


Abstract
Heterochromatin is a specialized form of chromatin that restricts access to DNA and inhibits genetic processes, including transcription and recombination. In Neurospora crassa, constitutive heterochromatin is characterized by trimethylation of lysine 9 on histone H3, hypoacetylation of histones, and DNA methylation. We explored whether the conserved histone demethylase, lysine-specific demethylase 1 (LSD1), regulates heterochromatin in Neurospora, and if so, how. Though LSD1 is implicated in heterochromatin regulation, its function is inconsistent across different systems; orthologs of LSD1 have been shown to either promote or antagonize heterochromatin expansion by removing H3K4me or H3K9me respectively. We identify three members of the Neurospora LSD complex (LSDC): LSD1, PHF1, and BDP-1. Strains deficient for any of these proteins exhibit variable spreading of heterochromatin and establishment of new heterochromatin domains throughout the genome. Although establishment of H3K9me3 is typically independent of DNA methylation in Neurospora, instances of DNA methylation-dependent H3K9me3 have been found outside regions of canonical heterochromatin. Consistent with this, the hyper-H3K9me3 phenotype of Δlsd1 strains is dependent on the presence of DNA methylation, as well as HCHC-mediated histone deacetylation, suggesting that spreading is dependent on some feedback mechanism. Altogether, our results suggest LSD1 works in opposition to HCHC to maintain proper heterochromatin boundaries.
INTRODUCTION
The basic unit of chromatin, the nucleosome, consists of about 146 bp of DNA wrapped around a histone octamer. Histones possess unstructured N-terminal tails that are subject to various post-translational modifications, which reflect and/or influence the transcriptional state of the underlying chromatin. Methylation of lysines 4 and 36 of histone H3 (H3K4, H3K36), as well as hyperacetylation of histones, are associated with transcriptionally active euchromatin while methylation of lysines 9 and 27 of histone H3 (H3K9, H3K27) and hypoacetylation are associated with transcriptionally repressive heterochromatin (1).
One of the defining characteristics of heterochromatin is its capability to propagate along the chromosome, often guided through feedback of histone marks. Some ‘writers’ of heterochromatin marks such as the H3K9-specific methyltransferase Clr4 of Schizosaccharomyces pombe and the mammalian H3K9 methyltransferase Suv39 are also ‘readers’ capable of binding to their own mark, which in turn promotes further methylation on adjacent nucleosomes (2,3). If left unchecked, heterochromatin tends to ‘spread’ into neighboring gene-rich euchromatin, with potentially negative consequences, such as down-regulation of important genes (1). Thus, mechanisms to prevent inappropriate expansion of heterochromatin are necessary. Natural barriers include nucleosome-depleted regions (4,5), high levels of nucleosome turnover (6), and genetic elements such as tRNA genes (7,8). In addition, factors antagonistic to the spreading of heterochromatin can be directed to the boundaries of heterochromatin domains. In S. pombe, the JmjC domain protein, Epe1, a putative H3K9 demethylase, localizes to the edges of heterochromatin in a Swi6/HP1-dependent manner (9). Similarly in N. crassa, the JmjC domain protein, DNA Methylation Modulator 1 (DMM-1), is part of a complex recruited to borders of some heterochromatin domains in an HP1-dependent manner, where it limits spreading presumably by demethylating trimethylated lysine 9 of histone H3 (H3K9me3) (10).
Previous work has uncovered lysine-specific demethylase 1 (LSD1) as another important regulator of heterochromatin spreading, prompting us to examine its function in Neurospora crassa. LSD1 is highly conserved in eukaryotes from yeasts to humans, and regulates heterochromatin propagation, apparently by removing mono- or dimethylation from either H3K4 or H3K9 (11–14). S. pombe has two LSD1 paralogs and two associated PHD-domain proteins, Phf1 and Phf2, whose specific functions have yet to be determined but are essential for viability. The LSD complexes in S. pombe appear to function as dedicated H3K9 demethylases, and localize to the edges of pericentromeric heterochromatin and promoters of certain genes, all of which display hyper H3K9 trimethylation when one or both of the LSD1 homologs are compromised (12). The Drosophila homolog of LSD1, SU(VAR)3–3, plays an important role in promoting heterochromatin formation during embryogenesis. In vitro, SU(VAR)3–3 was shown to eliminate the inhibitory effect of H3K4me1 or H3K4me2 on H3K9 methyltransferase activity of SU(VAR)3–9 (13). In vivo, SU(VAR)3–3 colocalizes with, and demethylates, H3K4me2 in inter-band regions, facilitating the formation of H3K9me-marked heterochromatin.
In humans, LSD1 appears to be able to act both as a transcriptional repressor and as an activator by removing mono- and dimethylation from either H3K4 or H3K9, depending on its binding partners or splicing isoform (11,14). LSD1 removes H3K4me2 from targeted promoters and is a member of the Co-REST complex, which functions to silence neuron-specific genes in non-neuronal cells (15). LSD1 is also essential for proper neuron maturation, and a neuron-specific isoform (LSD1+8a) possesses intrinsic demethylation activity towards H3K9 but not H3K4. LSD1+8a localizes to target genes during neuronal differentiation and removes H3K9me to promote transcription (14). LSD1 was also found to associate with androgen receptor (AR) in androgen-sensitive tissues leading to its recruitment to AR target genes and changing the specificity of LSD1 from H3K4 to H3K9, resulting in the loss of H3K9me and increased expression (11).
The fact that LSD1 homologs and isoforms have different roles in heterochromatin regulation in different organisms provided motivation for our investigation of the possible role of Neurospora's single LSD1 homolog in control of heterochromatin. Here, we report the identification of the Neurospora LSD complex (LSDC), including an LSD1 homolog, a PHD-domain protein homologous to S. pombe Phf1, and the bromodomain protein, BDP-1. Loss of any LSDC members, or of LSD1 catalytic activity, increases heterochromatin spreading and results in the appearance of new heterochromatin loci throughout the genome. The spreading is variable and dependent on the presence of DNA methylation as well as histone deacetylation by the HP1, CDP-1, HDA-1, CHAP (HCHC) complex. We conclude LSD1 functions to prevent heterochromatin expansion and to restrict heterochromatin to its normal chromosomal locations in Neurospora.
MATERIALS AND METHODS
N. crassa strains and general methods
All N. crassa strains and primers used in this study are listed in Supplemental Tables S7 and S8, respectively. Strains were cultured, crossed, and maintained according to standard procedures (16). Linear growth was assayed with ‘race tubes’ at 32°C (17), except 25 ml plastic serological pipets (89130-912; VWR) were used instead of glass tubes. Deletion strains were obtained from the Neurospora knockout collection (18) or made by replacing the indicated gene with the nat-1 marker as previously described (19). Epitope-tagged strains were built as previously described (19).
Generation of 3xFLAG-tagged catalytic null LSD1 strain
To generate a catalytic mutant of LSD1 tagged with 3xFLAG, we amplified the middle of the lsd1 gene (NCU09120) with genomic DNA from strain N3752 using primers 6716 and 6717 to make a fragment with a 3′ overhang harboring the target mutation and using primers 6718 and 6720 to generate a fragment with a 5′ overhang harboring the target mutation and a 3′ overhang complementary to the 10x glycine linker sequence of plasmid pZero::3xFLAG::nat-1::loxP. These two fragments were then combined by ‘stitching-PCR’. The 3′ UTR region of lsd1 was amplified using primers 6721 and 6722 to generate a fragment with a 5′ overhang to the loxP site of pZero::3xFLAG::nat-1::loxP. The combined fragment bearing the target mutation and the 3xFLAG::nat-1 cassette was PCR-stitched using primers 6716 and 4883 to create a 5′ split marker construct as described (20). The 3′ UTR fragment and the 3xFLAG::nat-1 cassette were PCR-stitched using primers 4884 and 6722 to create a 3′ split marker construct as described (20). These fragments were simultaneously transformed into strain N2930 and nat-1+ transformants were verified by Southern hybridizations and crossed to strain N625 to generate homokaryotic progeny.
