

The anaerobic gram-positive bacterium, Clostridium botulinum, produces a family of neurotoxins (BoNTs A–G) that are responsible for the debilitating effects of botulism intoxication. In humans, such intoxication is characterized by peripheral neuromuscular blockade resulting in flaccid paralysis, and typically occurs after consuming contaminated food or by wound infection with spores from soil. The consequences of exposure are extremely severe and frequently fatal, as BoNTs are some of the most potent poisons known – 1 ng kg1 is a lethal dose in humans.1 The CDC has classified the BoNTs as category A bioterrorism agents due to their extraordinary toxicity and persistence, their potential for easy dissemination, and the need for special public health preparations in the event of mass exposure.2
Despite their potential to cause extensive harm, there are no medications commercially available, or even in clinical trials, for the reversal of BoNT intoxication. Current treatment relies on the administration of an antitoxin prior to BoNT entry into nerve endings within 24 hours of symptom onset. The limited supply of these antitoxins would severely limit their utility in the event of a bioterrorism attack.3 Furthermore, once nerve endings have been intoxicated, medical management is limited to supportive care, often including mechanical ventilation. The remarkable potency of BoNTs combined with the high toxin burdens can cause such supportive care to last for months.4
As soon as sequestration of BoNTs into nerve terminals has occurred, a series of biochemical events ensue that can eventually lead to muscle paralysis. In short, all BoNT serotypes consist of a 100 kDa heavy chain (HC) linked to a 50 kDa light chain (LC) metalloprotease via a disulfide bond.5 The HC domain is able to bind to the cholinergic nerve terminals, where it is then internalized by receptor-mediated endocytosis. Following internalization and disulfide bond cleavage, the LC undergoes translocation into the cytosol and cleaves one of three SNARE proteins.5 Destruction of any one of these SNARE proteins results in inhibition of acetylcholine release at the presynaptic nerve terminal, thus interrupting neurotransmission.
Due to their role in modulating such neuronal signaling, voltage-gated potassium (Kv) channels have been proposed as potential therapeutic targets for the treatment of a variety of autoimmune and neurological disorders.6 Blockade of Kv channels in nerve terminals results in an influx of calcium ions to the presynaptic terminal, allowing for restoration of normal neuronal action potential firing in nerve fibers – a mechanism of action well suited for the treatment of botulism intoxication. Indeed, our laboratory and others have shown that the cell permeable Kv channel blocker 3,4-diaminopyridine (3,4-DAP) successfully antagonizes muscle paralysis following BoNT/A intoxication in vitro and in vivo.7 Thus far, 3,4-DAP is the only non-protein based small molecule antagonist that has shown efficacy in the reversal of BoNT/A intoxication. This efficacy, combined with its relatively simple chemical structure and low cost, make 3,4-DAP a compound of substantial interest in the effort to find a treatment for BoNT/A intoxication that can rapidly be made available.
Unfortunately 3,4-DAP has both a short plasma half-life and can induce seizures when present at high concentrations, following penetration of the blood–brain barrier.8 These physiochemical liabilities have led to the conclusion that continuous administration of the drug may be required for the treatment of BoNT intoxication.9 Considering that the symptoms of BoNT intoxication can last for months, continuous intravenous access would present a significant risk to patients. While an oral formulation of 3,4-DAP phosphate (amifampridine/Firdapse®) has garnered an orphan drug designation for the treatment of Lambert–Eaton Myasthenic Syndrome (LEMS), this immediate release formulation of the drug must be taken in divided doses three to four times daily to maintain efficacy while avoiding rapid spikes in concentration that can lead to seizure activity.10 Preliminary attempts to use this formulation for treating BoNT intoxication in humans have shown modest results, attributed to the low blood concentrations achieved.11
In an attempt to address the pharmacokinetic and practical concerns associated with 3,4-DAP administration for BoNT intoxication, we employed a microencapsulation strategy. Such micro- and nanoencapsulation strategies for controlled drug release have previously been explored in the context of developing synthetic or semi-synthetic delivery materials.12 However, given the potential need for rapid translation and dissemination of a therapeutic measure for BoNT intoxication, we opted to explore the potential of a naturally occurring biopolymer for this application – the exine microcapsule (LEM) extracted from Lycopodium clavatum (club moss) spores. These LEMs are hollow sporopollenin structures with a highly uniform diameter (25 mM) ridged luminal wall permeated with multi-directional nanochannels (200–300 nm diameter) to allow the passage of nutrients, water, and genetic material into/out of the interior of the pollen grain.13
The use of LEMs as a delivery system for 3,4-DAP was attractive for several reasons. Firstly, Lycopodium spores can be treated to remove the cytoplasmic materials and inner layer of cellulose while leaving the exine to generate a non-allergenic and non-toxic material, as demonstrated by their use in human studies to deliver such components as eicosapentaenoic acid and taste mask cod liver oil and ibuprofen.14 Furthermore, the biopolymer that makes up the LEMs can withstand the harsh environment of the stomach, enabling oral administration and preferential drug release into the gastrointestinal (GI) tract.13,14 This enteric release would eliminate the risks associated with continuous IV access, and could also allow for higher 3,4-DAP oral doses to be administered by blunting the initial spike in concentration and distributing the total dose across a longer time interval. Additionally, LEMs offer the potential of targeted 3,4-DAP administration through bioadhesion and possibly persorption across the epithelial cells in the intestine, which is known to be the route taken by BoNTs following intoxication.14,15 Finally, Lycopodium clavatum spores are a widely available and inexpensive material, allowing for economical scaling in the event that large scale production is required in the face of a bioterrorism threat.
