


Abstract
Adaptive evolution has shaped major biological processes. Finding the protein-coding genes and the sites that have been subjected to adaptation during evolutionary time is a major endeavor. However, very few methods fully automate the identification of positively selected genes, and widespread sources of genetic innovations such as gene duplication and recombination are absent from most pipelines. Here, we developed DGINN, a highly-flexible and public pipeline to Detect Genetic INNovations and adaptive evolution in protein-coding genes. DGINN automates, from a gene's sequence, all steps of the evolutionary analyses necessary to detect the aforementioned innovations, including the search for homologs in databases, assignation of orthology groups, identification of duplication and recombination events, as well as detection of positive selection using five methods to increase precision and ranking of genes when a large panel is analyzed. DGINN was validated on nineteen genes with previously-characterized evolutionary histories in primates, including some engaged in host-pathogen arms-races. Our results confirm and also expand results from the literature, including novel findings on the Guanylate-binding protein family, GBPs. This establishes DGINN as an efficient tool to automatically detect genetic innovations and adaptive evolution in diverse datasets, from the user's gene of interest to a large gene list in any species range.
INTRODUCTION
Genetic innovation is a major adaptation process that has impacted genome structures and functions over millions of years in response to natural selection. Such changes have shaped key biological functions, such as reproduction, adaptation to a new environment, immunity, sensory-perception, host–pathogen interaction. Adaptation in protein-coding genes can take place through several mechanisms. They include, amongst others, positive selection on coding sequences, duplication events with subsequent divergence of the copies, as well as recombination (1). The first is caused by natural selection that increases the frequency of advantageous mutations, leading to an apparent excess of non-synonymous substitution rates over synonymous ones over evolutionary times. This notably leads to the accumulation of beneficial amino-acid changes at the location of functionally important residues, such as the interface of proteins involved in host-virus interactions. Gene duplication is another important source of genetic novelty, which notably allows to increase the general evolvability (2,3). The fixation of multiple copies enables diversification of gene function through subfunctionalization or neofunctionalization. Moreover, gene conversion, by recombination between alleles, allows for rapid divergence of the copies. Gene duplication and loss may further be a dynamic and rapid adaptation process (2–4).
These mechanisms fueling genetic novelty are all parts of the response of organisms to selective pressures and must therefore be analyzed as much as possible together to wholly apprehend the evolutionary history of genes. However, despite their frequency and their biological importance and relevance, these diverse evolutionary innovations are not accounted for in most tools and studies analyzing genes under adaptive evolution (5–7). Lastly, performing gold-standard and complete phylogenetic analyses is usually highly hand-curated. Our goal was therefore to design a tool that would incorporate all these mechanisms at the origin of genetic innovation in a robust end-to-end pipeline to identify and characterize new protein-coding genes with signatures of adaptive evolution.
Such a pipeline requires the automation of essential steps. Firstly, searching for homologous gene sequences and identifying orthologous relationships represent a time-consuming and difficult process. No existing tool include these steps, because they either remain essentially provided by the user (Hyphy suite (8), Selecton (9), IDEA (10), JcoDa (11), PoSeiDon (12) and POTION (13)), are restricted to specific vertebrate and prokaryotic species (PhyleasProg (14) and PSP (15)), or rely on published orthologous annotations (essentially from the NCBI HomoloGene) which may become imprecise on non-model species.
Secondly, correct codon alignments are necessary for the accurate detection of residues under positive selection. However, current pipelines rely on protein or nucleotide alignment softwares like ClustalW (16) or Muscle (17), although more recent ones, such as PRANK (18), have been repeatedly shown to provide high-quality codon alignments, thereby diminishing false positives during the detection of positive selection (19–22).
Thirdly, we identified the need to include within a single analysis the detection of positive selection signatures by different methods and models, to allow for more specificity and sensitivity of the results, as well as to help ‘ranking’ genes in a screening approach (23–28). Moreover, the inclusion of methods in which the experienced user has access to the parameterization of the maximum likelihood models is needed (29). Existing tools rely almost exclusively on PAML codeml (30), which has allowed the identification of numerous genes under positive selection, but offers limited options for parameterization.
Overall, there seemed to exist a void when it comes to pipelines which fully automate the search for adaptive evolution in protein-coding genes, from retrieving homologous sequences of a gene of interest in any species range, establishing orthologous relationships, reconstructing codon alignments and the corresponding phylogenies, to detecting different genetic innovations using gold-standard and diverse methods to ensure high-degree of confidence in the results. We thus developed an integrative pipeline, that we named DGINN (for Detection of Genetic INNovations) to satisfy those requirements. All scripts are freely available on GitHub and as a docker on DockerHub. We also focused on user-friendliness and flexibility, so that biologists can use with ease and one can use only parts of the workflow for various purposes. DGINN was developed as a one-gene workflow and can easily be up-scaled to screen large datasets of hundreds of genes. Finally, we performed an extensive validation of our pipeline, using published and highly hand-curated phylogenetic data on a set of nineteen primate genes with various evolutionary histories including genes involved in virus-host evolutionary arms-races (1,31). Through DGINN, we further identified previously uncharacterized signatures of genetic conflict in the primate Guanylate-binding protein (GBP) family, which plays important roles in cell-autonomous immunity against pathogens (32,33).
MATERIALS AND METHODS
Pipeline structure
The overall goal of the DGINN pipeline (overviewed in Figure 1) is to provide an easy, integrated and robust way of detecting genetic innovations from a gene sequence provided by the user on two scales, either on one specific gene for fine-tuned analyses or on large sets of genes of interest for screening purposes.
Figure 1.

Workflow diagram of DGINN. Phylogenetic steps (yellow) happen sequentially from the entry point of the pipeline (Steps 1–4). Each genetic innovation step (purple, Steps 5, 6 and 7) is optional. All red arrowheads denote possible entry points into the pipeline following file formats from Table 1.
DGINN is implemented in Python and uses numerous modules, including some from Biopython, as well as several independent softwares. The list of modules and external softwares is provided in the pipeline documentation. All scripts and documentation can be downloaded from GitHub. To enhance user-friendliness, options are handled through a parameter file, minimizing the complexity of the command line. Importantly, a Docker image is also available for local use without manual installation of the external required softwares. The Docker may also be used to screen large dataset using AWS Batch for example (https://aws.amazon.com/batch/). A specific script for the extraction of batch results, parseResults.py and a graphical interface to produce basic figures with them, have also been developed (see Availability).
The overall workflow of the DGINN pipeline is a succession of eight steps, described hereafter. DGINN is designed to be extremely flexible as to its uses. The user can enter the workflow at any step with the files resulting from their own analyses, as indicated in Table 1 and Figure 1. The name of the step reflects the very first step performed with the option. For example, starting DGINN at the ‘blast’ step will make it begin with the BLAST search, and then execute the whole pipeline. To allow maximum flexibility, the duplication, recombination and positive selection steps will not be performed if the user has not opted in for them.
Table 1.
Overview of the possible entry steps into DGINN. DGINN can be entered at different steps to enhance flexibility. If the user introduces the name of the proper entry step option and inputs the appropriate files for this option in the parameter file, DGINN will start at that step, ignoring the upstream steps. If users wish to perform the detection of duplication and orthologous groups, they have to provide a species tree through the parameter file (see Materials and Methods and GitHub readme for details)
| Step | Name of entry step option | Input files | Format |
|---|---|---|---|
| 0 | BLAST | CDS of the gene of interest | Fasta |
| 1 | accession | List of BLAST results | NCBI tabulated format |
| 2 | fasta | List of accession identifiers (one per line) | Text file |
| 2 | orf | mRNA sequences of homologs | Fasta |
| 3 | alignment | CDS sequences of homologs/orthologs | Fasta |
| 4 | tree | (codon) alignment of homologs/orthologs | Fasta |
| 5 | duplication | (codon) alignment, gene tree | Fasta, newick |
| 6 | recombination | (codon) alignment | Fasta |
| 8 | positiveSelection | codon alignment, gene tree | Fasta, newick |
(Step 1) Automated retrieval of homologous genes in species of interest
DGINN uses BLAST+ search (34) against the NCBI databases. The BLAST search can be done against a local database constructed by the user, or online against specific NCBI databases. This allows the user to limit the search to certain sequences, such as ESTs or certain species, by providing the proper Entry Query following the syntax used on the NCBI website, as described in their documentation (https://www.ncbi.nlm.nih.gov/books/NBK3837/#EntrezHelp.Entrez_Searching_Options). BLAST+ is used by providing the coding sequence of the gene of interest against a nucleotide databank (blastn). We decided not to use blastp (protein query against protein database) as it significantly complicated the recuperation of the nucleotide sequences afterwards, which are indispensable to the rest of the pipeline. Moreover, nucleotide databases include more sequences and thus allow for a more exhaustive search. The number and speed of requests against NCBI databases can be increased through the acquisition of an NCBI API key, available online. This ensures access to the largest possible number of sequences, including those not annotated as orthologous or paralogous sequences. The user may modify minimum e-value, coverage, and identity values to reflect the specificities of the database and the species set against which they are using BLAST+. Because we validated our pipeline on primate evolution, we set those with default values of 10−4, 50% and 70%, respectively, to retrieve a maximum of homologous sequences without too many unrelated sequences.