Nucleic acid manipulations and molecular analyses
DNA isolation, Southern hybridization, co-IP, western blotting and IP/MS analyses were performed as previously described (10,21–24). DNA for methylation-sensitive Southern hybridizations was digested with AvaII unless noted otherwise. The following antibodies were used: anti-FLAG-HRP (A8592; Sigma), anti-HA (M180–3; MBL), anti-H3K9me1 (CS-065-100; Diagenode), and anti-hH3 (ab1791; Abcam). Co-IP experiments used anti-FLAG conjugated to agarose beads (A2220; Millipore). Secondary antibodies were used as previously described (21). Chemiluminescence resulting from the treatment with SuperSignal West Femto Substrate (34095; Thermo Fisher Scientific) for anti-FLAG-HRP, anti-HA, anti-H3K9me1 and anti-hH3 was imaged using a LI-COR Odyssey Fc imaging system. Yeast two-hybrid strains and assays were built and carried out as previously described (21).
ChIP-qPCR
ChIP was performed as previously indicated (25) using anti-H3K4me3 (ab8580; Abcam) or anti-H3K9me3 (39161; Active Motif). For quantitative ChIP, real-time qPCR experiments on independent experimental replicates were performed in triplicate using PerfeCTa SYBR Green Fastmix ROX (Quantabio) with the listed primers (Supplemental Table S8) and were analyzed using a StepOnePlus Real-Time PCR system (Life Technologies). Relative enrichment of each modification was determined by measuring enrichment as a percent of the total input. The enrichment of the indicated loci was then scaled to relative enrichment at hH4.
ChIP-seq
H3K9me3 ChIP samples were prepared for sequencing as previously described (25). Sequencing was performed using an Illumina NextSeq 500 using single-end 75 nucleotide reads or using an Illumina HiSeq 4000 using single-end 100 nucleotide reads. Manipulations of the sequencing data were performed on the Galaxy platform (26). All sequences were mapped to the corrected N. crassa OR74A (NC12) genome (27) using Bowtie2 (28). For visualization, the mapped read enrichment was calculated over 25 bp bins and normalized by RPKM using bamCoverage from deepTools (29). The sequencing tracks are displayed as 25-nt window bigWig files with the Integrative Genomics Viewer (IGV) (30).
Bisulfite-seq
Bisulfite-sequencing sample preparation was performed using the NEBNext Ultra DNA Library Prep Kit for Illumina (E7370; NEB) and the EpiMark Bisulfite Conversion Kit (E3318; NEB). Data acquisition and processing were performed as previously described (20). To display the bisulfite-sequencing data, the average 5mC level was calculated for 25 bp windows across the genome using the MethPipe program (31). The resulting wig files were displayed on IGV.
RNA-seq
RNA-sequencing, data processing, and analyses were performed as previously described (32). For qRT-PCR analyses, RNA from biological triplicates was isolated as previously described (33). Equal levels of Qubit-RNA (Life Technologies) assayed RNA were DNase-treated (Invitrogen), and cDNA was synthesized using the Superscript III kit (Life Technologies) according to the manufacturer's protocol. 1:10 diluted cDNA was used for qRT-PCR experiments with PerfeCTa SYBR Green Fastmix ROX (Quantabio) and listed primers (Supplemental Table S8), and were analyzed using a StepOnePlus Real-Time PCR system (Life Technologies). Enrichment for each gene was normalized to enrichment of the housekeeping gene rpt-1 (NCU02840, primers 6271/6272; Supplemental Table S8) (34).
RESULTS
Loss of LSD1 results in heterochromatin spreading
As one approach to identify factors involved in regulating constitutive heterochromatin, we screened strains with knockouts of putative histone demethylases for defects in H3K9 methylation or DNA methylation. LSD1 was a likely candidate given previous studies on its function as a histone lysine demethylase, its importance in regulating heterochromatin in yeasts, flies, and humans, and its overall conservation in eukaryotes examined previously (11–14,35,36). Neurospora possesses one gene, NCU09120, that is homologous to genes encoding LSD1 in other eukaryotes. The predicted Neurospora LSD1 contains the conserved Swi3p, Rsc8p, and Moira (SWIRM) and amine oxidase domains typical of LSD1 homologs, as well as a C-terminal HMG domain that is present in both S. pombe paralogs but absent in human LSD1 (Supplemental Figure S1).
In Neurospora, DNA methylation is directed to heterochromatin via trimethylation of lysine 9 on histone H3 (H3K9me3) and therefore can serve as a proxy for the presence of H3K9me3 (21,37,38). To test whether LSD1 plays a role in regulating heterochromatin in Neurospora, we performed whole genome bisulfite sequencing (WGBS) on DNA from an LSD1 knockout strain (FGSC# 11964) (18,39). The Δlsd1 strain exhibited extensive spreading of DNA methylation at select loci as well as the appearance of about 200 new regions of DNA methylation distributed throughout the genome (Figure 1A and Supplemental Figure S2). The hypermethylation, whether expanding from a normal domain or in a new region, typically spread over 2–5 kb, with the largest hypermethylation spreading covering over 8 kb. The spreading did not preferentially affect specific genic elements (e.g. promoters); it appeared to indiscriminately cover chromatin in a contiguous manner (Figure 1A and Supplemental Figure S2). Despite extensive spreading of DNA methylation, total H3K9me3 levels in the Δlsd1 strain appeared similar to wild type by western blot analysis (Supplemental Figure S3, right), implying Δlsd1-dependent spreading of H3K9me3 has a negligible effect on bulk histones.
Figure 1.

Identification of LSDC in Neurospora crassa. (A) WGBS tracks displaying DNA methylation in WT (blue) and Δlsd1 (red) strains over LG VIIL (additional tracks for other chromosomes can be found in Supplemental Figure S2). NCU02455 is indicated with an asterisk. (B) Schematic representation of the identified members of LSDC with their predicted domains and length (amino acids) indicated. (C) LSDC knockouts exhibit hypermethylation. Genomic DNA from WT, a strain lacking DNA methylation (Δdim-2), and LSDC knockouts (Δlsd1, Δphf1 and Δbdp-1) were digested with the 5mC-sensitive restriction enzyme AvaII and used for Southern hybridizations with the indicated probes for a euchromatin control (his-3), an unaffected heterochromatin control (8:A6), and two Δlsd1-sensitive regions (Tel VIIL and NCU02455). Strains (left to right): N3753, N4711, N5555, N6411, N6221, N6414, N6220 and N6416.
