counts and chemistry panels. During the first 10 days, all patients displayed generalized desquamation of the skin, muscle weakness and myalgia which slowly improved [26]. Longer term effects following exposure to TGN1412 affected all patients in four main areas: cognitive and psychiatric, autoimmune and inflammatory, immune mucosal barrier function, and alterations in immune cell subsets and cytokines in peripheral whole blood (Table 1; Supplementary Fig. 3). Herein, we describe these effects from Day + 10 to 2 years post-infusion.
Table 1.
Persistent symptoms and signs over two years following TGN1412-induced cytokine storm
| Categories | Symptoms and immune correlates (# of patients) | CTCAE grade |
|---|---|---|
| Neurocognitive and psychological | Memory impairment (6) | 1–2 |
| Impairment in attentional processing (6) | 1–2 | |
| Mild-moderate depression (3) | 1–2 | |
| Post-traumatic stress disorder (2) | 2 | |
| Anxiety requiring psychotherapy (4) | 1–3 | |
| Headaches (5) | 1–2 | |
| Blurred vision (5) | 1 | |
| Autoimmune and inflammatory | Mild neutropenia (3) | 1 |
| Arthralgias—knees, hands, back (6) | 1 | |
| Positive auto-antibodies (4) | 1 | |
| Ischemic extremities (1) | 3 | |
| Raynaud’s phenomenon (1) | 1 | |
| Immune mucosal barrier function | Diarrhea and ↑ γδT cells (3) | 1–2 |
| Skin dryness and ↑sensitivity (3) | 1–2 | |
| Pruritus (1) | 1–2 | |
| Peripheral blood eosinophilia (3) | 1 | |
| ↑ Serum IgE (4) | 1 | |
| Benign lipomas/angiolipomas (1) | 2 | |
| Immune cell subsets and cytokines | Gradual recovery of DC over 1 month (6) | 1 |
| Low-normal total CD4+ T cells (5) | 1 | |
| Low-normal total CD8+ T cells (6) | 1 | |
| ↑ sustained naïve CD8+ T cells (5) | 1 | |
| ↓ naïve CD4+ T cells over time (5) | 1 | |
| ↓ memory CD4+ T cells over time (4) | 1 | |
| ↓ memory CD8+ T cells over time (4) | 1 | |
| Normal Tregs with circannual cycle (6) | 1 | |
| Vβ repertoire normal (6) | – | |
| Normal immune responses in vitro (6) | – | |
| ↑ sustained erythropoietin level for 3 months (1) | 1 | |
| ↑ cytokine response from 3 months (1) | 1 | |
| IL-17 differences (6) | 1 |
CTCAE common terminology criteria for adverse events, version 4.03
Cognitive and psychiatric effects
The most consistent symptoms in all patients following the SAE were in memory and concentration (Table 1, Supplementary Table 3 and Supplementary Fig. 3). All patients reported subjective concentration and day-to-day memory problems, particularly for names. Although all patients scored within normal range on the Mini Mental State Examination, bedside cognitive examination and formal neuropsychometry performed between 6 and 12 months after the SAE revealed common deficits in verbal recall of information (Supplementary Table 3). Three patients exhibited poor verbal fluency suggestive of executive dysfunction (Supplementary Table 3). The patients initially showed some improvement, plateauing approximately 12 months after the event. These changes, in addition to the SAE itself, generated significant anxiety; psychotherapy was required in four (Table 1). All patients underwent a comprehensive psychiatric assessment including a Structured Clinical Interview for Axis I Disorders (SCID-I). Three patients were diagnosed with mild-to-moderate depression; two of these, who required a prolonged stay in intensive care, were also diagnosed with post-traumatic stress disorder, (one also had associated panic disorder and agoraphobia; Table 1). Five patients were able to return to work within 2 years. However, all noted that their previous work and everyday capabilities were limited compared with pre-trial due to decreased concentration, reduced memory and difficulty retaining information, persisting over 2 years.
Five patients had intermittent headaches that started several days after stopping steroids on Days + 21–31 and became less frequent in the second year (Table 1; Supplementary Fig. 3). The headaches were sharp, short-lasting and severe, often affecting the vertex of the head. All patients had unremarkable EEG and brain MRI. Five patients had normal FDG-PET brain scans (one patient declined). The eldest (patient A), whose memory and cognitive problems were most severe, had a lumbar puncture one year post-event which showed oligoclonal bands matched between serum and cerebrospinal fluid. The patients with headaches also had mild blurred distance vision (Table 1); in patient C, whose symptoms were most severe, ophthalmologic testing did not reveal retinal disease or intraocular inflammation, although a change in contrast sensitivity of vision was noted; he declined further testing. Patient B had mild dry eyes and blepharitis with hypermetropia. Interestingly, patient D did not have headaches or blurred vision, yet was the most physically ill, requiring a prolonged stay in intensive care and a prolonged course of steroids.