(Step 2) Elimination of overly long sequences and isolation of Open Reading Frames (ORFs)
Because the user may want to cast a wide net in terms of homolog retrieval, and thus use low coverage and identity for the blastn search (Step 1), a variety of resulting hits are retrieved, including overly long sequences from whole contigs or chromosomes originating from whole genomes where annotations are still an ongoing process. Those sequences considerably increase the analysis time if not properly curated, and the process of automatically detecting in any species the corresponding ORF in a contig is a highly complex task that we did not include in this pipeline. In DGINN, we identify and remove such sequences based on the median length of all the retrieved sequences. If the median is longer than 10 000 nucleotides, any sequences longer than twice the median are taken out. Otherwise, any sequences longer than three times the median are deleted. Alternatively, the user can chose to eliminate sequences based on another factor of the median length, or to eliminate outliers based on the InterQuartile Range (IQR) approach. The remaining sequences are searched for ORFs using ORFinder from the EMBOSS package (35) to keep only the coding sequence of each gene. The longest detected ORF of each sequence is selected for further analysis.
(Step 3) Initial codon alignment
Positive selection analyses rely on identifying substitutions leading to amino-acid changes over those being silent. Therefore, a codon alignment of good quality is essential. However, very few softwares propose true codon-alignment modes. To date, the best codon aligners are PRANK (18) and MACSE (36). PRANK has been shown to produce the best alignments for positive selection analyses (19–22,37). From our observations, MACSE also produced high-quality codon alignments, but it was significantly slower than PRANK. We therefore selected the latter as the best solution for both quality alignments and lower computational time. To gain rapidity, we first perform an initial nucleotide alignment by MAFFT (38) with automatic settings (mafft -auto, v7.3) after which we added a quality control step to eliminate sequences that did not align properly, using Python homemade scripts, based on alignment coverage against the query (either the user-provided value or default of 50%). PRANK alignments are performed with the codon model and without forcing insertions to be skipped, and otherwise default settings (prank -F -codon; version 150803).
(Step 4) Construction of the initial phylogenetic gene tree
The gene's phylogenetic reconstruction is performed with PhyML v3.2 (39). We opted for a HKY+G+I model as default, because it offers the best combination of realistic phylogenies without being too time-consuming. As the produced trees are only intended for screening purposes at this step, we also opted to use approximate Likelihood Ratio Test (aLRT) for the statistical support of the branches (40). Users can provide their own options for PhyML through the parameter file should they wish to use other models and statistics.
(Step 5) Identification of duplication events and orthologous groups
As previous steps retrieved homologs without relying on synteny or gene annotation, we implemented two strategies to identify duplicated genes and to constitute orthologous groups necessary for the positive selection analyses. DGINN first identifies the overly ‘long branches’ within the gene tree. By default, we define a ‘long branch’ as a branch which length is at least 50 times longer than the mean of all branch lengths in the tree (i.e. the estimated number of substitutions per position is at least 50 times superior in the ‘long branch’ compared to the mean). Alternatively, the user can cut branches based on another factor of the overall mean of the substitution rate, or to eliminate outliers based on the InterQuartile Range (IQR) approach. These options are accessible in the parameter file. When ‘long branches’ are identified, the tree is cut along those ‘long branches’ and the groups of sequences subsequently constituted are re-aligned (back to step 3) and their trees recomputed separately (step 4). This constitutes a first method of separating highly divergent groups of genes, between which detection of positive selection may be ambiguous because of suspicion of paralogy and branch length saturation. However, for multigenic families that include paralogs that have recently diverged, the gene members cannot be separated solely based on the relative lengths of the tree branches. We therefore included a phylogenetic reconciliation method, Treerecs (41), to identify genes sharing a common evolutionary history in our species of interest. To identify duplication events, Treerecs reconciles each gene tree to the user-provided species tree or cladogram. From each reconciled tree, DGINN establishes groups of orthologs based on the inferred ancestral duplication events. Duplication events on nodes that do not have at least two species in common in the groups formed on either side of the node are considered dubious: the corresponding annotated events are then ignored by DGINN. Since interspecific positive selection analyses rely on the comparison of several orthologous sequences, orthologous groups resulting from very recent duplications may have too few sequences to be informative for those analyses. So, DGINN ignores duplication events that are not ancestral enough, by taking into account the minimal number of species represented downstream of the event. This number is user-determined. We decided on a default setting of a minimum of eight species to extract a duplication group from the original alignment, based on the results obtained by Anisimova et al. (42), and in primates specifically by McBee et al. (27). After extraction based on ancestral duplication events, the orthologous groups are realigned using PRANK as in Step 3.
To run this step, the user has to provide, through the parameter file, a valid species tree (cladogram) of the species of interest. The format is a newick file with the species names following DGINN’s nomenclature (speSpe). If this file is absent, DGINN does not separate the sequences into orthologous groups.
(Step 6) Identification of recombination events and splitting of alignments along the significant breakpoints
To account for recombination, DGINN includes GARD from HYPHY (43) with standard parameters. The breakpoints are then assessed for statistical significance using a likelihood ratio test (LRT) with P < 0.05 against a null hypothesis of no breakpoint at that position. If any breakpoint is found significant, the sequence alignment is cut longitudinally at the breakpoint(s) to produce non-recombinant sequence alignments (preserving the codon units). These non-recombinant alignments, as well as the original one, will become the input in the following steps (and named fragPos1-Pos2).
(Step 7) Construction of the final phylogenetic trees
Following the analyses of duplication and recombination events (steps 5–6), new codon-wise alignments using PRANK (same parameters as in step 3) and new phylogenies using PhyML (same parameters as in step 4) are built for groups of non-recombinant fragments (see step 6) of orthologous genes (see step 5). These final codon alignments and gene trees will further provide the input for the positive selection analyses.
(Step 8) Positive selection analyses
Numerous softwares exist to identify positive selection on coding sequences. DGINN includes several methods of positive selection analyses, which the user can chose to turn on or off independently. Those analyses make extensive use of three packages: HYPHY (8), PAML codeml (30) through the ETE toolkit (http://etetoolkit.org/) and Bio++ (44).
From the HYPHY package, we included two methods. First, we included BUSTED (Branch-Site Unrestricted Statistical Test for Episodic Diversification), a random effect model which allows for gene-wide detection of episodic positive selection (45). Results are considered positive in the DGINN pipeline for a P-value < 0.05 for the LRT of the models admitting versus not admitting positive selection. Second, we included MEME (Mixed Effects Model of Evolution), which detects individual sites subjected to episodic positive selection based on a mixed effects model (46). These models are complementary, as BUSTED evaluates positive selection at the gene level and MEME at the site level.
Contrary to BUSTED and MEME, the codon substitution models used in PAML codeml focus on pervasive positive selection and not episodic events. Briefly, the codon alignments are fitted to models that do not allow for positive selection, M1 (with two classes ω < 1 and ω = 1) or M7 (where the ω < 1 class is modeled as a gamma law of n classes, n = 5 as default in DGINN), and the corresponding models allowing for positive selection with one class of ω > 1 (M2 or M8, respectively). Statistical significance of positive selection is determined through a chi-squared test of the LRT of both associated models (M1 versus M2, and M7 versus M8) to derive P-values. Results are considered positive in the DGINN pipeline for a P-value < 0.05.
However, PAML codeml relies on the assumption of stationarity (i.e. that the base composition of sequences is at the equilibrium of the evolutionary process), which may impact the detection of selection (47). It is also limited with regards to its parameterization. Therefore, we also integrated the parameterizable Bio++ library to propose similar models but without stationarity assumption (Bio++ models M1NS versus M2NS, and M7NS versus M8NS). Similarly, DGINN considers significant positive selection if P-value < 0.05 of each model comparison.
If positive selection is determined with PAML or Bio++, the pipeline will proceed to the identification of the sites under positive selection, using the Bayes Empirical Bayes statistics (BEB) from the M2 and M8 in PAML codeml and the Bayesian Posterior Probabilities (PP) from the M2NS and M8NS models in Bio++. Sites are considered as under significant positive selection if BEB or PP > 0.95.
To detect specific branches/lineages under positive selection, DGINN uses Bio++ to include a method similar to the Free-Ratio test available in PAML codeml, called One Per Branch in DGINN (OPB). The ω ratio is calculated along the branches of the phylogenetic tree by using a M0 model where all parameters but ω are homogeneous. As this step is independent and the Bio++ parameter file is fully accessible, an experienced user can choose any model they wish, allowing for maximum flexibility.
Each of those methods can be opted in or out through the parameter file, so that users can run any subset they want.