Identification of members of the Neurospora LSD complex
To identify potential LSD1 complex (LSDC) members in Neurospora, we generated strains with C-terminal 3xFLAG-tagged LSD1 expressed at its endogenous locus, immuno-purified the bait and associated proteins, and analyzed the material by mass spectrometry. We identified peptides covering 46% of a PHD finger domain-containing protein previously identified in S. pombe as an LSD complex member called PHF1 (PHD Finger 1) (12) and 13% of a bromodomain-containing protein named BDP-1 (Bromodomain Protein 1) (Supplemental Table S1). A reciprocal pull-down with PHF1–3xFLAG yielded peptides covering 42.1% of LSD1 and 24% of BDP-1, supporting its interaction with LSD1 and BDP-1 (Supplemental Table S2). Immunoprecipitation of BDP-1–3xFLAG did not yield LSD1 or PHF1 peptides, however (Supplemental Table S3). To further test these interactions, we performed co-immunoprecipitation experiments in strains with PHF1–3xFLAG or BDP-1–3xFLAG, and LSD1-HA, in which we immunoprecipitated the FLAG-tagged protein and then assayed both epitopes by western blotting. Consistent with the mass spectrometric analyses, PHF1–3xFLAG readily pulled down LSD1-HA, but BDP-1–3xFLAG did not (Supplemental Figure S4A). To examine further the potential interaction between LSDC members, we performed yeast two-hybrid assays. We observed no evidence of direct interaction between LSD1 and PHF1 or LSD1 and BDP-1, potentially because of interference by the tag moieties. However, constructs containing either the N-terminus (amino acids and 2–140) or the C-terminus (amino acids 401–605) of the PHF1 PHD domain fused to the GAL4 activating domain showed interaction with full length BDP-1 fused to the GAL4 DNA binding domain, i.e. growth on selective medium (Supplemental Figure S4B). We also tested for an interaction of full length PHF1 with BDP-1 fragments and found that BDP-1 fragments containing the N-terminal BTB domain (amino acids 45–236), but not the C-terminal Bromo domain (amino acids 198–480), supported growth, implicating an PHF1–BDP-1 interaction that may be important for interaction with the rest of the complex (Supplemental Figure S4B). Altogether, these results indicate PHF1 and BDP-1 directly interact. We conclude LSDC in N. crassa includes LSD1, PHF1, and BDP-1 (Figure 1B).
To investigate whether strains with knockout mutations for the other members of LSDC exhibit phenotypes similar to that of Δlsd1, we digested genomic DNA from phf1 and bdp-1 knockout strains with the 5-methylcytosine-sensitive restriction endonuclease AvaII, and performed Southern hybridizations probing for regions that we found hypermethylated in the Δlsd1 strain. We found that knockouts of all three members exhibited more high-molecular mass bands than the wild-type control, with Δphf1 and Δbdp-1 phenocopying Δlsd1 (Figure 1C). These results support the idea that LSD1, PHF1, and BDP-1 form a complex and function in the same pathway.
Variability of DNA hypermethylation in LSDC knockout strains
Curiously, we found that Δlsd1 siblings from sexual crosses exhibited different extents of DNA hypermethylation. Similarly, independently generated Δlsd1 knockout strains also exhibited variable DNA hypermethylation. This phenotype was also observed with Δphf1 and Δbdp-1 siblings as well as independently generated knockouts of these genes (Figure 1C). In contrast, normally methylated regions exhibit consistent levels of DNA methylation in wild-type strains. The variable nature of Δlsd1 hypermethylation is not consistent genome-wide as some strains exhibit less hypermethylation at some loci and more hypermethylation at others (Figure 2A; Supplemental Figure S5A). Double knockouts of any combination between the three complex members did not present an additive phenotype, but rather one similar to a Δlsd1 or Δphf1 single knockout (Supplemental Figure S5B), further supporting the hypothesis that LSD1, PHF1, and BDP-1 function in the same pathway by forming the LSDC.
Figure 2.

Variable hypermethylation and growth rates in strains with knockouts of LSDC members. (A) DNA from asexually-propagated Δlsd1 strains exhibit variable hypermethylation. Genomic DNA from the FGSC knockout of lsd1 and nine asexually-propagated strains was tested for DNA methylation by Southern hybridization, as in Figure 1C. The pedigree for these strains, relative to the initial strain (marked with an asterisk), is displayed on the right, and the pedigree provides a key to the numbers above the lanes in the Southern hybridization and to the strains in panel C. WT strain: N3753; Δdim-2 strain: N4711. (B) Linear growth rates of strains with LSDC member knockouts at 32°C. Strains (left to right): N3753, N5555, N7979, N6221, N6222, N6219 and N6220. (C) Linear growth rates of asexually-propagated Δlsd1 strains, relative to the initial ‘parental’ strain (N5555; indicated by an asterisk) at 32°C. The quadruple asterisks indicate strains that grew significantly slower than the other strains (P ≤ 0.0001).
To test whether variability of hypermethylation also occurred in asexually propagated strains, we isolated and tested strains generated from microconidia, which are generally mononucleate (40) unlike macroconidia, which possess multiple, potentially variably hypermethylated nuclei. Results of Southern hybridizations on genomic DNA digested with AvaII showed that the asexually-propagated isolates also exhibited variable levels of DNA hypermethylation, and that further propagation continued to yield isolates with variable hypermethylation (Figure 2A and Supplemental Figure S5A). Replicate WGBS experiments with the same isolate revealed slight differences at hypermethylated regions. Perhaps not surprisingly, less variability was observed in a strain propagated with a bulk transfer of macroconidia (Supplemental Figure S5A).
We were curious whether variably hypermethylated Δlsd1 strains would show other variable phenotypes. In S. pombe, deletion of lsd1 results in reduced growth rate and lsd2 is essential for viability, highlighting the importance of these two enzymes (12). To test the importance of each identified member of LSDC for Neurospora growth, we measured the linear growth rates of two independent knockouts of each complex member with ‘race tubes’. All knockouts exhibited a modest reduction in growth rate (Figure 2B). We then assayed the growth rate of the single-microconidium-propagated strains illustrated in Figure 2A by race tubes, in triplicate. Most of the strains grew at slightly slower rates than wild type, though two grew at much slower rates (Figure 2C, strains 1 and 7). All the single-microconidium-propagated stains exhibited greater variance in growth rates between the replicates than wild type. It did not appear that the slower growing isolates exhibited noticeably higher degrees of hypermethylation than the faster growing isolates (Figure 2A and Supplemental Figure S5A). We realized that the variable nature of Δlsd1 strains could have resulted in a selection bias against ‘sicker’ strains by virtue of their slower growth after germination. Therefore, we tested ascospore germination rates generated from Δlsd1 heterozygous (crossed with wild type) and homozygous crosses. Ascospores from both Δlsd1 heterozygous and homozygous crosses had significantly lower germination rates than ascospores from a homozygous wild-type cross (Supplemental Figure S5C), suggesting that loss of LSD1 may impact N. crassa growth more than that observed in our growth rate experiments.
Δlsd1 strains exhibit DNA methylation-dependent spreading of H3K9me3
In Neurospora, the establishment of DNA methylation at constitutive heterochromatin is dependent on the presence of H3K9me3, while H3K9me3 establishment is independent of DNA methylation (21,37,38). However, loss of the heterochromatin limiting complex DMM results in DNA methylation-dependent spreading of H3K9me3 at some heterochromatin boundaries (10). We therefore tested if the heterochromatin spreading resulting from loss of LSD1 is also dependent on DNA methylation. As a first step to explore the relationship of DNA methylation and H3K9 methylation in abnormal heterochromatin formed in the absence of LSD1, we performed H3K9me3 chromatin immunoprecipitation followed by high-throughput DNA sequencing (ChIP-seq) to test for concomitant spreading of H3K9me3 with DNA methylation in a Δlsd1 strain. As expected, we observed H3K9 hypermethylation concomitant with DNA hypermethylation in the Δlsd1 strain (Figure 3A). These results were confirmed in triplicate ChIP-qPCR experiments, which showed increased levels of H3K9me3 at Δlsd1-sensitive loci (Supplemental Figure S6A). To further confirm concomitant increases of H3K9me3 genome-wide, we performed a metaplot analysis of H3K9me3 levels at all identified Δlsd1-sensitive loci, and observed higher enrichment in the Δlsd1 strain (Supplemental Figure S6B).
