

Abstract
Coronavirus Disease 2019 (COVID-19) is an infectious illness caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), originally identified in Wuhan, China (December 2019) and has since expanded into a pandemic. Here, we investigate metabolites present in several common spices as possible inhibitors of COVID-19. Specifically, 32 compounds isolated from 14 cooking seasonings were examined as inhibitors for SARS-CoV-2 main protease (Mpro), which is required for viral multiplication. Using a drug discovery approach to identify possible antiviral leads, in silico molecular docking studies were performed. Docking calculations revealed a high potency of salvianolic acid A and curcumin as Mpro inhibitors with binding energies of −9.7 and −9.2 kcal/mol, respectively. Binding mode analysis demonstrated the ability of salvianolic acid A and curcumin to form nine and six hydrogen bonds, respectively with amino acids proximal to Mpro's active site. Stabilities and binding affinities of the two identified natural spices were calculated over 40 ns molecular dynamics simulations and compared to an antiviral protease inhibitor (lopinavir). Molecular mechanics-generalized Born surface area energy calculations revealed greater salvianolic acid A affinity for the enzyme over curcumin and lopinavir with energies of −44.8, −34.2 and −34.8 kcal/mol, respectively. Using a STRING database, protein-protein interactions were identified for salvianolic acid A included the biochemical signaling genes ACE, MAPK14 and ESR1; and for curcumin, EGFR and TNF. This study establishes salvianolic acid A as an in silico natural product inhibitor against the SARS-CoV-2 main protease and provides a promising inhibitor lead for in vitro enzyme testing.
Keywords: Spices, Secondary metabolites, SARS-CoV-2 main protease, Molecular dynamics, Molecular docking
Graphical abstract

Highlights
-
•
32 natural spices were examined as inhibitors for SARS-CoV-2 Mpro using molecular docking and molecular dynamics.
-
•
Salvianolic acid A and curcumin exhibited higher binding affinities against Mpro over lopinavir.
-
•
Salvianolic acid A demonstrated higher complex stability than curcumin or lopinavir.
-
•
Salvianolic acid A and curcumin exhibited promising drug-likeness properties.
1. Introduction
Coronaviruses belong to the Coronaviridae family and are named for distinctive protein spikes covering the virus' outer membrane surface. Several members of the family are known to cause respiratory tract infections in humans ranging from mild common colds to severe SARS and MERS infections [1,2]. Coronavirus Disease 2019 (COVID-19) was first observed in Wuhan Province and identified by the Chinese Center for Disease Control and Prevention as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) [3,4]. The viral genome harbors 11 genes encoding 29 proteins and peptides; (www.ncbi.nlm.nih.gov/nuccore/NC_045512.2?report=graph). Four proteins constitute the viral structure, including the spike or S protein [5]. In SARS-CoV-2, the S protein binds to an angiotensin-converting enzyme 2 (ACE2), a necessary step for viral entry into the host cell. Studies thus far indicate that the virus' S protein binds stronger to ACE2 than the one of SARS-CoV, providing a rationale why COVID-19 so easily spreads and is highly infectious. Another group of SARS-CoV-2 proteins controls how the virus replicates as well as avoids the host's immune system. These non-structural proteins initially expressed as two large polyproteins are processed into 16 peptide components. The main protease (Mpro or 3CLpro), cleaves the polyproteins into 11 fragments, whose structures were recently elucidated and an inhibitor that blocks the Mpro catalytic activity identified [6]. From this work, Mpro appears to be a promising target for designing small molecule inhibitors.
COVID-19 rapidly spreads due to the global mobility of humans and is currently present in more than 200 countries. Patients mainly suffer from fever, dry cough, labored breathing, and bilateral lung infiltrates. The causative agent is diagnosed from throat or nasal swabs with nucleic acid sequence similarity. Rapid disease spreading coupled with high mortality rates makes COVID-19 a major global public health threat [7]. Recently, emergency use of remdesivir has been issued by the U.S. Food and Drug Administration for treatment of COVID-19. With few treatment options available, there is an urgent need to seek out effective strategies for prophylaxis for such viral outbreaks. Using experimental methods of drug discovery is time-consuming and costly. Therefore, structure-based computational modeling of ligand-receptor interactions can be used to identify potential Mpro inhibitors to block viral replication.
Herbal extracts and spices are natural immune boosters and/or anti-infective agents currently utilized in many parts of the world [8]. In traditional folk medicine, spices, botanical detoxifiers [9], antioxidants [10] and plant haematinics [11] are used as antiviral mediators to prevent/minimize disease. Low toxicity makes such metabolites well suited as drug leads for viral diseases such as COVID-19. In this study, selected spices with documented, biologically activity (e.g. cinnamon, clove, ginger, mustard and others) were exemplarily chosen to generate a metabolite library for the screening of Mpro-specific drug candidates with presumable effectiveness against COVID-19.