Pipeline parallelization
DGINN has been developed to analyze each gene independently, with parallelization over large datasets being handled in a cluster environment. This is done through user-made scripts (such as job arrays) and facilitated through configuration parameters that are specific to this use. -i/–infile allows for easier parallelization by eliminating the need to create parameter files for each analyzed gene. -host/–hostfile allows the user to indicate the cluster hostfile to avoid conflicts when starting mpi processes.
Also, if the query genes are from the human genome, a separate script is provided for downloading their CCDS sequences prior to using DGINN itself. This script, called CCDSquery.py and available on the GitHub, only requires a table as its entry, with HUGO Gene Nomenclature Committee (HGNC) approved symbols in one column and the corresponding CCDS accessions in another. This table can be obtained through the HGNC biomart (http://biomart.genenames.org/).
Results extraction
An independent script, parseResults.py, is provided to extract the essential results after running the pipeline. This script outputs a table (described in DGINN’s documentation) which compiles, for each analyzed gene, the results regarding duplication and recombination events, and the different methods of positive selection detection used (including significance of each method and sites identified). This script only requires the path to the directory containing DGINN’s results as input.
An R Shiny App (see Availability) has been further designed to help the user visualize the results quickly. It only necessitates the file produced by parseResults.py. This app outputs the figures in the same format as those shown in Figures 3 and 4.
Figure 3.

DGINN results on the validation dataset. The nineteen primate genes studied are color-coded according to their selection profile category (Table 2). Left panel, number of methods detecting significant positive selection for each alignment; each method is color-coded (embedded legend). Right panel, percentage of positively selected sites (by at least one method) over the length of the query coding sequence. Genes are ordered by descending number of methods detecting positive selection then descending percentage of positively selected sites.
Figure 4.

Positive selection patterns on nineteen primate genes. The genes are color-coded according to their selection profile category (Table 2) and follow the same order as in Figure 3. Genes without positively selected sites were excluded from this representation. Positively selected sites are represented as a spike at their position on the alignment. Height of the peak is proportional to the number of methods that have identified the site as being under positive selection (posterior probabilities > 0.95 for Bio++ and PAML codeml M2 and M8 models, and P-value < 0.10 for MEME), with each method being represented by a different color (embedded legend). HYPHY MEME sites were only mapped if the gene was detected as under positive selection by BUSTED (P < 0.05). For each gene, alignment coverage is represented under the line, which itself represents the length of the alignment in light gray.
Validation dataset and method
To test our pipeline, we used a dataset of nineteen primate genes, for which evolutionary histories and positive selection profiles are either known and described in the literature or have been established within our laboratory in the past years (Table 2). We grouped those genes in three categories based on the clusters described by Murrell et al. (48): ‘canonical arms-race genes’ such as APOBEC3G and SAMHD1 (Table 2, red column), ‘genes described as presenting various selection profiles’ (Table 2, green column), such as HERC5 or SERINC3, either regarding the methods employed to detect positive selection or the strength of the detected signal, and ‘genes under no positive selection pressure’ such as GADD45A and RHO/rhodopsin (Table 2, blue column).
Table 2.
Validation dataset of nineteen primate genes with various evolutionary histories. Genes are categorized according to their selection profiles as reported in the literature. An asterisk (*) denotes a gene presenting at least 50% reciprocal identity with a paralog in primates. The corresponding literature reference for each gene of the validation dataset is indicated in the second column of each category (23,48,54,70–73,75,77,78). (Of note: Although there have been some contradictory reports on FOXP2 recent evolution in humans, this gene has been described under negative selection at the primate evolution scale (79))

|
The goal was to validate our automatic DGINN pipeline using data and findings from highly hand-curated phylogenetic and evolutionary analyses, and if possible to enrich them. To assess the pertinence of our detection of duplication events, we included nine genes belonging to multigene families (annotated with an asterisk in Table 2). A gene was considered as part of a multigene family if it had at least one paralog with over 50% reciprocal identity amongst primates (according to Ensembl). A member of the APOBEC3 gene family was also included as an extreme example of genes involved in virus-host evolutionary arms-races and that have undergone numerous genetic innovations (49–52). Another example of multigene family member included is HERC5, which exhibits antiviral activity (53) and described in the literature as evolving under positive selection (54). In this latter case, the analyses were performed on a limited number of primate species (seven species), which may bias the signatures of positive selection. Therefore, HERC5 was included in the ‘various’ category rather than in the ‘canonical’ one.
The primate species tree used to assess for duplication events is based on the one established by Perelman et al. (55) and updated by Pecon-Slattery (56), with minor modifications: species’ names according to the six-letter naming system nomenclature that is used in DGINN (and is similar to UCSC genome's nomenclature: the first three characters of the organism's genus and species classification in the format gggSss; e.g. Homo sapiens becomes homSap), species names were updated (e.g. Tarsius syrichta was replaced with carSyr for Carlito syrichta), Rhinopithecus bieti (rhiBie) and Rhinopithecus roxellana (rhiRox) were added as the closest relatives of Rhinopithecus brelichi (rhiBre). This modified tree is available on DGINN’s GitHub (see Availability). In the validation presented in this study, we used PRANK for both the initial (step 3) and the second alignments.
Reconstruction of the evolutionary history of primate Guanylate-binding protein (GBP) family
Homologs for human GBP4 and GBP6 were retrieved online through Blastn (https://blast.ncbi.nlm.nih.gov/) against the nr database limited to primates (taxid:9443). Sequences were manually selected to span as many primate species as available. Their accession numbers were added to the list of accession numbers previously obtained from the DGINN run from the human GBP5 query, then DGINN was run from the accession step to the duplication step (steps 2–5) to determine the new orthologous relationships and reconstruct the different gene trees.
Resources
DGINN was run on the nineteen genes in a cluster environment (PSMN, http://www.ens-lyon.fr/PSMN/) in two stages. The first one ran from BLAST step against the NCBI non-redundant nucleotide nr/nt database circumscribed to primate species, with default settings (2 CPUs for each gene) until the identification of recombination events (steps 1–7, Figure 1). The second stage focused solely on positive selection analyses (step 8, 1 CPU for each alignment). Running times are summarized in Table 3 and Supplementary Table S2.
Table 3.
Running times on the DGINN validation dataset. For each gene, the running time of ‘Steps 1–7’ and of ‘Step 8’ (Figure 1) is shown. For Step 8 (positive selection analyses), the running time of each method is further shown in Supplementary Table S2. Times for Step 8 are only shown for the query genes of the validation dataset following attribution of orthologous groups (Table 4). The last column of the Table corresponds to the option that best balances running times, sensitivity and specificity; i.e. phylogenetics (Steps 1–7) and positive selection analyses (Step 8) using Bio++ M1NS versus M2NS and M7NS versus M8NS only.
| Steps 1 – 7 | Step 8 | Balance speed and results (steps 1–7, step 8: Bio++ only) | |
|---|---|---|---|
| APOBEC3F | 12:18:39 | 04:11:18 | 14:56:01 |
| FOXP2 | 05:26:33 | 5 days, 23:28:04 | 08:38:01 |
| GADD45A | 01:24:26 | 02:17:59 | 01:57:40 |
| GBP5 | 14:04:30 | 06:43:06 | 15:23:34 |
| GMPR | 03:51:52 | 07:50:42 | 04:43:41 |
| HERC5 | 04:03:01 | 15:36:40 | 07:24:32 |
| IFI16 | 05:45:16 | 5 days, 8:01:45 | 08:35:56 |
| ISG20 | 01:34:50 | 08:01:08 | 02:14:25 |
| MX1 | 02:34:42 | 1 day, 18:16:17 | 07:07:03 |
| NT5C3A | 00:53:48 | 4 days, 20:17:21 | 2 days, 02:23:19 |
| RB1 | 00:14:09 | 14:16:03 | 02:22:44 |
| RHO | 00:06:30 | 02:05:53 | 00:46:35 |
| RSAD2 | 01:11:31 | 18:40:09 | 02:54:29 |
| SAMHD1 | 00:51:44 | 3 days, 13:26:08 | 04:22:11 |
| SERINC3 | 01:21:46 | 11:55:16 | 04:36:03 |
| SHH | 01:54:44 | 06:12:50 | 03:02:32 |
| SMC6 | 02:48:41 | 2 days, 20:34:48 | 06:29:27 |
| TREX1 | 01:01:04 | 09:43:10 | 03:09:30 |
| ZC3HAV1 | 02:38:52 | 2 days, 13:27:12 | 08:39:59 |
Availability
All scripts and documentation are freely available on GitHub and as a Docker on DockerHub. All links are available at: http://bioweb.me/DGINN-github. Example files to test DGINN are available to the users on GitHub. A specific script for the extraction of batch results, parseResults.py, is also available on the same GitHub. A graphical interface, which uses the file produced by parseResults.py as input and produces basic figures from the results (as in Figures 3 and 4), can be accessed through the same link.