Figure 3.

LSD1 prevents DNA methylation-dependent hyper H3K9me3. (A) H3K9me3 ChIP-seq and WGBS tracks showing H3K9me3 enrichment and DNA methylation respectively at two unaffected heterochromatin regions (Cen IIIL and 8:A6) and two Δlsd1-sensitive regions (NCU02455 and Tel VIIL) in the indicated strains. dim-2* denotes the C926A catalytic null mutation in dim-2 (42). NCU02455 is indicated with an asterisk. (B) Linear growth rates in WT, Δlsd1, and Δlsd1 strains bearing a deletion (Δ) or catalytic null (*) mutation in dim-2. Strains (left to right): N3753, N5555, N7979, N6337, N8081, N8082, N6679, N8083 and N8084. All Δlsd1; Δdim-2 and Δlsd1; dim-2* strains are siblings from separate crosses. (****) P ≤ 0.0001; (**) P ≤ 0.01. (C) Loss of LSD1 catalytic activity causes hyper DNA methylation. (Top panel) Sequence alignments of human, S. pombe, and Neurospora LSD1 homologs centered on a lysine essential for catalytic activity (highlighted). The NK982,983AA mutation introduced to create the presumptive catalytic null lsd1 Neurospora strains is displayed above. (Bottom panel) DNA methylation-sensitive Southern hybridization analysis (as in Figure 1C) on 3xFLAG-tagged and catalytic null (indicated by *) 3xFLAG-tagged LSD1 strains. Strains (left to right): N3753, N4711, N5555, N6300 and N7899.
We next investigated whether Δlsd1-induced spreading of H3K9me3 is dependent on DIM-2, as in dmm strains (10). To do so, we performed H3K9me3 ChIP-seq on a Δlsd1; Δdim-2 strain as well as a strain with catalytically inactive DIM-2 (dim-2C926A) (41) in a Δlsd1 background. In both cases, the H3K9me3 spreading observed in Δlsd1 single knockouts was absent; instead, the distribution of H3K9me3 was equivalent to the distribution in lsd1+ strains (Figure 3A; confirmation by ChIP-qPCR presented in Supplemental Figure S6A). This indicates that, in contrast to native constitutive heterochromatin domains, the abnormal H3K9me3 found in Δlsd1 strains depends on DNA methylation, as in DMM mutants, and as induced by convergent transcription of disiRNA at disiRNA locus DNA methylation (DLDM) regions (10,42).
Because loss of DNA methylation rescued the heterochromatin spreading phenotype observed in Δlsd1, we asked if the reduction in growth rate observed in LSDC knockout strains would also be affected. Curiously, triplicate growth assays of Δlsd1; Δdim-2 and Δlsd1; dim-2C926A strains showed that both strains were actually slower than wild-type and Δlsd1 single knockout strains (Figure 3B). This contrasts the situation with DMM mutants where loss of dim-2 rescues the growth defect phenotype of Δdmm-1 strains, and was surprising given the modest growth defects observed in Δlsd1 and Δdim-2 single knockout strains (21).
Considering that LSD1 is a presumptive H3K9 demethylase, it was of interest to test if mutation of conserved residues of the putative catalytic domain would result in spreading of H3K9me3 and DNA methylation, as in Δlsd1 strains. We therefore constructed a presumptive catalytic null 3xFLAG-tagged LSD1 strain by mutating a conserved lysine thought to be critical for catalytic activity in such proteins (Figure 3C) (43). Results of Southern hybridizations showed that wild-type 3xFLAG-tagged LSD1 strains had a modest hypermethylation phenotype (Supplemental Figure S7A) while the presumptive catalytic null strains exhibited increases of DNA methylation and H3K9me3 comparable to that of Δlsd1 strains (Supplemental Figure S7A and B). Altogether, these data support a model in which the H3K9 demethylase activity of LSDC is required to prevent heterochromatin spreading (Figure 3C) (11,12,14).
H3K9me1 persists in Δdim-5 strains
The catalytic mechanism of LSD1 is thought to restrict it to demethylating mono- or dimethylated lysines; trimethylated residues should not be susceptible (15). Since the principle form of histone methylation associated with constitutive heterochromatin in Neurospora is H3K9me3, it is not obvious how LSD1 affects heterochromatin spreading. Although ChIP-seq and western blotting experiments have not revealed H3K9me2 in Neurospora (37), low levels of this modification were detected by mass spectroscopy (25), consistent with the possibility that this could be a substrate for LSD1. Mass spectroscopy of Neurospora histone H3 also revealed H3K9me1, even though this modification was not been detected by ChIP (25). We obtained signal for H3K9me1 in western blots (Supplemental Figure S3, left panel) but curiously, nearly identical levels of H3K9me1 signals were found in wild-type, Δlsd1 and Δdim-5 strains, whereas control cell lysate from S. cerevisiae, which is thought to have no H3K9 methylation (44) did not yield a signal. These findings raised the possibility of an unknown H3K9 methyltransferase in Neurospora.
In an attempt to identify the hypothetical methyltransferase responsible for H3K9me1, we screened knockouts of every predicted SET domain protein as well as homologs of non-SET domain lysine methyltransferases, all in a Δdim-5 background. The apparent H3K9me1 signal persisted in every strain tested (for complete list of screened KOs, see Supplemental Table S4). These findings are comparable to observations in S. pombe clr4Δ strains (45) and leave open the possibility of an unrecognized H3K9 methyltransferase in N. crassa.
Gene expression in the presence of H3K9me3 and DNA methylation
Constitutive heterochromatin, such as that resulting from RIP operating on duplicated sequences, has been shown to cause gene silencing in Neurospora (46), at least in part because DNA methylation can inhibit transcriptional elongation (33). Prior work also showed that heterochromatin spreading into neighboring euchromatin in Neurospora strains deficient for DMM causes down-regulation of underlying genes (10). Considering that Δlsd1 strains also show spreading of heterochromatin over gene-rich euchromatin, we wished to test if this is associated with silencing. We therefore performed poly-A+ RNA-seq on wild-type and Δlsd1 strains and compared their expression profiles. Setting a 4-fold difference threshold, we identified 118 downregulated and 17 upregulated genes in the Δlsd1 strain. Curiously, only five of the downregulated genes and none of the upregulated genes were observed to be hypermethylated, as assayed by WGBS of the same Δlsd1 strain used in the RNA-seq analysis (Supplemental Tables S5 and S6). To test directly whether Δlsd1-induced hypermethylation silences genes, we examined four hypermethylated genes by RT-qPCR. All four genes (NCU00911: polysaccharide synthase, cps-1; NCU02455: FKBP-type peptidyl-prolyl cis-trans isomerase, fkr-5; NCU06512: methionine synthase, met-8; and NCU02437: histone H2A, hH2A) are within the top expression quartile in wild type. Surprisingly, no significant change in expression was detected for any of the four genes (Supplemental Figure S8), indicating that the spreading of H3K9me3 and DNA methylation resulting from the deletion of lsd1 was insufficient to silence underlying sequences. To address the possibility that increased levels H3K4me3, possibly resulting from loss of LSD1, are responsible for maintaining wild-type levels of expression, we performed H3K4me3 ChIP-seq and observed no differences in enrichment (Supplemental Figure S8). These results suggest that the observed expression changes in Δlsd1 are due to a LSD1 function besides its role regulating heterochromatin spreading. This finding is consistent with our observations that Δlsd1 strains are ‘healthier’ than Δdmm-1 strains and that growth of Δlsd1 strains is not improved by loss of DNA methylation, unlike the case for dmm mutants (10).