Due to the nanochannels that run through the LEM shell, we predicted that 3,4-DAP could be sequestered into a diffusion limited depot within the LEM, providing an enteric controlled release platform to expand its therapeutic window.13 In order to promote passage of the loaded LEMs through the low pH environment of the stomach and prevent a bolus release of active drug, we decided to incorporate a protective excipient to co-encapsulate with 3,4-DAP into the LEMs’ interior cavity.
Shellac was selected as the co-encapsulant of choice due to its wide availability, low cost, listing as a “generally regarded as safe” (GRAS) compound by the FDA, and its frequent use as an additive in food products. Furthermore, due to its high dissolution pH, shellac has previously been used to improve passage through the low pH environment in the stomach and enhance drug delivery in the distal portions of the GI tract.16
We first measured the release of 3,4-DAP from shellac-coencapsulated LEMs in either simulated gastric fluid (SGF) or phosphate buffered saline (PBS) to study whether it varied with pH; as a surrogate for gastric fluid, SGF has a lower pH (pH = 1.5) compared to PBS or the terminal ileum (pH = 7.4). Consistent with the known dissolution properties of shellac, our data indicate that 3,4-DAP is released slowly and to limited extent when the shellac-co-encapsulated LEMs are incubated at a lower pH (Fig. 1A). Indeed, when the co-encapsulated LEMs are subjected to SGF for up to 8 h they retain 72.2 ± 4.0% of the active drug. Conversely, the co-encapsulated LEMs exhibited substantial 3,4-DAP release upon incubation in PBS (pH 7.4). No alterations to the overall external LEM morphology were observed following sequential exposure to these pH conditions (Fig. S1. ESI†). This result indicated that we could expect preferential release of 3,4-DAP only after the LEMs leave the stomach to pass through the more basic environments in the mid and distal small bowel.17
