

Abstract
T cells are central to the immune response against various pathogens and cancer cells. Complex networks of transcriptional and post-transcriptional regulators, including microRNAs (miRNAs), coordinate the T cell activation process. Available miRNA datasets, however, do not sufficiently dissolve the dynamic changes of miRNA controlled networks upon T cell activation. Here, we established a quantitative and time-resolved expression pattern for the entire miRNome over a period of 24 h upon human T-cell activation. Based on our time-resolved datasets, we identified central miRNAs and specified common miRNA expression profiles. We found the most prominent quantitative expression changes for miR-155-5p with a range from initially 40 molecules/cell to 1600 molecules/cell upon T-cell activation. We established a comprehensive dynamic regulatory network of both the up- and downstream regulation of miR-155. Upstream, we highlight IRF4 and its complexes with SPI1 and BATF as central for the transcriptional regulation of miR-155. Downstream of miR-155-5p, we verified 17 of its target genes by the time-resolved data recorded after T cell activation. Our data provide comprehensive insights into the range of stimulus induced miRNA abundance changes and lay the ground to identify efficient points of intervention for modifying the T cell response.
INTRODUCTION
T cells play a central role within the adaptive immune defense. They fulfill a broad range of functions reaching from regulating the activity of other immune cells and eliminating pathogen infected or abnormal cells (1), to forming a pathogen specific immunological memory (2,3). T cell activation is induced by cellular interactions with antigen presenting cells resulting in T cell proliferation and effector cell differentiation (4–6). A strict regulation of T cell activity is essential for an effective immune response and it is usually altered in context with autoimmunity or the development of cancer (7,8). There is increasing evidence that miRNAs play a prominent role in the regulation of T cell activity (9–11). MiRNAs are small regulatory ribonucleic acids that exert their function via a RNA-induced silencing complex (RISC) leading to a down regulation of targets by a sequence specific binding of the miRNA's seed region to a 3′UTR target sequence (12–14). Changes in miRNA expression and subsequently in their targeting are of special interest to understand the gene regulatory processes that are induced upon T cell activation (11,15,16). Furthermore, miRNAs may allow the manipulation of specific T cell properties in context of immunotherapies and cancer treatment (17). A detailed understanding of the complex dynamics and consequences of miRNA expression changes upon T cell activation will facilitate the application of miRNAs in a therapeutic context. While most analyses on miRNA expression in T cells are focusing on specific time points, only a few longitudinal studies analyzed a time window between one and several days after T cell activation (11). During the initial 24 h of T cell activation the cells undergo the transition from the resting to the proliferative stage, accompanied by pivotal changes of signaling pathways (18–21). MiRNA expression profiles within the initial 24 h of T cell activation are, however, rarely described and limited to the analysis of individual time points (11,22). Here, we report a time-resolved overall RNA expression profiling of early human CD4+ T cell activation with a particular focus on the quantification of miRNA molecules and the dynamic interplay between the most prominent miRNA expression changes as well as the regulation of gene expression. We identify miRNAs that could become potent candidates for manipulative interventions in T cells. We also provide quantitative information about stimulus induced miRNA expression changes that can serve as a reference to improve future miRNA transfection approaches.
MATERIALS AND METHODS
Isolation of untouched peripheral human CD4+ T cells
Venous blood samples were obtained from volunteers, who were matched for age and gender (female). Cells from two donors (donor 1: age 26 years; donor 2: age 23 years) were used for the initial time-course analysis by microarray experiments. Cells from four additional donors (donor 3: age 27 years; donor 4: age 24 years; donor 5: age 25 years; donor 6: age 28 years) were used for the time-course validation experiments. The blood cell experiments were approved by the ethics committee of the Saarland University (Approval ID: 121/18). Written informed consents were obtained from all donors. Samples for subsequent CD4+ cell analysis were collected using lithium heparin containing collection tubes (S-Monovette®, Sarstedt AG& Co. KG, Numbrecht, Germany). PBMCs were isolated by Ficoll density gradient centrifugation. To avoid pre-activation of the T cells by any inadvertent receptor interactions, CD4+ T cells were isolated by negative selection (Human CD4+ T cell Isolation Kit, Miltenyi Biotech, Bergisch Gladbach, Germany). Cells were resuspended and cultured in RPMI 1640 medium (Life Technologies GmbH, Darmstadt, Germany), supplemented with 10% heat inactivated fetal bovine serum (Biochrom GmbH, Berlin, Germany), penicillin (100 U/mL) and streptomycin (100 μg/ml). Remaining CD4+ cells of donor 1 and donor 6 that were not utilized to study T cell activation over time were cryo-conserved at maximum 13 months before further use (Extraction of background RNA for standard curve generation in context with miRNA quantification analyses). Isolated cells used for the microarray analyses were quantified by flow cytometric analysis of 100 000 cells after staining with αCD4-APC (Cat#561840, BD Biosciences, Heidelberg, Germany).
T cell activation and time-course sampling
3.5 × 105 per well of the freshly isolated CD4+ cells were resuspended in 100 μl of medium and seeded into 96-well plates by using a multichannel pipette. Cells were subsequently activated in separate wells by αCD2/αCD3/αCD28 MACSiBead™ particles (T cell activation/expansion kit, human, Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Samples for microarray analyses were taken over a total time period of 24 h, collecting cells of each donor from three separate wells at intervals of 2 h. Samples for the time-course validation by TaqMan assays were collected at different time points after T cell activation (0, 2, 4, 8, 12 and 24 h) from four donors (3, 4, 5 and 6), each. As a control we used cells that were collected after 24 h of incubation without MACSiBead™ particles. Cells were pelleted and lysed immediately by QIAzol® Lysis Reagent from miRNeasy Micro Kit (Qiagen, Hilden, Germany). Samples were frozen (−70°C) for <2 months before RNA extraction.
RNA extraction and quality control
Total RNA extraction was performed in a semi-automated procedure, using QIAcube robot systems and following the manufacturers' instructions for miRNeasy Micro Kit (Qiagen, Hilden, Germany). The RNA was quantified by Qubit® Fluorometer using Qubit® RNA HS Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). RNA integrity was verified using Agilent 2100 Bioanalyzer instrument and RNA Pico Kit (Agilent Technologies, Santa Clara, CA, USA).
Time-resolved analysis of miRNA and mRNA expression profiles by microarray
To determine miRNA expression profiles, 75 ng total RNA of each sample was utilized for miRNA labeling and hybridization to Human SurePrint G3 Unrestricted miRNA 8 × 60K arrays (Release 21.0, G4872A, Agilent Technologies, Santa Clara, CA, USA) following the instructions of miRNA Complete Labeling and Hyb Kit. To determine mRNA expression profiles, 25 ng total RNA of each sample was utilized within mRNA One-Color Microarray-Based Gene Expression analysis. The synthesis of labeled cRNA was performed using Low Input Quick Amp Labeling Kit (One-Color) (Agilent Technologies, Santa Clara, CA, USA). The cRNA samples were purified by RNeasy Mini Kit (Qiagen, Hilden, Germany) and concentrations of the purified samples were determined by NanoDrop™ 2000c Spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) following the instructions of the labeling kit. Hybridization was performed using the Gene Expression Hybridization Kit and Human SurePrint G3 Gene Expression Microarrays (V3, G4851C; Agilent Technologies, Santa Clara, CA, USA) following the manufacturer's protocol. Arrays were scanned with a resolution of 3 μm. Raw data was extracted using Feature Extraction software (Agilent Technologies, Santa Clara, CA, USA). We then used the limma R-package (23) for background correction (method = normexp, offset = 16), quantile normalization and log2 transformation.
Examination of time-resolved miRNA expression profiles
For the statistical evaluation of miRNA expression changes that were detected by microarray, the R programming language was utilized. In order to identify the most relevant miRNAs for further inspection, we conducted the following analyses. The time points of maximum and minimum expression during the 24 h time-course were determined for each microRNA based on the median log2 expression data obtained from the measurement of three independently activated samples per time point. To identify the significantly changed miRNAs, a paired t-test was performed on the triplicate analysis of the respective time points, assuming a normal distribution of the data. P-values were adjusted by the Benjamini-Hochberg method (24). Fold changes (FC) between the maximum and minimum expression were then calculated based on the corresponding median log2 expression. Significantly changed miRNAs that held FC ≥1.5 within both donors were considered as miRNA candidates for further analyses. The miRNA candidates were clustered using a two-stage clustering. In a first step, initial clusters of highly similar miRNAs were created using a graph-based clustering algorithm based on the ILP formulation by Grötschel and Wakabayashi (25). In this approach, each miRNA is represented as a vertex in a graph. Two vertices in this graph are connected by an edge if their similarity is above a given threshold. Clusters were then calculated as maximal cliques in this graph. We used a threshold of 0.895 to create the initial clusters. These were then combined using standard hierarchical clustering with Ward's minimum variance method (26). In both clustering steps, we use the Euclidean distance for the gradients of two consecutive time points as a distance measure (27).
Microarray-based quantification of miRNA expression
Microarrays were calibrated using synthetic miRNA oligonucleotides (Supplementary Table S1). The lyophilized single stranded RNA oligonucleotides were purchased, synthesized and quality controlled by mass spectrometry (MALDI-TOF) from Eurofins Genomics (Ebersberg, Germany). MiRNA oligonucleotides were resolved to a concentration of 100 pmol/μl using RNAse free water, according the manufacturers' instructions. For the calibration assay of the relative quantification, different amounts of RNA oligonucleotides (50, 500 and 1000 amol), corresponding to the mature miRNA sequences, were pooled and spiked-in to total RNA from inactive CD4+ T cells. The total RNA (75 ng) was extracted from CD4+ T cells of donor 1 (cryo-conserved at 0 h of activation). Raw expression signals of microarray analysis were corrected from array noise. These noise-corrected signals are in the following referred to as Δ raw expression signals. In addition, we analyzed the total RNA from CD4+ T cells of donor 1, in the following referred to as background RNA, in a separate array reaction without oligonucleotides. The Δ raw expression signals of the background RNA analysis were subtracted from the Δ raw expression signals with the spike-ins. The resulting normalized data are referred to as ΔΔ raw expression signals. To determine a calibration curve for each miRNA, the ΔΔ raw expression signals of each miRNA were then related to the corresponding amounts of the spiked miRNA oligonucleotides.