Spreading of heterochromatin in Δlsd1 strains is dependent on HCHC
The HCHC histone deacetylase complex is involved in promoting some heterochromatin spreading in Neurospora (24,47), and similarly in S. pombe, spreading requires the recruitment of the histone deacetylase (HDAC) Clr3 (48,49). We noticed considerable overlap (∼55%) between regions that lose DNA methylation in HCHC-defective strains and those that gain DNA methylation in Δlsd1 strains (Figure 4A). Moreover, previous findings with tethered heterochromatin components demonstrated the importance of HCHC activity in nucleation of ectopic DNA methylation (50). We therefore tested whether HCHC catalytic activity is involved in Δlsd1-induced heterochromatin spreading. To do so, we generated Δlsd1 strains with catalytic null alleles of hda-1 (hda-1D263N), the catalytic subunit of HCHC (47), and performed DNA methylation-sensitive Southern hybridizations probing for Δlsd1-sensitive regions as well as H3K9me3 ChIP-seq. We observed loss of DNA methylation in hda-1D263N at these regions to levels even below that of a wild-type strain, and the level of DNA methylation in Δlsd1; hda-1D263N strains was identical to that in the hda-1D263N strain (Figure 4B). Furthermore, the spreading of H3K9me3 observed in Δlsd1 strains was also reduced to levels equivalent to, or below, that of wild-type, reflecting the observed levels of DNA methylation at these loci (Figure 4C). Altogether, these data suggest that histone deacetylation by HCHC is necessary for Δlsd1-induced spreading of heterochromatin.
Figure 4.
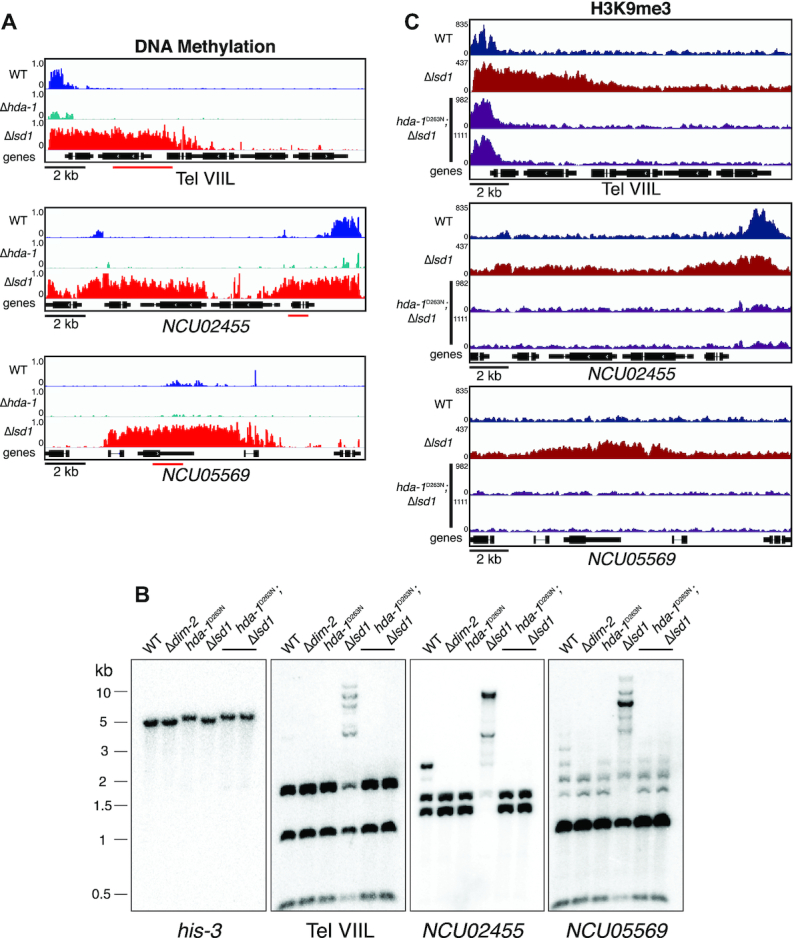
HCHC catalytic activity is necessary for Δlsd1-induced hyper DNA methylation. (A) WGBS tracks displaying DNA methylation in wild-type (WT), Δhda-1, and Δlsd1 strains at selected Δlsd1-sensitive regions. (B) DNA methylation-sensitive Southern hybridizations probing the regions illustrated in panel A showing the loss of Δlsd1-induced hypermethylation in a hda-1 catalytic-null background in sibling strains. The Southern probes used are indicated by a red bar in panel A. Strains (left to right): N3753, N4711, N3998, N6412, N8089 and N8090. (C) ChIP-seq tracks showing H3K9me3 enrichment in WT, Δlsd1 and Δlsd1; hda-1D263N strains at selected Δlsd1-sensitive regions.
DISCUSSION
LSDC-mediated demethylation in Neurospora
In Neurospora, H3K9me3 is the predominant form of H3K9 methylation associated with constitutive heterochromatin, presumably resulting from processive addition of methyl groups by DIM-5, which is thought to be responsible for all H3K9 methylation in Neurospora (23,37,51). As LSD1 is thought to be only able to act on mono- or dimethylated substrates (15), it is puzzling how LSD1 affects heterochromatin spreading. It is notable that in S. pombe, loss of LSD1 catalytic activity results in a larger increase of H3K9me3 than H3K9me2, suggesting that LSD1 functions to balance the levels of these two marks (12). Potentially, LSD1 actively removes H3K9me concurrently as it is catalyzed by the DIM-5/-7/-9, CUL4, DDB1 complex (DCDC), actively countering establishment of the mark at susceptible chromatin before it is trimethylated and ‘locked into place’ by virtue of no longer being a substrate of LSD1. Other, not necessarily mutually exclusive, possibilities are that H3K9me is catalyzed by DCDC and an additional, hypothetical histone methyltransferase that catalyzes mono- or dimethylation of H3K9 (Figure 5B), or that LSDC cooperates with a JmjC domain protein, which demethylates mono-, di-, and trimethylated lysines (52), in order to limit heterochromatin spreading. Neurospora LSD1 could also regulate heterochromatin through the direct demethylation of heterochromatin machinery proteins themselves. LSD1 has been shown to promote DNA methylation genome-wide in mouse embryonic stem cells by demethylating the maintenance DNA methyltransferase, Dmnt1, resulting in increased protein stability (36).
Figure 5.