Evidence of autoimmune phenomena and inflammation
Three patients (A, B, F) had mild and intermittent neutropenia, relative to normal clinical lab reference ranges (Table 1; Fig. 1a). These lower counts were not accompanied by infection. All patients reported intermittent arthralgias in knees, hands and back (Table 1; Supplementary Fig. 3). In four, symptoms were associated with low-titer anti-nuclear antibody (ANA; patient D), anti-cardiolipin antibody (ACA; patients A, D, E, F), intermittent equivocal rheumatoid factor (RF; patient A) or a positive anti-nuclear cytoplasmic antibody (ANCA) in perinuclear staining pattern directed against PR3 (patient A; also displayed mild neutropenia). These antibodies lasted less than 5 months at a time and did not correlate with changes in cell subsets or cytokine measurements. Complement levels were normal in all patients.
Fig. 1.

Two-year follow-up of relevant clinical parameters in the six patients Absolute neutrophil (a) and eosinophil counts (with values within the first 3 months highlighted in the panel to the right) (b) followed over the 2-year period since TGN1412-induced cytokine storm show that three of the patients had mild intermittent neutropenia and three had intermittent eosinophilia. The latter three patients also had elevated levels of IgE (c), not always correlating with the elevated eosinophil counts. Horizontal dashed lines indicate the normal control reference ranges for each parameter. Normal reference ranges were determined for a healthy population in the clinical pathology accredited hematology laboratory using standard operating procedures
Patient D was the most physically ill following TGN1412 infusion and suffered ischemic hands and feet of unclear cause (Table 1; Fig. 2a,b). With improvement in his overall clinical condition, the extent of ischemia also improved. He underwent bilateral transmetatarsal amputation and removal of the terminal phalanges of both hands (one on the right and two on the left). Histopathologic examination showed no features of primary vasculopathy. The fingers of both hands displayed decreased pigmentation and, 10 months post-infusion, Raynaud’s phenomenon was noted in both hands that worsened during winter of the second year (18–24 months; Table 1). Autoimmune antibody testing was consistently negative in this patient except for expression of a weak anti-nuclear antibody (ANA; 1:40 titer) in speckled pattern at 7 and 8 months and a low-titer anti-cardiolipin IgG antibody at 8 and 24 months following infusion.
Fig. 2.
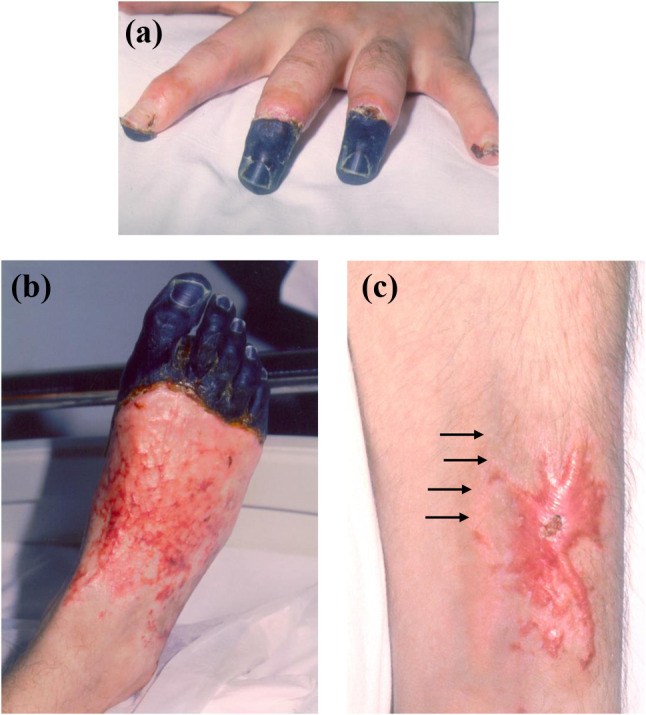
Ischemic and dermatologic changes in Patient D at three months following TGN1412-induced cytokine storm. Areas of dry gangrene became fully demarcated at 2–3 months following the insult with vascular skin changes in the areas that became revigorated. Shown are changes prior to amputation of the ischemic digits in the (a) left hand and (b) right foot. Following amputations, he had persistent pain in both feet, some of which could be ascribed to phantom-limb pain, and had a sensory deficit which followed a glove-and-stocking distribution, consistent with the areas originally affected by ischemia during his critical illness. (c) During the patient’s critical care phase, an arterial line had been placed in the left radial artery and with recovery, a hyperkeratotic scar formed, 7 cm × 4 cm in maximum dimensions, with faint satellite amelanotic lesions (1–3 mm dimension, arrows). These lesions continued to improve with time, with regression of the scar and disappearance of the white satellite spots
Altered mucosal and immune barrier function
Patients A, B and E had new gastrointestinal symptoms (diarrhea or frequent bowel motions) manifesting as intolerance to spicy foods and associated with a rise of γδT-cells in the blood (in press; Table 1 and Supplementary Fig. 3). As the intolerance improved, this T-cell subset also declined (in press). Because of the extent of symptoms in patient B, a full gastrointestinal work-up was undertaken including lactose intolerance test (normal), esophagogastroduodenoscopy, and colonoscopy; a 5 mm sessile polyp in the proximal ascending colon showed inflammatory change. A cause for diarrhea was not found on random biopsies.