Figure 1.
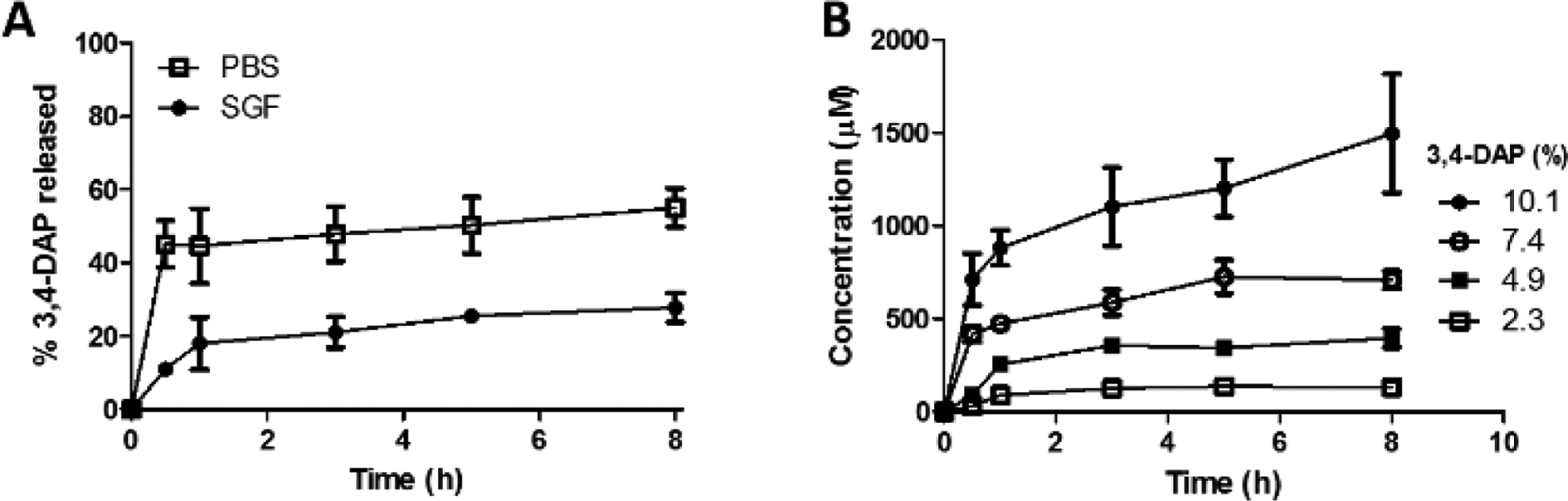
(A) Comparison of average percent 3,4-DAP released in PBS (pH 7.4) or SGF (pH 1.5), (n = 6). (B) Concentration of 3,4-DAP released from 2.3, 4.9, 7.4, and 10.1% 3,4-DAP loaded LEMs with 40% shellac in PBS (pH 7.4; n = 6).
Following this confirmation of pH-dependent release by the LEMs, it was pertinent to test whether this drug delivery platform could provide access to a broad concentration range of the active drug. As such, we loaded different percentages (w/w) ranging from 2.3–10.1% 3,4-DAP into the LEMs, and measured 3,4-DAP release in PBS (pH 7.4). Gratifyingly, all loading percentages released the drug in a time-dependent manner, demonstrating that increases in 3,4-DAP loading percentage can result in an improved in vitro release profile (Fig. 1B). Furthermore, our results indicate that this LEM platform can provide access to drug concentrations spanning more than an order of magnitude.
We next undertook a comparison of scanning electron micrographs to reveal how loaded LEMs can act as an effective controlled release technology. An image of the unloaded broken LEMs showed that the inner cavity was completely hollow (Fig. 2A) prior to the addition of active drug. However, when the LEMs were loaded with 3,4-DAP (Fig. 2B), the drug accumulated inside.
Figure 2.
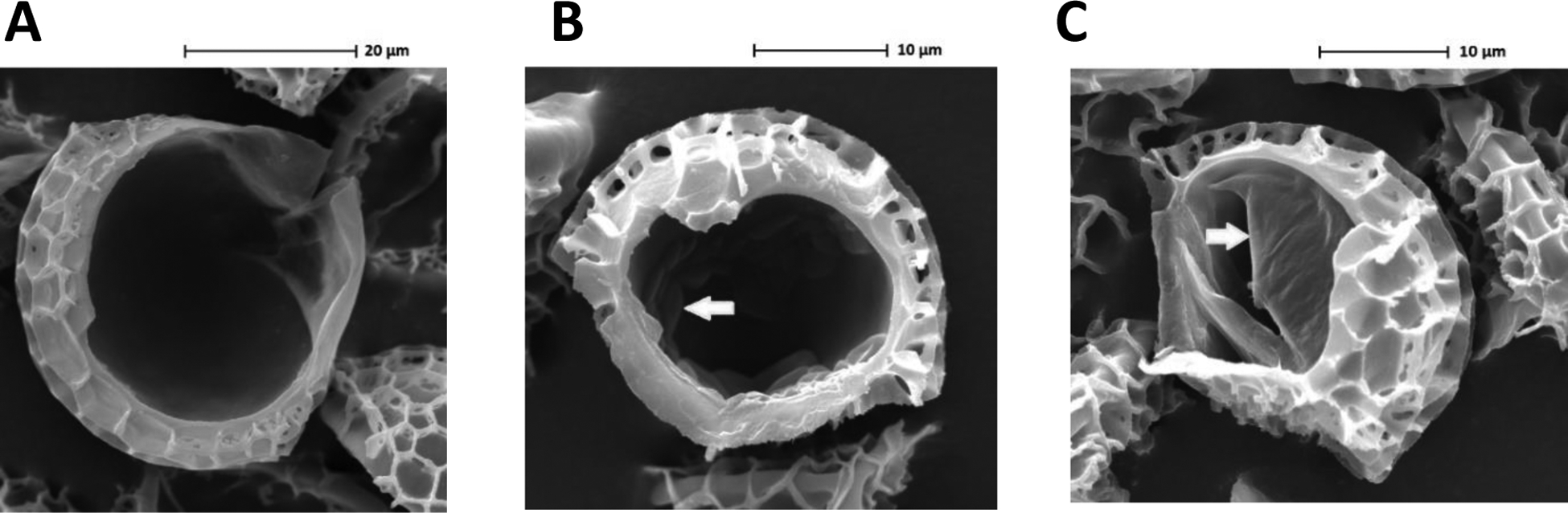
Scanning electron micrographs of A) unloaded broken LEM, B) broken LEM loaded with 10.1% 3,4-DAP, and C) broken LEM loaded with 10.1% 3,4-DAP and 49.9% shellac. Arrows indicate sites of 3,4-DAP encapsulation.