RESULTS AND DISCUSSION
1- Presentation and novelties of the DGINN pipeline
The DGINN pipeline presents an end-to-end solution for the phylogenetic and automated detection of genetic innovations on protein-coding genes that are suspected to have undergone adaptive evolution. It automates the search for homologous sequences, their codon alignment and the reconstruction of phylogenetic histories. This is followed by the identification of marks of genetic innovations: (i) duplication events (also allowing for the identification of orthologous groups), (ii) recombination events (also limiting bias in subsequent positive selection analyses), (iii) positive selection through different methods.
The detailed presentation of the steps is found in the Materials and Methods section.
Key novelties of the DGINN pipeline include a major focus on its flexibility of use: as such, it is possible to enter at any step in the pipeline without deep knowledge of the command line. The possibility to search with a single pipeline for diverse mechanisms of genetic innovations and to use different methods for positive selection analyses translates to saved time compared to independent performance of each analysis. Moreover, though DGINN is designed to screen large datasets, it can also be used to perform gold-standard analyses on a single gene of interest with ease. For example, in the analyses of Lahaye et al. (57), positive selection analyses on the NONO gene were performed through the use of DGINN to determine the evolutionary history of this newly discovered sensor of the human immunodeficiency virus (HIV) capsid. Finally, DGINN includes key features detailed hereafter which are novel in such pipelines and allow for a more versatile use than just the detection of positive selection.
Automatic retrieval of homologous sequences and constitution of orthologous groups by tree reconciliation
The first important step for the identification of genetic innovations in a protein-coding gene is the retrieval of orthologous sequences of this gene, in as many species as possible in a given range, clade or family of interest to the user. Automating this step is a challenge as the evolutionary characteristics of orthologous genes vary a lot (between organisms, between copies in different species, according to different molecular clocks or environmental constraints). Usually, this step is time consuming and demands high manual curation. This is even more true for genes that have rapidly evolved. Most available tools for the detection of positive selection rely on user-provided alignments or are limited to fixed input species such as in PosiGene (7). To circumvent these limits, DGINN uses BLAST against the NCBI online databases (see Materials and Methods – steps 1 and 2). This approach makes the search for homologs simpler and relies on a widely-used and well-known tool, BLAST, which can be parameterized by the user. As true orthologous genes are identified through a subsequent reconciliation step, the user can cast a wide net by tuning parameters in terms of minimum coverage, e-value, identity and species included.
From a set of homologous sequences, true orthologous groups are identified through a reconciliation software, Treerecs (41) and additional homemade scripts (steps 3–5). Using tree reconciliation instead of annotations or tools such as OMA or Eggnogg (58,59) may be advantageous when working with non-model species, unknown genes, and recent duplication events. By separating the two phases of homolog retrieval and ortholog identification, we ensure that the user can change BLAST parameters without compromising the validity of the subsequent positive selection analyses.
DGINN detects gene duplication events, which may themselves be hallmarks of genetic innovation
While tools for the detection of positive selection abound, they often leave aside the detection of other hallmarks of genetic innovations, such as duplication (2). Very often, duplicated genes are even taken out of the analysis entirely to avoid bias during the detection of positive selection (5). However, this may lead to missing potential genes of interest and dismissing gene copies that have been under adaptive evolution. On the contrary, DGINN looks for duplication events as signals of potential genetic innovation, as well as to identify relevant groups of orthology for further analyses. Similarly, tools which perform orthologous assignments from annotations cannot be trusted to detect either recent duplications or ancient ones on non-model species. To our knowledge this is the first time this feature is included in an automated pipeline searching for genetic innovation. The importance of accounting for those events is shown through the numerous genes involved in genetic conflicts which have undergone duplications and subsequent diversification (2). For example, many antiviral effectors, also called restriction factors, belong to multigene families, where duplicated copies have evolved varied antiviral functions and/or virus-host interfaces/determinants, such as the Mx (Myxovirus resistance) Dynamin Like GTPases Mx1 and Mx2 (60), the guanylate-binding proteins GBPs (61,62), the primate APOBEC3 gene family (49–51,63) or the genes from the TRIM family (26).
Accounting for recombination allows for the detection of an important source of genetic innovation, while also avoiding bias in subsequent positive selection analyses
DGINN uses GARD to detect significant recombination breakpoints along the aligned sequences. As previously mentioned, recombination and gene conversion may be major sources of genetic innovations (in particular in the context of large gene families), and are widely ignored in existing pipelines. One example is the TRIMcyp gene present in some primate species, which results from the recombination and fusion of a cypA gene with the antiviral TRIM5 gene, leading to a change of antiviral specificity (26). Moreover, recombination may also itself bias phylogenetic reconstruction and positive selection analyses (64,65), as exemplified by the multiple recombination and gene conversion events that occurred in the Mx gene family during mammalian evolution (66). To date, only the PSP (15) and PoSeiDon (12) pipelines account for such events in their workflow. In DGINN, detecting recombination events thus serves two purposes: identifying one possible hallmark of genetic innovation and avoiding bias in positive selection analyses.
DGINN integrates numerous methods for the detection of positive selection
The detection of signatures of positive selection is a key part of the pipeline. Indeed, very few pipelines include different models than the ones from PAML (9,15). In DGINN, we decided to implement various methods with different underlying models, so the results obtained are more robust and can be balanced between methods. It also helps to ‘rank’ the importance of signatures on genes when a large dataset is screened. The methods and models are described in the Method section, step 8. In addition to the widely used PAML codeml, we included Bio++ bppml with similar but non-stationary models. Of note, after LRT on our validation dataset, Bio++ bppml consistently calculated better likelihoods than PAML codeml (Supplementary Table S1). Moreover, because of its versatility, Bio++ allows for more parameterization and the easy declaration of many modelings that would permit to detect positive selection under user-defined scenarios (e.g. using non-homogeneous mixture models, or other kinds of models such as allowing amino-acid specificity or simultaneous substitutions (67,68)).
Lastly, HYPHY is a good complement in those analyses, as shown in various studies (23,25–28). We thus decided to include two methods from the HYPHY package: one that considers the impact of positive selection at the level of the gene itself, using a branch-site model (BUSTED (45)), and another one which detects episodic positive selection at the site level (MEME (46)).
However, codon models have long running times, and users may not want to run all of these methods in one go if they prefer fast answers. Running times of Bio++ non-stationary models outperformed PAML codeml models in almost every instance in the validation dataset presented hereafter: 17 out of 19 analyses were faster in either M1NS versus M2NS and M7NS versus M8NS, compared with codeml M1 versus M2 and M7 versus M8 (Table 3 and Supplementary Table S2). Moreover, Bio++ parameter files can be easily modified to accelerate the modeling even further. As such, we would suggest the use of Bio++ only for such users for whom time is of the essence.
2- Validation
We tested our pipeline on nineteen primate genes selected for their various evolutionary histories and positive selection profiles (Table 2). These genes were grouped in three categories based on the clusters described in Murrell et al. (48): ‘canonical arms-race genes’ such as MX1 and SAMHD1, ‘genes described as presenting various selection profiles’ such as HERC5 or SERINC3, and ‘genes under no positive selection pressure’ such as GADD45A and RHO/rhodopsin (Table 2). The intermediate category was attributed on the basis of the methods employed to detect positive selection or the strength of the detected signal (see Method section).
An overview of the complete execution of DGINN on a protein-coding gene
A brief overview of DGINN’s workflow on a specific gene, HERC5, is presented in Figure 2. The BLAST search returned 71 primate homologous sequences, of which twelve were eliminated by the subsequent filters, yielding to 59 sequences. As a duplication event was detected by Treerecs, these 59 sequences were then automatically (and correctly) split into two groups: one with 32 sequences corresponding to HERC5 and one with 27 sequences corresponding to HERC6. No recombination event was identified and the positive selection analyses then followed. All methods found highly significant evidence of positive selection on the complete alignment of 59 mixed HERC5-HERC6 sequences, with P-values ranging from 2.24e−05 to 2.27e−13 for PAML and Bio++ models. However, after separating the two paralogs into orthologous groups, it appeared that most of this signal was driven by the very high positive selection of HERC6 (P-values of 4.38e−11 to 8.10e−15 for PAML and Bio++ models). Indeed, the signal on HERC5 sequences was present but much more modest (P-values, 0.030–0.004), with BUSTED even returning a non-significant P-value for positive selection on that alignment. For a query on the HERC5 gene, keeping the initial mixed HERC5-HERC6 alignment could have caused a mistaken conclusion that primate HERC5 has been under very strong positive selection, though the signal was mostly driven by HERC6. Moreover, the sites identified as under positive selection on that alignment would also be erroneous. This strongly highlights the necessity to properly separate paralogs from each other prior to performing the analyses.
Figure 2.

Example of workflow on the HERC5 primate gene. The workflow follows the diagram from Figure 1. Using human HERC5 CDS as the starting point in DGINN gave results for both HERC5 and HERC6. The number of sequences (seq) retrieved or left after each step is indicated. In the bottom panel, each colored circle represents the results from one of the five methods to detect positive selection at the gene level, with red representing significant evidence of positive selection and blue no significant evidence. P-values are indicated below the colored circles. Gp, orthologous group.