2. Materials and methods
2.1. Mpro preparation
The resolved crystal structure of the main protease (Mpro) of SARS-CoV-2 in complex with N3 inhibitor (PDB code: 6LU7 [12]) was used for molecular docking as well as molecular dynamics calculations. Water and spectator ions were deleted. H++ server was used to study the protonation state of Mpro and to add all missing hydrogen atoms [13]. In H++ calculations, the following physical conditions were applied: pH = 6.5, internal dielectric = 10, external dielectric = 80 and salinity = 0.15.
2.2. Inhibitor preparation
The chemical structures of the 32 investigated natural spices were retrieved from the PubChem database and their 3D structures were generated using Omega2 software [14,15]. All generated structures were minimized using Merck Molecular Force Field 94 (MMFF94S) with the assistance of available software (SZYBKI) [16]. The 2D chemical structures of the investigated compounds are illustrated in Table 1 .
Table 1.
Chemical structures, plant sources, docking scores, and binding features for 32 natural spices against SARS-CoV-2 main protease (Mpro).
| Compound Name |
Chemical Structure | Plant Source |
Docking Score (kcal/mol) | Binding Featuresa |
|---|---|---|---|---|
| Salvianolic acid A | 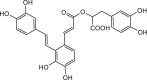 |
Salvia officinalis (Sage) | −9.7 | GLU166 (2.24, 2.15 Å), PHE140 (2.09, 2.21 Å), GLN189 (2.74, 2.06 Å), TYR54 (3.01 Å), THR190 (1.87, 1.86 Å) |
| Curcumin | 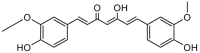 |
Curcuma longa (Turmeric) | −9.2 | HIS163 (1.90 Å), CYS145 (2.72 Å), GLY143 (2.85 Å), SER144 (1.97, 2.01 Å), LEU141 (1.94 Å) |
| Crocetin | Crocus sativus (Saffron) | −8.9 | ASP189 (1.84 Å), TYR54 (2.10 Å), CYS44 (1.79 Å), GLU166 (1.73 Å) | |
| Salvianolic acid B | 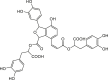 |
Salvia officinalis (Sage) | −8.5 | GLU166 (2.87, 2.33 Å), THR190 (2.27, 1.93, 1.81 Å), MET49 (2.38 Å), HIS41 (2.05 Å), GLY143 (2.67 Å) |
| Quercetin | 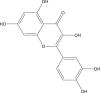 |
Crocus sativus (Saffron) | −8.3 | THR190 (1.82 Å), GLU166 (2.07, 2.18 Å), ASP187 (2.05 Å) |
| Piperine | 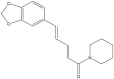 |
Piper nigrum(Black pepper) | −8.2 | GLN189 (3.07 Å), GLY143 (2.15 Å) |
| Picrocrocin | 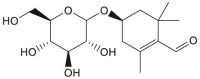 |
Crocus sativus (Saffron) | −8.2 | CYS145 (2.48 Å), GLU166 (2.56 Å), SER144 (3.09 Å), LEU141 (2.78, 2.17 Å), SER144 (2.19 Å) |
| Mahanine | 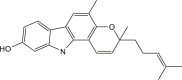 |
Murraya koenigii(Curry leaf) | −8.0 | MET165 (2.51 Å), THR190 (1,83 Å) |
| Capsanthin |  |
Capsicum annum (Sweet pepper) | −8.0 | TYR26 (2.60 Å), SER144 (2.79 Å), CYS145 (1.88 Å) |
| Capsaicin | 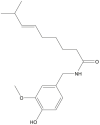 |
Capsicum annuum (Chili pepper) | −8.0 | THR190 (2.25 Å), GLU166 (2.10, 2.10 Å) |
| Carnosol |  |
Rosmarinus officinalis (Rosemary) | −7.9 | GLU166 (2.21 Å) |
| Tanshinone I |  |
Salvia officinalis (Sage) | −7.8 | GLU166 (1.95 Å) |
| Kaempferol |  |
Crocus sativus (Saffron) | −7.8 | THR190 (1.96 Å), ASP187 (1.95 Å), HIS164 (2.22 Å) |
| Baicalin |  |
Rosmarinus officinalis (Rosemary) | −7.6 | ASN142 (2.54 Å), GLY143 (2.14 Å), HIS163 (2.10 Å) |
| Cryptotanshinone |  |
Salvia officinalis (Sage) | −7.6 | GLU166 (1.92 Å) |
| Girinimbine |  |
Murraya koenigii(Curry leaf) | −7.5 | MET165 (2.80 Å), ARG188 (2.10 Å) |
| Shogaols |  |
Zingiber officinale (Ginger) | −7.4 | THR190 (2.27 Å), GLU166 (2.01 Å) |
| Carnosic acid |  |
Rosmarinus officinalis (Rosemary) | −7.3 | GLN189 (2.18 Å) |
| Gingerols | 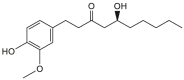 |
Zingiber officinale (Ginger) | −7.1 | THR190 (2.21 Å), GLU166 (2.01 Å), HIS164 (1.80 Å) |