We first subtracted the array noise from the time-resolved miRNA expression data to obtain the corresponding Δ raw expression signals. From these data, we further subtracted the expression values from the inactive CD4+ cells (analyzed at 0 h time point) to determine the ΔΔ raw expression signals. The above determined calibration curve of each miRNA was then applied to time-resolved miRNA expression data. For each miRNA we obtained a corresponding amount [amol], representing its change relative to the inactive T cell state within each of the time-course analyses. Using Avogadro′s constant we then calculated the numbers of molecules for each miRNA at each time point analysis. Since this calculation was based on the amount of RNA within a single array reaction (75 ng RNA), we next extrapolated the corresponding values for the total amount of each RNA sample. We obtained the number of miRNA molecules representing the total result of all analyzed cells for each time-course sample. Molecules per cell were then calculated, based on the assumption that the initial cell count stayed unchanged.
For the calibration assay of the absolute quantification, different amounts of RNA oligonucleotides (50, 500, 1000, 2000 and 5000 amol), corresponding to the mature miRNA sequences, were pooled and spiked-in to total RNA (75 ng) from bacteriophage MS2 (Roche, Basel, Switzerland). Raw expression signals of microarray analysis were again corrected from array noise (Δ raw expression signals). To determine an average calibration curve for all miRNA, the average Δ raw expression signals of all tested miRNAs were then related to the corresponding amounts of the spiked miRNA oligonucleotides. The calibration curve was applied to the Δ raw expression signals of the spike-in test samples and the time course samples. Molecules/cell were calculated as described above.
Validation of time-resolved miRNA expression patterns
To validate time-resolved miRNA expression patterns, RNA samples of the donors 3, 4, 5 and 6 were analyzed by TaqMan assays. RNA of donor 2 was included as a reference for the initial microarray analyses. TaqMan assays were conducted using TaqMan™ MicroRNA Reverse Transcription (RT) Kit and TaqMan™ Fast Advanced Master Mix in combination with RT primers and specific assay probes for hsa-let-7b-5p (Assay ID: 002619), hsa-miR-17-5p (Assay ID: 002308), hsa-miR-20a-5p (Assay ID: 000580), hsa-miR-21-5p (Assay ID: 000397), hsa-miR-26a-5p (Assay ID: 000405), hsa-miR-132-3p (Assay ID: 000457), hsa-miR-155-5p (Assay ID: 002623), hsa-miR-223-3p (Assay ID: 002295) and RNU48 (Assay ID: 001006). Materials were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, United States) and used following the manufacturers' instructions (TaqMan™ Small RNA Assay user guide). 5 ng of RNA was used for miRNA specific reverse transcription and 3 μl of the resulting cDNA were used for the assay reactions. The samples were assayed on a StepOnePlus™ real-time PCR instrument (Thermo Fisher Scientific). The Cq (quantitation cycle) threshold of the amplification curves was set to 0.2. ΔCq was determined by subtraction of the Cq from houskeeping gene (RNU48). The time points of maximum and minimum expression were determined for each microRNA based on the median ΔCq expression (donors 3, 4, 5 and 6). A paired t-test was performed, comparing the respective time points under the assumption of a normal distribution of the data.
Validation of the miRNA quantification
For the validation of the microarray based miRNA quantification, relative miRNA expression changes were quantified by calibrated TaqMan assays. The TaqMan assay protocol was conducted as described above ('Independent validation of time-resolved miRNA expression patterns' section). RT primers and assay probes were used for hsa-miR-155-5p (Assay ID: 002623), hsa-miR-221-3p (Assay ID: 000524), hsa-miR-132-3p (Assay ID: 000457) TaqMan™ microRNA assays and RNU48 (Assay ID: 001006) TaqMan™ microRNA control assay. Each cDNA was assayed three times. TaqMan™ analyses were performed on one of the three replicates of donor 1 and donor 2 using the same time course RNA samples for each miRNA. Due to the low amount of RNA, remaining from former microarray analyses, not all of the time points were tested for all of the miRNA assays. ΔCq was determined by subtraction of the Cq from houskeeping gene, RNU48, and ΔΔCq was calculated by subtraction of ΔCq from inactive state (0 h).
For the quantification of the miRNA changes measured by TaqMan assay approaches, we used calibration curves analogous to those of the microarray analyses. In detail, 50, 500 and 1000 amol of the respective oligonucleotides were added to 75 ng of background RNA, also extracted from CD4+ T cells of donor 1. 5 ng of these RNA samples were utilized for miRNA specific cDNA syntheses, respectively. The resulting Cq values were normalized by subtraction of the corresponding housekeeping gene (RNU48) signal (ΔCq) and the oligonucleotide caused changes were determined by the subtraction of CD4+ background signal without accessory oligonucleotides (ΔΔCq). Calibration curves were then determined as a result of exponential increase in -ΔΔCq (abscissa: −ΔΔCq; ordinate: oligonucleotide concentration added to 75 ng RNA). The trend line equations were applied to the time-resolved TaqMan data as described for microarray based quantification.
Regulator analysis of miR-155
Transcription factor (TF) binding sites, extracted from chromatin immunoprecipitation sequencing experiments, within the regulatory regions of the miR-155 encoding gene (MIR155HG) were identified by the in silico tool RegulatorTrail (28). Binding motifs of transcription factor complexes were extracted from literature (29). We used a custom python script to find matches of these motifs in regulatory regions. In detail, the script reads reference sequences of regulatory regions of the TF binding motif and searches for exact matches of the motif within the reference sequences that were gathered from the GENCODE basic annotation of GRCh38 (V30) (30). Promotor regions were defined as windows surrounding the transcription start side (TSS) of each gene (TSS ± 1000 bp). Enhancer regions were extracted from the GeneHancer database (31).
Prediction and validation of putative miR-155-5p targets by dual luciferase reporter gene assays
In silico target prediction was performed utilizing miRWalk 2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/index.html), choosing all available databases and a minimum seed binding of 6 nt within the 3′UTRs of the predicted targets (32). Results were filtered for a prediction by at least five databases. The resulting list of predicted miR-155-5p targets was checked for an inverse correlation of the corresponding mRNA profile with time-resolved miR-155-5p profile. By requiring a Pearson's (PCC) and/or Spearman′s correlation coefficient (SCC) of at least −0.8 and a different range in fold decrease compared to the inactive state, we chose 19 putative miR-155-5p targets. 3′UTR sequences of the chosen genes were cloned (Supplementary Tables S2 and S3a) into pMIR-RNL-TK dual luciferase reporter plasmid (33). The pMIR-ZFP36 3′UTR plasmid was carried on from a former publication (34). Mutagenesis of miR-155-5p binding sites was conducted by overlap extension PCRs (35) (Supplementary Table S3b).
The reporter constructs were then tested with an automated liquid handling system based dual luciferase reporter assay. HEK293T cells were seeded to a count of 2.4–2.6 × 104 cells/ well in 96-well plates using the liquid handling system epMotion 5075 (Eppendorf, Hamburg, Germany). The following day, cells were transfected with 200 ng of empty pSG5 (Agilent Technologies Inc., CA, USA) or pSG5-miR-155 expression plasmid, in combination with either 50 ng 3′-UTR reporter construct or empty pMIR-RNL-TK reporter plasmid by the liquid handling system. The pSG5-miR-155 plasmid was kindly provided and former described by Grässer et al. (36). Transfection was performed following the protocol of Polyfect™ transfection reagent (Qiagen N.V., Hilden, Germany). Cells were lysed 48 h after transfection, using the liquid handling system and cell lysates were prepared according the protocol of Promega Dual Luciferase® Reporter Assay System (Mannheim, Germany). Luminescence was measured using a GlowMax navigator microplate luminometer (Promega, Madison, USA). Firefly luciferase activity was standardized by constitutively expressed renilla luciferase (pMIR-RNL-TK encoded) and by relative luciferase activity of the respective 3′UTR construct without an ectopic miR-155 expression (empty pSG5 vector). Results were statistically evaluated in comparison to the empty reporter plasmid in combination with ectopic miR-155 expression by unpaired t testing (two-side), assuming a normal distribution of the data. P-values were adjusted for multiple comparisons by the Benjamini and Hochberg procedure (24). A positive control was carried out for all measurements by testing a plasmid containing the FOS 3′UTR; (Chr.14: 75 281 425–75 281 901), which was described as a target of miR-155 by Dunand-Sauthier et al. (37). Further information about the results of empty reporter control as well as positive control measurements is included within the supplement (Supplementary Figure S1).
Analysis of miRNA−Target interaction networks
MiRNA-Target interaction networks were generated by miRTargetLink (38). To this end, we used the ‘Search by microRNA’ function of the tool with a strong evidence type for the experimentally validated target interactions. The function of shared target genes, was determined by GeneTrail2 enrichment analysis (39), choosing the top 5 results of the category ‘Gene Ontology (GO): Biological Process’ sorted by hits (Over-representation analysis, FDR adjustment (40)).