Overall, the complete DGINN analyses with HERC5 as query took less than 20 h (Table 3, 4h03 for the data mining and phylogenetics, and 15h36 for the detection of genetic innovations per se).
Detection of ancestral duplications allows for proper assignation of orthologous groups
We identified genes as belonging to multigene families if at least one member had over 50% reciprocal identity with our gene query according to ENSEMBL annotations (Table 2). Given this definition, we were able to retrieve multiple family members for the majority of the genes belonging to such families, when performing BLAST with the minimum coverage (50%) and identity (70%) values. The sole exception was SERINC3, for which no paralog was returned through our BLAST search. Two additional exceptions were observed, first with HERC5, for which the BLAST search also returned HERC6 sequences, though reciprocal identity between the two paralogs was below our threshold. The second case concerned TREX1, for which the BLAST search also returned sequences annotated as ATRIP, an adjacent gene. Given that read-through transcription of TREX1-ATRIP occurs naturally and yields a non-coding transcript, it is probable that those sequences annotated ATRIP actually represents the non-coding transcript and not the mRNA of the ATRIP gene. This explains the retrieval of ATRIP-annotated genes through BLAST despite the two genes not being strictly homologous.
DGINN efficiently reconstructed orthologous groups (Table 4). Indeed, in the case of multigene families (from two to five paralogs retrieved here), we were able to properly reconstruct orthologous groups for our genes of interest, without mixture with other paralogs. Our approach allowed us to separate the different family members retrieved through BLAST in groups which did not mix paralogous sequences through long branch partition (LB) and/or through reconciliation (Treerecs). For example, using the human CCDS sequence of FOXP2 as input in DGINN, we retrieved sequences from both FOXP2 and its paralog FOXP1. The tree reconstructed from the initial alignment featured a branch over 50 times longer than the mean length of the tree's branches. By automatically splitting the sequences separated by that branch, we were able to reconstitute two groups corresponding to the paralogs. However, paralogs from other families may not have diverged enough for long branch partition to be able to properly discriminate them into different groups. We resolved those through Treerecs, reconciling the tree obtained from the Blast-retrieved sequences with the primate species tree. This is the case, for example, of the immune sensor IFI16, which was properly assigned to a different group than MNDA through our Treerecs-based approach.
Table 4.
Groups of orthologs reconstructed by DGINN, using long-branch partition and Treerecs for identification of duplication events. For each gene of the validation dataset, are represented the orthologous groups that were identified, the number of sequences per group, the orthologs present in the group and the method used to separate the groups (long branch (LB) partition or TreeRecs-based). Groups kept for subsequent analyses are highlighted in yellow.
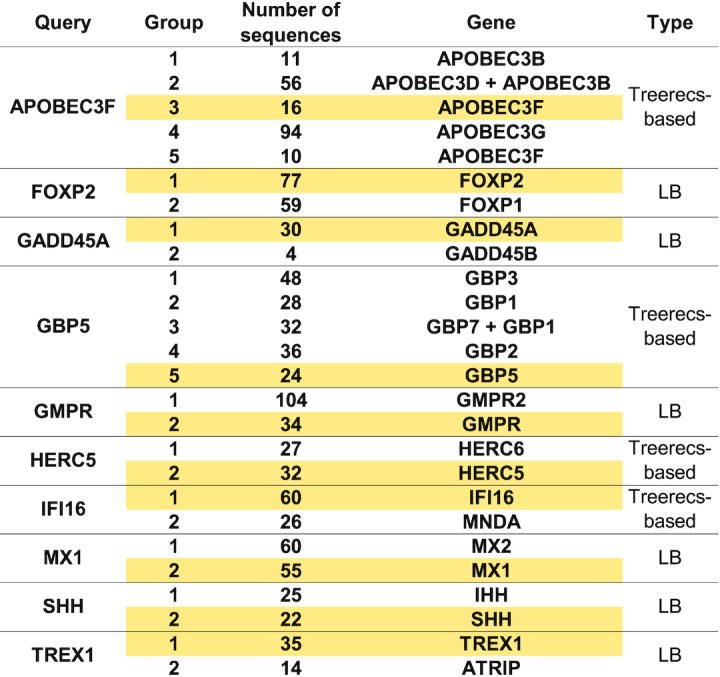
|
Non-annotated sequences (such as those referred as LOCXXX in databases) were also assigned to groups through this process, showing that this method of attributing orthologous relationships might help with non-annotated sequences in the databases.
Of our nineteen genes of interest, only one presented some inaccuracies in the distribution of sequences to ortholog groups. With an APOBEC3F query, DGINN erroneously divided APOBEC3F itself in two different groups (groups 3 and 5, Table 4). By further analyzing all the retrieved paralogs, we observed two mixes: in the APOBEC3F query, group 2 contained APOBEC3D and APOBEC3B sequences and APOBEC3B was split in two groups, and a similar pattern occurred in the GBP5 query, with GBP1 in groups 2 and 3 (Table 4). These errors could be explained by the particularly complicated evolutionary histories of those two expanded gene families during primate evolution (49,51,63). This highlights a need to improve the management of the detection of duplication events in further versions of DGINN. Importantly, because such genes would be tagged by DGINN with ‘detected duplication events’, these cases would anyway not be missed by the user and the gene of interest could be reanalyzed through DGINN after curation.
Using several positive selection methods together allows for more sensitivity and specificity and a ‘ranking’ of genes’ positive selection status during screening
Positive selection results were analyzed according to two different aspects. The first aspect focused on how many methods found a gene with significant evidence of positive selection (Figure 3, left panel—produced using the Shiny app openly available). The methods considered at this point were those on which a LRT could be performed: HYPHY BUSTED, the M1 versus M2 and M7 versus M8 models of PAML Codeml, and the M1NS versus M2NS and M7NS versus M8NS models of Bio++ bppml. Genes were ranked according to the number of positive results. This allowed us to compare the results obtained for the three categories of genes (Table 2). The canonical arms-race genes were all detected under positive selection by all five methods, with the exception of RSAD2/Viperin which was detected by four methods (Figure 3). Genes which presented variable signs of positive selection in the literature (green category, Table 2) also fell into a middle category in the DGINN screen. Genes without signs of positive selection in previous studies (blue category, Table 2) displayed low signs of positive selection: detected by less than two methods in DGINN. Two genes were detected by two methods: FOXP2 and RHO. FOXP2 was detected by both PAML M7 versus M8 and Bio++ M7NS versus M8NS, but both the mean omega and the very low number of sites detected under positive selection (n = 1) suggested artefactual results. Similarly, RHO was detected by BUSTED and Bio++ M7NS versus M8NS, but only two sites were detected. Therefore, our DGINN screen efficiently recapitulated results from published studies.
These results further highlight the advantage of using different methods within a single analysis to confirm results and discriminate for false positives. Doing this validation also showed that amongst those methods, BUSTED and M7 versus M8 in PAML codeml and Bio++ appeared the least conservative methods to detect positive selection.
Overall, if one would run less methods because of time constraint, our validation results indicate that running Bio++ methods would best balance running times, sensitivity and specificity (Table 3).
The second aspect taken into account focused on the percentage of positively-selected sites. Overall, the arms-race genes displayed higher proportions of positively selected sites (2.4–8.8%) compared to other genes (Figure 3, right side). However, this does not represent a hard rule, as some of those arms-race genes show rather low percentages, such as MX1 (∼3.2%). This suggests that ranking genes by the number of significant methods rather than the proportion of positive selection sites, as in Figure 3, is a better proxy for positive selection status.
DGINN recapitulates and expands the findings from previously published profiles of positively selected sites along genes
To identify the domains that have evolved under positive selection, we mapped every positively selected site detected by DGINN by a peak along the alignment (Figure 4, using the Shiny app). The height of the peak is proportional to the number of methods detecting that site under significant positive selection amongst five methods: M2 and M8 results of PAML codeml, M2NS and M8NS results of Bio++ bppml, and HYPHY MEME (Figure 4). Overall, we observed similar patterns as described in the literature, especially on the canonical arms-race genes. For example, in the case of SAMHD1, we found most positively selected sites at the N- and the C-termini (Figure 4). This is in accordance with the findings that the N-ter and C-ter domains both play a role in the antiviral/escape determinants of primate SAMHD1 and that rapid evolutions at these sites are adaptive as a result of lentiviral selective pressure (69–71). In the case of ZC3HAV1/ZAP, we found the positively selected sites cluster at both extremities of the alignment (Figure 4). However, the middle portion without positively selected sites corresponds to a gap-enriched region in the alignment linked to the different possible isoforms of the gene. Interestingly, this shows that the maintenance of these gap regions in the alignment did not lead to an excess of false positive detection in DGINN. If we now consider the main ORF (with the gap-enriched region ignored), it appears that the positively selected sites are spread over the whole length of the gene (Figure 4). Previous results established that the C-ter domain in particular was under significant positive selection (72). In contrast, the N-ter domain was not detected, probably because we used more methods and had more species/sequences available for analyses.The differences between our results and the published ones for APOBEC3F (48) were mainly due to the sequences used for the positive selection analyses. Indeed, our analyses excluded four species that were correctly retrieved in the early steps of DGINN but were erroneously assigned by Treerecs to another group, so the detection of positive selection was only performed on a subset of primate sequences, spanning solely Old World monkeys. However, we have included the solution to such problems in DGINN thanks to its high flexibility. The user may retrieve the gene sequences (here APOBEC3F) from the different groups and re-enter DGINN at step 3/alignment (Figure 1 and Table 2) to obtain the complete evolutionary history and positive selection analyses.