| Tanshinone IIA | 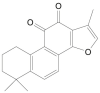 |
Salvia officinalis (Sage) | −6.7 | ---b |
| Marliolide | 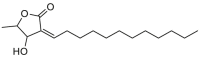 |
Cinnamomum verum (Cinnamon) | −6.2 | THR190 (2.03 Å) |
| Zingerone | 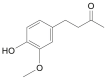 |
Zingiber officinale (Ginger) | −5.7 | CYS44 (2.74 Å), GLU166 (2.18 Å) |
| Acetyleugenol | 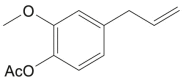 |
Zingiber officinale (Ginger) | −5.3 | CYS145 (1.95 Å) |
| Thymoquinone | 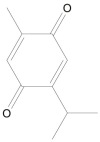 |
Nigella sativa (Black seeds) | −5.2 | ---b |
| Safranal | 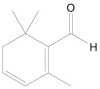 |
Crocus sativus (Saffron) | −5.2 | ---b |
| Eugenol | 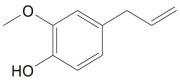 |
Syzygium aromaticum (Cloves) | −5.1 | GLU166 (1.99 Å) |
| S-Allyl cysteine | 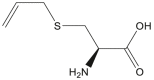 |
Allium sativum (Garlic) and/or Allium cepa (Onion) | −4.4 | ARG188 (2.14 Å), THR190 (1.92 Å), GLN192 (2.34 Å), GLU166 (1.85) |
| Di-allyl trisulfide | 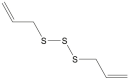 |
Allium sativum (Garlic) and/or Allium cepa (Onion) | −4.1 | ---b |
| Dipropyl disulfide | Allium sativum (Garlic) and/or Allium cepa (Onion) | −3.7 | ---b | |
| Di-allyl disulfide | Allium sativum (Garlic) and/or Allium cepa (Onion) | −3.7 | ---b | |
| Dipropyl sulfide | Allium sativum (Garlic) and/or Allium cepa (Onion) | −3.6 | ---b | |
| Di-allyl sulfide | Allium sativum (Garlic) and/or Allium cepa (Onion) | −3.5 | ---b |
Conventional hydrogen bond only is listed. For the other interactions, see Fig. S1.
No hydrogen bond was observed.
2.3. Molecular docking
For molecular docking calculations, AutoDock4.2.6 software was utilized [17]. The pdbqt file of SARS-CoV-2 Mpro was prepared according to the AutoDock protocol [18]. In AutoDock4.2.6, default parameters were employed, except the numbers of genetic algorithm (GA) run and energy evaluations (eval). GA and eval were set to 250 and 25, 000, 000, respectively. The grid was defined to cover the active site of the SARS-CoV-2 Mpro. The grid size and spacing value were 60 Å × 60 Å × 60 Å and 0.375 Å, respectively. The grid center coordinates were −13.069, 9.740, 68.490 (XYZ assignments, respectively). The atomic charges of studied natural spices were assigned using the Gasteiger method [19]. The predicted binding poses for each compound were processed by the built-in clustering analysis (1.0 Å RMSD tolerance), with the conformation of the lowest energy with respect to the largest cluster selected as representative.
2.4. Molecular dynamics simulations
AMBER16 software was utilized to conduct molecular dynamics (MD) simulation for the natural spices in complex with SARS-CoV-2 Mpro [20]. The details of the employed MD simulations are described in Ref. [21,22]. In brief, general AMBER force field (GAFF) [23] and AMBER force field 14SB [24] were applied to describe spices compounds and Mpro, respectively. Restrained electrostatic potential (RESP) approach [25] was utilized to assign the atomic partial charges of the natural spices using Gaussian09 software [26]. Docked spice-Mpro complexes were water solvated with 15 Å distances between the box edge and atoms of the spice-Mpro complexes. Solvated spice-Mpro complexes were minimized by 5000 steps and afterward smoothly heated from 0 K to 300 K over a brief interval (50 ps). Using periodic boundary conditions and NPT ensemble, the spice-Mpro systems were simulated for 10 ns of equilibration and 40 ns of production. All molecular dynamics simulations were carried out with pmemd. cuda implemented in AMBER16. All molecular docking and molecular dynamics calculations were performed on CompChem GPU/CPU cluster (hpc.compchem.net).