RESULTS
Quality control of time-resolved RNA profiling data
To determine RNA expression profiles upon CD4+ T cells activation, we isolated CD4+ T cells from the peripheral blood of two young adult female donors (Donor 1 and donor 2). Complete blood counts of the two donors verified that red and white blood cell content, hemoglobin, and hematocrit values were within healthy reference ranges (41,42). Flow cytometric determination of CD4+ surface expression on the isolated cells of both donors showed high purity of CD4+ T cells of 95.0 % and 83.7 % for donor1 and donor 2, respectively (Supplementary Figure S2a). Freshly isolated T cells were in vitro activated by bead coupled antibodies. To allow a time-resolved RNA expression profiling, we collected samples at intervals of 2 h from 0 h-24 h. Harvesting the cells form three separate wells for each time point and each donor resulted in a collective of 78 samples (Schematic Figure 1A). A high RNA integrity was confirmed by RNA integrity numbers ranging from 8.2 to 10.0. Total RNA yields were within a range of 130–540 ng and showed a slight increase at the later time points (Figure 1B). Analysis of time-resolved mRNA profiles (Figure 1C) confirmed T cell activation by significant increases in related pathways. An increased CD28 mRNA expression upon T-cell activation as compared to the inactive cells (0 h) argues against an in vitro aging. Likewise, the significant changes of genes involved in the mitotic cell cycle, preferentially at the later time points of the 24 h analysis, speak against this effect (43,44) (Supplementary Figure S2b and c).
Figure 1.

Time-resolved RNA profiling within 24 h of CD4+ T cell activation. (A) Schematic overview on the experimental setup. Peripheral CD4+ T cells were isolated from the blood of two healthy, age and gender matched donors. The cells were in vitro activated by antibody-coupled beads. RNA samples were collected from three independently activated samples per time point and donor at intervals of 2 h and over a total time period of 24 h (n = 78 samples). Expression courses of miRNAs and mRNAs were determined by microarray-based profiling. (B) The total RNA yield of each time-course sample is shown for each time point. (C) T cell activation pathways (GO terms) were significantly upregulated, comparing the time-resolved mRNA expression data after T-cell activation to the expression values at 0 h (P-values adjusted by Benjamini−Hochberg). At each time point, three samples were analyzed for each donor.
Detection of discrete time-resolved miRNA expression patterns
In total, 815 miRNAs were detected by miRNA specific time course analyses. By comparing the maximal and minimal expression of each miRNA during the 24 h time course, we found 140 significantly changed miRNAs for donor 1 and 231 miRNAs for donor 2 with 73 miRNAs found for both donors. Out of these, there were 39 miRNAs with median log2 fold changes of at least 1.5 for the two donors. We considered these miRNAs as the most promising candidates relevant for the regulation of early CD4+ T cell activation (Supplementary Table S4; Figure 2A). To test the statistical significance of the concordance between the detected miRNA expression patterns, we calculated how high the probability has to be to obtain 39 or more common miRNAs, when drawing the significantly changed miRNAs for each donor independently and at random (without replacements). The results of this hypergeometric distribution analysis verified a highly significant overlap of 39 (P = 1.46e−41).
Figure 2.

Overview on the detected miRNA expression changes within early CD4+ T cell activation. (A) Overview on the number of miRNAs that were detected by time-resolved expression profiling. Significantly changed miRNAs were identified for each donor. Out of these, 39 miRNAs showed a median fold change larger 1.5 in both donors and were defined as potential candidates for the general regulation of early T cell activation signaling. (B) The identified 39 miRNA candidates showed a high correlation for a donor-wise comparison between the time-resolved miRNA expression data (log2) of the three separate T cell activation reactions per time point (denoted as A, B and C) and for the comparison between the median results of the two donors (n = 507 comparisons each). Regression equations and coefficients (R2) are indicated. (C) Grouping of the 39 miRNAs into classes of similar time-resolved expression patterns The analysis was based on the median result of all RNA samples from donor 1 and 2 (n = 6) per time point. Clusters were calculated using the ILP formulation by Grötschel and Wakabayashi in combination with a hierarchical clustering (B and A). The red line indicates the cut off, used to create the final clustering. The heatmap (C) shows scaled log2 expression values for each miRNA with the resulting classes of time-resolved expression patterns (D).
Comparing the time-resolved log2 expression data of these miRNA candidates revealed a very high concordance between the three independently activated samples at each time point and also between the two donors. When donor-wise plotting the log2 expression data of the replicates for the complete time course, the slopes of regression lines were in a range of 0.9988–1.0019 and coefficients of determination (R2) were between 0.9934–0.9978 (Figure 2B). Correlating the median log2 expression data between both donors resulted in a regression line slope of 0.999 and a R2 of 0.9921 (Figure 2B). This consistency of time-resolved expression courses allowed to group the 39 miRNAs into distinct classes, characterized by a similarity of time-resolved miRNA changes. Initial clusters of highly similar miRNAs were determined by using a graph-based clustering algorithm based on the ILP formulation by Grötschel and Wakabayashi (25). These clusters were then refined using standard hierarchical clustering with Ward's minimum variance method (26). The clustering analyses resulted in the detection of 11 distinct classes of time-resolved expression patterns (Figure 2C and Supplementary Figure S3). The largest class (class 1) contained 10 miRNAs that were characterized by steady and moderately decreasing expression levels. The second largest class (class 4) vice versa contained eight miRNAs with steady and moderately increasing expression levels. Four miRNAs (miR-223-3p, miR-155-5p, miR-132-3p and miR-21-3p), however, showed unique expression courses and were therefore grouped into separate classes, each (classes 2, 9, 10 and 11). Out of all miRNAs, miR-155-5p yielded the most prominent expressional increase over time of all miRNAs. We found steady increases or decreases of miRNAs as well as expression curves with a plateau like course. None of the expression patterns showed an alternating course with a change of increasing and decreasing expression levels.
MiRNAs of the same family often showed similar expression patterns, e.g. the miR-15 family members miR-15a-5p and miR-15b-5p in class 1. Likewise, miRNAs with a different strand bias (45) also showed similar expression patterns, e.g. the non-star strands of miR-17 and miR-18a were contained in class 3 and star strands of miR-155 and miR-29b-1 in class 8. The 3p- and 5p-strands of the same miRNA duplex were frequently found in different classes, e.g. miR-21-5p in class 4 and -3p in class 11.
Validation of time-resolved miRNA expression patterns
To confirm the validity of the time-resolved miRNA expression patterns, the time-course experiments were reproduced with cells that were isolated from four additional age and gender matched donors (donor 3, 4, 5 and 6). Time-course samples were collected at time-points 0, 2, 4, 8, 12 and 24 h after activation. Since the 0 h RNA sample of donor 6 was lost during its processing, this sample was replaced by RNA that was extracted from a cyro-conserved aliquot of the corresponding non-activated cells. To control for in vitro aging during the 24 h time period, we included non-activated cells, that were analyzed after 24 h. RNA integrity was confirmed by RNA integrity numbers ranging from 8.6 to 9.6. Time-resolved miRNA expression patterns were analyzed by TaqMan assays for eight miRNAs that were representative for different expressional ranges and shapes of expression courses.
We found a striking similarity between the results of donor 2, which was also tested in the initial experiment, and the four additional donors. Both, the ΔCq ranges and the shapes of the expression curves of donor 2, were confirmed by the results of the donors 3–6 (Figure 3). The RNA sample derived from the cryo-conserved cells (donor 6, 0 h), however, revealed some variance in ΔCq, which may be due to the storage process. All of the analyzed miRNAs showed significant expressional changes over time for the additional 4 donors, supporting the general involvement of these miRNAs in the regulation of early human CD4+ T cell activation. The non-activated control cells showed no significant miRNA expression changes, with exception of miR-26a-5p that showed a slight change, which was, however, oppositely from the change found in activated cells.
Figure 3.

Validation of time-resolved miRNA expression patterns by TaqMan assays for independent donors. To confirm the validity of the time-resolved miRNA expression patterns of the microarray analyses (donors 1 and 2), the time-course experiments were reproduced with cells of four independent donors (donors 3, 4, 5 and 6). Time-course RNA samples were collected at 0, 2, 4, 8, 12 and 24 h after activation. As control, RNA samples was collected from non-activated cells after 24 h. Time-resolved miRNA expression patterns were analyzed by TaqMan assays. Donor 2 served as a reference of the microarray time-courses. A paired t-test was performed, comparing the time points of minimum and maximum expression that were determined based on the median results of the donors 3–6, assuming a normal distribution of the data.
Relative quantification of miRNA expression changes in context with T cell activation
We next quantified the miRNA changes observed during the time course. Therefore, we selected 5 miRNAs, each with a different fold change over the 24 h period. MiR-30c-5p, miR-132-3p, miR-155-5p and miR-221-3p were significantly changed while miR-182-5p stayed largely unchanged. Different amounts of RNA oligonucleotides, corresponding to the mature miRNA sequences, were pooled and spiked-in to a cellular background RNA from inactive CD4+ T cells (RIN: 7.8). The resulting samples were analyzed by microarrays. Background RNA without the addition of any oligonucleotides was analyzed in a separate array reaction and utilized to subsequently normalize the results of the spike-in reactions (Supplementary Figure S4). The normalized array signals were then related to the corresponding amounts of accessory miRNA oligonucleotides. We obtained standard curves with regression coefficients (R2) between 0.9952 and 0.9997 (Supplementary Figure S5a−e). To ensure that the standard curves could be applied to time-resolved miRNA data, the background RNA signaling of the calibration array was compared to the array signals of the time course samples (0 h). The results were in good agreement with less than 20 % difference between the samples of the calibration assay and the time course analysis (Supplementary Figure S5f).