HCHC catalytic activity and DNA methylation become necessary for heterochromatin formation with increasingly GC-rich DNA. (A) Metaplot displaying the averaged profile of H3K9me3 enrichment in WT and Δhda-1 strains as determined by ChIP-seq and averaged %GC-content over all constitutive heterochromatin domains in wild type. (B) Model showing interactions between factors involved in establishing heterochromatin with decreasing DNA AT-content. (Left) In the absence of HCHC catalytic activity, the significant AT-richness of the interior of heterochromatin domains is sufficient to recruit the histone lysine methyltransferase complex, DCDC, to establish H3K9me3 and subsequent methylation of underlying DNA. However, the abruptly decreasing AT content at heterochromatin borders is insufficient for DCDC-induced heterochromatin, and without HCHC, heterochromatin is unable to properly spread over the canonical domain. (Middle) In a wild-type scenario, HCHC is able to localize to heterochromatin boundaries through binding of H3K9me3 catalyzed by DCDC, as well as through AT-hook domains in the CHAP subunit. Histone deacetylation activity by HCHC is able to recruit DCDC to further mark H3K9me3 on neighboring chromatin and establish a propagating feedback loop capable of spreading heterochromatin across the entirety of the canonical domain. Here, factors such as LSD1 that limit heterochromatin spreading act to keep the expansion in check within proper limits of what should be heterochromatin. (Right) In the absence of such limiting factors where heterochromatin spreads over its boundary into euchromatin, or in DLDM where convergent transcription induces H3K9me3 and DNA methylation, heterochromatin is established over DNA with AT-content well below the level for RIP-induced DNA methylation. Typically, DNA methylation is dependent on the H3K9me3 mark and loss of DNA methylation has no impact on H3K9me3 (21,37,38). In instances of heterochromatin over low (∼50%) AT-richness, DNA methylation now becomes essential for H3K9me3 and further spreading.
Though the exact mechanism of LSDC recruitment is unknown, it seems likely both PHD1 and BDP-1 play important roles as PHD and bromo domains are known readers of histone modifications, binding to lysine acetylation and methylation, respectively. These domains were also dispensable for interactions detected in our yeast two-hybrid assays, consistent with the possibility that they fulfill other roles outside of complex association (Supplemental Figure S4B) (53). It is possible the SWIRM domain of LSD1 itself assists in localization. SWIRM domains are present in many chromatin remodeling or histone modifying proteins and have been shown to bind double-stranded DNA and dinucleosomes in vitro (54).
H3K9me1 in Neurospora
It was recently proposed that, in S. pombe, H3K9me1 persists in clr4Δ strains, suggesting there is an additional enzyme catalyzing H3K9me (45). We too observed, by western blotting, the persistence of H3K9me1 in Δdim-5 strains (Supplemental Figure S3, left). Consistent with this observation, a previous middle-down hybrid chromatography/tandem mass spectrometry (LC-MS/MS) analysis of wild-type Neurospora histones detected both H3K9me1 and H3K9me3 (25). Despite screening through knockouts strains for all SET domain proteins as well as non-SET domain lysine methyltransferases, all in a Δdim-5 background, we have yet to identify a hypothetical methyltransferase. H3K9me1-specific histone methyltransferases have been previously shown to be critical for heterochromatin regulation. Indeed, work by Pinheiro and colleagues revealed the importance of H3K9me1 in heterochromatin establishment and integrity in mammals, where cytoplasmic histone H3 is marked with H3K9me1 by the redundant H3K9me1-specific methyltransferases Prdm3 & Prdm16 (55). Knock-downs of these enzymes prevent Suv39h-dependent H3K9me3 and induce the disintegration of heterochromatin foci that are largely maintained after loss of H3K9me3 in Suv39h and Suv39h/Eset knock-down experiments (55). Neurospora does not show homologs of Prdm3 or Prdm16, but the observation of only modest Hi-C pattern changes in Δdim-5 strains, suggests that H3K9me3 is mostly dispensable for proper maintenance of chromatin architecture (27). Conceivably, H3K9me1 is more important for maintaining proper chromatin conformation in Neurospora, as it appears to be in mammalian contexts, and is the target substrate of LSDC as its catalytic activity is restricted to mono- or dimethylated lysines.
Variable heterochromatin spreading in Neurospora
The variable spreading of heterochromatin displayed by LSDC mutants is particularly interesting. Although enigmatic, it is reminiscent of the variability seen in classic ‘position-effect variegation’ (PEV), first discovered in flies showing mottled eye color due to chromosomal rearrangements with breakpoints within heterochromatin (56). Interestingly, PEV is partly alleviated by adding extra heterochromatin, e.g. in the form of additional Y chromosomes, consistent with the possibility that heterochromatin machinery can be ‘titrated away’ by additional substrate (57). This suggests spreading is partially driven by a ‘surplus’ of heterochromatin elements, providing a need for regulation by factors such as LSD1 (1).
LSDC might remove H3K9me at the edges of heterochromatin domains or in other regions susceptible to aberrant heterochromatin formation, potentially creating a local chromatin environment less conducive for heterochromatin establishment and spreading, leaving the heterochromatin machinery sequestered at its normal regions. In the absence of LSD1, competition for factors might result between regions as heterochromatin reinforces itself via feedback mechanisms, stochastically leading to greater spreading at some regions than others, i.e. the observed variability. During the reestablishment of heterochromatin throughout cell divisions, this unstable balance of machinery may be tipped towards other, less methylated Δlsd1-sensitive regions, and titrate away machinery from previously hypermethylated regions, causing the observed shifts in spreading throughout the genome.
DNA methylation-dependent H3K9me3
Our studies provide a new example of DNA methylation-dependent H3K9me3, which is uncommon in Neurospora (10,42). We note that the DNA methylation-dependent H3K9me3 resulting from defects in LSDC, as well as that previously discovered associated with dmm mutants and at DLDM loci, occurs in chromatin with a higher GC-content than in natural constitutive heterochromatin, which is largely comprised of AT-rich relicts of transposons altered by RIP (38,58). Indeed, it appears that as heterochromatin spreads outside of the normal boundaries of heterochromatin associated with AT-rich DNA, additional heterochromatin-associated factors become necessary for propagation across chromatin. In wild-type Neurospora, both the H3K9 methyltransferase complex, DCDC, and the HDAC complex, HCHC, are recruited to AT-rich heterochromatin regions, where the respective activities of these complexes are able to further reinforce the recruitment of the other (23,24,47,50). In HCHC mutants, DCDC is still recruited to heterochromatin via AT-rich DNA, but in the absence of HCHC activity, heterochromatin fails to spread out to the boundaries of these domains, which show steeply decreasing AT-richness (47) (Figure 5A, B left). HCHC can be directed to heterochromatin through the AT-hook domains of the CHAP subunit, as well as through binding of HP1 to H3K9me3, and subsequent deacetylation is able to recruit DCDC-mediated methylation, leading to further establishment of H3K9me3 (50) (Figure 5B middle). Through feedback between the respective activities of these two complexes, heterochromatin is able to propagate across domains. Interestingly, only in contexts where heterochromatin forms or spreads over non-RIP’d DNA in which the base composition is neutral (i.e. ∼50%), such as in LSDC mutants, dmm mutants (10), and at DLDM loci (42), is DNA methylation required for H3K9me3 (Figure 5B right). While the possible requirement of HCHC activity for spreading in a DMM mutant or for DLDM has not been tested, this would be consistent with what we found for Δlsd1-induced heterochromatin spreading. In a study examining the sufficiency of tethered heterochromatin factors to induce H3K9 and DNA methylation at a euchromatic locus, it was observed that loss of DNA methylation resulted in ∼50% less induced H3K9me3 with tethered HP1 (50). It is possible DNA methylation assists the localization of other heterochromatin machinery or contributes to a more conducive environment for its activity. How DNA methylation affects heterochromatin propagation remains unclear, however. No DNA methylation-binding factor that affects heterochromatin has been yet identified in Neurospora (21,37,38).