2.5. MM-GBSA binding energy
The binding energies of the investigated spices compounds with SARS-CoV-2 Mpro were estimated using molecular mechanical-generalized Born surface area (MM-GBSA) approach with modified GB model (igb = 2) implemented in AMBER16 software [27]. For the MM-GBSA calculations, uncorrelated snapshots were collected over the production run, and a single-trajectory approach was employed, in which compound, receptor, and complex coordinates were retrieved from a single trajectory. The binding energy (ΔG binding) was estimated as follows:
where the energy term (G) is estimated as:
E vdw and E ele are van der Waals and electrostatic energies, respectively. The electrostatic solvation free energy (G GB) was calculated from the generalized Born (GB) equation. The nonpolar energy (G SA) was estimated with the solvent-accessible surface area (SASA). For all investigated natural spices, entropy contributions were neglected.
2.6. Drug likeliness
The physicochemical parameters of the most promising natural spices as SARS-CoV-2 Mpro inhibitors were predicted using the online Molinspiration cheminformatics software (http://www.molinspiration.com). The predicted parameters included the number of rotatable bonds (nrotb), number of hydrogen bond acceptors (nON), number of hydrogen bond donors (nOHNH), n-octanol/water partition coefficient (P), molecular weight (MWt), molecular volume (MVol), topological polar surface area (TPSA) and percent absorption (% ABS). %ABS was estimated as follows [28]:
| %ABS = 109 − [0.345 × TPSA] |
2.7. Protein-protein interaction
The online web-based tools of SwissTargetPredicition (http://www.swisstargetprediction.ch) were applied to predict the biological targets for the most promising natural spices as SARS-CoV-2 Mpro inhibitors. The DisGeNET online database (https://www.disgenet.org) was utilized to collect the available database for SARS diseases. Venn Diagram was designed using InteractiVenn online tool [29]. Protein-protein interaction (PPI) network was generated using a STRING functional database for top predicted targets [30]. Cytoscape 3.8.0 was employed to investigate target-function relation based on the network topology [31].
3. Results and discussion
Lack of treatments against COVID-19 pinpoints a critical need to systematically screen and identify compounds that can block viral reproduction. Since the main protease of SARS-CoV-2 (Mpro) plays a critical role in the viral replication process, structure-based computational modeling of ligand-receptor interactions and molecular dynamics has been used to screen metabolites from common spices as potential Mpro inhibitors. Indeed several herbal plants have already been reported as antiviral entities against hepatitis B, respiratory syncytial virus and influenza [32].
3.1. Molecular docking
Molecular docking technique was utilized to predict binding modes and affinities of 32 natural products towards SARS-CoV-2 Mpro. Compounds were prepared and docked into the virus Mpro active site using AutoDock4.2.6 software. Docking scores were estimated and the corresponding binding features were analyzed (Table 1). Most docked natural products shared the same binding modes, forming hydrogen bonds with key amino acid residues in the active site such as THR190, GLY143, CYS145, and GLU166.2D binding modes for each of the compounds are displayed in Fig. S1. A wide range of docking scores was observed ranging from −3.5 to −9.7 kcal/mol. Salvianolic acid A, a phenolic acid obtained from sage (Salvia officinalis) showed the highest binding affinity towards SARS-CoV-2 Mpro with a docking score of −9.7 kcal/mol. Curcumin, phenolic compound separated from turmeric (Curcuma longa), exhibited the second highest binding affinity with a docking score of −9.2 kcal/mol. The surpass potentialities of salvianolic acid A and curcumin as Mpro inhibitors are attributed to their ability to form multiple hydrogen bonds, van der Waals interactions as well as hydrophobic and pi-based interactions with the key amino acids within the active site (Fig. 1 ). Specifically, salvianolic acid A forms nine hydrogen bonds with GLU166, PHE140, GLN189, TYR54, and THR190 amino acid with bond lengths ranging from 1.86 to 3.01 Å (Fig. 1). Compared to salvianolic acid A, curcumin formed fewer hydrogen bonds with (six hydrogen bonds, interacting with HIS163, CYS145, GLY143, SER144 and LEU141 with bond lengths ranging from 1.90 to 2.85 Å, Fig. 1). In contrast, some natural products were not capable of similar active site bonding such as tanshinone IIA, thymoquinone, safranal, diallyl trisulfide, dipropyl disulfide, diallyl disulfide, dipropyl sulfide and diallyl sulphide. Absence of such hydrogen bonding resulted in weak natural product-Mpro binding affinity.
Fig. 1.

(a) 3D and (b) 2D representations of interactions of (i) salvianolic acid A, (ii) curcumin and (iii) lopinavir with amino acid residues of SARS-CoV-2 main protease (Mpro).