All patients had skin erythema followed by desquamation to varying degrees and hair thinning in the days and weeks immediately following TGN1412 infusion. Increased skin dryness and sensitivity to sun exposure, chemicals and soaps was noted in three patients (A, C, D; Table 1). Patient B suffered from ongoing pruritus over all skin areas; cetirizine could not be discontinued over 2 years due to recurrence of debilitating symptoms (Table 1 and Supplementary Fig. 3). He had a persistently increased level of IgE, and intermittent mild eosinophilia without other associated signs or symptoms of allergy (Fig. 1b,c). Three other patients (D, E, F) also had persistently elevated IgE, without specific symptoms or seasonal correlation, two of whom also showed mild eosinophilia (E, F; Fig. 1b,c). Mast cells were absent from blood and gastrointestinal biopsies of patient B. Mobile, nontender and rubbery subcutaneous lumps were noted on the arms, and thorax of patient C (Table 1). Three of these lumps were biopsied on two occasions, 14 months apart and were found to be benign lipomas or angiolipomas. Immune cells were not identified in these biopsies and a CT scan did not show significant adenopathy or lesions elsewhere.
Recovery kinetics of immune cell subsets and cytokines in peripheral blood
Both CD11c+ conventional and CD11c− plasmacytoid dendritic cells (DCs) were initially depleted from the blood and recovered slowly over the first month towards normal (Fig. 3, Supplementary Fig. 4–6, a&b). Peripheral blood monocytes had recovered to normal by Day + 10, unrelated to the recovery kinetics of DCs [26]. Naïve CD4+ and CD8+ T-cells were detectable in low-to-normal range with a cyclical recovery pattern to normal over the first 5 months. Thereafter, naïve CD8+ T-cells persisted above normal and naïve CD4+ T-cells gradually declined (Fig. 3, Supplementary Fig. 4–6, c&d). Memory CD4+ and CD8+ T-cells were detected at higher-than-normal values and remained so until 9–12 months post-infusion, followed by a gradual decline in both to subnormal levels (Fig. 3, Supplementary Fig. 4–6, e&f). Activated CD4+ and CD8+ T-cells were initially high, returned to normal range over 6 months (Fig. 3, Supplementary Fig. 4–6, i&j) and followed the recovery pattern of DCs and the slow decrease of memory T-cells (Supplementary Fig. 7).
Fig. 3.