Applying the calibration equations to time-resolved data (microarray datasets), the miRNA expression changes (molecules/cell) were calculated relative to their expression at time point 0 h (inactive cell). Resulting amounts of all miRNA changes were within the calibrated range. According changes in molecule numbers were determined based on the total yield of isolated RNA and the assumption that the initial cell count stayed unchanged. As shown in Figure 4, the median decrease of miR-30c-5p was between 13 and 17 molecules/cell after 24 h for donor 2 and donor 1, respectively. For miR-132-3p the expression changes in the period of 2–24 h, were within a median range of 4–7 molecules/cell in donor 1 and 4–9 molecules/cell in donor 2 samples. MiR-155-5p increased with a high rate of ∼60 molecules/h after the initial 2 h (Supplementary Figure S5g), resulting in a total increase of up to 1300 and 1357 molecules/cell (donors 1 and 2). MiR-182-5p showed almost no variability during the time course of activation with median changes in molecules per cell between 1 and −1. For miR-221-3p there were no median changes for donor 1 and minimal changes for donor 2 (0–1 molecules/cell) during the first 4 h. Over the entire time course, there was a median increase of 2 and 6 molecules/cell in donor 1 and donor 2, respectively. The results of the quantification were validated by a TaqMan assay based quantification (Supplementary Figure S6a-c). The TaqMan assay confirmed the relative changes found by the array analysis for miR-132-3p, miR-155-5p and miR-221-3p (Supplementary Figure S6d−f). In summary, we were able to quantify miRNA expression changes relative to the inactive cell state by microarray calibration.
Figure 4.

Changes of miRNA molecules per cell upon activation of CD4+ T cells. MiRNA expression changes [molecules/cell] related to the 0 h time-point (non-activated cells) were exemplary determined for selected miRNAs by the application of corresponding microarray calibration curves to the time-resolved expression data. Results of the represented miRNAs are shown as median (line) of the three separate T cell activation reactions per time point and donor. The expressional ranges of the three activation reactions are shown for each time point by filled areas.
Time-resolved absolute miRNA abundances in context with T cell activation
To quantify the absolute expression status of all detected miRNAs, we repeated the array calibration assay using a viral background RNA (MS2 bacteriophage carrier RNA) that had only a minor impact on the calibration curves. Upon hybridization, we determined the averaged signals related to specific amounts of the various spike-in miRNAs. The resulting average calibration curve (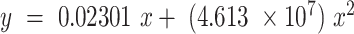 ) assigned microarray signals to the corresponding miRNA amounts [amol] (Supplementary Figure S7a).
) assigned microarray signals to the corresponding miRNA amounts [amol] (Supplementary Figure S7a).
We next tested the performance of the calibration curve by using different miRNA oligonucleotide spike-ins. The employed spike-in amounts of miR-15a-5p, miR-17-5p, miR-150–5p and miR-223-3p corresponded on average to the amounts calculated by the standard curve (Supplementary Figure S7b). These results confirm the general applicability of standard curve to quantify absolute miRNA amounts.
MiRNA molecules/cell of the time-resolved miRNA expression patterns were determined based on the total RNA yield and the assumption of no change of the initial cell count. A quantitative overview of all detected miRNAs over time is given in Supplementary Table S5. For miR-7975 and miR-7977 the quantified amounts were beyond the range of the calibration curve. Quantitative data about these two miRNAs are provided under the assumption that the calibration curve is valid beyond the calibrated range. The results of our absolute quantification were also in good agreement with the results of the microarray based relative quantification (Supplementary Figure S7c−g).
The total amount of all miRNA molecules/cell showed an increase from 2.51 × 105 molecules (0 h) to 5.06 × 105 miRNA molecules/cell after 24 h of activation (Figure 5A). Out of the 815 detected miRNAs we found 78 miRNAs with expression values of 20–99 molecules/cell and 54 miRNAs with expression maximum of ≥100 molecules/cell. The maximal expression values for most of the remaining miRNAs were <20 molecules/cell during the observed time (Figure 5B). Comparing the values of the 54 highly expressed miRNAs between 0, 12 and 24 h (Figure 5C), we found 31 miRNAs that were constantly expressed within the same order of magnitude. In detail, 16 miRNAs stayed at ≥100 molecules/cell throughout that time course, 7 miRNAs at ≥500 molecules/cell and 8 miRNAs at ≥1000 molecules/cell. As for the miRNAs with altered expression levels, we found one miRNA (miR-155-5p) that increased from a low expression (≤100 molecules) to ≥1000 molecules/cell within 24 h. We found 11 miRNAs that showed expression changes reaching from low (<100 molecules/cell) to high expression (≥100 molecules/cell). Among the highly expressed miRNAs, we found miR-20a-5p with an expression increase by two orders of magnitude. We also found 11 highly expressed miRNAs that changed their expression by one order of magnitude.
Figure 5.

Quantification of absolute miRNA molecules per cell in context with CD4+ T cell activation. Absolute miRNA expression [molecules/cell] was determined for all detected miRNAs by the application of a microarray calibration curve to the time-resolved expression data. The quantitative and time-resolved miRNA patterns were evaluated based on the median result of all RNA samples from donors 1 and 2 (n = 6 per time point). (A) Total miRNA expression was determined by the sum of all detected miRNAs at 0 and 24 h after activation (n = 815). (B) The maximum expression of each miRNA was determined for the total 24 h time frame. (C) The highest expressed miRNAs were compared between the 0, 12 and 24 h time points. The increase of miR-451a was during 7–8 h after activation and is therefore not represented by the displayed time points.
Identification of miR-155 regulators
Due to its prominent expression changes amongst the detected miRNAs and its pivotal role for T cell activation capability (46), we chose miR-155-5p to further capture its regulatory network in context with T cell activation. We first analyzed transcription factors (TFs) that could be involved in the upstream regulation of miR-155. We identified 80 potential regulators by the in silico tool RegulatorTrail (28). We then condensed the set of TFs by considering only genes that showed increased expression immediately after the CD4+ T cell activation, i.e. genes with an absolute log2-fold-change of at least 1 between the first time point (0 h) and the three consecutive time points (2, 4 or 6 h) in both donors. This resulted in 24 potential regulators of miR-155. We next measured the association between these TFs and miR-155-5p by calculating different distance correlations (47): (i) between our mRNA data of the MIR155HG and all potential regulators, (ii) between the expression values of miR-155-5p in our miRNA data set and corresponding time points of all potential regulators in the mRNA data set, and (iii) between the GTEx tissue atlas expression data of MIR155HG and all potential regulators (48). An overview of all distance correlation measurements between the TFs and miR-155 is given in Supplementary Table S6. Figure 6A shows the top 10 regulators based on the average distance correlation of the three comparisons. The average distance correlation indicated Interferon Regulatory Factor 4 (IRF4) as a promising candidate to regulate miR-155, a finding that is consistent with previous reports on IRF4 binding to regulatory regions of the encoding MIR155HG gene (49). The role of IRF4 as a central regulator of miR-155 was further supported by our finding that SPI1 and BATF were also amongst the top 10 regulators of miR-155. Since several binding motifs (EICE, ISRE, and AICE) have been described for IRF4 homodimers and its heterodimer complexes with SPI1 and BATF (29) (see Figure 6B), we performed a sequence analysis and found all three motifs in the upstream enhancer region of the MIR155HG gene. We additionally found the ISRE motif in the promotor region of MIR155HG, which indicates that IRF4 homodimers might be the predominant regulator complex. This complex primarily forms, when IRF4 is highly abundant (50). The time course expression data verified a high increase of IRF4 mRNA with a plateau approached after 4 h of T cell activation and a consecutive expressional increase of MIR155HG gene (pre-miR-155) expression with a plateau approached after 6 h (Figure 6c).
Figure 6.

Potential upstream regulators of miR-155 in context with T cell activation. (A) Top 10 list of potential regulators of MIR155HG gene sorted by their mean distance correlation on all investigated data sets. (B) Network of miR-155 and its potential upstream regulators IRF4, SPI1, BATF and annotation of corresponding binding motifs of their complexes within the regulatory regions of MIR155HG gene. (C) Time-resolved expression patterns (Median results of the mRNA data) of IRF4 and pre-miR-155 (MIR155HG) within 24 h of CD4+ T cell activation. (D) Scatter plot between the expression (TPM: transcripts per million) of IRF4 and MIR155HG in the GTEx data set.
We next analyzed the GTEx tissue atlas, which summarizes expressional datasets of different tissues (48) and found a dependency between the expression of IRF4 and the expression of miR-155, i.e. increased expression values of miR-155-5p were found almost exclusively within a discrete expression range of IRF4 (Figure 6d). Overall, the in silico analyses of time-resolved expression data highlight a central role of IRF4 and its complexes with SPI1 and BATF for the transcriptional regulation of miR-155 in context with T cell activation.
Identification of miR-155-5p target genes
To further analyze the miR-155-5p regulatory network in context of T cell activation, we expanded our analyses to the downstream effects of miR-155-5p. For miRNA-target identification, we performed in silico prediction of miR-155-5p targets by miRWalk 2.0, which combines the information of 13 independent databases (32). This analysis identified 42 554 putative miR-155-5p−target interactions. To condense the number of potential targets, we limited our analysis to targets that were identified by at least five databases (5305 targets). As the next selection criterion, we utilized the time-resolved mRNA expression data. In detail, we chose targets with mRNA expression levels inversely correlated to miR-155-5p with Pearson′s and/or Spearman′s correlation coefficients of ≤−0.8 in at least one donor. From the remaining 535 genes, we exemplarily selected 19 putative targets showing either strong, average and less pronounced mRNA fold decreases and different expression ranges (Supplementary Table S7; Figure 7A).
Figure 7.