For MX1, we were first surprised that we did not detect such a high signal of positive selection in the L4 loop as described previously (Figure 4) (73). However, we found that this was mainly due to differences in the alignments, because PRANK (as opposed to ClustalX used previously) introduced many gaps in the L4 loop region due to the extremely-high divergence of the region. Whether MX1 adaptation to viral countermeasures has occurred by accumulation of non-synonymous changes and/or by indels in the L4 loop remains to be determined.
In the case of HERC5, four methods detected the gene as under positive selection during primate evolution (Figures 2 and 3), but only one site was identified as positively selected (Figure 4). These results differ from the ones reported previously (54), which found a much larger number of residues under positive selection (n = 50). This discrepancy, however, can be explained by the fact that the previous study identified positive selection on an alignment that included six non-primate species and only seven primate species, while ours focused exclusively on primates and included twenty species. It is therefore possible that a stronger selective pressure has occurred in placental mammals outside of primate evolution. Interestingly, in DGINN, our BLAST search with HERC5 as query also automatically retrieved HERC6 sequences (Figure 2). The latter were then correctly assigned to a different orthologous group than HERC5. As previously reported in mammals (74), we identified strong evidence of positive selection on primate HERC6 (with five methods, Figure 2). This could mean that while both HERC5 and HERC6 have been evolving under positive selection in mammals, they have been subjected to different evolutionary constraints in primates, with a lower selective pressure on primate HERC5 vs HERC6. It further shows that DGINN is an efficient tool to screen not only the query genes but also the evolutionary history of their closest gene relatives, which may have themselves evolved under positive selection and would be missed by most analyses.
Identification of the loss of GBP5 during primate evolution using DGINN
The positive selection results obtained through DGINN screening for GBP5 showed strong positive selection (identified by five methods). This is in accordance with previous results from McLaren et al. (75). By analyzing the phylogenetic tree generated by DGINN for all the homologs retrieved with the GBP5 query (after step 4, Figure 5A), we found that no sequence from Old World monkeys were retrieved for GBP5 through our BLAST search. This absence was confirmed in the tree reconstructed with only GBP5 sequences after ortholog group attribution (step 5, Figure 5B). However (and as expected), the entire GBP gene family was not retrieved by DGINN using human GBP5 as query (with blastn 70% identity and 50% coverage); in particular, GBP4 and GBP6 were too divergent to be retrieved by DGINN. To reconstruct GBP family's evolutionary history, we independently retrieved primate sequences of GBP4 and GBP6 by blastn and added the new sequences to a large GBP family sequence file. This served as input to DGINN steps 2–5 to automatically perform alignments, phylogenies, and duplication/orthologous group detection. The final tree confirmed that GBP5 is absent in Old World Monkeys (Figure 5C). This might also be the case for GBP4, for which we did not retrieve sequences from Old World Monkeys; with the exception of two sequences from Papio anubis and Mandrillus leucophoeus that were annotated as ‘GBP4’ but did not follow a typical orthologous phylogeny, possibly due to recombination/gene conversion events (Figure 5C). Genomic analyses of the GBP locus in several primates confirmed that GBP5 was lost in the ancestor of Old World monkeys during primate evolution (Figure 5D). This finding was also reported by a study from Kohler et al. during the revision of this work (76). Phylogenetic and genomic analyses of GBP4 suggest that its evolution in the Old World monkeys is more complex and could involve gene loss and recombination with GBP7. Further analyses and genomic sequences in the locus would allow to precisely determine its evolutionary history. Overall, these results show that GBP5 has been subjected to strong positive selection during primate evolution, but has also entirely been lost in the Cercopithecinae. Whether part of this has been driven by pathogens such as lentiviruses (33) or bacteria (32) should be investigated.
Figure 5.

Evolutionary history of the primate GBP family. (A) Maximum-likelihood phylogeny established through DGINN based on a run on the GBP5 query (step 4). The four main primate lineages are identified by color-coding: Old World monkeys, blue; Hominoids, green; New World monkeys, orange; prosimians, purple/pink. Asterisks (*) denote nodes that are statistically supported by aLRT > 0.90. The GBP5 group, which lacks Old World monkey sequences, is boxed in yellow. The scale bar represents the number of nucleotide substitutions per site and the tree was midpoint rooted. (B) Maximum-likelihood phylogeny of the GBP5 group of primate orthologs established through DGINN screen (step 7). (C) Maximum-likelihood phylogeny of the whole GBP family performed in DGINN after manual addition of primate GBP4 and GBP6 sequences. (D) Diagram of the genomic locus of the GBP gene family in seven simian primate species. The reference genomes from the NCBI used were: papAnu (Papio anubis): Panu_3.0, macMul (Macaca mulatta): Mmul10, chlSab (Chlorocebus sabaeus): Chlorocebus_sabeus 1.1, homSap (Homo sapiens): GRCh38.p13, gorGor (Gorilla gorilla): gorGor4, saiBol (Saimiri boliviensis): saiBol1.0. All alignments and phylogenies for panel A, B and C (referred as 5A_aln, 5A_tree etc.) can be found on the GitHub (see Availability).
CONCLUSION
We have developed DGINN, an integrative pipeline for the automatic detection of genetic innovations, and made it freely available through both GitHub and Docker. DGINN was validated for screening usage against nineteen primate genes (all results are available on GitHub -see Availability). It automates and streamlines those analyses, allowing the user to simply provide the coding sequence of their gene of interest and a parameter file to launch the whole workflow, from retrieval of homologous sequences to the detection of orthology relationships, recombination events and positive selection.
Through our validation, we confirmed and expanded on results previously established in the literature. Genes described as engaged in arms-races with viruses were found under strong positive selection by all five methods included in DGINN. Our analyses allowed us to establish clearer profiles for the genes belonging to the ‘varied’ category, owing to our inclusion of different methods for positive selection: this way, we were able to establish that some genes previously thought to present moderate signs of positive selection presented stronger signs than suspected. Little evidence of positive selection was found on the genes belonging to ‘no positive selection’ category, in accordance with the literature.
An important feature of DGINN is its flexibility, which allows usage beyond its screening capacity. Indeed, in cases of dubious results, the possibility remains for the user to curate their input files and perform the appropriate analyses by entering DGINN at any of the downstream steps. This also means that the ‘positive selection’ part might be of primary interest to scientists wishing to perform gold-standard positive selection analyses on their favorite gene, because they could enter their curated alignment and phylogeny and obtain results of positive selection analyses from five methods in a single query.
Using DGINN to analyze nineteen primate genes also allowed us to enrich some findings, notably on the importance of detecting duplications and properly ascribing ortholog groups, as exemplified by the case of HERC5 and its paralog HERC6 in primates. The ability to check multiple members of a query's gene family is a major advantage of DGINN, as it allows the user to automatically identify related genes with signs of genetic innovations. Improving the constitution of ortholog groups will remain an objective in future versions of DGINN.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Stéphanie Jacquet for her comments on the manuscript. We also thank Bastien Boussau, Marie Cariou, Hélène Dutartre, Laurent Modolo, Xavier Morelli and Guy Perrière for helpful discussions on this project. We gratefully acknowledge support from the PSMN (Pôle Scientifique de Modélisation Numérique) of the ENS de Lyon for the computing resources, and the PRABI (Pôle Rhône-Alpes de BioInformatique) for further bioinformatics support. We thank all the contributors of publicly available genome sequences, as well as the scientists who developed the methods included in DGINN.
Author contributions: Conceptualization and supervision: L.E., L.G. Formal analysis: L.P., L.E., L.G. Pipeline development: L.P., L.G., Q.G. Funding acquisition: L.E., L.G. Investigation: L.P., Q.G., O.A., A.C., L.G., L.E. Methodology: L.P., L.G., L.E. Project administration: L.E., L.G. Resources: A.C., L.E., L.G. Writing – original draft: L.P., L.E., L.G. Writing – review and editing: All the authors.
Contributor Information
Lea Picard, CIRI - Centre International de Recherche en Infectiologie, Univ Lyon, Inserm U1111, Université Claude Bernard Lyon 1, CNRS UMR5308, ENS de Lyon, Lyon, France; Laboratoire de Biologie et Biométrie Evolutive, CNRS UMR 5558, Université Claude Bernard Lyon 1, Villeurbanne, France.