Time course of changes in immune cell subsets during the first two years following infusion of TGN1412. Ongoing monitoring of T-cell and DC subsets in the peripheral blood have shown changes over time since the start of the monitoring period, 10 days following infusion of TGN1412. After the 4-month time-point, significant disparity in numbers of certain T-cell subsets was observed, mostly due to shorter handling times for the samples and a resultant decrease in cell death. The data have been separated by a vertical dashed line to indicate this change; the entire 2-year monitoring period is shown in a continuous time-course, but with a split in the data after the 4-month change in protocol. The cell subsets measured were: (a) HLA-DR+/Lin−/CD11c+ conventional (“myeloid”) and (b) CD11c− plasmacytoid dendritic cells, (c) CD45RA+/CD45RO− naive CD4+ helper and (d) CD8+ cytotoxic T-cells, (e) CD45RA−/CD45RO+ memory CD4+ helper and (f) CD8+ cytotoxic T-cells, CD25+/CD28+ “T-regulatory” CD4+ (g) and CD8+ T-cells (h) and CD69+ activated CD4+ (i) and CD8+ (j) T-cells (with the first 4 months shown in the inset for clarity of early events). Total CD3+ T-cells (k) indicates that although the total number of T-cells remained in the normal or high–normal range, the cell subsets making up the total changed over time. Whereas naïve CD8+ T-cells remained in the high–normal or higher range at the 2-year follow-up, all other T-cells were below normal, especially in memory subsets and CD25+/CD28+ subsets which included Tregs. However, these low values at the two-year point were found at the expected trough of the circannual cycling pattern and may be normal. CD45RA−/CD45RO− T-cells were not observed at any time point during immune recovery. CD45RA+/CD45RO+ T-cells were observed intermittently throughout recovery in all patients and controls, albeit in small numbers (data not shown). Median and interquartile ranges for the cohort are shown for each time-point. Median and interquartile ranges for the normal controls (n = 24) drawn at the same time points are shown separately for the first 4 months and the remaining 18, indicated by the horizontal dashed lines on each figure. Total CD3+ (l), CD4+ (m) and CD8+ (n) T-cell subsets were also evaluated in the clinical laboratory by flow cytometry at the same time-points and served as an internal control. The total CD3+ cells correlated well between the research laboratory (k) and the clinical laboratory (l) and the total CD4+ and CD8+ T-cells remained in the low–normal range over 2 years. Conventional units are shown in the y-axes (cells/mm3 = cells/μl) and is equivalent to 106cells/L in SI units
TGN1412 was a CD28 superagonist and preclinical studies indicated preferential stimulation of Tregs. The CD25+/CD28+ T-cell subsets, which include the CD4+ Tregs and corresponding CD8+ “Treg” population, were in normal range when monitoring started, decreased to low-to-normal levels over months 1 and 2, and continued to cycle circannually within the normal range over the 2-year period (Fig. 3, Supplementary Fig. 4–6, g&h). This circannual cycle was also observed in healthy controls. Interestingly, the first peak of the CD8+ “Treg” subset occurred three months prior to that of the CD4+ “Treg” peak, but thereafter cycled together. At 15 months following infusion, the number of FoxP3+ Tregs was similar to that in healthy controls (data not shown). In vitro correlates of immune function indicated that T-cell proliferative responses to PPD antigen presentation, an antigen against which all six patients had previously been immunized, were similar to that of normal controls at 7 months following infusion. T-cell Vβ repertoires were also normal in the total T-cell populations at 8.6 and 12.1 months following infusion (data not shown).
Despite these similarities, differences in immune cell subsets and cytokines were observed: Patients A and D showed different cell subset numbers and longitudinal follow-up versus those of the other four patients (Fig. 3; p < 0.001), especially regarding low numbers of CD4+ naïve T-cells and “Tregs” over time. They demonstrated no clinical similarities beyond those shared with the other patients. Comparison of intracellular CD4+ and CD8+ T-cell cytokines (spontaneous and stimulated IL-4, IL-10 and IFNγ) indicated that patients B and D were similar to each other and different from patients A, C, E and F (p < 0.001). These patients also shared no unique clinical features from the other four. Although the clinical relevance of these groupings is currently unclear, it is notable that patient D, who had several operations and recurrent infections of his wounds during the 2-year follow-up, shared similar cellular and cytokine profiles with those of patients A and B, respectively. These similarities may indicate potentially common cellular/immune mechanisms underlying different clinical problems found in these patients. All patients showed a type 1 cytokine skew with significantly higher intracellular IFNγ production in both CD4 + and CD8 + T-cells upon stimulation (data not shown). Patient B also had higher serum IFNγ (p < 0.001) starting from 2.5 months, and a different cytokine response in general when compared with that of the other patients (Fig. 4; p < 0.001). He had a sustained increase in erythropoietin from Day + 10 following infusion until 3 months when IFNγ, IL-1β, IL-12p70, IL-8, IL-4, IL-5 and IL-17 all started to increase with another peak in these cytokines noted at month nine. He also had a worsening of his memory and cognitive symptoms and pruritus at month three following TGN1412 infusion. IL-6, IL-17 and TNFα levels in patients A, D, E and F increased during the last 6 months of monitoring, and coincided with declining CD4+/CD45RA+, CD4+/CD45RO+ and CD8+/CD45RO+ T-cell and Treg numbers during this time. The significance of this is unclear.
Fig. 4.