CONCLUDING REMARKS
Our investigation of LSD1 provides further evidence of its function as a regulator of heterochromatin and describes an example of variable heterochromatin spreading in N. crassa. Though the target substrate of LSDC in Neurospora remains uncertain, our results indicate that LSD1 functions to prevent HCHC-driven heterochromatin expansion. Our data and previous studies on S. pombe suggest fungal LSD1 functions to limit heterochromatin spreading (12), contrasting with studies from mammalian models, which show LSD1 can promote or antagonize heterochromatin depending on splicing isoform or associated factors. In the future, it would be of interest to investigate thoroughly the individual contributions of PHF1 and BDP-1 in LSDC function. Altogether, our results are in line with the conserved role of LSD1 and provide further insight on regulation of heterochromatin.
DATA AVAILABILITY
Complete WGBS, ChIP-seq, RNA-seq files, gene expression values, and ChIP-seq intensity values have been deposited in NCBI’s Gene Expression Omnibus (GEO; http://ncbi.nlm.nih.gov/geo) and are accessible through GEO Series accession number GSE137018 and as part of previously reported series GSE82222 (59) and GSE81129 (47).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Proteomics Facility at the OHSU and the Genomics Core Facility at the University of Oregon for carrying out the mass spectrometry and the high-throughput DNA sequencing and Andrew Klocko, Jordan Gessaman, Elizabeth Wiles, Kevin McNaught, and Ken-ichi Noma for comments and discusssion. We also thank the Nolen laboratory (University of Oregon) for the S. cerevisiae lysate and Eri Shimada for the technical assistance of yeast two-hybrid assay.
Notes
Present address: Vincent T. Bicocca, Convergent Genomics, 425 Eccles Ave, South San Francisco, CA 94080, USA.
Present address: Michael R. Rountree, Nzumbe, Inc., 3439 NE Sandy Blvd. #330, Portland, OR 97232, USA.
Present address: Tereza Ormsby, Institute of Organic Chemistry and Biochemistry, Flemingovo náměstí 542/2, 166 10 Praha 6, Czech Republic.
Contributor Information
William K Storck, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA.
Vincent T Bicocca, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA.
Michael R Rountree, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA.
Shinji Honda, Faculty of Medical Sciences, University of Fukui, Fukui 910-1193, Japan.
Tereza Ormsby, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA.
Eric U Selker, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [GM035690, R35GM127142 to E.U.S.]; KAKENHI [24870012 to S.H.]; Competitive Funds in Program to Disseminate Tenure Tracking System, MEXT, Japan); V.T.B. and W.K.S. were supported in part by an NIH postdoctoral fellowship [CA180468]; NIH T32 training grant [GM007413]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Allshire R.C., Madhani H.D.. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2018; 19:229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang K., Mosch K., Fischle W., Grewal S.I.S.. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat. Struct. Mol. Biol. 2008; 15:381–388. [DOI] [PubMed] [Google Scholar]
- 3. Al-Sady B., Madhani H.D., Narlikar G.J.. Division of labor between the chromodomains of HP1 and Suv39 methylase enables coordination of heterochromatin spread. Mol. Cell. 2013; 51:80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coveney J., Woodland H.R.. The DNase I sensitivity of Xenopuslaevis genes transcribed by RNA polymerase III. Nature. 1982; 298:578–580. [DOI] [PubMed] [Google Scholar]
- 5. DeLotto R., Schedl P.. Internal promoter elements of transfer RNA genes are preferentially exposed in chromatin. J. Mol. Biol. 1984; 179:607–628. [DOI] [PubMed] [Google Scholar]
- 6. Aygün O., Mehta S., Grewal S.I.S.. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat. Struct. Mol. Biol. 2013; 20:547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scott K.C., Merrett S.L., Willard H.F.. A heterochromatin barrier partitions the fission yeast centromere into discrete chromatin domains. Curr. Biol. 2006; 16:119–129. [DOI] [PubMed] [Google Scholar]
- 8. Raab J.R., Chiu J., Zhu J., Katzman S., Kurukuti S., Wade P.A., Haussler D., Kamakaka R.T.. Human tRNA genes function as chromatin insulators. EMBO J. 2012; 31:330–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ayoub N., Noma K. -i., Isaac S., Kahan T., Grewal S.I.S., Cohen A.. A novel jmjC domain protein modulates heterochromatization in fission yeast. Mol. Cell. Biol. 2003; 23:4356–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Honda S., Lewis Z.A., Huarte M., Cho L.Y., David L.L., Shi Y., Selker E.U.. The DMM complex prevents spreading of DNA methylation from transposons to nearby genes in Neurosporacrassa. Genes Dev. 2010; 24:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Metzger E., Wissmann M., Yin N., Müller J.M., Schneider R., Peters A.H.F.M., Günther T., Buettner R., Schüle R.. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005; 437:436–439. [DOI] [PubMed] [Google Scholar]
- 12. Lan F., Zaratiegui M., Villén J., Vaughn M.W., Verdel A., Huarte M., Shi Y., Gygi S.P., Moazed D., Martienssen R.A. et al.. S.pombe LSD1 homologs regulate heterochromatin propagation and euchromatic gene transcription. Mol. Cell. 2007; 26:89–101. [DOI] [PubMed] [Google Scholar]
- 13. Rudolph T., Yonezawa M., Lein S., Heidrich K., Kubicek S., Schäfer C., Phalke S., Walther M., Schmidt A., Jenuwein T. et al.. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3-3. Mol. Cell. 2007; 26:103–115. [DOI] [PubMed] [Google Scholar]
- 14. Laurent B., Ruitu L., Murn J., Hempel K., Ferrao R., Xiang Y., Liu S., Garcia B.A., Wu H., Wu F. et al.. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol. Cell. 2015; 57:957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi Y., Lan F., Matson C., Mulligan P., Whetstine J.R., Cole P.a., Casero R.a., Shi Y.. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004; 119:941–953. [DOI] [PubMed] [Google Scholar]
- 16. Davis R. Neurospora: Contributions of a Model Organism. 2000; Oxford: Oxford University Press. [Google Scholar]
- 17. Davis R.H., de Serres F.J.. Genetic and microbiological research techniques for Neurospora crassa. Methods in Enzymology. 1970; 17:79–143. [Google Scholar]
- 18. Colot H.V., Park G., Turner G.E., Ringelberg C., Crew C.M., Litvinkova L., Weiss R.L., Borkovich K.A., Dunlap J.C.. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:10352–10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Honda S., Selker E.U.. Tools for fungal proteomics: multifunctional Neurospora vectors for gene replacement, protein expression and protein purification. Genetics. 2009; 182:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klocko A.D., Rountree M.R., Grisafi P.L., Hays S.M., Adhvaryu K.K., Selker E.U.. Neurospora Importin α is required for normal heterochromatic formation and DNA methylation. PLoS Genet. 2015; 11:e1005083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Honda S., Selker E.U.. Direct interaction between DNA methyltransferase DIM-2 and HP1 is required for DNA methylation in Neurosporacrassa. Mol. Cell. Biol. 2008; 28:6044–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lewis Z.A., Adhvaryu K.K., Honda S., Shiver A.L., Selker E.U.. Identification of DIM-7, a protein required to target the DIM-5 H3 methyltransferase to chromatin. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:8310–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lewis Z.A., Adhvaryu K.K., Honda S., Shiver A.L., Knip M., Sack R., Selker E.U.. DNA methylation and normal chromosome behavior in Neurospora depend on five components of a histone methyltransferase complex, DCDC. PLoS Genet. 