Interestingly, when shellac was added as a co-encapsulant, the loaded contents appeared to be peeling away from the wall of the cavity (Fig. 2C). It was of particular note that neither extraneous 3,4-DAP nor shellac could be seen on the external surface topological features of the LEMs upon loading; this demonstrated the creation of an internal depot of 3,4-DAP, analogous to previous findings for both fluorescent tracer compounds and therapeutic protein conjugates.18 This finding highlights the generalizability of the formation of an internal depot in the LEMs following vacuum loading with a molecule small enough to traverse the LEM nanochannels.
Next, to follow up on the promising in vitro release profile for the loaded LEMs, we examined the pharmacokinetics of 3,4-DAP release in mice. IV administration of 3,4-DAP indicated that it had a plasma half-life of about 1 hour in this model system (Fig. 3A). A comparison of IV 3,4-DAP with oral 3,4-DAP phosphate, which is the clinically used salt, indicated that the oral formulation had about 57% bioavailability (F) in mice. Moreover, this clinical formulation also exhibited a rapid spike in plasma concentrations, particularly when compared to the LEM encapsulated form, which had a broader and right-shifted peak (Fig. 3B). Yet, the bioavailability of this 7.4% 3,4-DAP LEM formulation was much lower, around 17%. Fortunately, as predicted by our in vitro data, this signal was substantially increased when the LEM loading percentage of 3,4-DAP was increased from 7.4 to 10.1%. Intriguingly, the bioavailability of this formulation also appeared to be much higher, indicating that alteration of the ratios of the LEM platform components could potentially impact the overall amount of drug delivery.
Figure 3.
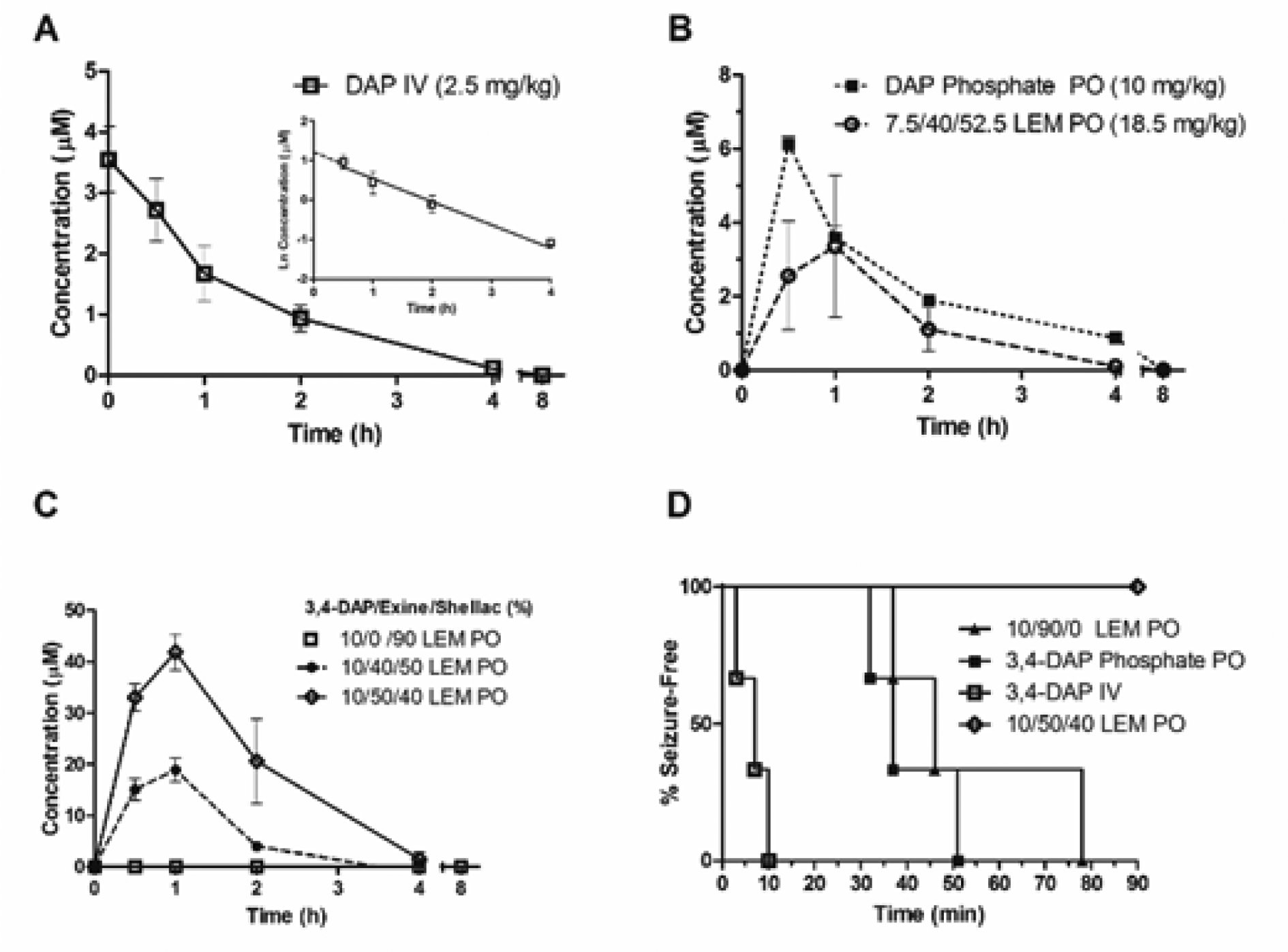
A) Pharmacokinetic profile of 3,4-DAP in CD-1 mouse plasma following IV administration (n = 3). B) Oral administration of 3,4-DAP phosphate or 3,4-DAP loaded LEMs. (n = 3). C) Pharmacokinetic profile of 3,4-DAP release with varying 3,4-DAP: LEM: shellac ratios (25 mg/kg; n = 3). D) Seizure-free survival curve following administration of varying 3,4-DAP formulations (25 mg/kg; n = 3).