MiR-155-5p and its target genes. (A) Overview on the time-resolved expression patterns of miR-155-5p and of its potential target genes (mRNA). Time courses (log2 expression) are donor-wise represented as median result of the three activation reactions per time point. Putative targets were selected based on an in silico prediction and an inverse correlation between their time-resolved mRNA profile and the corresponding miR-155-5p expression. (B) The interactions of miR-155-5p with its putative target genes were analyzed by dual luciferase assays. 3′UTR sequences of putative target genes were cloned into luciferase reporter plasmids (pMIR-RNL-TK) and tested in presence of miR-155 expression plasmid (pSG5-miR-155) or (empty) pSG5 control, respectively. The KDM5B 3′UTR was subdivided into two reporter constructs. Firefly and Renilla luciferase activities were measured 48 h after transfection of HEK293T cells. Results were standardized based on the transfection efficiency (determined by Renilla luciferase activity) and the basic activity of the 3′UTR-plasmid (empty pSG5 co-transfection). Results are shown in relation to the activity of an empty reporter control (pMIR without 3′UTR with miR-155 co-transfection ≡ 100%) as the average (line) with range (bars) of three independent experiments (conducted in duplicates). Statistical evaluation was performed in comparison to empty reporter control. P-values were adjusted by Benjamini–Hochberg. (C) For the exemplary validation of the luciferase assays, putative miR-155 binding sites in the 3′UTRs of four positively tested target genes were mutated and tested in comparative luciferase assays with both the wild type and corresponding mutated reporter constructs.
Twenty 3′UTR sequences of these potential targets were cloned into pMIR-RNL-TK reporter plasmid to simultaneously test them in combination with miR-155 overexpression by dual luciferase reporter assay. The 3′UTR of KDM5B was subdivided into two constructs, due to its length and two miR-155 binding sites. The results confirmed the functional miR-155-5p binding for 17 of the 19 tested genes (Figure 7B), which corresponds to a validation rate of 89.47 %. Only RXRA and RASA3 showed no significant changes in relative luciferase activity. Compared to empty reporter control, a significantly decreased relative luciferase activity was detected upon miR-155 overexpression for 3′UTR reporter constructs of ADD3, CYP2U1, DDX17, EZH1, HERC3, IFT80, JMY, KDM5B, LAT2, LDLRAP1, S100B, SH3BP4, SORL1, STARD8, TADA2B, TAF7 and ZFP36. Out of these, ADD3, LAT2, S100B and STARD8 were chosen for the validation of the results. We mutated the putative miR-155 binding sites and performed comparative luciferase assays with both the wild type and corresponding mutated reporter constructs (Figure 7C). By this mutation the significant effect of an ectopic miR-155 expression on relative luciferase activity was reverted for all four targets, confirming the effects on wild type constructs of being based on miR-155 binding.
Concerning the functions of the identified miR-155 target genes, we performed an over-representation analysis (FDR adjusted (40)) using the online pathway enrichment analysis tool Genetrail 3.0 (27). Six out of the 17 identified targets (DDX17, EZH1, JMY, KDM5B, TADA2B, TAF7) were significantly enriched in the GO molecular function term ‘transcription coregulator activity’ (P = 6.89E−04). Four of them (DDX17, JMY, TADA2B, TAF7) were also significantly enriched in the term ‘transcription coactivator activity’ (P = 9.52E−03).
Literature investigation of further identified targets (Table 1) highlight their implication in other basic cellular processes, such as the regulation of endocytosis, cytoskeleton and subcellular localization (LDLRAP1, SH3BP4, SORL1 and STARD8), RNA and protein degradation (HERC3 and ZFP36), calcium signaling (ADD3, S100B), and the general modulation of immune cell signaling (CYP2U1 and LAT2). IFT80 has originally been described as a flagellar transport protein in ciliated cells (51,52). Ciliary transport processes have been implicated in the process of T cell activation (53).
Table 1.
Overview on the functions of miR-155-5p target genes that were identified by dual luciferase reporter assays
| Putative target gene | Function | Reference |
|---|---|---|
| ADD3 | Membrane skeleton protein; regulator of spectrin-actin network; interaction with proteinkinases and Ca2+ binding calmodulin | (54) |
| CYP2U1 | Cytochrome P450 family member; suggested modulation of signal transduction in immune cells | (55,56) |
| DDX17 | RNA helicase; broad impact on RNA metabolism | (57) |
| EZH1 | Component of the Polycomb repressive complex 2; direct negative regulator of transcription | (58) |
| HERC3 | Ubiquitin ligase; regulator of NF-κB signaling | (59) |
| IFT80 | Flagellar transport protein | (51,52) |
| JMY | Transcriptional co-activator in context with p53 | (60) |
| KDM5B | Histone demethylase; negative regulator of transcription | (61) |
| LAT2 | Transmembrane adaptor; fine regulator of lymphocyte activation | (62) |
| LDLRAP1 | Low density lipoprotein receptor adaptor protein; regulator of endocytosis and subcellular localization | (63) |
| S100B | Calcium binding protein of the S100 family; calcium induced signal transduction | (64) |
| SH3BP4 | SH3 domain binding protein; regulator of clathrin endocytosis | (65) |
| SORL1 | Sortilin related receptor; regulator of protein subcellular localisation | (66) |
| STARD8 | Rho GTPase activating protein; negative regulator of Rho-ROCK signaling | (67,68) |
| TADA2B | Transcriptional adaptor protein; transcriptional coactivator | (69) |
| TAF7 | TATA-box binding protein associated factor; regulator of transcriptional initiation | (70) |
| ZFP36 | RNA-binding protein; inductor of targeted mRNA degradation | (71) |
Identification of potential cooperative miRNA pairs
Since the identification of cooperative miRNA pairs is a central request of current miRNA research (72), we utilized the absolute miRNA expression data (Supplementary Table S5) in context with miRNA-target networks to highlight presumable miRNA cooperativity. Amongst the former defined miRNA candidates (see also Figure 2), we considered miRNAs with high abundance changes of >20 molecules/cell. We obtained a list of 23 miRNAs, including 15 miRNAs with increasing and 8 miRNAs with decreasing expression changes (Figure 8A). Considering only the miRNAs with increasing expression changes, we performed an interaction network analysis by miRTargetLink (38). Since miRTargetLink analysis is based on the availability of data of formerly identified miRNA targets, the analysis did not include miR-1260a, miR-4286, miR-1273g-3p, miR-6085, miR-21–3p, miR-29b-1–5p. We identified multiple shared targets common to both miR-17–5p and miR-20a-5p (Figure 8B). These 31 targets are involved in central cellular processes such as cell cycle regulation, Notch signaling and cell maturation (Figure 8C). Likewise, common target genes were shared by miR-155-5p and miR-21-5p. The eight targets are mostly involved in the regulation of immune cell activation pathways (Figure 8D). Considering the miRNAs with decreasing expression changes, miRTargetLink identified nine shared targets for let-7b-5p and miR-26a-5p. These targets are involved in the regulation of cellular senescence and the negative regulation of metabolism and translation (Figure 8e and f).
Figure 8.

MiRNA-Target network analysis of prominently changed miRNA candidates. (A) Based on the quantitative and time-resolved miRNA patterns, 23 of the miRNA candidates showed abundance changes of more than 20 molecules/cell. (B) A miRNA-Target network was determined for those of the 23 miRNAs with increasing expression levels. (C, D) Multiple shared targets were detected between miR-17–5p and miR-20a-5p and between miR-21-5p and miR-155-5p. Major functional involvements of the shared target genes are indicated. (B) A miRNA-Target network was determined for those of the 23 miRNAs with decreasing expression levels. (F) Multiple shared targets were detected between let-7b-5p and miR-26a-5p. Major functional involvements of the shared target genes are indicated. (The network images in B−F were exported from miRTargetLink (38).)
DISCUSSION
MicroRNAs play a central role in the regulation of T cell functions (9,10,73) and the application of miRNAs in immune therapeutics is envisaged (15–17). We performed time-resolved RNA profiling during the initial 24 h of human CD4+ T cell activation to address imminent challenges in T cell related miRNA research, including the identification of miRNA candidates for future immune manipulation approaches (17,74), the specification of common profiles in miRNA processing (75) and the characterization of miRNAs in context with T cell functionality.
We found 39 miRNAs, which showed consistent and significant changes for both of the initially tested donors. The time-curves of eight selected miRNAs were reproduced for four additional donors. These data indicate that the identified miRNAs are most likely involved in the regulation of T cell activation and may serve as potent candidates for T cell manipulation at the early stages of activity. Former long-term analyses of miRNA expression in mouse and human reported comparable expressional changes for several of these miRNAs upon T cell activation (11), supporting their central involvement in T cell regulation. Grouping of these miRNAs into classes with similar expression patterns yielded a surprisingly limited number of time-resolved expression patterns, considering the large number of theoretically possible time courses. Commonly grouped miRNAs may therefore share common regulatory mechanisms. This hypothesis is further supported by the finding that classes frequently contained miRNAs from the same family or of the same strand type, e.g. star and non-star strand. The 3p-strand and 5p-strands of the same miRNA duplex frequently showed different expression pattern e.g. have been grouped into different classes. This difference between 3p- and 5p-strand of the same miRNA duplex may result from different miRNA turnover rates (76,77). We found striking concordance in the ranges and shapes of the time-resolved miRNA expression patterns. The high degree of consistency indicates a rather low susceptibility to inter-individual immune variability (78). Since the blood donors shared the same sex and were nearly of the same age, analyzing CD4+T cells from elderly, male or diseased donors may yield different miRNA profiles due to differences in immune functionality (79). This has to be taken into account towards a possible future use of miRNA time-courses related to T cell activation as biomarker.
Regarding the overall pattern changes, we found that decreasing miRNAs showed less pronounced fold changes than the miRNAs, which increased during T cell activation. This may be due to the generally long half-lives of miRNAs or the speed in processing the precursors and may be even more evident with prolonged observation time (80–82). Additionally, miRNA decay may be impacted by the abundance of mRNA targets, which can affect the miRNA stability either in a positive or a negative way (76,83). In general, declining miRNAs may not only be a leftover from the quiescent T cell state, but may also actively contribute to the down-regulation of target genes.