The peptidomimetic molecule lopinavir, which functions as an antiretroviral protease inhibitor against HIV was used as a positive control [33,34] as it has recently been clinically investigated as an anti-COVID-19 drug [35,36]. Lopinavir exhibited high binding affinity (−9.8 kcal/mol) forming four hydrogen bonds with HIS164, SER144, LEU141, and GLY143 with bond lengths of 2.62, 3.09, 1.96 and 2.01 Å, respectively (Fig. 1). A docking comparison of lopinavir with salvianolic acid A and curcumin revealed competing binding affinities suggesting the in silico potentiality of the three compounds as Mpro inhibitors.
3.2. MD simulation and binding energy calculations
Since the reliability of ligand-enzyme binding energies using molecular docking scores have been questioned due to complicating environmental factors such as a lack of ligand-receptor flexibility, solvent effects, and dynamics [37,38], molecular dynamics (MD) simulations are employed to increase the reliability of predicted ligand-enzyme binding energies. Salvianolic acid A and curcumin were further investigated by MD over 40 ns simulation time. Based on collected compound-Mpro snapshots over the production stage of 40 ns, the binding energies (ΔG binding) were estimated using MM-GBSA approach and summarized in Table 2 . Salvianolic acid A and curcumin displayed robust binding affinities (ΔG binding) with values of −44.8 and −34.2 kcal/mol, respectively. Compared with lopinavir (ΔG binding = −34.8 kcal/mol), the MM-GBSA binding affinity of curcumin is similar to that of lopinavir, while salvianolic acid A, in fact, showed a significantly higher binding energy.
Table 2.
Calculated average MM-GBSA binding energies and the corresponding energy components for lopinavir and the two identified potent natural spices components in complex with SARS-CoV-2 main protease (Mpro) over 40 ns MD simulations.
| Compound Name | Calculated MM-GBSA binding energy (kcal/mol) |
||||||
|---|---|---|---|---|---|---|---|
| ΔEVDW | ΔEele | ΔEGB | ΔESUR | ΔGgas | ΔGsolv | ΔGbinding | |
| Salvianolic acid A | −45.4 | −65.5 | 72.5 | −6.3 | −111.0 | 66.1 | −44.8 |
| Curcumin | −47.5 | −19.8 | 39.2 | −6.1 | −67.4 | 33.1 | −34.2 |
| Lopinavir | −46.8 | −26.1 | 43.9 | −5.9 | −72.8 | 38.0 | −34.8 |
MM-GBSA binding energies were decomposed to identify the nature of the predominant interactions. The estimated energy components for salvianolic acid A-, curcumin- and lopinavir-Mpro complexes are listed in Table 2. For salvianolic acid A, binding energy was dominated by E ele interactions with an average value of −65.5 kcal/mol which was three times higher than that of lopinavir and curcumin, with an average value of −26.1 and −19.8 kcal/mol, respectively. This is attributed to a higher number of hydrogen bonds for salvianolic acid A with the key amino acids inside Mpro's active site, compared to lopinavir or curcumin (Table 1). E vwd interactions were the dominant force in the binding affinity of lopinavir and curcumin with an average value of −46.8 and −47.5 kcal/mol, respectively while salvianolic acid had an average value of −45.4 kcal/mol. Together these results provide quantitative data of the binding affinities of salvianolic acid A and curcumin as SARS-CoV-2 Mpro inhibitors.
3.3. Post-MD analyses
While molecular docking and MD combined with MM-GBSA binding energy calculation revealed the potentiality of salvianolic acid A and curcumin as SARS-CoV-2 Mpro inhibitors, additional MD-based analyses would be required to demonstrate structural and energetic stabilities for ligand-enzyme interactions. The structural and energetical analyses included binding energy per-frame, center-of-mass (CoM) distance, and root-mean-square deviation (RMSD).
MM-GBSA binding energy per-frame for salvianolic acid A and curcumin were evaluated and compared to lopinavir over 40 ns MD simulations (Fig. 2 ). As can be seen from data in Fig. 2, overall stabilities for salvianolic acid A-Mpro, curcumin-Mpro, and lopinavir-Mpro complexes were observed throughout the MD simulation with average binding energies (ΔG binding) of −44.8, −34.2 and −34.8 kcal/mol, respectively. These results indicated satisfactory stabilities of the ligand-enzyme complexes.
Fig. 2.

Estimated MM-GBSA binding energies (in kcal/mol) for salvianolic acid A, curcumin and lopinavir, with SARS-CoV-2 main protease (Mpro) during 40 ns MD simulation.
Center-of-mass (CoM) distance between an inhibitor and an essential amino acid residue would give a deeper insight into the stability of ligand-enzyme complex over the MD simulation. Therefore, CoM distances between salvianolic acid A, curcumin and lopinavir and Mpro GLY143 were measured (Fig. 3 ). The CoM distances were more narrow-fluctuated for salvianolic acid A compared to curcumin or lopinavir complexes, with average CoM distances of 10.7, 11.2 and 11.1 Å, respectively. These findings indicated that salvianolic acid A bounds more tightly with Mpro complex compared to curcumin and lopinavir.