Thus, we next wanted to probe the effect of the LEM: shellac ratio on the release rate of 3,4-DAP in vivo. As a comparison to the initial formulations that contained approximately 40% LEM/50% shellac, we prepared a formulation where the LEM: shellac ratio was increased to 50% LEM/40% shellac for the 10.1% 3,4-DAP loaded LEMs. Upon dose-matched oral administration to mice, this reformulation dramatically increased the 3,4-DAP Cmax and AUC (Fig. 3C). In contrast, a dose-matched preparation of 3,4-DAP in shellac alone was unable to generate any measurable concentration of 3,4-DAP in mouse plasma, even at the earliest measured time points, while a formulation containing 3,4-DAP in LEMs without shellac led to uncontrollable seizure activity in the first 30 minutes after administration (Fig. 3D). This seizure activity was also seen in animals receiving an equivalent dose of free 3,4-DAP, and was even seen in one mouse dosed orally with 3,4-DAP phosphate at 10 mg kg−1. Taken together, these results demonstrate the central importance of both the LEM component and shellac component to prolong drug release and blunt the initial concentration spike following administration.
Analysis of the pharmacokinetic parameters associated with each LEM formulation revealed that increasing 3,4-DAP loading concentrations led to substantial effects on Cmax and F, while there were no noticeable changes in the rate of elimination between these groups (Table S1. ESI). Remarkably, altering the LEM: shellac ratio also led to additional substantial increases in Cmax and F. Although the absolute F of the optimized formulation could not be accurately calculated, likely due to an oversimplification of an initial phase of IV distribution at higher doses, this finding highlights the utility of the LEM in safely allowing access to a greater dose range than currently available formulations (uniform lethality at higher IV doses prevents a more direct dose comparison). Previous studies indicate that 10–15 mM 3,4-DAP in circulation is required per hour for potassium channel blockade and acetylcholine release at the synapse.19 Using the improved formulation, 3,4-DAP remains above this threshold concentration for over three hours after oral administration; in contrast, due to seizure activity at and above 10 mg kg−1 dosing, this concentration cannot be safely and sustainably achieved with immediate release formulations of 3,4-DAP.