MiR-155-5p, which has been extensively described in context with T cell functionality (46,73), was the most prominent miRNA within the time-resolved analyses and it yielded an exceptional abundance increase. Potential upstream regulators of miR-155 in the context of T cell activation are IRF4 together with SPI1 and BATF. The regulatory impact of IRF4 has also been indicated by former findings showing that IRF4 homodimers are capable to bind to ISRE motives at the MIR155HG locus preferentially in context of high IRF4 expression rates as found after T cell receptor activation (50). In detail, the IRF4 mRNA shows a high increase within the early hours of T cell activation followed by increase in miR-155 expression. IRF4 can form complexes with SPI1 and BATF, an involvement of which has been described in context with B lymphocyte activation (29). It is conceivable that these TF complexes also play a role in context of the activation of T lymphocytes. There is also evidence for a regulation of SPI1 by miR-155 (84) and a feedback loop between SPI1 and miR-155 (85).
While most studies of miRNA functions are based on analyses at supraphysiological miRNA concentrations (86), we quantitatively analyzed stimulus induced miRNA abundance changes. Knowing the magnitude (∼103 molecules/cell) and the rate of expressional changes, which was e.g. for miR-155 in the range of about 60 molecules/h helps to advance future miRNA manipulation approaches and miRNA target analyses. Our quantitative time-course data provide a physiological reference that can help to avoid toxicity and off-target effects, which may occur due to supraphysiological doses in miRNA transfection (86).
Overall, the quantified range of total miRNA content is consistent with former quantitative analyses on different cell types and organisms. These studies report a total miRNA content within the magnitude of ∼105 molecules/cell (87–89). Likewise, the overall range of the most abundant miRNAs (∼103–105 molecules/cell) is according with a former quantification approach of human cell lines (90). Moreover, the finding of only a limited number of highly abundant miRNAs within specific cell types is consistent with the literature (88,91).
Formerly, a threshold in miRNA expression level has been suggested for a sufficient target regulation (92). Consistent with this hypothesis, we assume that those miRNAs, which become highly abundant upon the activation stimulus play a prominent role within the miRNA regulated networks of early T cell activation. The list of miRNA candidates for manipulating immune cells was condensed to 23 candidates by using the quantification data. Notwithstanding the dominant role of highly changed miRNAs, miRNAs with a rather constant expression and low or moderate abundance changes may also play a role in the overall miRNA target network via high target affinities and altered miRNA-target ratios (81).
There is evidence for a repeated activity of single miRNA molecules. This mechanism may further increase the efficiency of miRNAs to regulate a wide spectrum of targets via multiple rounds of targeting (83,93). In addition, miRNAs may exhibit cooperative functions, which could further increase their effects on specific signaling pathways (94). By miRNA-target network analysis of miRNAs with high abundance changes, we found putative miRNA pairs that share both the basic expression courses as well as multiple common targets. Amongst the pairs of miRNAs with an increasing expression, we identified miR-17-5p and miR-20a-5p, which show high homology and also share the same seed sequence. We cannot exclude a competition for target binding sites between these two miRNAs (95). There is however evidence by studies on other cell types, where both miR-17 and miR-20a promote the cell cycle progression and the cell proliferation (96,97). For the second miRNAs pair with increasing expression i.e. miR-21 and miR-155 there is evidence for a cooperative regulation of the ubiquitin conjugating enzyme E2K in context with T cell activity (98). MiR-21 and miR-155 are both involved in the regulation of IFN-gamma signaling and the differentiation of specific T cell subtypes (99,100). Other targets of miR-21 and miR-155 suggest that the miRNA pair could balance the activation signaling (101,102). The basic functions of the miR-155 target genes that we identified in our study further support the central role of this miRNA for the modulation of the T cell signaling.
Among the decreasing miRNAs, we identified let-7b-5p and miR-26a-5p, which possibly act in a cooperative manner to inhibit cell cycle progression and metabolic processes (103–106). The down-regulation of these two miRNAs in context with T cell stimulation qualifies them as promising candidates for inhibitory T cell manipulation approaches.
Overall, the combination of our time-resolved expression analysis with an absolute quantification of miRNA expression changes offers new opportunities to unravel miRNA regulatory networks and highlights the functional dominance of specific miRNAs within the early T cell activation.
DATA AVAILABILITY
Microarray data of the time-resolved mRNA profiles (#GSE136625) and the time-resolved miRNA profiles (#GSE136626) are available at the GEO database. The source code for the GeneTrail C++ library as well as the clustering approach are publicly available on https://github.com/unisb-bioinf.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Dr Friedrich Grässer from the Institute of Virology (Saarland University, Medical Center, Homburg, Germany) for providing the pSG5-miR-155 plasmid.
Author contributions: C.D., E.M. T.K., N.L., M.S., A.K., B.W.R., H.P.L. and M.H. designed the study. C.D. performed the experiments with support from S.R., L.K., S.P., D.S., T.T. and M.H.; T.K., C.D., K.L., M.H. and H.P.L. analyzed the data; C.D., M.H. and T.K. visualized the data; C.D., M.H. and E.M. wrote the manuscript; all authors edited and approved the final manuscript.
Contributor Information
Caroline Diener, Institute of Human Genetics, Saarland University, 66421 Homburg, Germany.
Martin Hart, Institute of Human Genetics, Saarland University, 66421 Homburg, Germany.
Tim Kehl, Center for Bioinformatics, Saarland Informatics Campus, Saarland University, 66123 Saarbrücken, Germany.
Stefanie Rheinheimer, Institute of Human Genetics, Saarland University, 66421 Homburg, Germany.
Nicole Ludwig, Institute of Human Genetics, Saarland University, 66421 Homburg, Germany.
Lena Krammes, Institute of Human Genetics, Saarland University, 66421 Homburg, Germany.
Sarah Pawusch, Institute of Human Genetics, Saarland University, 66421 Homburg, Germany.
Kerstin Lenhof, Center for Bioinformatics, Saarland Informatics Campus, Saarland University, 66123 Saarbrücken, Germany.
Tanja Tänzer, Institute of Virology and Center of Human and Molecular Biology, Saarland University, 66421 Homburg, Germany.
David Schub, Department of Transplant and Infection Immunology, Saarland University, 66421 Homburg, Germany.
Martina Sester, Department of Transplant and Infection Immunology, Saarland University, 66421 Homburg, Germany.
Barbara Walch-Rückheim, Institute of Virology and Center of Human and Molecular Biology, Saarland University, 66421 Homburg, Germany.
Andreas Keller, Chair for Clinical Bioinformatics, Saarland University, 66123 Saarbrücken, Germany; Department of Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford, CA 94305, USA.
Hans-Peter Lenhof, Center for Bioinformatics, Saarland Informatics Campus, Saarland University, 66123 Saarbrücken, Germany.
Eckart Meese, Institute of Human Genetics, Saarland University, 66421 Homburg, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work has received no external funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gaudino S.J., Kumar P.. Cross-talk between antigen presenting cells and T cells impacts intestinal homeostasis, bacterial infections, and tumorigenesis. Front. Immunol. 2019; 10:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marshall J.S., Warrington R., Watson W., Kim H.L.. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018; 14:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mond J.J., Takahashi T., Thorbecke G.J.. Surface antigens of immunocompetent cells. 3. In vitro studies of the role of B and T cells in immunological memory. J. Exp. Med. 1972; 136:663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pollizzi K.N., Powell J.D.. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 2014; 14:435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Golubovskaya V., Wu L.. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel). 2016; 8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bierer B.E., Greenstein J.L., Sleckman B., Ratnofsky S., Peterson A., Seed B., Burakoff S.J.. Functional analysis of CD2, CD4, and CD8 in T-cell activation. Ann. N. Y. Acad. Sci. 1988; 532:199–206. [DOI] [PubMed] [Google Scholar]
- 7. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer. 2005; 5:263–274. [DOI] [PubMed] [Google Scholar]
- 8. Wang Z., Chang C., Lu Q.. Epigenetics of CD4+ T cells in autoimmune diseases. Curr. Opin. Rheumatol. 2017; 29:361–368. [DOI] [PubMed] [Google Scholar]
- 9. Cobb B.S., Hertweck A., Smith J., O’Connor E., Graf D., Cook T., Smale S.T., Sakaguchi S., Livesey F.J., Fisher A.G. et al.. A role for Dicer in immune regulation. J. Exp. Med. 2006; 203:2519–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muljo S.A., Ansel K.M., Kanellopoulou C., Livingston D.M., Rao A., Rajewsky K.. Aberrant T cell differentiation in the absence of Dicer. J. Exp. Med. 2005; 202:261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rodriguez-Galan A., Fernandez-Messina L., Sanchez-Madrid F.. Control of immunoregulatory molecules by miRNAs in T cell activation. Front. Immunol. 2018; 9:2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–297. [DOI] [PubMed] [Google Scholar]
- 13. Gregory R.I., Chendrimada T.P., Cooch N., Shiekhattar R.. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005; 123:631–640. [DOI] [PubMed] [Google Scholar]