Cytokine levels in patient sera over two years following TGN1412-induced cytokine storm. Cytokine bead array or ELISA was used to measure cytokines (a) IFNγ, (b) IL-1β, (c) IL-12p70, (d) IL-8, (e) IL-4, (f) IL-5, (g) IL-17, (h) erythropoietin, (i) IL-2, (j) IL-10, (k) IL-6, (l) TNFα, (m) IL-11, (n) IL-15, (o) IL-23 and (p) sCD28 in all six patients for the 2-year clinical follow-up. In comparison with the other five patients, patient B was clearly different in the cytokine response (p < 0.001). The level of IL-17 in the serum of patients over time was found to be different compared with that of matched controls (p < 0.001) and with that of the serum concentrations in the same patients of IL-11, erythropoietin, IL-15, IL-23, sCD28, IFNγ, IL-8, IL-6 and IL-10 (p < 0.001). This IL-17 signal suggests a role for cells secreting the cytokine in the immune reconstitution following cytokine storm. There is no statistical difference between the patient data for IL-15, IL-11 and IL-23 and those of the normal controls. Statistical comparisons were done using three-way ANOVA
Discussion
Intravenous infusion of the CD28 superagonist antibody, TGN1412, resulted in a cytokine storm heralded by high serum TNFα levels within an hour of infusion, with fever, delirium, headache, nausea, vomiting and diarrhea, early and rapidly progressive lung involvement with hypoxemia and disseminated intravascular coagulation. Early peripheral blood depletion of lymphocytes and monocytes was observed with slow and specific recovery kinetics, shared by all affected individuals [26]. All volunteers who received TGN1412 became patients who, on longer term follow-up, had evidence of cognitive dysfunction. Some also had psychological difficulties, headaches, autoimmune and mucosal barrier dysregulation in addition to immune cell subset and cytokine irregularities in peripheral blood. These features are now recognized in patients treated with checkpoint blockade and other cancer immunotherapies or in those who have suffered from CRS. However, it is usually unclear if the irAEs are related to other premorbid pathology or due to targeting of the underlying disease for which they required treatment [1, 3–6, 22–24]. In contrast, the significance of the cohort given TGN1412 is that they were all young and healthy, having been screened extensively for a first-in-man clinical trial, and had an immune stimulus that resulted in CRS simultaneously. The patients received similar and concurrent treatment, which enabled monitoring for irAEs and immunological biomarker assessment for clinical–pathologic correlation.
The cognitive symptoms were the most consistent feature shared by all patients. Where psychometric testing was performed, this confirmed initial deficits in recall and learning. The relationship of the headaches to this cognitive problem is unclear, although headache is now a well-described irAE [9]. The first headaches in all patients appeared within 90 min of infusion, coincident with delirium and an early rise in TNFα [30, 31]. In pre-clinical testing of TGN1412, specific fibrillary staining was seen in the cerebrum, cerebellum, spinal cord and pituitary gland of both humans and cynomolgus monkeys [32]. Since no adverse neurological observations were reported, it was concluded that this cross-reactivity with central nervous system (CNS) tissues may not be of major clinical relevance and that TGN1412 was not expected to adversely affect the CNS in humans. It is unclear whether the described difficulties in cognition and memory related to specific antibody targeting of the CNS, to the CRS that ensued [9, 17, 30], to the immune dysregulation that resulted [31, 33], or to the psychological impact of the events thereafter.
Autoimmune colitis, vitiligo, and autoantibody production are now included in CTCAE grading of irAEs due to immunotherapy [1, 4, 6, 20, 22, 34]. It is still unknown if the patients presented here suffered from CRS due to ligation of CD28 on T-cells or another mechanism, such as monocyte or endothelial activation in the gastrointestinal tract or lungs, the first organs to be affected after intravenous infusion [11–15]. The etiology of autoimmune or inflammatory phenomena remains uncertain. It is notable that following the initial disappearance of blood mononuclear cells (coincident with CRS) there were different recovery kinetics of monocytes and DCs, consistent with the distinct lineages of these populations. The Raynaud’s phenomenon in patient D was directly related to the areas of resolved vascular injury, although the cause of the amelanotic areas of skin was less clear and may have been autoimmune in nature. Three patients with gastrointestinal symptoms had an associated increase in blood γδT-cells though without evidence of colitis on gut biopsies. All patients had IL-17 levels during immune reconstitution higher than those of normal controls, and a high level of IgE was found in four patients. These observations suggest a role for Th17-cells following TGN1412-induced CRS, and are consistent with other reports of irAEs due to immunotherapy [23, 35, 36]. One patient with elevated IgE and debilitating pruritus was distinct in that he also had marked elevation of type-1 and -2 cytokines starting from three months post-infusion. This cytokine rise coincided with the decline in serum erythropoietin and, in the setting of hemoglobin levels and renal function similar to those of the other patients, may have indicated that erythropoietin was immunomodulatory in this setting [37, 38].