Fig. 3.

Center-of-mass (CoM) distances (in Å) between salvianolic acid A, curcumin and lopinavir and GLY143 of SARS-CoV-2 main protease (Mpro) during 40 ns MD simulation.
Root-mean-square deviation (RMSD) for the complex backbone atoms was estimated to inspect the structural changes in the Mpro. For salvianolic acid A-Mpro and lopinavir-Mpro complexes, RMSD was observed to be below 0.25 nm while curcumin-Mpro exhibited slightly lower stability (Fig. 4 ). Consistency of these energetic and structural measurements, salvianolic acid A is ranked as having higher complex stability than curcumin or lopinavir.
Fig. 4.

Root-mean-square deviation (RMSD) of the backbone atoms from the initial structure for salvianolic acid A, curcumin, and lopinavir with the SARS-CoV-2 main protease (Mpro) over 40 ns MD simulation.
3.4. Drug-likeliness
Drug-likeness is a qualitative measure utilized in drug discovery to evaluate pharmacokinetic properties such as oral bioavailability. Physicochemical parameters were evaluated using Molinspiration cheminformatics (http://www.molinspiration.com), online software calculation toolkit. The predicted parameters are summarized in Table 3 . The permeability across the cell membrane, as measured by the milog P value, was less than five (2.9 and 3.0 for salvianolic acid A and curcumin, respectively) indicating that these components have satisfactory membrane permeability. Moreover, their molecular weights of 494 and 370, for salvianolic acid A and curcumin, respectively, should be readily transferred, diffused and absorbed. Another parameter indicating of molecular bioabsorption is the topological polar surface area (TPSA) calculated as a surface sum of polar atoms or molecules, including oxygen, nitrogen and attached hydrogens. Molecules with a TPSA greater than 140 Å squared tend to be poor at permeating cell membranes, whereas a TPSA less than 90 Å squared is usually highly favorable. Salvianolic acid A and curcumin TPSAs of 185 and 116 Å, respectively, indicate an intermediate cell membrane permeability and oral bioavailability level.
Table 3.
Predicted physiochemical parameters of the two identified natural spices as putative SARS-CoV-2 main protease (Mpro) inhibitors and their different structural descriptors.
| Compound name | miLog P | TPSA | nON | nOHNH | Nrotb | MVol | MWt | %ABS |
|---|---|---|---|---|---|---|---|---|
| Salvianolic acid A | 3.0 | 185 | 10 | 7 | 9 | 418 | 494 | 45% |
| Curcumin | 2.9 | 116 | 7 | 4 | 7 | 323 | 370 | 69% |
3.5. Molecular target prediction and network analysis
Salvianolic acid A and curcumin protein targets were predicted and classified using a SwissTargetPrediction (Fig. S2). One hundred and seventeen genes were identified using DisGeNET online tools for Severe Acute Respiratory Syndrome diseases (SARS, C1175175). Utilizing Venn diagram comparison analysis, commonly shared genes for salvianolic acid A included ACE, CASP3, CASP1, ESR1 and MAPK14, and for curcumin TNF, EGFR and ADAM17 (Fig. 5 ). Angiotensin-converting enzyme 2 (ACE2) is a host protein and the receptor for SARS-CoV-2 entry [39]. MAPK14 inhibition is predicted to block the ACE2 signaling pathway, and in turn, reduce cell internalization of SARS-CoV-2. For SARS-S ADAM17-dependent shedding of ACE2, a process coupled with TNF-α production, reduced viral reproduction [40]. Salvianolic acid A and curcumin predicted genes targets were also analyzed via a STRING PPI network and visualized by Cytoscape 3.8.0. The top 10 scored genes for salvianolic acid A included ACE, MAPK14 and ESR1 and for curcumin EGFR and TNF (Table S1).
Fig. 5.

(i) Venn diagram analysis for salvianolic acid A and curcumin and SARS disease genes, and (ii) STRING PPI network for the top 10 targets for (a) salvianolic acid A and (b) curcumin as potent SARS-CoV-2 main protease (Mpro) inhibitors.