- 14. Friedman R.C., Farh K.K., Burge C.B., Bartel D.P.. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009; 19:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fernandes Q. MicroRNA: defining a new niche in Leukemia. Blood Rev. 2017; 31:129–138. [DOI] [PubMed] [Google Scholar]
- 16. Narayan N., Morenos L., Phipson B., Willis S.N., Brumatti G., Eggers S., Lalaoui N., Brown L.M., Kosasih H.J., Bartolo R.C. et al.. Functionally distinct roles for different miR-155 expression levels through contrasting effects on gene expression, in acute myeloid leukaemia. Leukemia. 2017; 31:808–820. [DOI] [PubMed] [Google Scholar]
- 17. Ji Y., Hocker J.D., Gattinoni L.. Enhancing adoptive T cell immunotherapy with microRNA therapeutics. Semin. Immunol. 2016; 28:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hess K., Yang Y., Golech S., Sharov A., Becker K.G., Weng N.P.. Kinetic assessment of general gene expression changes during human naive CD4+ T cell activation. Int. Immunol. 2004; 16:1711–1721. [DOI] [PubMed] [Google Scholar]
- 19. Jelley-Gibbs D.M., Lepak N.M., Yen M., Swain S.L.. Two distinct stages in the transition from naive CD4 T cells to effectors, early antigen-dependent and late cytokine-driven expansion and differentiation. J. Immunol. 2000; 165:5017–5026. [DOI] [PubMed] [Google Scholar]
- 20. London C.A., Lodge M.P., Abbas A.K.. Functional responses and costimulator dependence of memory CD4+ T cells. J. Immunol. 2000; 164:265–272. [DOI] [PubMed] [Google Scholar]
- 21. Ullman K.S., Northrop J.P., Verweij C.L., Crabtree G.R.. Transmission of signals from the T lymphocyte antigen receptor to the genes responsible for cell proliferation and immune function: the missing link. Annu. Rev. Immunol. 1990; 8:421–452. [DOI] [PubMed] [Google Scholar]
- 22. Bronevetsky Y., Villarino A.V., Eisley C.J., Barbeau R., Barczak A.J., Heinz G.A., Kremmer E., Heissmeyer V., McManus M.T., Erle D.J. et al.. T cell activation induces proteasomal degradation of Argonaute and rapid remodeling of the microRNA repertoire. J. Exp. Med. 2013; 210:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B (Methodological). 1995; 57:289–300. [Google Scholar]
- 25. Grötschel M., Wakabayashi Y.. A cutting plane algorithm for a clustering problem. Math. Program. 1989; 45:59–96. [Google Scholar]
- 26. Ward J.H. Hierarchical grouping to optimize an objective function. J. Am. Statist. Assoc. 1963; 58:236–244. [Google Scholar]
- 27. Gerstner N., Kehl T., Lenhof K., Muller A., Mayer C., Eckhart L., Grammes N.L., Diener C., Hart M., Hahn O. et al.. GeneTrail 3: advanced high-throughput enrichment analysis. Nucleic Acids Res. 2020; 48:W515–W520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kehl T., Schneider L., Schmidt F., Stockel D., Gerstner N., Backes C., Meese E., Keller A., Schulz M.H., Lenhof H.P.. RegulatorTrail: a web service for the identification of key transcriptional regulators. Nucleic Acids Res. 2017; 45:W146–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ochiai K., Maienschein-Cline M., Simonetti G., Chen J., Rosenthal R., Brink R., Chong A.S., Klein U., Dinner A.R., Singh H. et al.. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity. 2013; 38:918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fishilevich S., Nudel R., Rappaport N., Hadar R., Plaschkes I., Iny Stein T., Rosen N., Kohn A., Twik M., Safran M. et al.. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017; 2017:bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dweep H., Gretz N.. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat. Methods. 2015; 12:697. [DOI] [PubMed] [Google Scholar]
- 33. Beitzinger M., Peters L., Zhu J.Y., Kremmer E., Meister G.. Identification of human microRNA targets from isolated argonaute protein complexes. RNA Biol. 2007; 4:76–84. [DOI] [PubMed] [Google Scholar]
- 34. Hart M., Walch-Ruckheim B., Krammes L., Kehl T., Rheinheimer S., Tanzer T., Glombitza B., Sester M., Lenhof H.P., Keller A. et al.. miR-34a as hub of T cell regulation networks. J. Immunother. Cancer. 2019; 7:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R.. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989; 77:51–59. [DOI] [PubMed] [Google Scholar]
- 36. Barth S., Pfuhl T., Mamiani A., Ehses C., Roemer K., Kremmer E., Jaker C., Hock J., Meister G., Grasser F.A.. Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic Acids Res. 2008; 36:666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dunand-Sauthier I., Santiago-Raber M.L., Capponi L., Vejnar C.E., Schaad O., Irla M., Seguin-Estevez Q., Descombes P., Zdobnov E.M., Acha-Orbea H. et al.. Silencing of c-Fos expression by microRNA-155 is critical for dendritic cell maturation and function. Blood. 2011; 117:4490–4500. [DOI] [PubMed] [Google Scholar]
- 38. Hamberg M., Backes C., Fehlmann T., Hart M., Meder B., Meese E., Keller A.. MiRTargetLink–miRNAs, genes and interaction networks. Int. J. Mol. Sci. 2016; 17:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Backes C., Keller A., Kuentzer J., Kneissl B., Comtesse N., Elnakady Y.A., Muller R., Meese E., Lenhof H.P.. GeneTrail–advanced gene set enrichment analysis. Nucleic Acids Res. 2007; 35:W186–W192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Benjamini Y., Yekutieli D.. The control of the false discovery rate in multiple testing under dependency. Ann. Statist. 2001; 29:1165–1188. [Google Scholar]
- 41. George-Gay B., Parker K.. Understanding the complete blood count with differential. J. Perianesth. Nurs. 2003; 18:96–114. [DOI] [PubMed] [Google Scholar]
- 42. Krause J.R., Boggs D.R.. Search for eosinopenia in hospitalized patients with normal blood leukocyte concentration. Am. J. Hematol. 1987; 24:55–63. [DOI] [PubMed] [Google Scholar]
- 43. Onyema O.O., Njemini R., Bautmans I., Renmans W., De Waele M., Mets T.. Cellular aging and senescence characteristics of human T-lymphocytes. Biogerontology. 2012; 13:169–181. [DOI] [PubMed] [Google Scholar]
- 44. Effros R.B., Dagarag M., Valenzuela H.F.. In vitro senescence of immune cells. Exp. Gerontol. 2003; 38:1243–1249. [DOI] [PubMed] [Google Scholar]
- 45. Kozomara A., Birgaoanu M., Griffiths-Jones S.. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019; 47:D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rodriguez A., Vigorito E., Clare S., Warren M.V., Couttet P., Soond D.R., van Dongen S., Grocock R.J., Das P.P., Miska E.A. et al.. Requirement of bic/microRNA-155 for normal immune function. Science. 2007; 316:608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Szekely G.J., Rizzo M.L., Bakirov N.K.. Measuring and testing dependence by correlation of distances. Ann. Stat. 2007; 35:2769–2794. [Google Scholar]
- 48. Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Escobar T.M., Kanellopoulou C., Kugler D.G., Kilaru G., Nguyen C.K., Nagarajan V., Bhairavabhotla R.K., Northrup D., Zahr R., Burr P. et al.. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve polycomb-mediated repression. Immunity. 2014; 40:865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krishnamoorthy V., Kannanganat S., Maienschein-Cline M., Cook S.L., Chen J., Bahroos N., Sievert E., Corse E., Chong A., Sciammas R.. The IRF4 gene regulatory module functions as a read-write integrator to dynamically coordinate T helper cell fate. Immunity. 2017; 47:481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang R., Deng X., Yuan C., Xin H., Liu G., Zhu Y., Jiang X., Wang C.. IFT80 improves invasion ability in gastric cancer cell line via ift80/p75NGFR/MMP9 signaling. Int. J. Mol. Sci. 2018; 19:3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J. et al.. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007; 39:727–729. [DOI] [PubMed] [Google Scholar]
- 53. Stephen L.A., ElMaghloob Y., McIlwraith M.J., Yelland T., Castro Sanchez P., Roda-Navarro P., Ismail S.. The ciliary machinery is repurposed for T cell immune synapse trafficking of LCK. Dev. Cell. 2018; 47:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kiang K.M., Leung G.K.. A review on adducin from functional to pathological mechanisms: future direction in cancer. Biomed. Res. Int. 2018; 2018:3465929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dhers L., Ducassou L., Boucher J.L., Mansuy D.. Cytochrome P450 2U1, a very peculiar member of the human P450s family. Cell. Mol. Life Sci. 2017; 74:1859–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chuang S.S., Helvig C., Taimi M., Ramshaw H.A., Collop A.H., Amad M., White J.A., Petkovich M., Jones G., Korczak B.. CYP2U1, a novel human thymus- and brain-specific cytochrome P450, catalyzes omega- and (omega-1)-hydroxylation of fatty acids. J. Biol. Chem. 2004; 279:6305–6314. [DOI] [PubMed] [Google Scholar]
- 57. Xing Z., Ma W.K., Tran E.J.. The DDX5/Dbp2 subfamily of DEAD-box RNA helicases. Wiley Interdiscip. Rev. RNA. 2019; 10:e1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Margueron R., Li G., Sarma K., Blais A., Zavadil J., Woodcock C.L., Dynlacht B.D., Reinberg D.. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell. 2008; 32:503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hochrainer K., Pejanovic N., Olaseun V.A., Zhang S., Iadecola C., Anrather J.. The ubiquitin ligase HERC3 attenuates NF-kappaB-dependent transcription independently of its enzymatic activity by delivering the RelA subunit for degradation. Nucleic Acids Res. 2015; 43:9889–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Adighibe O., Pezzella F.. The role of JMY in p53 regulation. Cancers (Basel). 2018; 10:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zheng Y.C., Chang J., Wang L.C., Ren H.M., Pang J.R., Liu H.M.. Lysine demethylase 5B (KDM5B): A potential anti-cancer drug target. Eur. J. Med. Chem. 2019; 161:131–140. [DOI] [PubMed] [Google Scholar]