To examine the therapeutic relevance of the loaded LEMs containing 3,4-DAP we used a mouse lethality assay that measures mouse life-span following BoNT administration, using time to death as the primary end point. Our findings revealed that neither LEMs alone (182 ± 43 min) nor the maximum safe orally deliverable dose of 3,4-DAP alone (225 ± 24 min) could significantly increase the time to death following toxin administration (216 ± 29 min). However, when the 10/50/40 3,4-DAP/LEM/shellac formulation was administered at 25 mg kg−1 the time to death was 302 ± 26 min – a 40% increase as compared to toxin alone (Fig. 4, p ≤ 0.001). Crucially, this optimized LEM formulation also led to significantly prolonged life-span as compared to the maximum safe oral 3,4-DAP dose + toxin (p ≤ 0.01). This demonstrates that LEMs can be used to increase the therapeutic window for oral 3,4-DAP treatment, thus enabling administration of more efficacious doses following BoNT/A exposure.
Figure 4.
Time to death following administration of 5 LD50 BoNT/A in female CD-1 mice (n = 6). Empty and loaded LEM groups – 25 mg/kg of LEMs per mouse. Loaded LEMs were formulated with 10% 3,4-DAP / 50% LEM / 40% shellac. 3,4-DAP alone administered at 10 mg/kg. * = p ≤ 0.01; ** = p ≤ 0.001
In the course of these studies, we have developed a generalizable two-component LEM delivery platform from L. clavatum spores for the improved delivery of aminopyridines. We have demonstrated that these prepared LEMs can release an encapsulated active drug in a pH dependent manner in vitro, that modifications to the 3,4-DAP/LEM/shellac ratio can result in an improved drug release profile, and that inclusion of both components is needed to achieve effective controlled release.
As a proof-of-concept, we used this LEM platform to provide controlled release of a Kv channel blocker with physiochemical liabilities and adverse events that limit its therapeutic value for the treatment of BoNT intoxication. Critically, we established that using a 10.1% 3,4-DAP/50% LEM/39.9% shellac LEM formulation can alter the rate of 3,4-DAP delivery in vivo to improve its therapeutic window and efficacy in a BoNT/A lethality assay – a goal that could not be safely achieved in the absence of the LEM.
Because the components of this platform are inexpensive, widely available, and safe for human consumption, rapid scalability of the technology is practically and economically feasible – an important concern in the event of an attack with BoNT/A. Therefore, these studies identify a viable pathway toward the oral delivery of aminopyridines to humans for the reversal of BoNT intoxication of nerve terminals, a condition for which there are currently no treatment options.
Supplementary Material
Acknowledgements
The authors acknowledge Amanda Roberts (TSRI), Michael Goodnough (Metabiologics), Garry S. Robinson (U. Hull) and Tony Sinclair (U. Hull) for technical support. We thank the Skaggs Institute of Chemical Biology for generous funding. ADT receives funding from Sporomex Ltd, which holds patents on the use of sporopollenin exines as drug delivery systems.