4. Conclusions
The COVID-19 pandemic has had a catastrophic impact on human health and global economies. SARS-CoV-2 main protease (Mpro) may well prove to be the Achilles heel of viral replication. Using molecular docking and molecular dynamics approaches, 32 natural products were screened as possible competitive inhibitors of Mpro. Molecular docking calculations revealed the high binding affinities of salvianolic acid A and curcumin towards Mpro with docking scores of −9.7 and −9.2 kcal/mol, respectively. The two compounds when subjected to MD simulations demonstrated promising binding affinities with Mpro (calculated MM-GBSA binding energies of −44.8 and −34.2 kcal/mol). Post-dynamics analyses were consistent with ligand-enzyme affinity and stability. Physicochemical parameters also exhibited promising drug-likeness properties. The results of the current study reveal two promising natural products, salvianolic acid A and curcumin as potential inhibitors of Mpro. Due to the limitation of experimental test, further in vitro and/or in vivo investigation of the potent natural metabolites under study is highly recommended as a promising starting point for the development of natural drugs targeting SARS-CoV-2 Mpro.
Author contributions
Conceptualization, Mahmoud Ibrahim and Mohamed Elamir F. Hegazy; Data curation, Alaa Abdelrahman and Esraa Badr; Formal analysis, Alaa Abdelrahman; Investigation, Alaa Abdelrahman and Mohamed Elamir F. Hegazy; Methodology, Mahmoud Ibrahim; Project administration, Mahmoud Ibrahim and Mohamed Elamir F. Hegazy; Resources, Mahmoud Ibrahim; Software, Mahmoud Ibrahim; Supervision, Mahmoud Ibrahim; Visualization, Alaa Abdelrahman; Writing – original draft, Alaa Abdelrahman, Taha Hussien, Esraa Badr and Tarik Mohamed; Writing – review & editing, Mahmoud Ibrahim, Hesham El-Seedi, Paul W. Paré, Thomas Efferth and Mohamed Elamir F. Hegazy. All authors have read and agreed to the published version of the manuscript.
Funding
The computational work was completed with resources supported by the Science and Technology Development Fund, STDF, Egypt, Grants No. 5480 & 7972 (Granted to Mahmoud Ibrahim).
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
Prof. Mohamed Hegazy gratefully acknowledges the financial support from Alexander von Humboldt Foundation “Georg Foster Research Fellowship for Experienced Researchers”. Prof. Hesham.R. El-Seedi is very grateful to the Swedish Research Council VR (grants 2015–05468 and 2016–05885).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2020.104046.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Woo P.C., Huang Y., Lau S.K., Yuen K.Y. Coronavirus genomics and bioinformatics analysis. Viruses. 2010;2:1804–1820. doi: 10.3390/v2081803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin Y., Wunderink R.G. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology. 2018;23:130–137. doi: 10.1111/resp.13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W., China novel coronavirus, T. Research A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization WHO director-general’s remarks at the media briefing on 2019-nCoV on 11 February. 2020. https://www.who.int/dg/speeches/detail/who-director-general-s-remarks-at-the-media-briefing-on-2019-ncov-on-11-february-2020 2020 [cited 2020 Apr 2], Available from:
- 5.Liu Z., Xiao X., Wei X., Li J., Yang J., Tan H., Zhu J., Zhang Q., Wu J., Liu L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020;92:595–601. doi: 10.1002/jmv.25726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coronavirus Update (Live) 2,418,429 cases and 165,739 deaths from COVID−19 virus pandemic—worldometer. 2020. https://www.worldometers.info/coronavirus/ Available online. accessed on 20 April.
- 8.Gbadamosi I.T. Stay safe: helpful herbal remedies in COVID-19 infection. Afr. J. Biomed. Res. 2020;23:131–133. [Google Scholar]
- 9.Gbadamosi I.T. 2019. Hidden Treasures of Ethnobotanical Medicine. A Faculty Lecture Delivered at the Faculty of Science, University of Ibadan on 26th June. 2019. [Google Scholar]
- 10.Gbadamosi I. A.J.J.o.A.B. Afolayan, in vitro anti-radical activities of extracts of Solanum nigrum (L.) from South Africa. J. App. Biosci. 2016;98:9240–9251. [Google Scholar]
- 11.Gbadamosi I., Moody J. A.J.E.J.o.M.P. Yekini, Nutritional composition of ten ethnobotanicals used for the treatment of anaemia in Southwest Nigeria. Eur. J. Med. Plants. 2012;2:140–150. [Google Scholar]
- 12.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 13.Gordon J.C., Myers J.B., Folta T., Shoja V., Heath L.S., Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawkins P.C.D., Skillman A.G., Warren G.L., Ellingson B.A., Stahl M.T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the protein databank and cambridge structural database. J. Chem. Inf. Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.OMEGA OpenEye Scientific Software, Santa Fe, NM, USA, 2013.
- 16.SZYBKI OpenEye Scientific Software, Santa Fe, 2016.