The initial appearance of increased activated T-cells and memory subsets was consistent with generalized immune activation immediately following the cytokine storm. The relatively few naïve T-cells during early reconstitution is consistent with programmed recovery in patients following chemotherapy [39, 40], immune checkpoint blockade [20, 24], or after infection [41, 42]. Contrary to that observed in response to viral infections and irAEs [20], the immune recovery following TGN1412-induced CRS was predominantly a CD4+ T-cell response. Since the patients are adults, the increase in naïve CD8+ T-cells over 2 years may suggest thymic-independent recovery [39], or the generation of a stable CD45RA+ memory cell subset from the CD45RO+ pool [43], also described in patients receiving checkpoint blockade [24]. This latter point may partially explain declining memory CD45RO+ T-cells over time. However, in light of the low-to-normal total CD4+ and CD8+ T-cells, this apparent decline could also be explained by the low point of normal T-cell circannual kinetics at the 2-year follow-up [44].
Over 2 years, T-cell subsets displayed circannual kinetics [44, 45], especially noted in the CD25+/CD28+ subsets, incorporating Tregs. The Treg populations were found in normal numbers in patients relative to control values, and the kinetics of cell recovery over 2 years indicates that Treg numbers found in blood or tissues on single-point testing should be interpreted cautiously. Although TGN1412 was intended to target Tregs, the Tregs in the patients who received the antibody were normal in number.
Physical, cognitive and immune abnormalities were observed in previously fit and healthy young men following infusion of TGN1412. CD28 on T-cells was the intended antibody target, yet the actual in vivo target in humans after intravenous infusion is unclear; these human data suggest that primary activation of monocytes or endothelium in the gut or lung were the primary targets. Clinical–pathologic correlation in these individuals resulted in valuable observations that may be instructive in understanding mechanisms of immune-induced pathologies, including those of checkpoint inhibitors, CAR-T cells, and infections such as COVID-19, known to target mucosal tissues and cause severe CRS [1, 5, 8, 18, 19]. These observations made in previously healthy individuals may also provide a template for long-term monitoring strategies that could be used for patients affected by irAEs and CRS specifically.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We are grateful for the clinical support of staff and services in the National Health Service at Northwick Park Hospital and for the technical support of Elizabeth Mann and Nichola Gellatly in the Antigen Presentation Research Group, Imperial College London. Financial support for some of the work was provided by The North West London Hospitals NHS Trust incorporating Northwick Park Hospital, Cancer Research UK, and The Northwick Park Hospital Leukemia Research Trust Fund. Above all, we thank the patients who have given consent for presentation of their personal data.
Authors’ Contributions
NP coordinated the study, carried out the clinical follow-up, coordinated experimental design, interpreted results and wrote the paper. NEM and AJS coordinated experimental design, carried out the immune monitoring, interpreted the results and assisted in manuscript preparation. CLP and HOA assisted with experiments, compilation of data and with manuscript preparation. MH, CM, NA and DG carried out subspecialist clinical follow-up, interpreted clinical findings and assisted in manuscript preparation. MK and AM coordinated ELISA experimental design, interpreted ELISA results, performed the statistical analysis for all immune monitoring and assisted in manuscript preparation. SCK assisted in experimental design, interpretation of the results and manuscript preparation.
Funding
The North West London Hospitals NHS Trust; Cancer Research UK; The Northwick Park Hospital Leukemia Research Trust Fund.
Data availability
As this is a clinical cohort follow-up, and not data provided on a clinical trial, the data are unavailable due to personal privacy protections.
Compliance with ethical standards
Conflicts of Interest
None of the authors declare a financial conflict of interest. NP, SCK, MH, DG, CLP, HOA, MK, and AM declare no conflicts of interest. NEM is supported by a Career Development Award from The Medical Research Council (Grant Ref: MR/R008302/1) and is in receipt of a project grant from Bart’s and The London Charity (MGU0465). He has also received consultancy fees and funding for research from ImCheck Therapeutics SAS. AJS research is supported by grants from Gilead Sciences AbbVie, The Medical College of St Bartholomew's Hospital Trust, Bowel & Cancer Research and Bart's Charity. CLM has received consultancy fees from Roche and Biogen for clinical trials in Alzheimer's disease, not relevant to the work presented here. NA has received honoraria from Janssen and Pfizer and also consulting for Janssen. She also has an affiliation with Imperial College London. SCK, NEM and AJS have done contract work for Parexel pre-dating the work described in this report. At the time of this work and report, Parexel Clinical Trials Unit had a short-term contract with the Antigen Presentation Research Group (APRG) to use a Class II cabinet within the laboratory. The APRG has also been contracted to perform immunological studies by a pharmaceutical company, the tissue specimens for which were supplied on behalf of that company via Parexel which is located adjacent to the APRG department. There is no conflict of interest involved.