Quentin Ganivet, Laboratoire de Biologie et Biométrie Evolutive, CNRS UMR 5558, Université Claude Bernard Lyon 1, Villeurbanne, France.
Omran Allatif, CIRI - Centre International de Recherche en Infectiologie, Univ Lyon, Inserm U1111, Université Claude Bernard Lyon 1, CNRS UMR5308, ENS de Lyon, Lyon, France.
Andrea Cimarelli, CIRI - Centre International de Recherche en Infectiologie, Univ Lyon, Inserm U1111, Université Claude Bernard Lyon 1, CNRS UMR5308, ENS de Lyon, Lyon, France.
Laurent Guéguen, Laboratoire de Biologie et Biométrie Evolutive, CNRS UMR 5558, Université Claude Bernard Lyon 1, Villeurbanne, France; Swedish Collegium for Advanced Study, Uppsala, Sweden.
Lucie Etienne, CIRI - Centre International de Recherche en Infectiologie, Univ Lyon, Inserm U1111, Université Claude Bernard Lyon 1, CNRS UMR5308, ENS de Lyon, Lyon, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
ANR LABEX ECOFECT [ANR-11-LABX-0048 of Université de Lyon, within the program ‘Investissements d’Avenir’ (ANR-11-IDEX-0007) operated by the French National Research Agency to L.E. and L.G.]; L.E. is supported by the CNRS and by grants from the amfAR (Mathilde Krim Phase II Fellowship 109140-58-RKHF); ‘Fondation pour la Recherche Médicale’ [FRM ‘Projet Innovant’ ING20160435028]; FINOVI (‘recently settled scientist’ grant); ANRS [ECTZ19143, ECTZ118944]; JORISS incubating grant; L.G. is supported by the Université Claude Bernard Lyon 1 and the Swedish Center of Advanced Study; A.C. is supported by the CNRS and by grants from the ANRS, Sidaction and the ENS-L.
Conflict of interest statement. None declared.
REFERENCES
- 1. Daugherty M.D., Malik H.S.. Rules of engagement: molecular insights from host-virus arms races. Annu. Rev. Genet. 2012; 46:677–700. [DOI] [PubMed] [Google Scholar]
- 2. Daugherty M.D., Zanders S.E.. Gene conversion generates evolutionary novelty that fuels genetic conflicts. Curr. Opin. Genet. Dev. 2019; 58–59:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kondrashov F.A. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc. R. Soc. B. 2012; 279:5048–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McLaughlin R.N., Malik H.S.. Genetic conflicts: the usual suspects and beyond. J. Exp. Biol. 2017; 220:6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kosiol C., Vinař T., da Fonseca R.R., Hubisz M.J., Bustamante C.D., Nielsen R., Siepel A.. Patterns of positive selection in six mammalian genomes. PLoS Genet. 2008; 4:e1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hawkins J.A., Kaczmarek M.E., Müller M.A., Drosten C., Press W.H., Sawyer S.L.. A metaanalysis of bat phylogenetics and positive selection based on genomes and transcriptomes from 18 species. Proc. Natl Acad. Sci. U.S.A. 2019; 116:11351–11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sahm A., Bens M., Platzer M., Szafranski K.. PosiGene: automated and easy-to-use pipeline for genome-wide detection of positively selected genes. Nucleic Acids Res. 2017; 45:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pond S.L.K., Frost S.D.W., Muse S.V.. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005; 21:676–679. [DOI] [PubMed] [Google Scholar]
- 9. Stern A., Doron-Faigenboim A., Erez E., Martz E., Bacharach E., Pupko T.. Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 2007; 35:W506–W511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Egan A., Mahurkar A., Crabtree J., Badger J.H., Carlton J.M., Silva J.C.. IDEA: interactive display for evolutionary analyses. BMC Bioinformatics. 2008; 9:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Steinway S.N., Dannenfelser R., Laucius C.D., Hayes J.E., Nayak S.. JCoDA: a tool for detecting evolutionary selection. BMC Bioinformatics. 2010; 11:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fuchs J., Hölzer M., Schilling M., Patzina C., Schoen A., Hoenen T., Zimmer G., Marz M., Weber F., Müller M.A. et al.. Evolution and antiviral specificities of Interferon-Induced Mx proteins of bats against ebola, influenza, and other RNA viruses. J. Virol. 2017; 91:e00361-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hongo J.A., de Castro G.M., Cintra L.C., Zerlotini A., Lobo F.P.. POTION: an end-to-end pipeline for positive Darwinian selection detection in genome-scale data through phylogenetic comparison of protein-coding genes. BMC Genomics. 2015; 16:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Busset J., Cabau C., Meslin C., Pascal G.. PhyleasProg: a user-oriented web server for wide evolutionary analyses. Nucleic Acids Res. 2011; 39:W479–W485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Su F., Ou H.-Y., Tao F., Tang H., Xu P.. PSP: rapid identification of orthologous coding genes under positive selection across multiple closely related prokaryotic genomes. BMC Genomics. 2013; 14:924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thompson J.D., Higgins D.G., Gibson T.J.. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994; 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loytynoja A., Goldman N.. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science. 2008; 320:1632–1635. [DOI] [PubMed] [Google Scholar]
- 19. Fletcher W., Yang Z.. INDELible: a flexible simulator of biological sequence evolution. Mol. Biol. Evol. 2009; 26:1879–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Privman E., Penn O., Pupko T.. Improving the performance of positive selection inference by filtering unreliable alignment regions. Mol. Biol. Evol. 2012; 29:1–5. [DOI] [PubMed] [Google Scholar]
- 21. Jordan G., Goldman N.. The effects of alignment error and alignment filtering on the sitewise detection of positive selection. Mol. Biol. Evol. 2012; 29:1125–1139. [DOI] [PubMed] [Google Scholar]
- 22. Markova-Raina P., Petrov D.. High sensitivity to aligner and high rate of false positives in the estimates of positive selection in the 12 Drosophila genomes. Genome Res. 2011; 21:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abdul F., Filleton F., Gerossier L., Paturel A., Hall J., Strubin M., Etienne L.. Smc5/6 antagonism by HBx is an evolutionarily conserved function of hepatitis B virus infection in mammals. J. Virol. 2018; e00769-18.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elde N.C., Child S.J., Geballe A.P., Malik H.S.. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature. 2009; 457:485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shultz A.J., Sackton T.B.. Immune genes are hotspots of shared positive selection across birds and mammals. eLife. 2019; 8:e41815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Malfavon-Borja R., Sawyer S.L., Wu L.I., Emerman M., Malik H.S.. An evolutionary screen highlights canonical and noncanonical candidate antiviral genes within the primate TRIM gene family. Genome Biol. Evol. 2013; 5:2141–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McBee R.M., Rozmiarek S.A., Meyerson N.R., Rowley P.A., Sawyer S.L.. The effect of species representation on the detection of positive selection in primate gene data sets. Mol. Biol. Evol. 2015; 32:1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rowley P.A., Ho B., Bushong S., Johnson A., Sawyer S.L.. XRN1 is a species-specific virus restriction factor in Yeasts. PLoS Pathog. 2016; 12:e1005890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van der Lee R., Wiel L., van Dam T.J.P., Huynen M.A.. Genome-scale detection of positive selection in nine primates predicts human-virus evolutionary conflicts. Nucleic Acids Res. 2017; 45:10634–10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007; 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- 31. Duggal N.K., Emerman M.. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat. Rev. Immunol. 2012; 12:687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim B.-H., Shenoy A.R., Kumar P., Bradfield C.J., MacMicking J.D.. IFN-Inducible GTPases in host cell defense. Cell Host Microbe. 2012; 12:432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krapp C., Hotter D., Gawanbacht A., McLaren P.J., Kluge S.F., Stürzel C.M., Mack K., Reith E., Engelhart S., Ciuffi A. et al.. Guanylate Binding Protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe. 2016; 19:504–514. [DOI] [PubMed] [Google Scholar]
- 34. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L.. BLAST+: architecture and applications. BMC Bioinformatics. 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rice P. EMBOSS: the european molecular biology open software suite. Trends Genet. 2000; 16:276–277. [DOI] [PubMed] [Google Scholar]
- 36. Ranwez V., Harispe S., Delsuc F., Douzery E.J.P.. MACSE: multiple alignment of coding sequences accounting for frameshifts and stop codons. PLoS One. 2011; 6:e22594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schneider A., Souvorov A., Sabath N., Landan G., Gonnet G.H., Graur D.. Estimates of positive darwinian selection are inflated by errors in sequencing, annotation, and alignment. Genome Biol. Evol. 2009; 1:114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Katoh K., Misawa K., Kuma K., Miyata T.. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002; 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O.. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010; 59:307–321. [DOI] [PubMed] [Google Scholar]