- 17.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools 4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forli S., Huey R., Pique M.E., Sanner M.F., Goodsell D.S., Olson A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gasteiger J., Marsili M. Iterative partial equalization of orbital electronegativity - a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 20.Case D.A., Betz R.M., Cerutti D.S., Cheatham T.E., Darden T.A., Duke R.E., Giese T.J., Gohlke H., Goetz A.W., Homeyer N., Izadi S., Janowski P., Kaus J., Kovalenko A., Lee T.S., LeGrand S., Li P., Lin C., Luchko T., Luo R., Madej B., Mermelstein D., Merz K.M., Monard G., Nguyen H., Nguyen H.T., Omelyan I., Onufriev A., Roe D.R., Roitberg A., Sagui C., Simmerling C.L., Botello-Smith W.M., Swails J., Walker R.C., Wang J., Wolf R.M., Wu X., Xiao L., Kollman P.A., AMBER . University of California; San Francisco: 2016. 2016. [Google Scholar]
- 21.Ibrahim M.A.A., Abdelrahman A.H.M., Hegazy M.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1791958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibrahim M.A.A., Abdeljawaad K.A.A., Abdelrahman A.H.M., Hegazy M.F. Natural-like products as potential SARS-CoV-2 M(pro) inhibitors: in-silico drug discovery. J. Biomol. Struct. Dyn. 2020:1–13. doi: 10.1080/07391102.2020.1790037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 24.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. ff14SB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayly C.I., Cieplak P., Cornell W., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 26.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas Ö., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. 2009. Gaussian 09, Gaussian inc., wallingford CT, USA. [Google Scholar]
- 27.Massova I., Kollman P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000;18:113–135. [Google Scholar]
- 28.Zhao Y.H., Abraham M.H., Le J., Hersey A., Luscombe C.N., Beck G., Sherborne B., Cooper I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. (N. Y.) 2002;19:1446–1457. doi: 10.1023/a:1020444330011. [DOI] [PubMed] [Google Scholar]
- 29.Heberle H., Meirelles G.V., da Silva F.R., Telles G.P., Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinf. 2015;16:169–175. doi: 10.1186/s12859-015-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li R., Ma X., Song Y., Zhang Y., Xiong W., Li L., Zhou L. Anti-colorectal cancer targets of resveratrol and biological molecular mechanism: Analyses of network pharmacology, human and experimental data. J. Cell. Biochem. 2019;120:11265–11273. doi: 10.1002/jcb.28404. [DOI] [PubMed] [Google Scholar]
- 31.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin L.T., Hsu W.C., Lin C.C. Antiviral natural products and herbal medicines. J. Tradit. Complementary Med. 2014;4:24–35. doi: 10.4103/2225-4110.124335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandwani A., Shuter J. Lopinavir/ritonavir in the treatment of HIV-1 infection: a review. Therapeut. Clin. Risk Manag. 2008;4:1023–1033. doi: 10.2147/tcrm.s3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee G.A., Seneviratne T., Noor M.A., Lo J.C., Schwarz J.M., Aweeka F.T., Mulligan K., Schambelan M., Grunfeld C. The metabolic effects of lopinavir/ritonavir in HIV-negative men, AIDS. 2004;18:641–649. doi: 10.1097/00002030-200403050-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., Li X., Xia J., Chen N., Xiang J., Yu T., Bai T., Xie X., Zhang L., Li C., Yuan Y., Chen H., Li H., Huang H., Tu S., Gong F., Liu Y., Wei Y., Dong C., Zhou F., Gu X., Xu J., Liu Z., Zhang Y., Li H., Shang L., Wang K., Li K., Zhou X., Dong X., Qu Z., Lu S., Hu X., Ruan S., Luo S., Wu J., Peng L., Cheng F., Pan L., Zou J., Jia C., Wang J., Liu X., Wang S., Wu X., Ge Q., He J., Zhan H., Qiu F., Guo L., Huang C., Jaki T., Hayden F.G., Horby P.W., Zhang D., Wang C. A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19, new engl. J. Med. 2020;382:1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye X.T., Luo Y.L., Xia S.C., Sun Q.F., Ding J.G., Zhou Y., Chen W., Wang X.F., Zhang W.W., Du W.J., Ruan Z.W., Hong L. Clinical efficacy of lopinavir/ritonavir in the treatment of Coronavirus disease 2019. Eur. Rev. Med. Pharmacol. Sci. 2020;24:3390–3396. doi: 10.26355/eurrev_202003_20706. [DOI] [PubMed] [Google Scholar]
- 37.Pantsar T., Poso A. Binding affinity via docking: fact and fiction. Molecules. 2018;23:1899–1909. doi: 10.3390/molecules23081899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pagadala N.S., Syed K., Tuszynski J. Software for molecular docking: a review. Biophys. Rev. 2017;9:91–102. doi: 10.1007/s12551-016-0247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., Muller M.A., Drosten C., Pohlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palau V., Riera M., Soler M.J. ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol. Dial. Transplant. 2020;35:1071–1072. doi: 10.1093/ndt/gfaa093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.