2010; 6:e1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Honda S., Lewis Z.A., Shimada K., Fischle W., Sack R., Selker E.U.. Heterochromatin protein 1 forms distinct complexes to direct histone deacetylation and DNA methylation. Nat. Struct. Mol. Biol. 2012; 19:471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jamieson K., Wiles E.T., Mcnaught K.J., Sidoli S., Leggett N., Shao Y., Garcia B.A., Selker E.U.. Loss of HP1 causes depletion of H3K27me3 from facultative heterochromatin and gain of H3K27me2 at constitutive heterochromatin. Genome Res. 2016; 26:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Afgan E., Baker D., van den Beek M., Blankenberg D., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Eberhard C. et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016; 44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Galazka J.M., Klocko A.D., Uesaka M., Honda S., Selker E.U., Freitag M.. Neurospora chromosomes are organized by blocks of importin alpha-dependent heterochromatin that are largely independent of H3K9me3. Genome Res. 2016; 26:1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thorvaldsdóttir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song Q., Decato B., Hong E.E., Zhou M., Fang F., Qu J., Garvin T., Kessler M., Zhou J., Smith A.D.. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS One. 2013; 8:e81148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bicocca V.T., Ormsby T., Adhvaryu K.K., Honda S., Selker E.U.. ASH1-catalyzed H3K36 methylation drives gene repression and marks H3K27me2/3-competent chromatin. eLife. 2018; 7:e41497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rountree M.R., Selker E.U.. DNA methylation inhibits elongation but not initiation of transcription in Neurosporacrassa. Genes Dev. 1997; 11:2383–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hurley J.H., Dasgupta A., Andrews P., Crowell A.M., Ringelberg C., Loros J.J., Dunlap J.C.. A tool set for the genome-wide analysis of Neurosporacrassa by RT-PCR. G3 (Bethesda). 2015; 5:2043–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wissmann M., Yin N., Müller J.M., Greschik H., Fodor B.D., Jenuwein T., Vogler C., Schneider R., Günther T., Buettner R. et al.. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007; 9:347–353. [DOI] [PubMed] [Google Scholar]
- 36. Wang J., Hevi S., Kurash J.K., Lei H., Gay F., Bajko J., Su H., Sun W., Chang H., Xu G. et al.. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009; 41:125–129. [DOI] [PubMed] [Google Scholar]
- 37. Tamaru H., Zhang X., McMillen D., Singh P.B., Nakayama J., Grewal S.I., Allis C.D., Cheng X., Selker E.U.. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurosporacrassa. Nat. Genet. 2003; 34:75–79. [DOI] [PubMed] [Google Scholar]
- 38. Lewis Z.A., Honda S., Khlafallah T.K., Jeffress J.K., Freitag M., Mohn F., Schu D., Selker E.U.. Relics of repeat-induced point mutation direct heterochromatin formation in Neurosporacrassa. Genome Res. 2009; 19:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCluskey K., Wiest A., Plamann M.. The fungal genetics stock center: a repository for 50 years of fungal genetics research. J. Biosci. 2010; 35:119–126. [DOI] [PubMed] [Google Scholar]
- 40. Ebbole D., Sachs M.S.. A rapid and simple method for isolation of Neurosporacrassa homokaryons using microconidia. Fungal Genet. Rep. 1990; 37:17–18. [Google Scholar]
- 41. Kouzminova E., Selker E.U.. dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J. 2001; 20:4309–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dang Y., Li L., Guo W., Xue Z., Liu Y.. Convergent transcription induces dynamic DNA methylation at disiRNA loci. PLoS Genet. 2013; 9:e1003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee M.G., Wynder C., Cooch N., Shiekhattar R.. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005; 437:432–435. [DOI] [PubMed] [Google Scholar]
- 44. Rusche L.N., Kirchmaier A.L., Rine J.. The establishment, inheritance, and function of silenced chromatin in Saccharomycescerevisiae. Annu. Rev. Biochem. 2003; 72:481–516. [DOI] [PubMed] [Google Scholar]
- 45. Jih G., Iglesias N., Currie M.A., Bhanu N.V., Paulo J.A., Gygi S.P., Garcia B.A., Moazed D.. Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature. 2017; 547:463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Irelan J.T., Selker E.U.. Cytosine methylation associated with repeat-induced point mutation causes epigenetic gene silencing in Neurosporacrassa. Genetics. 1997; 146:509–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Honda S., Bicocca V.T., Gessaman J.D., Rountree M.R., Yokoyama A., Yu E.Y.. Dual chromatin recognition by the histone deacetylase complex HCHC is required for proper DNA methylation in Neurosporacrassa. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E6135–E6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamada T., Fischle W., Sugiyama T., Allis C.D., Grewal S.I.S.. The nucleation and maintenance of heterochromatin by a histone deacetylase in fission yeast. Mol. Cell. 2005; 20:173–185. [DOI] [PubMed] [Google Scholar]
- 49. Fischer T., Cui B., Dhakshnamoorthy J., Zhou M., Rubin C., Zofall M., Veenstra T.D., Grewal S.I.S.. Diverse roles of HP1 proteins in heterochromatin assembly and functions in fission yeast. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:8998–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gessaman J.D., Selker E.U.. Induction of H3K9me3 and DNA methylation by tethered heterochromatin factors in Neurosporacrassa. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E9598–E9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang X., Tamaru H., Khan S.I., Horton J.R., Keefe L.J., Selker E.U., Cheng X.. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell. 2002; 111:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Klose R.J., Kallin E.M., Zhang Y.. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006; 7:715–727. [DOI] [PubMed] [Google Scholar]
- 53. Yun M., Wu J., Workman J.L., Li B.. Readers of histone modifications. Cell Res. 2011; 21:564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qian C., Zhang Q., Li S. De, Zeng L., Walsh M.J., Zhou M.M.. Structure and chromosomal DNA binding of the SWIRM domain. Nat. Struct. Mol. Biol. 2005; 12:1078–1085. [DOI] [PubMed] [Google Scholar]
- 55. Pinheiro I., Margueron R., Shukeir N., Eisold M., Fritzsch C., Richter F.M., Mittler G., Genoud C., Goyama S., Kurokawa M. et al.. Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell. 2012; 150:948–960. [DOI] [PubMed] [Google Scholar]
- 56. Schultz J. Variegation in Drosophila and the inert chromosome regions. Proc. Natl. Acad. Sci. U.S.A. 1936; 22:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dimitri P., Pisano C.. Position effect variegation in Drosophila melanogaster: relationship between suppression effect and the amount of Y chromosome. Genetics. 1989; 122:793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tamaru H., Selker E.U.. A histone H3 methyltransferase controls DNA methylation in Neurosporacrassa. Nature. 2001; 414:277–283. [DOI] [PubMed] [Google Scholar]
- 59. Klocko A.D., Ormsby T., Galazka J.M., Leggett N.A., Uesaka M., Honda S., Freitag M., Selker E.U.. Normal chromosome conformation depends on subtelomeric facultative heterochromatin in Neurosporacrassa. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:15048–15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Complete WGBS, ChIP-seq, RNA-seq files, gene expression values, and ChIP-seq intensity values have been deposited in NCBI’s Gene Expression Omnibus (GEO; http://ncbi.nlm.nih.gov/geo) and are accessible through GEO Series accession number GSE137018 and as part of previously reported series GSE82222 (59) and GSE81129 (47).