Ethics approval
Ethics approval had been obtained for the TGN1412 trial (by the investigators—none of the authors of this report were involved in the clinical trial). At the time of the trial-related serious adverse event, clinical and immune monitoring ensued as a matter of standard clinical care; no studies were done outside what was required for clinical care of the patients. Discussions between the Ethics Committee, MHRA and Expert Scientific Group set-up by the Minister of Health (UK) at the time in order to investigate the trial outcome unanimously concluded that the monitoring (as outlined in this report) should continue for standard of care, and that specific ethics approval was not required due to the extraordinary circumstances.
Consent to Participate
Patients consented to clinical follow-up and immune monitoring. None of the authors of this work were involved with the conduct of the clinical trial or any of the pre-clinical testing of TGN1412. The patient cohort had consented to the TGN1412 first-in-man clinical trial that resulted in the cytokine storm serious adverse event. At the time of the start of sample collection for the current report, the patients had been removed from the trial and were being treated based on clinical need, rather than trial protocol.
Consent for publication
Patients have provided written informed consent to the publication of the clinical follow-up and immune monitoring data.
Footnotes
The work was done at the Antigen Presentation Research Group and the Department of Hematology, Imperial College London, Northwick Park and St. Mark’s Campus, London UK.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23(5):540–547. doi: 10.1038/nm.4321. [DOI] [PubMed] [Google Scholar]
- 2.Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood. 2018;131(1):58–67. doi: 10.1182/blood-2017-06-741033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanjanapan Y, Day D, Butler MO, et al. Delayed immune-related adverse events in assessment for dose-limiting toxicity in early phase immunotherapy trials. Eur J Cancer. 2019;107:1–7. doi: 10.1016/j.ejca.2018.10.017. [DOI] [PubMed] [Google Scholar]
- 4.Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378:158–168. doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- 5.Cousin S, Italiano A. Molecular pathways: immune checkpoint antibodies and their toxicities. Clin Cancer Res. 2016;22(18):4550–4555. doi: 10.1158/1078-0432.CCR-15-2569. [DOI] [PubMed] [Google Scholar]
- 6.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teachey DT, Lacey SF, Shaw PA, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6(6):664–679. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santomasso BD, Park JH, Salloum D, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018;8:958–971. doi: 10.1158/2159-8290.CD-17-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wick W, Hertenstein A, Platten M. Neurological sequelae of cancer immunotherapies and targeted therapies. Lancet Oncol. 2016;16:e529–e541. doi: 10.1016/S1470-2045(16)30571-X. [DOI] [PubMed] [Google Scholar]
- 10.Cuzzubbo S, Javeri F, Tissier M, et al. Neurological adverse events associated with immune checkpoint inhibitors: review of the literature. Eur J Cancer. 2017;73:1–8. doi: 10.1016/j.ejca.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Gust J, Hay KA, Hanafi L-A, et al. Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7(12):1404–1419. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giavridis T, van der Stegen SJC, Eyquem J, et al. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24:731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Fang Y, Chen X, et al. Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020 doi: 10.1126/sciimmunol.aax7969. [DOI] [PubMed] [Google Scholar]
- 15.Staedtke V, Bai R, Kim K, et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature. 2018;564:273–277. doi: 10.1038/s41586-018-0774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agarwal S, June CH. Harnessing CAR T cell insights to develop treatments for hyperinflammatory responses in COVID-19 patients. Cancer Discov. 2020 doi: 10.1158/2159-8290.CD-20-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hay KA, Hanafi L-A, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood. 2017;130(21):2295–2306. doi: 10.1182/blood-2017-06-793141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chua RL, Lukassen S, Trump S, et al. COVID-19 severity correlates with airway epithelium-immune cell interaction identified by single-cell analysis. Nat Biotechnol. 2020 doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 19.Kuri-Cervantes L, Pampena MB, Meng W, et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol. 2020 doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subudhi SK, Aparicio A, Gao J, et al. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc Nat Acad Sci. 2016;113(42):11919–11924. doi: 10.1073/pnas.1611421113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hartmann FJ, Babdor J, Gherardini PF, et al. Comprehensive immune monitoring of clinical trials to advance human immunotherapy. Cell Reports. 2019;28(3):P819–831.e4. doi: 10.1016/j.celrep.2019.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh DY, Cham J, Zhang L, et al. Immune toxicities elicited by CTLA-4 blockade in cancer patients are associated with early diversification of the T-cell repertoire. Cancer Res. 2017;77(6):1322–1330. doi: 10.1158/0008-5472.CAN-16-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. 2016;22(8):1865–1874. doi: 10.1158/1078-0432.CCR-15-1507. [DOI] [PubMed] [Google Scholar]