- 62. Fuller D.M., Zhu M., Ou-Yang C.W., Sullivan S.A., Zhang W.. A tale of two TRAPs: LAT and LAB in the regulation of lymphocyte development, activation, and autoimmunity. Immunol. Res. 2011; 49:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sirinian M.I., Belleudi F., Campagna F., Ceridono M., Garofalo T., Quagliarini F., Verna R., Calandra S., Bertolini S., Sorice M. et al.. Adaptor protein ARH is recruited to the plasma membrane by low density lipoprotein (LDL) binding and modulates endocytosis of the LDL/LDL receptor complex in hepatocytes. J. Biol. Chem. 2005; 280:38416–38423. [DOI] [PubMed] [Google Scholar]
- 64. Donato R., Sorci G., Riuzzi F., Arcuri C., Bianchi R., Brozzi F., Tubaro C., Giambanco I.. S100B’s double life: intracellular regulator and extracellular signal. Biochim. Biophys. Acta. 2009; 1793:1008–1022. [DOI] [PubMed] [Google Scholar]
- 65. Tosoni D., Puri C., Confalonieri S., Salcini A.E., De Camilli P., Tacchetti C., Di Fiore P.P.. TTP specifically regulates the internalization of the transferrin receptor. Cell. 2005; 123:875–888. [DOI] [PubMed] [Google Scholar]
- 66. Schmidt V., Subkhangulova A., Willnow T.E.. Sorting receptor SORLA: cellular mechanisms and implications for disease. Cell. Mol. Life Sci. 2017; 74:1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moon S.Y., Zheng Y.. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003; 13:13–22. [DOI] [PubMed] [Google Scholar]
- 68. Braun A.C., Olayioye M.A.. Rho regulation: DLC proteins in space and time. Cell. Signal. 2015; 27:1643–1651. [DOI] [PubMed] [Google Scholar]
- 69. Barlev N.A., Emelyanov A.V., Castagnino P., Zegerman P., Bannister A.J., Sepulveda M.A., Robert F., Tora L., Kouzarides T., Birshtein B.K. et al.. A novel human Ada2 homologue functions with Gcn5 or Brg1 to coactivate transcription. Mol. Cell. Biol. 2003; 23:6944–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gegonne A., Devaiah B.N., Singer D.S.. TAF7: traffic controller in transcription initiation. Transcription. 2013; 4:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Guo J., Qu H., Chen Y., Xia J.. The role of RNA-binding protein tristetraprolin in cancer and immunity. Med. Oncol. 2017; 34:196. [DOI] [PubMed] [Google Scholar]
- 72. Lai X., Eberhardt M., Schmitz U., Vera J.. Systems biology-based investigation of cooperating microRNAs as monotherapy or adjuvant therapy in cancer. Nucleic. Acids. Res. 2019; 47:7753–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kroesen B.J., Teteloshvili N., Smigielska-Czepiel K., Brouwer E., Boots A.M., van den Berg A., Kluiver J.. Immuno-miRs: critical regulators of T-cell development, function and ageing. Immunology. 2015; 144:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Giri B.R., Mahato R.I., Cheng G.. Roles of microRNAs in T cell immunity: implications for strategy development against infectious diseases. Med. Res. Rev. 2019; 39:706–732. [DOI] [PubMed] [Google Scholar]
- 75. Mehta A., Baltimore D.. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016; 16:279–294. [DOI] [PubMed] [Google Scholar]
- 76. Winter J., Diederichs S.. Argonaute proteins regulate microRNA stability: increased microRNA abundance by Argonaute proteins is due to microRNA stabilization. RNA Biol. 2011; 8:1149–1157. [DOI] [PubMed] [Google Scholar]
- 77. Guo Y., Liu J., Elfenbein S.J., Ma Y., Zhong M., Qiu C., Ding Y., Lu J.. Characterization of the mammalian miRNA turnover landscape. Nucleic. Acids. Res. 2015; 43:2326–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Brodin P., Davis M.M.. Human immune system variation. Nat. Rev. Immunol. 2017; 17:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kverneland A.H., Streitz M., Geissler E., Hutchinson J., Vogt K., Boes D., Niemann N., Pedersen A.E., Schlickeiser S., Sawitzki B.. Age and gender leucocytes variances and references values generated using the standardized ONE-study protocol. Cytometry A. 2016; 89:543–564. [DOI] [PubMed] [Google Scholar]
- 80. Marzi M.J., Ghini F., Cerruti B., de Pretis S., Bonetti P., Giacomelli C., Gorski M.M., Kress T., Pelizzola M., Muller H. et al.. Degradation dynamics of microRNAs revealed by a novel pulse-chase approach. Genome Res. 2016; 26:554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bosson A.D., Zamudio J.R., Sharp P.A.. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol. Cell. 2014; 56:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bail S., Swerdel M., Liu H., Jiao X., Goff L.A., Hart R.P., Kiledjian M.. Differential regulation of microRNA stability. RNA. 2010; 16:1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Baccarini A., Chauhan H., Gardner T.J., Jayaprakash A.D., Sachidanandam R., Brown B.D.. Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr. Biol. 2011; 21:369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vigorito E., Perks K.L., Abreu-Goodger C., Bunting S., Xiang Z., Kohlhaas S., Das P.P., Miska E.A., Rodriguez A., Bradley A. et al.. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007; 27:847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stopka T., Vargova K., Kokavec J., Pospisil V., Curik N., Burda P., Skoultchi A.I., Zavadil J.. Mutual regulatory loop between miR-155 and PU.1 is a candidate pathogenesis factor in CLL. Blood. 2007; 110:1130–1130. [Google Scholar]
- 86. Rupaimoole R., Slack F.J.. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017; 16:203–222. [DOI] [PubMed] [Google Scholar]
- 87. Calabrese J.M., Seila A.C., Yeo G.W., Sharp P.A.. RNA sequence analysis defines Dicer's role in mouse embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:18097–18102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Janas M.M., Wang B., Harris A.S., Aguiar M., Shaffer J.M., Subrahmanyam Y.V., Behlke M.A., Wucherpfennig K.W., Gygi S.P., Gagnon E. et al.. Alternative RISC assembly: binding and repression of microRNA-mRNA duplexes by human Ago proteins. RNA. 2012; 18:2041–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Reichholf B., Herzog V.A., Fasching N., Manzenreither R.A., Sowemimo I., Ameres S.L.. Time-resolved small RNA sequencing unravels the molecular principles of MicroRNA homeostasis. Mol. Cell. 2019; 75:756–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Song Y., Kilburn D., Song J.H., Cheng Y., Saeui C.T., Cheung D.G., Croce C.M., Yarema K.J., Meltzer S.J., Liu K.J. et al.. Determination of absolute expression profiles using multiplexed miRNA analysis. PLoS One. 2017; 12:e0180988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kuchen S., Resch W., Yamane A., Kuo N., Li Z., Chakraborty T., Wei L., Laurence A., Yasuda T., Peng S. et al.. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity. 2010; 32:828–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Brown B.D., Gentner B., Cantore A., Colleoni S., Amendola M., Zingale A., Baccarini A., Lazzari G., Galli C., Naldini L.. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 2007; 25:1457–1467. [DOI] [PubMed] [Google Scholar]
- 93. Hutvagner G., Zamore P.D.. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002; 297:2056–2060. [DOI] [PubMed] [Google Scholar]
- 94. Schmitz U., Lai X., Winter F., Wolkenhauer O., Vera J., Gupta S.K.. Cooperative gene regulation by microRNA pairs and their identification using a computational workflow. Nucleic Acids Res. 2014; 42:7539–7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Koscianska E., Witkos T.M., Kozlowska E., Wojciechowska M., Krzyzosiak W.J.. Cooperation meets competition in microRNA-mediated DMPK transcript regulation. Nucleic Acids Res. 2015; 43:9500–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhuo W., Ge W., Meng G., Jia S., Zhou X., Liu J.. MicroRNA20a promotes the proliferation and cell cycle of human osteosarcoma cells by suppressing early growth response 2 expression. Mol. Med. Rep. 2015; 12:4989–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Luo H., Zou J., Dong Z., Zeng Q., Wu D., Liu L.. Up-regulated miR-17 promotes cell proliferation, tumour growth and cell cycle progression by targeting the RND3 tumour suppressor gene in colorectal carcinoma. Biochem. J. 2012; 442:311–321. [DOI] [PubMed] [Google Scholar]
- 98. Sandberg R., Neilson J.R., Sarma A., Sharp P.A., Burge C.B.. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008; 320:1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lu T.X., Hartner J., Lim E.J., Fabry V., Mingler M.K., Cole E.T., Orkin S.H., Aronow B.J., Rothenberg M.E.. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-gamma pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J. Immunol. 2011; 187:3362–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Banerjee A., Schambach F., DeJong C.S., Hammond S.M., Reiner S.L.. Micro-RNA-155 inhibits IFN-gamma signaling in CD4+ T cells. Eur. J. Immunol. 2010; 40:225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Carissimi C., Carucci N., Colombo T., Piconese S., Azzalin G., Cipolletta E., Citarella F., Barnaba V., Macino G., Fulci V.. miR-21 is a negative modulator of T-cell activation. Biochimie. 2014; 107:319–326. [DOI] [PubMed] [Google Scholar]
- 102. Lind E.F., Ohashi P.S.. Mir-155, a central modulator of T-cell responses. Eur. J. Immunol. 2014; 44:11–15. [DOI] [PubMed] [Google Scholar]
- 103. Schultz J., Lorenz P., Gross G., Ibrahim S., Kunz M.. MicroRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res. 2008; 18:549–557. [DOI] [PubMed] [Google Scholar]
- 104. Rippo M.R., Olivieri F., Monsurro V., Prattichizzo F., Albertini M.C., Procopio A.D.. MitomiRs in human inflamm-aging: a hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol. 2014; 56:154–163. [DOI] [PubMed] [Google Scholar]
- 105. Qian H., Yang C., Yang Y.. MicroRNA-26a inhibits the growth and invasiveness of malignant melanoma and directly targets on MITF gene. Cell Death Discov. 2017; 3:17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chen B., Liu Y., Jin X., Lu W., Liu J., Xia Z., Yuan Q., Zhao X., Xu N., Liang S.. MicroRNA-26a regulates glucose metabolism by direct targeting PDHX in colorectal cancer cells. BMC Cancer. 2014; 14:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data of the time-resolved mRNA profiles (#GSE136625) and the time-resolved miRNA profiles (#GSE136626) are available at the GEO database. The source code for the GeneTrail C++ library as well as the clustering approach are publicly available on https://github.com/unisb-bioinf.