- 40. Anisimova M., Gascuel O.. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 2006; 55:539–552. [DOI] [PubMed] [Google Scholar]
- 41. Comte N., Morel B., Hasic D., Guéguen L., Boussau B., Daubin V., Penel S., Scornavacca C., Gouy M., Stamatakis A. et al.. Treerecs: an integrated phylogenetic tool, from sequences to reconciliations. Bioinformatics. 2019; btaa615. [DOI] [PubMed] [Google Scholar]
- 42. Anisimova M., Bielawski J.P., Yang Z.. Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol. Biol. Evol. 2002; 19:950–958. [DOI] [PubMed] [Google Scholar]
- 43. Kosakovsky Pond S.L., Posada D., Gravenor M.B., Woelk C.H., Frost S.D.W.. GARD: a genetic algorithm for recombination detection. Bioinformatics. 2006; 22:3096–3098. [DOI] [PubMed] [Google Scholar]
- 44. Guéguen L., Gaillard S., Boussau B., Gouy M., Groussin M., Rochette N.C., Bigot T., Fournier D., Pouyet F., Cahais V. et al.. Bio++: efficient extensible libraries and tools for computational molecular evolution. Mol. Biol. Evol. 2013; 30:1745–1750. [DOI] [PubMed] [Google Scholar]
- 45. Murrell B., Weaver S., Smith M.D., Wertheim J.O., Murrell S., Aylward A., Eren K., Pollner T., Martin D.P., Smith D.M. et al.. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015; 32:1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Murrell B., Wertheim J.O., Moola S., Weighill T., Scheffler K., Kosakovsky Pond S.L.. Detecting individual sites subject to episodic diversifying selection. PLos Genet. 2012; 8:e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guéguen L., Duret L.. Unbiased estimate of synonymous and nonsynonymous substitution rates with nonstationary base composition. Mol. Biol. Evol. 2018; 35:734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murrell B., Vollbrecht T., Guatelli J., Wertheim J.O.. The evolutionary histories of antiretroviral proteins SERINC3 and SERINC5 do not support an evolutionary arms race in primates. J. Virol. 2016; 90:8085–8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nakano Y., Aso H., Soper A., Yamada E., Moriwaki M., Juarez-Fernandez G., Koyanagi Y., Sato K.. A conflict of interest: the evolutionary arms race between mammalian APOBEC3 and lentiviral Vif. Retrovirology. 2017; 14:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Etienne L., Bibollet-Ruche F., Sudmant P.H., Wu L.I., Hahn B.H., Emerman M.. The role of the antiviral APOBEC3 gene family in protecting chimpanzees against lentiviruses from monkeys. PLoS Pathog. 2015; 11:e1005149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Desimmie B.A., Delviks-Frankenberrry K.A., Burdick R.C., Qi D., Izumi T., Pathak V.K.. Multiple APOBEC3 restriction factors for HIV-1 and one vif to rule them all. J. Mol. Biol. 2014; 426:1220–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sawyer S.L., Emerman M., Malik H.S.. Ancient adaptive evolution of the primate antiviral DNA-Editing enzyme APOBEC3G. PLoS Biol. 2004; 2:e275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kluge S.F., Sauter D., Kirchhoff F.. SnapShot: antiviral restriction factors. Cell. 2015; 163:774.E1. [DOI] [PubMed] [Google Scholar]
- 54. Woods M., Tong J., Tom S., Szabo P., Cavanagh P., Dikeakos J., Haeryfar S., Barr S.. Interferon-induced HERC5 is evolving under positive selection and inhibits HIV-1 particle production by a novel mechanism targeting Rev/RRE-dependent RNA nuclear export. Retrovirology. 2014; 11:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Perelman P., Johnson W.E., Roos C., Seuánez H.N., Horvath J.E., Moreira M.A.M., Kessing B., Pontius J., Roelke M., Rumpler Y. et al.. A molecular phylogeny of living primates. PLos Genet. 2011; 7:e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pecon-Slattery J. Recent advances in primate phylogenomics. Annu. Rev. Anim. Biosci. 2014; 2:41–63. [DOI] [PubMed] [Google Scholar]
- 57. Lahaye X., Gentili M., Silvin A., Conrad C., Picard L., Jouve M., Zueva E., Maurin M., Nadalin F., Knott G.J. et al.. NONO detects the nuclear HIV capsid to promote cGAS-Mediated innate immune activation. Cell. 2018; 175:488–501.E22. [DOI] [PubMed] [Google Scholar]
- 58. Altenhoff A.M., Glover N.M., Train C.-M., Kaleb K., Warwick Vesztrocy A., Dylus D., de Farias T.M., Zile K., Stevenson C., Long J. et al.. The OMA orthology database in 2018: retrieving evolutionary relationships among all domains of life through richer web and programmatic interfaces. Nucleic Acids Res. 2018; 46:D477–D485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huerta-Cepas J., Szklarczyk D., Forslund K., Cook H., Heller D., Walter M.C., Rattei T., Mende D.R., Sunagawa S., Kuhn M. et al.. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016; 44:D286–D293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Haller O., Staeheli P., Schwemmle M., Kochs G.. Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015; 23:154–163. [DOI] [PubMed] [Google Scholar]
- 61. Tretina K., Park E.-S., Maminska A., MacMicking J.D.. Interferon-induced guanylate-binding proteins: guardians of host defense in health and disease. J. Exp. Med. 2019; 216:482–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang S., Meng Q., Maminska A., MacMicking J.D.. Cell-autonomous immunity by IFN-induced GBPs in animals and plants. Curr. Opin. Immunol. 2019; 60:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Münk C., Willemsen A., Bravo I.G.. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol. Biol. 2012; 12:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Anisimova M., Nielsen R., Yang Z.. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics. 2003; 164:1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Posada D., Crandall K.A.. The effect of recombination on the accuracy of phylogeny estimation. J. Mol. Evol. 2002; 54:396–402. [DOI] [PubMed] [Google Scholar]
- 66. Mitchell P.S., Young J.M., Emerman M., Malik H.S.. Evolutionary analyses suggest a function of MxB immunity proteins beyond lentivirus restriction. PLoS Pathog. 2015; 11:e1005304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Weber C.C., Whelan S.. Physicochemical amino acid properties better describe substitution rates in large populations. Mol. Biol. Evol. 2019; 36:679–690. [DOI] [PubMed] [Google Scholar]
- 68. Zaheri M., Dib L., Salamin N.. A generalized mechanistic codon model. Mol. Biol. Evol. 2014; 31:2528–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fregoso O.I., Ahn J., Wang C., Mehrens J., Skowronski J., Emerman M.. Evolutionary toggling of Vpx/Vpr specificity results in divergent recognition of the restriction factor SAMHD1. PLoS Pathog. 2013; 9:e1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Laguette N., Rahm N., Sobhian B., Chable-Bessia C., Münch J., Snoeck J., Sauter D., Switzer W.M., Heneine W., Kirchhoff F. et al.. Evolutionary and functional analyses of the interaction between the myeloid restriction factor SAMHD1 and the lentiviral Vpx protein. Cell Host Microbe. 2012; 11:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lim E.S., Fregoso O.I., McCoy C.O., Matsen F.A., Malik H.S., Emerman M.. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe. 2012; 11:194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kerns J.A., Emerman M., Malik H.S.. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet. 2008; 4:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mitchell P.S., Patzina C., Emerman M., Haller O., Malik H.S., Kochs G.. Evolution-guided identification of antiviral specificity determinants in the broadly acting Interferon-Induced innate immunity factor MxA. Cell Host Microbe. 2012; 12:598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Paparisto E., Woods M.W., Coleman M.D., Moghadasi S.A., Kochar D.S., Tom S.K., Kohio H.P., Gibson R.M., Rohringer T.J., Hunt N.R. et al.. Evolution-Guided structural and functional analyses of the HERC family reveal an ancient marine origin and determinants of antiviral activity. J. Virol. 2018; 92:e00528-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McLaren P.J., Gawanbacht A., Pyndiah N., Krapp C., Hotter D., Kluge S.F., Götz N., Heilmann J., Mack K., Sauter D. et al.. Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology. 2015; 12:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kohler K.M., Kutsch M., Piro A.S., Wallace G.D., Coers J., Barber M.F.. A rapidly evolving polybasic motif modulates bacterial detection by guanylate binding proteins. mBio. 2020; 11:e00340-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cagliani R., Forni D., Biasin M., Comabella M., Guerini F.R., Riva S., Pozzoli U., Agliardi C., Caputo D., Malhotra S. et al.. Ancient and recent selective pressures shaped genetic diversity at AIM2-Like nucleic acid sensors. Genome Biol. Evol. 2014; 6:830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lim E.S., Wu L.I., Malik H.S., Emerman M.. The function and evolution of the restriction factor viperin in primates was not driven by lentiviruses. Retrovirology. 2012; 9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Atkinson E.G., Audesse A.J., Palacios J.A., Bobo D.M., Webb A.E., Ramachandran S., Henn B.M.. No evidence for recent selection at FOXP2 among diverse human populations. Cell. 2018; 174:1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.