- 24.Weide B, Di Giacomo AM, Fonsatti E, Zitvogel L. Immunologic correlates in the course of treatment with immunomodulating antibodies. Semin Oncol. 2015;42(3):448–458. doi: 10.1053/j.seminoncol.2015.02.016. [DOI] [PubMed] [Google Scholar]
- 25.Lim SY, Lee JH, Gide TN, et al. Circulating cytokines predict immune-related toxicity in melanoma patients receiving anti-PD-1-based immunotherapy. Clin Cancer Res. 2019;25(5):1557–1563. doi: 10.1158/1078-0432.CCR-18-2795. [DOI] [PubMed] [Google Scholar]
- 26.Suntharalingam G, Perry M, Ward S, et al. Cytokine Storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 27.Beyersdorf N, Gaupp S, Balbach K, et al. Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J Exp Med. 2005;202(3):445–455. doi: 10.1084/jem.20051060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panoskaltsis N, Reid CDL, Knight SC. Quantification and cytokine production of circulating lymphoid and myeloid cells in acute myelogenous leukemia (AML) Leukemia. 2003;17:716–730. doi: 10.1038/sj.leu.2402835. [DOI] [PubMed] [Google Scholar]
- 29.Maxwell S, Delaney H. Designing experiments and analysing data: a model comparison perspective. New Jersey: Lawrence Erlbaum Associates; 2003. [Google Scholar]
- 30.Mrak R, Griffin W. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Kipnis J, Derecki N, Yang C, Scrable H. Immunity and cognition: what do age-related dementia, HIV-dementia and "chemo-brain" have in common? Trends Immunol. 2008;29(10):455–463. doi: 10.1016/j.it.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 32.TGN1412 Investigator's Brochure. TeGenero Immunotherapeutics. (Accessed 5 May, 2006, at https://www.mhra.gov.uk/home/idcplg?IdcService=GET_FILE&dDocName=CON2023518&RevisionSelectionMethod=LatestReleased.)
- 33.Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci USA. 2004;101(21):8180–8185. doi: 10.1073/pnas.0402268101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beck K, Blansfield J, Tran K, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24(15):2283–2289. doi: 10.1200/JCO.2005.04.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volpe E, Sevant N, Zollinger R, et al. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nat Immunol. 2008;9(6):650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 36.Stummvoll G, DiPaolo R, Huter E, et al. Th1, Th2, and Th17 effector T cell-induced autoimmune gastitis differs in pathological pattern and in susceptibility to suppression by regulatory T cells. J Immunol. 2008;181:1908–1916. doi: 10.4049/jimmunol.181.3.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shang Y, Li X, Prasad P, et al. Erythropoietin attenuates lung injury in lipopolysaccharide treated rats. J Surgical Res. 2009;155:104–110. doi: 10.1016/j.jss.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 38.Yuan R, Maeda Y, Li W, et al. Erythropoietin: a potent induces of peripheral immuno/inflammatory modulation in autoimmune EAE. PLoS ONE. 2008;3(4):e1924. doi: 10.1371/journal.pone.0001924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackall C, Fleisher T, Brown M, et al. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. 1997;89(10):3700–3707. doi: 10.1182/blood.V89.10.3700. [DOI] [PubMed] [Google Scholar]
- 40.Mackall C, Fleisher T, Brown M, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N Engl J Med. 1995;332(3):143–149. doi: 10.1056/NEJM199501193320303. [DOI] [PubMed] [Google Scholar]
- 41.Kaech S, Wherry E, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 42.Homann D, Teyton L, Oldstone M. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med. 2001;7(8):913–919. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- 43.Faint J, Annels N, Curnow S, et al. Memory T cells constitute a subset of the human CD8+CD45RA+ pool with distinct phenotypic and migratory characteristics. J Immunol. 2001;167:212–220. doi: 10.4049/jimmunol.167.1.212. [DOI] [PubMed] [Google Scholar]
- 44.Levi F, Canon C, Touitou Y, et al. Seasonal modulation of the circadian time structure of circulating T and natural killer lymphocyte subsets from healthy subjects. J Clin Invest. 1988;81:407–413. doi: 10.1172/JCI113333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levi F, Canon C, Dipalma M, et al. When should the immune clock be reset? From circadian pharmacodynamics to temporally optimized drug delivery. Ann N Y Acad Sci. 1991;618:312–329. doi: 10.1111/j.1749-6632.1991.tb27251.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
As this is a clinical cohort follow-up, and not data provided on a clinical trial, the data are unavailable due to personal privacy protections.