Notes and references
- 1.Schantz E and Johnson E, Microbiol. Rev, 1992, 56, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control. 2007, February 12 Bioterrorism Overview. Retrieved from http://www.emergency.cdc.govon01/11/2016. [Google Scholar]
- 3.Arnon S, Schechter R, Inglesby T, Henderson D and Bartlett J, et al. , J. Am. Med. Assoc, 2001, 285, 1059. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control. 2013, March 1 Botulism: Treatment Overview for Clinicians. Retrieved from http://www.bt.cdc.govon01/11/2016. [Google Scholar]
- 5.(a) Oguma K, Fujinaga Y and Inoue K, Microbiol. Immunol, 1995, 39, 161; [DOI] [PubMed] [Google Scholar]; (b) Willis B, Eubanks L, Dickerson Tand Janda K, Angew. Chem., Int. Ed, 2008, 47, 8360; [DOI] [PubMed] [Google Scholar]; (c) Schiavo G, Shone C, Rossetto O, Alexander Fand Montecucco C, J. Biol. Chem, 1993, 268, 11516; [PubMed] [Google Scholar]; (d) Yamasaki S, Binz T, Hayashi T, Szabo Eand Yamasaki N, et al. , Biochem. Biophys. Res. Commun, 1994, 200, 829. [DOI] [PubMed] [Google Scholar]
- 6.Wulff H, Castle N and Pardo L, Nat. Rev. Drug Discovery, 2009, 8, 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Adler M, Scovill J, Parker G, Lebeda F, Piotrowski J and Deshpande S, Toxicon, 1995, 33, 527; [DOI] [PubMed] [Google Scholar]; (b) Simpson L, Infect. Immun, 1986, 52, 858; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mayorov A, Willis B, Di Mola A, Adler Dand Borgia J, et al. , ACS Chem. Biol, 2010, 5, 1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemeignan M, Millart H, Lamiable D, Molgo J and Lechat P, Brain Res., 1984, 304, 166. [DOI] [PubMed] [Google Scholar]
- 9.Adler M, Capacio B and Deshpande S, Toxicon, 2000, 38, 1381. [DOI] [PubMed] [Google Scholar]
- 10.Firdapses ® [package insert] London. Biomarin Europe Ltd; 2010. [Google Scholar]
- 11.Friggeri A, Marcon F, Marciniak S, Lemaire-Hurtel A and Seydi A, J. Crit. Care, 2013, 17, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Zhang N, Li J and Jiang W, et al. , Int. J. Pharm, 2010, 393, 212; [DOI] [PubMed] [Google Scholar]; (b) Ma G, J. Controlled Release, 2014, 193, 324; [DOI] [PubMed] [Google Scholar]; (c) Ahmed RZ, Patil Gand Zaheer Z, Drug Dev. Ind. Pharm, 2013, 39, 1263; [DOI] [PubMed] [Google Scholar]; (d) Costa RR, Alatorre-Meda Mand Mano JF, Biotechnol. Adv, 2015, 33, 1310; [DOI] [PubMed] [Google Scholar]; (e) Musyanovych Aand Landfester K, Macromol. Biosci, 2014, 14, 458. [DOI] [PubMed] [Google Scholar]
- 13.(a) Wittborn J, Rao KV, El-Ghazaly G and Rowley JR, Ann. Bot, 1998, 82, 141; [Google Scholar]; (b) Barrier S, Diego-Taboada A, Thomasson M, Madden Land Pointon J, et al. , J. Mater. Chem, 2011, 21, 975; [Google Scholar]; (c) Mackenzie G, Beckett Sand Atkin SL, Patent WO2009077749A1, 2009; [Google Scholar]; (d) Diego-Taboada A, Beckett S, Atkin SL and Mackenzie G, Pharmaceutics, 2014, 6, 80; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Diego-Taboada A, Cousson Pand Raynaud E, et al. , J. Mater. Chem, 2012, 22, 9767. [Google Scholar]
- 14.(a) Wakil A, Mackenzie G, Diego-Taboada A, Bell J and Atkin SL, Lipids, 2010, 45, 645; [DOI] [PubMed] [Google Scholar]; (b) Diego-Taboada A, Maillet L, Banoub J, Lorch Mand Rigby A, et al. , J. Mater. Chem. B, 2013, 1, 707; [DOI] [PubMed] [Google Scholar]; (c) Barrier S, Rigby A, Diego-Taboada A, Thomasson M, Mackenzie Gand Atkin SL, LWT – Food Sci. Technol, 2010, 43, 73. [Google Scholar]
- 15.Simpson L, Toxicon, 2013, 68, 40. [DOI] [PubMed] [Google Scholar]
- 16.(a) Farag Y and Leopold C, Eur. J. Pharm. Sci, 2011, 42, 400; [DOI] [PubMed] [Google Scholar]; (b) Hamad SA, Stoyanov SDand Paunov VN, Soft Matter, 2012, 8, 5069; [Google Scholar]; (c) Hamad SA, Stoyanov SDand Paunov VN, Phys. Chem. Chem. Phys, 2013, 15, 2337. [DOI] [PubMed] [Google Scholar]
- 17.Evans D, Pye G, Bramley R, Clark A, Dyson T and Hardcastle J, Gut, 1988, 29, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atwe S, Ma Y and Gill H, J. Controlled Release, 2014, 194, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris T, Lowery C, Hixon Mand Janda K, ACS Chem. Neurosci, 2014, 5, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.